
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in porphyrin- and phthalocyanine-containing perovskite solar cells
Yutaka Matsuo *abc,
Keisuke Ogumiad,
Il Jeonb,
Huan Wangc and
Takafumi Nakagawab
*abc,
Keisuke Ogumiad,
Il Jeonb,
Huan Wangc and
Takafumi Nakagawab
aInstitute of Materials Innovation, Institutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: yutaka.matsuo@chem.material.nagoya-u.ac.jp
bDepartment of Mechanical Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
cHefei National Laboratory for Physical Science at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China
dTokyo Metropolitan Industrial Technology Research Institute, 2-4-10 Aomi, Koto-ku, Tokyo 135-0064, Japan
First published on 3rd September 2020
Abstract
In this review, we summarize the application of porphyrins and phthalocyanines in perovskite solar cells to date. Since the first porphyrin- and phthalocyanine-based perovskite solar cells were reported in 2009, their power conversion efficiency has dramatically increased from 3.9% to over 20%. Porphyrins and phthalocyanines have mostly been used as the charge selective layers in these cells. In some cases, they have been used inside the perovskite photoactive layer to form two-dimensional perovskite structures. In other cases, they were used at the interface to engineer the surface energy level. This review gives a chronological introduction to the application of porphyrins and phthalocyanines for perovskite solar cells depending on their role. This review article also provides the history of porphyrin and phthalocyanine derivative development from the perspective of perovskite solar cell applications.
1. Introduction
Solar energy is so abundant that every hour the Sun provides enough energy to meet humankind's annual energy consumption (4.6 × 1020 J).1 The energy provided by the sun in just 1.5 days is equivalent to all the world's fossil fuel resources.2 For the past three billion years, nature has taken advantage of the vast amount of solar energy through photosynthesis.3 At the heart of photosynthesis is the organometallic pigment chlorophyll, which absorbs green light and plays key roles in solar energy conversion. There are two archetypal planar heterocyclic macrocyclic chemicals, porphyrins (Fig. 1b) and phthalocyanines (Pcs), both of which resemble naturally occurring chlorophyll. Porphyrins are composed of four modified pyrrole subunits interconnected at their α carbon atoms via methine bridges (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–). On the other hand, Pcs have nitrogen instead of CH at the meso-position of porphyrin. Both porphyrins and Pcs can form organometallic compounds (metal coordination complexes) just like chlorophyll. Porphyrins and Pcs exhibit excellent thermal and chemical stability, and they also have a wide range of optical and electronic properties, which can be tuned by synthetic modifications, such as attaching functional groups to the periphery of the molecule.4 Therefore, the application of porphyrins and Pcs in photovoltaics has been actively researched in both organic solar cells and dye-sensitized solar cells. Tang and Albrecht introduced organic solar cells using chlorophyll in 1975.5 Later in 1986, Tang used CuPc as the p-type layer in a two-layer p–n junction system. Long-wavelength absorption and good thermal stability of CuPc led to a PCE of 1%.6 Various other porphyrins and Pcs have subsequently been evaluated in organic solar cells.7 Kay and Grätzel also reported the application of different porphyrins including chlorophyll in dye-sensitized solar cells.8,9 This was followed by numerous reports on the application of porphyrins10,11 and Pcs.12–14
CH–). On the other hand, Pcs have nitrogen instead of CH at the meso-position of porphyrin. Both porphyrins and Pcs can form organometallic compounds (metal coordination complexes) just like chlorophyll. Porphyrins and Pcs exhibit excellent thermal and chemical stability, and they also have a wide range of optical and electronic properties, which can be tuned by synthetic modifications, such as attaching functional groups to the periphery of the molecule.4 Therefore, the application of porphyrins and Pcs in photovoltaics has been actively researched in both organic solar cells and dye-sensitized solar cells. Tang and Albrecht introduced organic solar cells using chlorophyll in 1975.5 Later in 1986, Tang used CuPc as the p-type layer in a two-layer p–n junction system. Long-wavelength absorption and good thermal stability of CuPc led to a PCE of 1%.6 Various other porphyrins and Pcs have subsequently been evaluated in organic solar cells.7 Kay and Grätzel also reported the application of different porphyrins including chlorophyll in dye-sensitized solar cells.8,9 This was followed by numerous reports on the application of porphyrins10,11 and Pcs.12–14
