
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot oxime ligation from peptides bearing thiazolidine and aminooxyacetyl groups†
Stéphane Duflocq a,
Jingjing Zhoua,
Florent Huguenota,
Michel Vidalab and
Wang-Qing Liu*a
a,
Jingjing Zhoua,
Florent Huguenota,
Michel Vidalab and
Wang-Qing Liu*a
aUniversité de Paris, CiTCoM, 8038 CNRS, U 1268 INSERM, F-75006 Paris, France. E-mail: wangqing.liu@parisdescartes.fr
bService Biologie du médicament, toxicologie, AP-HP, Hôpital Cochin, F-75014 Paris, France
First published on 6th May 2020
Abstract
One-pot oxime ligation under mild conditions using Pd(II) as a shared catalyst from an aldehyde precursor (Thz) and a protected aminooxyacetyl group (Proc-Aoa) is reported. Two complementary metal-free protocols using unmasked Aoa-peptide are also described. Acetoxime-peptide can proceed to the desired oxime through an additional transoximation step.
Oxime ligation is one of the hot topics in the field of bioconjugation of molecules due to its chemoselectivity and hydrolytic stability.1,2 It requires an electrophilic moiety such as a carbonyl group (aldehyde/ketone) and a nucleophilic aminooxy partner (H2N–O–R). Numerous methods have emerged over the past decade to insert these two components into peptide sequences.1,3 A critical step in oxime ligation is aldehyde/ketone formation. The functionalization strategies that have been developed using either chemical, enzymatic or bioengineering methods exhibit disparities in the selectivity and the facility of this electrophile formation.4
Thiazolidine (named Thz) was first used as an orthogonal cysteine protection strategy, especially in native chemical ligation (NCL) (Fig. 1a, right pathway, R = H).5 Such 4-carboxy thiazolidine can be introduced at the N-terminus of peptide by using commercially available protected thioproline (ThP) component. The cysteine residue can then be generated by treating the ThP-peptide with methoxyamine in buffer6,7 or by Pd(II) catalyst.8 Recent works used 2-carboxy thiazolidine to introduce a highly reactive α-oxo aldehyde for further bioconjugation.9,10 (Fig. 1a, left pathway, R′ = H). This second thiazolidine can be appended into peptides with the precursor Boc-Thz-OH (Fig. 1b), then hydrolyzed with a metal-assisted ring opening using either silver or palladium catalysts.9,10 The scope of thiazolidine in bioconjugation was recently expanded with the use of thiazolidin-5-imine as a new linkage method in protein and live cell labelling.11
 | ||
| Fig. 1 (a) 2-Carboxy thiazolidine as a new highly reactive aldehyde precursor (Thz, left pathway). 4-Carboxy thiazolidine as N-term cysteine protection (thioproline analogue (ThP), right pathway). (b) Boc protected 1,3-thiazolidine-2-carboxylic acid (Boc-Thz-OH) and propargyloxycarbonyl (Proc) protected aminooxyacetic acid (Proc-Aoa-OH). | ||
In oxime conjugation, the other component is the aminooxyacetyl peptide. Given the high nucleophilic reactivity of the aminooxy moiety that traps laboratory ambient traces of acetone, acetaldehyde or formaldehyde,12–14 Mezö et al. proposed to add 10-fold excess of free aminooxyacetic acid (Aoa-OH) as a carbonyl capture to overcome this undesired oxime bond.13 When this strategy is needed, it requires an additional purification step prior to oxime ligation to remove the excess of Aoa-OH. Thus, the use of an orthogonal protecting group for conventional Fmoc peptide chemistry can ease the purification and the storage of Aoa-peptides. Propargyloxycarbonyl (Proc) protecting group (Fig. 1b) was used to protect Aoa-OH in solid-phase peptide synthesis (SPPS) as it remains stable under harsh acidic condition like resin cleavage and can be selectively deprotected by several Pd(II) catalysts such as [Pd(allyl)Cl]2 and Pd(dba)2.8,15
Several oxime conjugation methods were reported using Pd(II) or Ag(I) to uncage of 2-carboxy thiazolidine (Fig. 2a).9,10 These strategies have been used to label peptide or protein with biotin or fluorophore but they were not applied to peptide–peptide ligation. Proc group has been used to protect N-terminus cysteine for NCL purpose (Fig. 2b).8 All of these procedures required additional purification steps to isolate the intermediates from the high amount of catalysts. The need of a precious care of the highly reactive functions α-oxo aldehyde and aminooxy groups before oxime conjugation led us to develop new methods.
 | ||
| Fig. 2 (a) 2 steps synthesis with thiazolidine deprotection mediated by either [Pd] or [Ag] complex followed by a purification step and oxime coupling with nude aminooxy tag (biotine or dansyl).9,10 (b) Proc removal with [Pd] complex in NCL.8 (c) Kinetic studies of Pd-mediated deprotection of Thz or Proc groups in pH 2 solution. % deprotection was calculated from HPLC peak areas. (d) One-pot method starting from both protected partners to reach oxime ligation at pH 2 with HPLC monitoring. Peptides were numbered as in Table 1. | ||
In our project to deliver a cyclic 17-mer peptide into cells, the oxime bond was adopted to append a cell penetrating peptide (CPP).16,17 Boc-Thz-OH (Fig. 1b) was rapidly prepared according to the reported method9 and was introduced during SPPS at N-terminus of a peptide, with a flexible spacer Ahx (6-amino hexanoic acid) between Thz and the linear peptide of sequence CPSDSTRRKGGRCGRRL. The peptide was then cleaved, totally deprotected and finally cyclized between the side chains of Cys residues through an o-xylene moiety18,19 to give peptide 1 (formula shown in ESI†), the precursor of peptide 2 (Table 1). Among the available vectors, antennapedia (Antp) is well known for its fast delivery of cargo peptides into cells.20,21 Proc-Aoa-OH was prepared by protection of free Aoa-OH with propagyl chloroformate in basic condition (ESI†) then similarly introduced at N-terminus of an antennapedia derived 16-mer of sequence RQIKIWFQNRRMKWKK22 to give peptide 3. Peptide 4 was prepared by using commercial Boc-Aoa-OH (Table 1).
| Compound | Sequence | Mass found (Da) |
|---|---|---|
| Peptide 1 | Thz-Ahx-C*PSDSTRRKGGRC*GRRL-NH2 | 2234 |
| Peptide 2 | CHO-(CO)-Ahx-C*PSDSTRRKGGRC*GRRL-NH2 | 2175 |
| Peptide 3 | Proc-Aoa-RQIKIWFQNRRMKWKK-NH2 | 2400 |
| Peptide 4 | Aoa-RQIKIWFQNRRMKWKK-NH2 | 2318 |
| Oxime 1 | Peptide 2–peptide 4 conjugate | 4473 |
| Oxime 2 | Acetoxime of peptide 4 | 2358 |
To better ease the conjugation between Thz-peptides and Aoa-peptides, we aimed to develop a convenient one-pot deprotection–conjugation procedure with both protected partners. Thiazolidine ring-opening can be mediated by Ag(I).10 Nonetheless, as Ag(I) cannot deprotect Proc group, we focused our attention on Pd(II) as a shared catalyst for both Thz and Proc uncaging. Several Pd(II) complexes including [Pd(allyl)Cl]2, Pd(OAc)2 and PdCl2 were first tested. In agreement with reported works,8,9 [Pd(allyl)Cl]2 and PdCl2 exhibited better results than Pd(OAc)2 (data not shown). Proc group was easily removed by both PdCl2 and [Pd(allyl)Cl]2, whereas the latter was known for giving an unidentified by-product during Proc removal.8 Taken together, these preliminary results focused our method on PdCl2 as a shared catalyst. Due to its limited aqueous solubility, a 1.25 mM PdCl2 stock solution in HPLC mobile phase (aqueous 0.1% TFA, pH 2, named solution A) was prepared for deprotection experiments. Proc and Thz deprotection were first investigated separately, using 0.5 and 1.0 equivalent of PdCl2 in solution A at room temperature with HPLC monitoring during 0–2 h (Fig. 2c). Due to the release of β-mercaptoethylamine, Pd can be captured and not proceed in catalytic amounts. 0.5–1.0 eq. of PdCl2 exhibited optimal Thz and Proc deprotection within few hours. Based on these results, a one-pot conjugation protocol was designed using 1.0 eq. PdCl2 and a slight excess of Proc-Aoa-peptide 3 (about 1.5 eq.) in solution A. This convenient approach requiring less steps and a minimum amount of Pd would provide a powerful tool for peptide–peptide ligation. In agreement with the kinetic studies, after 3 h Thz-peptide 1 showed almost complete deprotection into 2 in comparison with Proc-peptide 3, which was partially deprotected (Fig. 2d). 