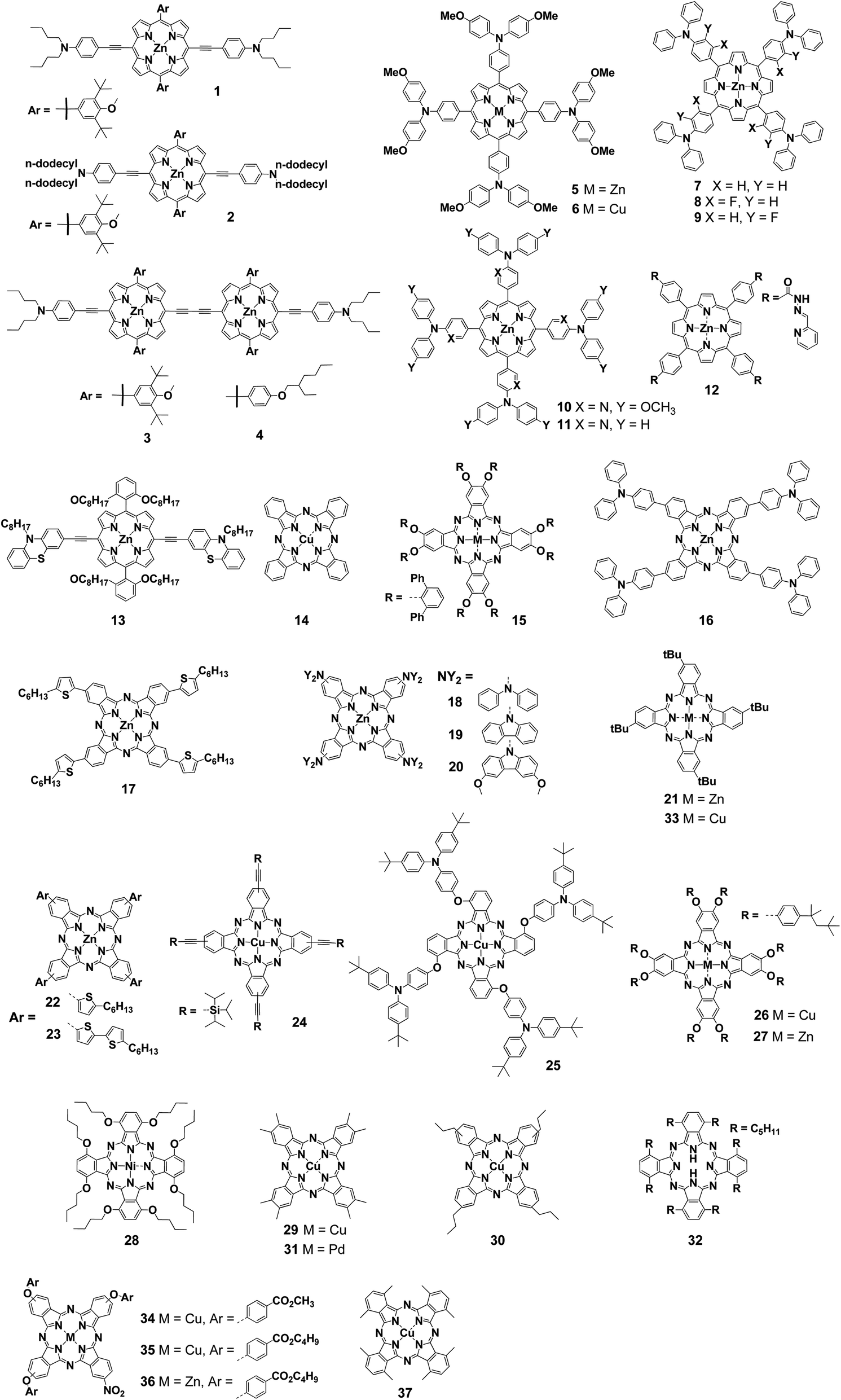 | ||
| Fig. 1 Structures of porphyrins and phthalocyanines for charge transport layers. | ||
In this review, we focus on recent progress in perovskite solar cells using porphyrins and Pcs as interface materials instead of polymer semiconductors, organic semiconductors, and inorganic oxides. We narrow our focus because research on solution-processed perovskite solar cells has expanded rapidly over the past 5 years.15 Also, porphyrins and Pcs with improved molecular design have gradually penetrated into perovskite solar cell research.16–18 Porphyrin and Pc interface materials have several advantages over their polymer counterparts. Firstly, the fact that they exhibit no molecular weight dependence means they have higher purity and less batch-to-batch variability. Secondly, they have higher charge carrier mobility due to their better structural organization. Lastly, they have greater flexibility in terms of chemical modifications, which means it is easier to tune their energy levels and control their solubility. A number of reviews have covered the application of porphyrins and Pcs in organic thin-film solar cells,19–21 so we give only a short summary of reported porphyrin- and Pc-based organic solar cells, moving on to the use of porphyrins and Pcs as charge selective layers in perovskite solar cells.
2. Application of porphyrins and phthalocyanines in photovoltaic devices
2.1 Porphyrin- and Pc-based organic solar cells
When porphyrins and Pcs were first used in photovoltaic devices, they had low solubility making it difficult to form uniform thin films and therefore had to be thermally evaporated.22 However, the development of a series of soluble tetrahedral-shaped oligothiophene molecular donors by Roncali and colleagues in 2007 enabled solution processing of porphyrin and Pc films.23 This was a watershed moment in the advancement of organic solar cells.Porphyrins were initially used in either side chains or in the backbone of polymer donors to improve light harvesting, but this approached had limited success.24,25 One of the authors of this review reported a p–i–n junction organic solar cells that used a soluble porphyrin precursor to tetrabenzoporphyrin with bis(dimethylphenylsilylmethyl)[60]fullerene (SIMEF).26 In 2012, the authors of this review firstly employed the strategy of using an ethynyl linker at the meso-position to maximize the conjugation effect.27 A major step forward came in 2013, when Peng and colleagues combined porphyrin with diketopyrrolopyrrole (DPP).28 An electron-withdrawing moiety, DPP is amenable to synthetic modification and substitution of various aromatic groups at the 2,5-position.29 DPP-based electron donor materials gave PCEs of less than 5%.30,31 By flanking DPP units with two thiophene (T) units, the solar cell performance improved.32–35 Then, porphyrin-integrated T–DPP–T led to 7.23% PCE, which was a breakthrough.36 In their molecular design, the porphyrin core had two T–DPP–T units a two ethynyl linkers. The substituents attached to the porphyrin unit were then modified to improve intermolecular π–π stacking of the porphyrin core by using a shorter alkyl chain. The morphology of the porphyrin-based bulk heterojunction with phenyl-C61-butyric acid methyl ester (PC61BM) was comprehensively studied by grazing incidence X-ray diffraction (GIXRD), resonant soft X-ray scattering, atomic force microscopy (AFM), and transmission electron microscopy for various fabrication conditions.37
The key feature of Peng's design strategy was the A–π–D–π–A configuration. Sharma, Langa, and co-workers also constructed analogous A–π–D–π–A systems with ethynylene linkers between Zn-porphyrin and thienylene vinylene thiophene with the ends capped by dicyanovinylene or 3-ethylrhodanine groups. These types of active layer materials also show outstanding PCEs of 7–10%.38 The authors of this review investigated the substituent effects of the aryl group on Mg-porphyrin with the A–π–D–π–A structure39 and used tetraethynyl Mg-porphyrin to maximize the intramolecular charge transfer effect, finding an optimized structure of D–(π–A)4.40 Zhu, Wong, and co-workers developed direct peripheral meso-alkyl substitutions onto the porphyrin ring, leading to improved solubility and charge transport property and ultimately to excellent PCEs.41
Because of their well-known electron-donating ability, porphyrin-based molecules had long been used only as donor materials. In 2017, Li and co-workers first developed a powerful non-fullerene acceptor based on Zn-porphyrin tethered to four perylene bisimide (PDI) units via ethynyl linkers.42 This spurred many follow-up works.43 To date, porphyrin-based organic solar cells have made great progress and this work has provided a solid foundation for other device applications, such as perovskite solar cells.