2 was observed with its hydrated from 2′ according to the reported work of Brabham et al. in Thz deprotection.9 We suppose through HSAB theory that the better Lewis base property of sulfur in Thz tends to faster its hydrolysis comparing to Proc group. A new peak corresponding to Oxime 1 appeared. After an overnight stirring (15 h), a great conversion of the α-oxoaldehyde 2 into oxime was achieved and the excess of 3 was fully deprotected. PdCl2 catalyst was then quenched by an aqueous solution of dithiothreitol (DTT) (50 eq.).23 As our Aoa-peptide 4 and the resulting Oxime 1 have similar retention times, the excess of free Aoa-peptide was easily converted into acetoxime derivative, named Oxime 2 (Table 1) upon acetone addition. This retention time increase alleviated Oxime 1 purification.
Although only 1.0 eq. of PdCl2 is needed to remove both Thz and Proc, metal ions might be caged by Cys/Met/His rich proteins and may induce undesired biological effects.24 Consequently, very stringent standards are applied in drug for limit content (EMA/CHMP/ICH/353369/2013). In our case, as a last step prior to cell assays, metal-free conditions might be preferable. Recent strategies have emerged to promote oxime ligation using other electrophile precursors such as N-phenylglycine peptides.25 Regarding the thiazolidine ring, metal-free ThP uncaging was described with a large excess of methoxyamine (>500 eq.) in 8–12 h under acidic conditions (Fig. 3a).6,7 The released formaldehyde was directly quenched into oxime by methoxyamine. Hence, further studies were carried out to investigate the propensity of our Aoa-peptide 3 to mimic this metal-free thiazolidine deprotection-quench. This first-in-class method would allow Aoa-peptide to deprotect and form the desired Oxime 1 whilst releasing the β-mercaptoethylamine moiety. Two short model peptides, Thz-GYRMHK 5 and Aoa-GYRMHK 6, were thus prepared for this metal-free ligation. Among the screened conditions, solution A and standard ammonium acetate buffer at pH 4.5 with or without aniline catalyst26 exhibited better results compared to conventional protein chemistry buffer PBS 1× (Table S1, ESI†). Additionally, a mild heat at 37 °C is strongly needed to complete the deprotection and thus the conjugation as recently described by Streefkerk et al.19 Numerous amine catalysts have been investigated to accelerate the reaction in oxime ligation.27,28 In our case, 10 mM aniline did not show catalyst property. More than 95% conversion was reached after 48 h treatment at 37 °C in solution A (Table S1, ESI†). Room temperature experiment gave 44% conversion in the same conditions (Table S1, ESI†). Taken together, these results exhibit the key roles played by both the low pH and the temperature in Thz uncaging and oxime formation without metal catalyst. We applied these conditions to our peptides of interest 1 and unprotected peptide 4 at 37 °C. After an overnight reaction, ∼60% of peptide 1 were converted into oxime and one additional night resulted in >95% neat conversion at pH 2 (Fig. 3c). It has been reported in a mechanism study that 2-substituted thiazolidine proceeds to ring opening at 50–90 °C through an iminium intermediate.29 Thz-peptide in solution A at 37 °C gave 45% and 65% of hydrolyzed α-oxo aldehyde after 24 h and 48 h respectively, meanwhile it remained stable at room temperature (data not shown). Combined, these results suggest that two pathways might be involved in this conjugation to reach >95% conversion. The slow acidic Thz hydrolysis releases the aldehyde that subsequently reacts with the aminooxy moiety to give the corresponding oxime (Scheme 1, pathway B). Interestingly, when comparing HPLC spectra in Fig. 2d and 3c, no aldehyde-peptide 2 was detected in the presence of free Aoa-peptide 4. Considering the slow rate of the Thz hydrolysis and the fact that aniline did not catalyze the process, we suppose that nucleophilic Aoa might assist the thiazolidine ring opening and directly proceed to oxime conjugation (Scheme 1, pathway A).