2.2 Porphyrins as hole-transport materials in perovskite solar cells
First, Chou et al. applicated porphyrin derivatives as hole transport materials (HTMs) for perovskite solar cells. They created two kinds of materials based on the porphyrin skeleton for use as new HTMs (1 and 2) (Fig. 1).44 In general, HTMs in perovskite solar cells should be transparent so as not to hinder light absorption by the perovskite layer. Although porphyrin derivatives were seemed to encumber the light absorption of perovskite layer, the absorption spectra of perovskite films with and without 1 revealed that porphyrin materials were able to work as HTMs instead of photosensitizers. While the device using 1 as HTMs showed a PCE of 16.6%, compound 2 bearing dodecyl groups gave a only 10.6% (Table 1). This deference was depended on the morphology of surface on HTM layer. Scanning electron microscopy (SEM) images explained that the pinholes were observed in the case of 2 and those reduced the photocurrent and FF due to the recombination.| Compound | HOMO [V] | LUMO [V] | μh [cm2 V−1 s−1] | Eg [eV] | VOC [V] | JSC [mA cm−2] | FF [%] | PCE [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | −5.22 | −3.39 | 2.04 × 10−4 | 1.83 | 0.99 | 22.82 | 73.34 | 16.60 | 44 |
| 2 | −5.21 | −3.32 | 1.53 × 10−5 | 1.89 | 1.01 | 17.80 | 58.69 | 10.55 | 44 |
| 1 | −5.22 | −3.45 | 3.20 × 10−5 | 1.77 | 1.09 | 22.62 | 73.02 | 17.93 | 45 |
| 3 | −5.14 | −3.54 | 4.20 × 10−4 | 1.60 | 1.10 | 22.60 | 78.52 | 19.44 | 45 |
| 4 | −5.11 | −3.53 | 9.30 × 10−5 | 1.58 | 1.04 | 22.97 | 74.75 | 17.84 | 45 |
| 5 | −5.29 | −3.35 | 3.06 × 10−4 | 1.94 | 1.10 | 22.69 | 71.3 | 17.78 | 46 |
| 6 | −5.37 | −3.40 | 2.89 × 10−4 | 1.97 | 0.71 | 21.60 | 66.3 | 15.36 | 46 |
| 7 | −5.20 | −3.26 | 3.37 × 10−4 | 1.94 | 1.10 | 21.07 | 70.4 | 16.37 | 48 |
| 8 | −5.40 | −3.41 | 3.91 × 10−4 | 1.99 | 1.12 | 22.21 | 75.4 | 18.85 | 48 |
| 9 | −5.29 | −3.34 | 3.54 × 10−4 | 1.95 | 1.11 | 21.86 | 72.7 | 17.70 | 48 |
| 9 | −5.13 | −3.23 | 3.51 × 10−4 | 1.90 | 1.03 | 19.76 | 61.0 | 12.40 | 49 |
| 7 | −5.20 | −3.26 | 3.38 × 10−4 | 1.94 | 1.03 | 19.35 | 60.0 | 11.96 | 49 |
| 10 | −5.23 | −3.37 | 3.85 × 10−4 | 1.86 | 1.04 | 20.28 | 64.0 | 13.52 | 49 |
| 11 | −5.36 | −3.46 | 4.10 × 10−4 | 1.90 | 1.05 | 20.74 | 65.0 | 14.11 | 49 |
| 12 | −5.35 | −2.55 | — | 2.80 | 1.09 | 22.29 | 73.12 | 17.82 | 50 |
| 13 | −5.05 | — | — | — | 1.000 | 19.66 | 57.3 | 11.26 | 51 |
| 14 | −5.2 | −3.5 | — | 1.7 | 0.75 | 16.3 | 44 | 5.0 | 52 |
| 15 | — | — | — | — | 0.797 | 16.35 | 50.3 | 6.7 | 53 |
| 16 | −5.20 | −3.40 | — | 1.80 | 0.67 | 22.10 | 40.0 | 5.60 | 54 |
| 17 | −5.19 | −3.43 | — | 1.72 | 0.98 | 17.15 | 72.0 | 12.30 | 56 |
| 18 | −4.96 | −3.24 | — | 1.72 | 1.01 | 16.67 | 68.1 | 11.75 | 57 |
| 19 | −5.39 | −3.57 | — | 1.82 | 1.00 | 10.69 | 59.8 | 6.65 | 57 |
| 20 | −5.18 | −3.39 | — | 1.79 | 1.03 | 17.43 | 61.0 | 11.44 | 57 |
| 21 | −5.31 | −3.49 | 1.82 | 0.89 | 16.79 | 43.7 | 5.16 | 58 | |
| 21 | −5.32 | −3.49 | — | 1.83 | 1.03 | 20.19 | 66.3 | 13.3 | 59 |
| 22 | −5.19 | −3.44 | — | 1.75 | 1.05 | 20.28 | 80.3 | 17.1 | 59 |
| 23 | −5.15 | −4.42 | — | 1.73 | 1.10 | 20.16 | 69.4 | 15.5 | 59 |
| 24 | −5.42 | −3.69 | — | 1.73 | 1.01 | 21.40 | 65.0 | 14.00 | 60 |
| 25 | −5.22 | −3.57 | — | 1.65 | 1.01 | 21.90 | 68.0 | 15.00 | 61 |
| 26 | −5.35 | −3.58 | — | 1.77 | 0.87 | 19.01 | 50.56 | 8.33 | 64 |
| 27 | −5.26 | −3.34 | — | 1.92 | 0.89 | 17.52 | 46.52 | 7.25 | 64 |
| 28 | −5.06 | −3.61 | 1.90 × 10−4 | 1.45 | 1.07 | 23.00 | 72.8 | 16.80 | 65 |
| 29 | −5.20 | −3.50 | 4.79 × 10−2 | 1.70 | 1.08 | 21.32 | 68.0 | 15.73 | 67 |
| 30 | −5.13 | −3.36 | 2.16 × 10−3 | 1.77 | 1.09 | 23.20 | 76.0 | 17.80 | 70 |
| 31 | −5.11 | −3.30 | 3.42 × 10−4 | 1.81 | 1.06 | 21.08 | 73.0 | 16.28 | 71 |
| 32 | −5.30 | −3.70 | 1.80 × 10−3 | 1.60 | 0.99 | 18.80 | 55.0 | 11.50 | 72 |
| 14 | −5.2 | −3.5 | 3.85 × 10−4 | 1.70 | 1.01 | 20.90 | 64.0 | 14.89 | 74 |
| 14 | −5.28 | — | — | — | 1.015 | 22.15 | 74.8 | 16.85 | 74 |
| 33 | — | — | — | — | 1.15 | 23.60 | 74.2 | 20.09 | 75 |
| 33 | −5.20 | −3.50 | — | 1.70 | 1.07 | 22.60 | 77.5 | 18.80 | 76 |
| 34 | −4.98 | −3.21 | 5.02 × 10−5 | 1.77 | 1.02 | 20.01 | 60.0 | 12.52 | 77 |
| 35 | −4.96 | −3.23 | 8.21 × 10−5 | 1.73 | 1.04 | 20.62 | 64.0 | 13.66 | 77 |
| 36 | −4.95 | −3.24 | 11.4 × 10−5 | 1.71 | 1.10 | 21.00 | 68.0 | 15.74 | 77 |
| 37 | −5.06 | −3.61 | 3.84 × 10−3 | 1.99 | 1.01 | 22.44 | 73.43 | 16.61 | 78 |
To achieve higher efficiency, Chou et al. also developed porphyrin HTMs (3 and 4). Those porphyrin dimers 3 and 4 gave a PCE of 19.4% and 17.8% respectively.45 The authors explained that the extended π-conjugation of the porphyrin dimers resulted in stronger intermolecular interaction than that of the corresponding monomers. This gave not only high hole mobility but also reduced pinholes during the film formation, which was advantageous for high-performance photovoltaic devices. They also investigated the stability toward humidity, light and thermal stress. The device using a porphyrin dimer 3 as HTMs was more stable than the conventional device using 2,2′,7,7′-tetrakis-(N,N-di-4-methoxyphenylamino)-9,9′-spirobifluorene (spiro-OMeTAD). In particular, 3-based devices were stable against humidity, maintaining at least 90% of their initial PCE even after 800 h, whereas the PCEs of spiro-OMeTAD based devices dropped to 80% or less of the initial level within 300 h. To explain this difference, they focused on the hydrophilicity of 3 and spiro-OMeTAD and measured the contact angle of each device. As a result, the contact angle of 103° using 3 was higher than that of spiro-OMeTAD (65°). This showed that 3 was highly hydrophobic and effective in protecting the perovskite layer from moisture in the atmosphere. However, those porphyrin compounds were required multistep synthesis and it was a commercial problem.