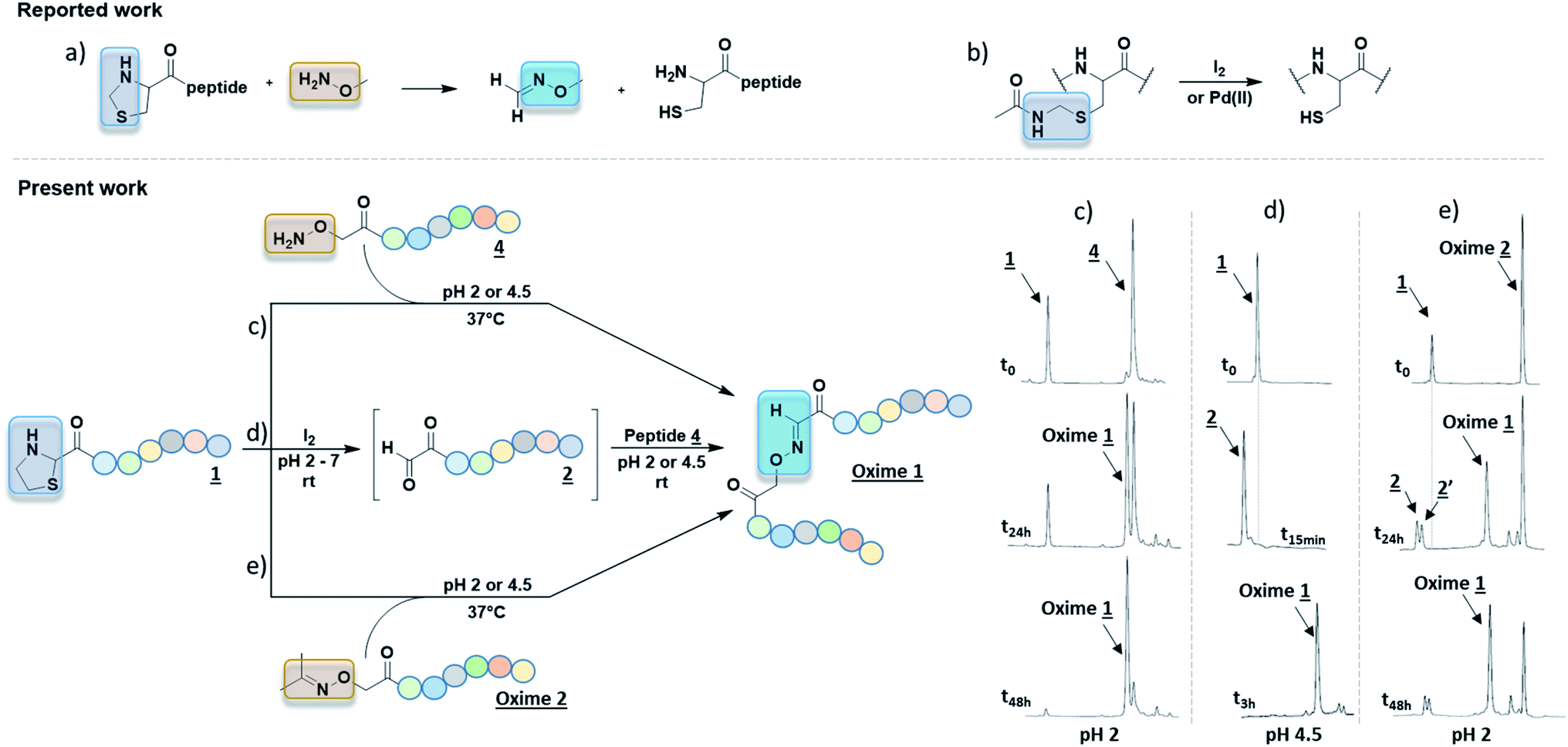 | ||
| Fig. 3 (a) 4-Carboxy thiazolidine deprotected by methoxylamine for further NCL.6,7 (b) Cys(Acm) deprotection mediated by iodine or Pd(II) complex.30 One-pot metal-free 2-carboxy thiazolidine deprotection using aminooxy assisted acidic hydrolysis at 37 °C (c); iodine at room temperature (d) or by transoximation (e) to reach peptide–peptide oxime bond. HPLC monitoring is depicted to illustrate these three different conditions. | ||
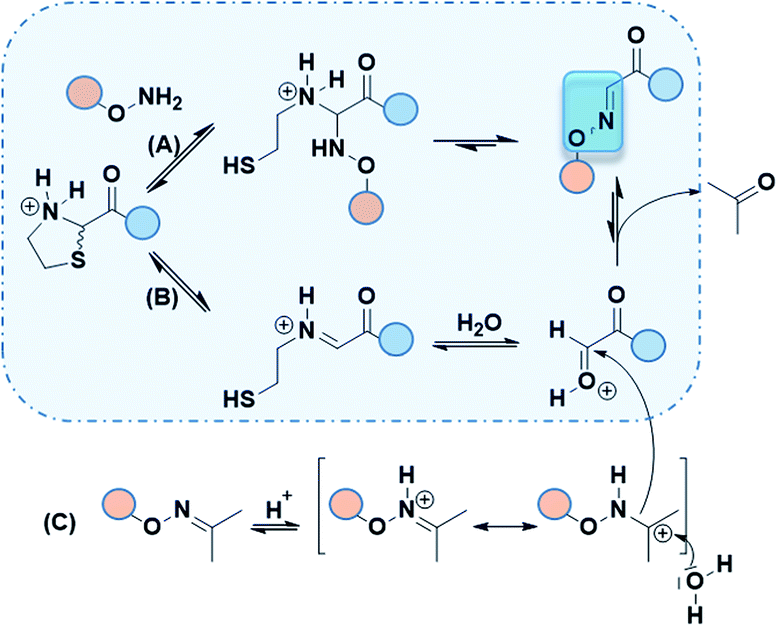 | ||
| Scheme 1 Suggested mechanism involved in Aoa-assisted Thz opening followed by oxime ligation (pathway A). Acetoxime peptide can also proceed to oxime coupling through an additional transoximation step (pathway B + C). | ||
As Thz opening remains the slowest step, we focused our interest on promoting this deprotection. Recent work described the use of Pd(II) complex to deprotect Cys(Acm) compared to conventional iodine method (Fig. 3b).30 Given the similarity between cysteine side chain protecting group acetamidomethyl (Acm) and thiazolidine function, we hypothesized that iodine could deprotect Thz as well as Acm. Thz-GYRMHK 5 was thus treated at room temperature with different amounts of iodine (0.2, 0.4, 0.6 and 1.0 eq.) at pH 2, 4.5 and 7 (Table S3, ESI†). 1.0 eq. iodine was found to achieve neat and rapid Thz deprotection within 15 min irrespective of the pH condition. In these conditions, peptide sequence was stable and Tyr was not iodinated. These conditions were used to afford Oxime 1 from the peptides of interest 1 and 4. As in the case of model peptides, full Thz deprotection was achieved by 1.0 eq. of iodine in water or in pH 2, 4.5 and 7 buffers at room temperature. After iodine quench upon sodium thiosulfate addition (1.2 eq.), complete conversion into Oxime 1 was obtained in 3 h at room temperature in pH 4.5 buffer (Fig. 3d). Interestingly, we could reach >85% conversion in pH 7 buffer containing 100 mM aniline after an overnight stirring at 37 °C (Fig. S1, ESI†).