Chen et al. synthesized symmetrical porphyrins (5 and 6) with triphenylamine-based substituents at four meso positions.46 Those porphyrin derivatives could be prepared on a gram scale from aldehydes and pyrroles in only two steps. The highest occupied molecular orbital (HOMO) levels of 5 and 6 were −5.29 eV and −5.37 eV, respectively, which were similar to that of spiro-OMeTAD (−5.22 eV). Therefore, effective hole transfer from the perovskite layer to a gold electrode could be expected. On the other hand, the lowest unoccupied molecular orbital (LUMO) levels of −3.35 eV in 5 and −3.40 eV in 6 were lower than that of spiro-OMeTAD, −2.28 eV. The authors explained that those lower LUMO levels prevented the inflow of electrons and suppress charge recombination in the HTM layer. To investigate the hole mobility of 5 and 6, they measured space-charge limited current (SCLC). Hole mobilities of 5 (3.06 × 10−4) and 6 (2.89 × 10−4) were higher than that of spiro-OMeTAD (1.58 × 10−4). Moreover, they calculated the hole reorganization energy (ER). The calculated values were 303 meV in 5, 374 meV in 6 and 495 meV in spiro-OMeTAD. Those ER values showed compounds 5 and 6 had advantages as HTMs. The PCEs of devices using 5 (17.8%) and 6 (15.4%) were comparable to the PCE of the spiro-OMeTAD system (18.6%) under the same conditions. At that time, the PCE of over 17% was the highest yet reported for perovskite solar cells with porphyrin- or Pc-based HTMs. The device using 5 also showed excellent stability. The spiro-OMeTAD systems require hygroscopic dopants,47 which results PCE decreasing by 45% after 30 days in the atmosphere (20–25 °C, 40–45%), whereas the devices using 5 showed a decrease of only 15%.
Azmi et al. synthesized three types of zinc porphyrins bearing triphenylamino groups (7–9) to investigate the effect of introducing fluorine, an electrophilic group, on the porphyrin substituents with the aim of improving electrochemical properties and device performance.48 The HOMO levels by CV measurement were −5.14 eV for 7, −5.37 eV for 8, and −5.24 eV for 9. Introducing more fluorine atoms or positioning a fluorine atom close to the porphyrin core lowered the HOMO level. A similar trend was also observed using ultraviolet photoelectron spectroscopy (UPS). The PCEs when lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and 4-tert-butylpyridine (t-BP) were used in the HTM layer were 16.4% for 7, 18.9% for 8, and 17.7% for 9. The difference of the HOMO levels clearly affected open-circuit voltage (VOC). To evaluate hole injection from the perovskite layer into the HTM layer, Azmi et al. performed photoluminescence (PL) measurement. While a strong peak was observed around 770–780 nm. In only perovskite layer, the PL peaks were remarkably small in the devices using 7–9. This observation reflected the transport of holes from the perovskite layer to the HTM layer. Among the three, 8 had the smallest peak. From this result, they considered that the introduction of fluorine not only lowers the HOMO levels but also facilitates effective hole transport between the perovskite layer and the HTM layer. Furthermore, 2D-GIXRD showed that 7–9 were layered with a face-on orientation with respect to the perovskite layer. Among them, compounds 8 and 9 were tightly stacked compared with compound 7, indicating that fluorine improved the intermolecular interactions. For the stability of the device in the atmosphere, the additive-doped compound 8 system was also more stable than the additive-doped spiro-OMeTAD system. After 40 days, the order of their PCEs was reversed (8: 17.0%; spiro-OMeTAD: 16.5%).
The same research group synthesized compounds 10 and 11, in which the phenyl group closest to the porphyrin core was replaced with a pyridine-based electron-deficient group. They investigated the effect on the performance of HTMs.49 In addition, by using ZnO instead of TiO2 as the electron-transport material (ETM) layer, a low-temperature process was demonstrated in which the temperature required for the entire device fabrication process was only 140 °C. Compounds 10 and 11 containing pyridine have lower HOMO levels than the triphenylamine-type compounds. As a result, the electron-donating property of the arylamine to the porphyrin core was suppressed. The trend of HOMO levels was reflected in VOC. Compound 11 having the lowest HOMO level gave the highest VOC (1.05 V). The PCE for 11 was 14.1%, which was close to that of the reference using spiro-OMeTAD (14.6%) prepared under the same conditions. The difference between short-circuit current density (JSC) obtained from the actual measurement and the external quantum efficiency (EQE) spectrum was less than 4% for all the compounds. 2D-GIXD measurements showed that pyridine-substituted compounds 10 and 11 had stronger face-on stacking than the triarylamine compounds. Because the pyridine-containing compounds were relatively superior in terms of hole mobility according to SCLC, the pyridine moiety was considered to promote the face-on stacking and consequently improve the hole mobility.
Because perovskite solar cells using doped spiro-OMeTAD as HTMs are unstable to the atmosphere and humidity, considerable efforts have been devoted to improving stability by various approach. In recent years, it has been found that at around 80 °C, CH3NH3+ and I− ions in the perovskite layer diffuse into the spiro-OMeTAD layer, lowering device efficiency. Lv et al. reported the use of a zinc porphyrin derivative (12) with acylhydrazone substituents as HTMs to improve the thermal stability of perovskite solar cells.50 Their previous studies have shown that acylhydrazone-substituted porphyrins exhibited excellent charge carrier mobility. To investigate the effect of 12 on the perovskite layer, Fourier-transform infrared spectroscopy was performed for the device with a structure of TiO2/perovskite/12, which exhibited an absorption peak derived from Pb–N vibration at 418 cm−1. This result showed that the acylhydrazone of 12 and the nitrogen atom of the pyridine site were effective for electronic passivation of Pb2+ in the perovskite layer. In addition, electrochemical impedance measurements showed that the device using 12 had a lower resistance to hole transport and a higher charge recombination resistance than the spiro-OMeTAD system. The device using 12 showed a high PCE of 17.8%. The thermal stability of the fabricated devices was investigated at 85 °C under a nitrogen atmosphere. After 100 h, the PCE of the spiro-OMeTAD-based device dropped to nearly 20% of the initial value, whereas the 12-based device showed almost no change, with PCE remaining above 90% of the initial value. This result suggested that 12 was useful for improving the thermal stability of perovskite solar cells.
Recently, Reddy et al. reported porphyrin 13 having a D–π–D configuration with N-octyl phenothiazine units.51 Interestingly, the device using 13 as HTMs was fabricated through a screen-printing process. This method enabled fabrication of large-area devices and minimized the waste of materials. The device using 13 without a dopant gave a good JSC of 19.66 mA cm−2 and a PCE of 11.26%. This was the highest reported efficiency at the time in a porphyrin hole-transport layer (HTL)-based perovskite solar cells using a carbon–graphene composite as a cathode.
2.3 Phthalocyanines as hole-transport materials in perovskite solar cells
Thus far, we have discussed examples in which not only porphyrins but also various Pcs were used as HTMs in perovskite solar cells. The use of Pcs as HTMs in perovskite solar cells was first reported in 2015. The first copper phthalocyanine (CuPc, 14) HTM was introduced into perovskite solar cells by vacuum thermal evaporation by Kumar et al.,52 who obtained a PCE of 5.0%. Then, Ramos et al. reported a non-aggregated Zn(II)octa(2,6-diphenylphenoxy)Pc (15) used as a solution-processable HTM for perovskite solar cells.53 A PCE of 6.7% was obtained with the use of LiTFSI and t-BP additives. The Lianos group reported a soluble zinc Pc with four triphenylamine groups (16) and a triphenylamine-based substituent for fabrication of devices through spin-coating.54 The obtained PCE achieved 5.60% under the optimized conditions. Subsequently, they increased the PCE to 13.65% by using a mixture of 16 and Al2O3 as the buffer layer.55Gao et al. synthesized a symmetric zinc Pc 17 bearing four thiophene groups with low bandgap characteristics as a HTM in perovskite solar cells.56 They observed the time-resolved PL decay at the perovskite/compound 17 interface and revealed that 17 exhibited the quick quenching as same as spiro-OMeTAD. A device with a structure of fluorine-doped tin oxide (FTO)/TiO2/meso-TiO2/CH3NH3PbI3/17/Au gave a PCE of 4.21%. This low PCE was attributed to the shunt path between the perovskite layer and the gold electrode. The low solubility of 17 resulted in insufficient film thickness and inhomogeneity of the HTM layer. The authors proved this inference by a cross-sectional SEM measurements of the devices. To address this problem, they inserted aluminum oxide as a buffer layer at the perovskite-HTM interface. Aluminum oxide increases the thickness of the HTM layer and prevents contact between the perovskite layer and the gold electrode. In the device with aluminum oxide (FTO/c-TiO2/meso-TiO2/CH3NH3PbI3/meso-Al2O3/17/Au), the conversion efficiency improved to 12.8%.
Compounds 18–20 are zinc phthalocyanine compounds bearing secondary amines.57 Computational studies showed that the introduction of secondary amines significantly affected the photochemical and electrochemical properties of Pcs. Compound 18–20 bearing amines as electron-donating group had high HOMO/LUMO levels and narrow bandgaps according to electrochemical measurements. The VOC of perovskite solar cells using these compounds were high, exceeding 1 V. The maximum PCE of 11.8% was recorded using 17.
Spiro-OMeTAD and poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA) are currently most famous HTMs, however these compounds are expensive because they require multistep synthesis and purification. For this reason, the development of alternative materials with reduced manufacturing costs is required for perovskite solar cells. The excellent stability and conductivity of Pcs make them a possible alternative to spiro-OMeTAD and PTAA. As of 2016, examples of using Pc complexes with copper as the metal center had been reported, but HTM layers using these complexes required a vacuum deposition process for device fabrication. Therefore, these compounds were not suitable for reducing the manufacturing cost. Wu et al. synthesized a zinc phthalocyanine compound (21) with improved solubility to enable the film formation by spin-coating.58 Cross-section SEM image indicated that 21 was coated on the perovskite layer sufficiently with uniformity. The PCE of the resulting device was 5.16%, and the fill factor (FF) was 0.341, which is rather low. The current density and voltage (J–V) curve showed significant hysteresis. Although the authors thought this modest device performance was derived from their immature device fabrication procedure, they concluded 21 was suitable for the solution process and had a potential for the low-cost HTMs.
Cho et al. selected three Pc derivatives with different bulkiness of substituent (21–23) to investigate the impact of steric effects and aggregation of Pc derivatives on charge-transfer properties.59 Compound 22 was a new compound, but the other two had already been reported. The PCEs were 13.3% for 21, 17.1% for 22, and 15.1% for 23. SEM images of the surfaces of Pc derivatives by spin-coating showed that the flower-type brunched micrometer-size aggregates were observed in the case of compound 22. They mentioned the aggregation enhanced the device performance. Compound 22 also showed excellent lateral conductivity of the HTM layer of 8.0 × 10−5 S cm−1 compared with 21 (5.0 × 10−7 S cm−1) and 23 (6.0 × 10−7 S cm−1).