Although free aminooxy is frequently used in recent works, its high reactivity towards volatile carbonyl compounds makes purification and storage not easy. Based on the reported transoximation observed during tandem oxime ligation,12,14 we investigated the equilibrium between two oxime states to appreciate the role of acetoxime by-product in peptide ligation. Hyodo et al. reported the catalyst role of several Brønsted acids including HClO4, HCO2H, AcOH, H2SO4, and TFA in small organic molecules transoximation.31 In their work, TFA gave sufficient transoximation rate. Moreover, among the oxime starting material that were described for transoximation, acetoxime was pleasantly ranked among the most reactive ones. The authors described transoximation protocol using 5–30 mol% HClO4 with 40 °C heat, conditions that are inappropriate for peptide chemistry. Yuen et al. described a transhydrazonation in small molecule for cellular aldehydes labelling32 with several amine-based catalyst to promote the reaction. Although ethyl acetimidate has been described as a slightly more reactive precursor in acid catalyzed transoximation,31 the formation of the starting oxime would not be as easy as in the case of acetoxime.33 As no evidences were found in transoximation between two peptides and dual Thz deprotection–transoximation (Scheme 1, pathway B + C), Aoa-GYRMHK 6 was beforehand converted upon acetone addition into acetoxime. When mixed with Thz-GYRMHK 5 in solution A, acetoxime peptide gave 9% transoximation at room temperature contrary to 60% in sealed vial and 71% in open vial experiments at 37 °C (Table S3, ESI†). Standard oxime buffer gave similar results with aniline at pH 4.5 while higher amount of acetoxime did not increase transoximation rate (Table S3, ESI†). Using peptide 1 and Oxime 2 (3.0 eq.) (Fig. 3e), 60% transoximation were obtained after an overnight reaction in solution A at 37 °C to give Oxime 1. As in the case of Thz deprotection mediated by Pd complex (Fig. 2d), the slow transoximation allows the formation of 2 and 2′ by acidic hydrolysis. These results show that it is unnecessary to remove acetoxime by-product as it can also perform oxime conjugation in acidic conditions with moderate heating.
In summary, three complementary methods to perform one pot oxime ligation have been demonstrated. The first one consisting of a 1.0 eq. PdCl2 catalyzed Thz and Proc removal and subsequent in situ oxime conjugation at room temperature in pH 2. The second with metal-free aminooxy-assisted Thz opening and oxime conjugation at 37 °C in pH 2 or pH 4.5. The third method using 1.0 eq. iodine allows the oxime ligation to be performed either at room temperature in pH 2–4.5 or at 37 °C in physiological pH with aniline catalyst. We also show that acetoxime-peptide can undergo a transoximation combined with Thz hydrolysis to reach the desired oxime at 37 °C in pH 2 or pH 4.5. The Pd(II) mediated conjugation using both stable protected partners can be run at room temperature while the metal-free ligation requires either a moderate heat (37 °C) or a treatment with iodine to enhance Thz opening. HPLC mobile phase can be used routinely as it remains efficient and convenient without any aromatic amine catalyst. These results were validated for small and medium-size peptides including a large array of amino acids. As far as we know, these methods have never been reported.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr François Guillonneau from Cochin Hospital for his assistance with the MALDI-TOFF mass analysis. This work was supported by “Université de Paris”, CNRS France, INSERM France and the “Agence Nationale de la Recherche” project (ANR-2015-CE17-0005-04). Stephane Duflocq thanks the PhD scholarship funded by “Ecole doctorale Médicament Toxigologie Chimie Imagerie” (MTCI).Notes and references
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem.–Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed.
- S. M. Agten, P. E. Dawson and T. M. Hackeng, J. Pept. Sci., 2016, 22, 271–279 CrossRef CAS PubMed.
- R. J. Spears and M. A. Fascione, Org. Biomol. Chem., 2016, 14, 7622–7638 RSC.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- M. Villain, J. Vizzavona and K. Rose, Chem. Biol., 2001, 8, 673–679 CrossRef CAS PubMed.
- V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2007, 46, 1667–1670 CrossRef CAS PubMed.
- M. Jbara, S. K. Maity, M. Seenaiah and A. Brik, J. Am. Chem. Soc., 2016, 138, 5069–5075 CrossRef CAS PubMed.