A copper phthalocyanine (24) with triisopropylsilyl groups for improving the solubility and enabling solution processability has been reported.60 According to DFT calculations, 24 had a lower recombination energy than that of spiro-OMeTAD (24: 239 meV; spiro-OMeTAD: 495 meV). The authors, Jiang et al., employed a low-cost carbon electrode for the device with 24. The device performance using 24 as HTMs without dopant gave a PCE of 14.0%. According to the stability test in the dark, the device using 24 maintained 90% of its PCE for 30 days, while that of the spiro-OMeTAD device decreased to 80% or less. This difference in stability was attributed to the hydrophobicity of the HTM layer. The water contact angles of the surface of compound 24 and spiro-OMeTAD were 104.5° and 82.5°, respectively. That indicated that the thin film of 24 was highly hydrophobic.
To improve the solubility, Jiang et al. reported a copper phthalocyanine derivative (25) with tert-butyl triphenylamine groups.61 Although the hole mobility of 25 was as low as 5 × 10−7 S cm−1, it was improved to 8.9 × 10−6 S cm−1 by adding the p-type dopant 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ; 6% w/w). The authors employed carbon electrode to enable solution-processing for all fabrication process owing to avoidance of vacuum deposition.62 PCE was 10.5% using 25 compared to 9.3% in absence of HTL. This PCE was further increased to 15.0% when 25 was used with F4-TCNQ (6% w/w). They thought compound 25 layer was prevented direct contact between the perovskite and the carbon electrode. The HTL using 25 consequently restrained charge recombination and extract a hole.63 To further investigate the electrochemical properties, they measured electrochemical impedance measurements (EIS) and showed that the HTM-free device had higher hole transport resistance and the device using 25 had high charge recombination resistance at any voltage.
Calio et al. also synthesized 26 and 27 with 4-tert-octylphenoxy substituents for spin-coating deposition.64 Spin-coating process could control the thickness of HTL by the concentration of HTMs. The authors found the thinnest HTL with 10 mM gave the best PCE of 8.33%, a VOC of 0.87 V, JSC of 19.01 mA cm−2 and FF of 50.56%. They concluded this result was caused from a decrease in series resistance. The series resistance decreased from 211.22 to 71.55 Ω cm2 for 30 mM to 10 mM concentrations.
Cheng et al. reported the use of a new nickel-centered Pc derivative, 28.65 Although the device using 28 gave a PCE of 9.9%, there was a problem of a low VOC. The factor affecting VOC generally is considered from the HOMO level of the HTMs, however, the author focused on the morphology of the HTL.66 SEM images showed that the spiro-OMeTAD layer (80 mg mL−1) uniformly covered the crystals of the perovskite layer, whereas the crystals of the perovskite layer were visible on the surface of 28 (30 mg mL−1). Thus, they thought the recombination of holes and electrons might decreased VOC and FF. To solve this problem, they considered two methods. One was the increasing of the film thickness of the HTL, and the other was the introducing an additional buffer layer between the HTL and the gold electrode. Although the former was simple because it could be accomplished by increasing the HTM concentration, an additive was required to prevent the FF from decreasing with increasing film thickness. The PCE increased to 17.0% when the concentration of 28 was increased to 50 mg mL−1 and LiTFSI and t-BP were used as additives. On the other hand, a buffer layer did not require the use of additives. Vanadium oxide, a p-type metal oxide, was employed as the buffer layer and it gave a PCE of 17.6%. In this system, the HTL using 28 was fabricated from a 30 mg mL−1 solution, and no additives were required. SEM images confirmed that no crystals of the perovskite layer were visible on the surface of the vanadium oxide layer. Since the efficiency of the device using only vanadium oxide as the HTM was about 10.9%, they concluded that the combination of the 28 as the HTL with the inorganic buffer was important for improving performance. In stability testing, the device using 28 with vanadium oxide showed slower deterioration of PCE compared with the spiro-OMeTAD system. The order of the PCEs of the two systems reversed within 10 days at 20–25 °C and 40–45% relative humidity. This was derived from the low hygroscopicity of the 28 and vanadium oxide combination. The device surface did not change after 30 days for the 28 and vanadium oxide system, whereas the doped spiro-OMeTAD system showed deterioration of the perovskite layer.
The effect of orientational differences during the formation of Pc films was investigated by Yang et al. They compared unmodified copper phthalocyanine (CuPc) with a methylated CuPc derivative (29).67 GIXRD showed that pristine CuPc had an edge-on orientation and 29 had a face-on orientation on the perovskite layer. Time-resolved PL measurements of each device indicated that the system with 29 had a shorter PL lifetime. This suggested that the face-on orientation of 29 was superior to the edge-on orientation of the pristine CuPc system for the extracting of hole from the perovskite layer. The PCEs were 12.6% for the pristine CuPc system and 15.7% for the compound 29 system. The pristine system had hole mobility of 7.25 × 10−4 cm2 V s−1 while that of the 29 system was 4.79 × 10−2 cm2 V s−1. The latter value was also superior to that of the spiro-OMeTAD system. This result came from the strong π–π stacking of 29 in the face-on orientation. The difference of orientation also greatly influenced the device stability.68 AFM measurement of HTL showed the particle sizes of the surface were about 100 nm using pristine CuPc and 20 nm using 29. The water contact angles on the surface also were 81.2° for the pristine CuPc and 119.6° for 29. The difference of contact angle was attributed to the additional hydrophobicity of the methyl group as well as the difference in orientation.69 In stability testing at 25 °C and 50% humidity in air, the PCE after 2000 h kept over 95% in the HTL using 29. Therefore, it is presumed that the face-on orientation contributes greatly to the stability of these perovskite solar cells.