- R. L. Brabham, R. J. Spears, J. Walton, S. Tyagi, E. A. Lemke and M. A. Fascione, Chem. Commun., 2018, 54, 1501–1504 RSC.
- X. Bi, K. K. Pasunooti, J. Lescar and C.-F. Liu, Bioconjugate Chem., 2017, 28, 325–329 CrossRef CAS PubMed.
- X. Bi, J. Yin, C. Rao, S. Balamkundu, B. Banerjee, D. Zhang, D. Zhang, P. C. Dedon and C.-F. Liu, Org. Lett., 2018, 20, 7790–7793 CrossRef CAS PubMed.
- C. Buré, D. Lelièvre and A. Delmas, Rapid Commun. Mass Spectrom., 2000, 14, 2158–2164 CrossRef.
- G. Mezö, I. Szabó, I. Kertész, R. Hegedüs, E. Orbán, U. Leurs, S. Bösze, G. Halmos and M. Manea, J. Pept. Sci., 2011, 17, 39–46 CrossRef PubMed.
- D. Lelièvre, C. Buré, F. Laot and A. Delmas, Tetrahedron Lett., 2001, 42, 235–238 CrossRef.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed.
- A. Borrelli, A. Tornesello, M. Tornesello and F. Buonaguro, Molecules, 2018, 23, 295 CrossRef PubMed.
- T. Nishihara, H. Kitada, D. Fujiwara and I. Fujii, Pept. Sci., 2016, 106, 415–421 CrossRef CAS PubMed.
- D. E. Streefkerk, M. Schmidt, J. H. Ippel, T. M. Hackeng, T. Nuijens, P. Timmerman and J. H. van Maarseveen, Org. Lett., 2019, 21, 2095–2100 CrossRef CAS PubMed.
- D. Cussac, M. Vidal, C. Leprince, W. Liu, F. Cornille, G. Tiraboschi, B. P. Roques and C. Garbay, FASEB J., 1999, 13, 31–38 CrossRef CAS PubMed.
- J. Dourlat, W.-Q. Liu, F. Sancier, T. Edmonds, P. Pamonsinlapatham, F. Cruzalegui and C. Garbay, Biochimie, 2009, 91, 996–1002 CrossRef CAS PubMed.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CAS.
- A. Krȩżel, W. Leśniak, M. Jeżowska-Bojczuk, P. Młynarz, J. Brasuñ, H. Kozłowski and W. Bal, J. Inorg. Biochem., 2001, 84, 77–88 CrossRef.
- J.-F. Gaucher, M. Reille-Seroussi, N. Gagey-Eilstein, S. Broussy, P. Coric, B. Seijo, M.-B. Lascombe, B. Gautier, W.-Q. Liu, F. Huguenot, N. Inguimbert, S. Bouaziz, M. Vidal and I. Broutin, PLoS One, 2016, 11, e0167755 CrossRef PubMed.
- Q. A. E. Guthrie and C. Proulx, Org. Lett., 2018, 20, 2564–2567 CrossRef CAS PubMed.
- A. Dirksen and P. E. Dawson, Bioconjugate Chem., 2008, 19, 2543–2548 CrossRef CAS.
- M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93–101 CrossRef CAS PubMed.
- M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, Bioconjugate Chem., 2013, 24, 333–342 CrossRef CAS PubMed.
- T. H. Fife, R. Natarajan, C. C. Shen and R. Bembi, J. Am. Chem. Soc., 1991, 113, 3071–3079 CrossRef CAS.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed.
- K. Hyodo, K. Togashi, N. Oishi, G. Hasegawa and K. Uchida, Green Chem., 2016, 18, 5788–5793 RSC.
- L. H. Yuen, N. S. Saxena, H. S. Park, K. Weinberg and E. T. Kool, ACS Chem. Biol., 2016, 11, 2312–2319 CrossRef CAS PubMed.
- V. Duléry, O. Renaudet and P. Dumy, Tetrahedron, 2007, 63, 11952–11958 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03235b |
| This journal is © The Royal Society of Chemistry 2020 |