Furthermore, the same research group reported orientation studies using soluble compounds which could be employed for solution processes.60,70 They used a new compound with propyl chains (30) that had excellent solubility. GIXRD explained that 30 showed an edge-on orientation on the FTO glass surface, whereas the mixture of edge- and face-on orientation on the perovskite layer. Hole mobility on the face-on orientation was superior to that of the edge-on orientation. PL measurements showed that the luminescence peak of the perovskite layer/30 was weaker than that of the perovskite layer/spiro-OMeTAD. Similarly, in time-resolved PL measurement, the carrier lifetime of the perovskite layer/30 was shorter than that of the perovskite/spiro-OMeTAD. Excellently, the best PCE using 30 reached 17.8% compared with a PCE of 17.5% using spiro-OMeTAD. The authors also verified that covering the surface of the perovskite layer using 30 was effective for ensuring device stability. In stability testing at 25 °C and 75% humidity, over 94% of an initial PCE was maintained even after 800 h.
To reduce charge recombination, Zheng et al. synthesized compound 31 with a palladium atom at the Pc core.71 Compared with the octamethylated CuPc complex 29,67 the triplet state of heavy elements, such as palladium metal, has been shown to increase carrier diffusion length (LD). Transient absorption spectroscopy showed that 31 had a carrier lifetime of 7.7 ns. This lifetime was longer than that of 3.4 nm using corresponding copper complex 29. Although a long charge lifetime was likely to lead to charge recombination, compound 31 showed an increase in the hole mobility due to the strong spin–orbit interaction of a palladium atom. As a result, the LD of 31 was 26.00 nm, which was an improvement from the LD of 29 (20.45 nm). The employment of 31 gave a PCE of 16.28%.
Dao et al. investigated the effect of annealing of HTL to the device performance.72 They fabricated a device using a metal-free Pc derivative (32) and confirmed a significant improvement in PCE from 6.1% to 11.5% after treatment under the optimum annealing conditions at 130 °C for 10 min compared with the case without annealing. They then compared other indicators in the case of pristine/annealed devices. In the EQE spectrum, the yield at 480 nm was improved from 78% to 84%, and the emission suppression of the perovskite layer in the PL spectra also increased from 65% to 72% after annealing. Furthermore, the hole mobility obtained by photo-charge extraction by linearly increasing voltage (photo-CELIV) measurements was nearly 10 times for the annealed device compared with the unannealed device (1.8 × 10−3 cm2 V s−1 versus 2.8 × 10−4 cm2 V s−1). Those results demonstrate that annealing is effective for improving device performance.
The influence of the temperature during vacuum deposition of the HTL to device performance was also investigated using a generic CuPc (14).73 As a result, PCE was improved from 7.80% at 30 °C to 14.9% at 100 °C. This value was superior to the efficiency achieved with spiro-OMeTAD produced under the same conditions (13.4%). When SEM images of the HTL formed at 30 °C and 100 °C were compared, needle-like aggregates were observed at 100 °C. It was speculated that the acicular aggregates provided an ideal interface with the perovskite layer and were effective for the extracting of hole. Lei et al. also manufactured a flexible device using the same compound CuPc 14 on polyethylene terephthalate (PET)-ITO substrates.74 CuPc was inserted between a vanadium oxide layer and the electrode as in their previous report.64 They achieved a high PCE of 16.9% in a device using 14 and vanadium oxide as the HTL on a glass substrate. Notably, they also fabricated devices using PET-ITO substrates, and even in this system, a High PCE of 14.4% was obtained. It was slightly lower than that of the device on a glass substrate owing to the large series resistance and the low transparency of the PET substrate. However, the fabrication of flexible devices using PET-ITO substrates did not require high temperature more than 60 °C throughout the all process. This was advantageous for improving the manufacturing process in perovskite solar cell research.
With respect to device stability, Duong et al. studied the morphology of HTL for high thermal stability.75 In the HTL using Pc or porphyrin derivatives stacked by spin coating, cracks were obtained. Those cracks led to short-circuiting and charge recombination due to the direct contact between the perovskite layer and the metal electrode. The authors produced a device using a commercially available and soluble CuPc derivative with four tert-butyl groups (33) and confirmed that cracks appeared in the HTM layer. To address this problem, they proposed a thermal treatment at 85 °C for 20 h in nitrogen atmosphere. This thermal treatment improved the PCE from 7.5% to 18.7% on average. Notably, the champion efficiency with this approach reached to 20.1%. SEM images explained that the cracks were observed on the HTL before thermal treatment while those cracks disappeared and gold particles of Au electrode were formed after the annealing. The authors explained that the gold particles at the contact area migrated and were rearranged between the perovskite layer and the gold electrode by the thermal treatment. Those particles thereby shut the cracks and suppressed short-circuiting and charge recombination. GIXRD and UPS measurements did not show changes on the surface of the HTL before and after thermal treatment. However, in the case of thin Au layer of about 5 to 10 nm, the work function changed after the thermal treatment. From this change, the authors speculated that the gold nanoparticles penetrated into the HTM layer during the thermal treatment.
Regarding the stability of perovskite solar cells, many papers have discussed device stability against humidity and light irradiation, but there have been few reports on thermal stability. Duong et al. studied the thermal stability of devices76 using 33 as HTM.75 The fabricated device gave a PCE of 18.8% under the optimal conditions. The rate of decrease of PCE by the thermal treatment in a nitrogen atmosphere was tested. The devices using general spiro-OMeTAD or PTAA as HTMs exhibited a sudden decrease at 85 °C. Then, at 130 °C, the efficiency had decreased by more than 60% compared with that of devices kept at room temperature. However, for the devices using 33, there was almost no decrease in PCE even after thermal treatment at 130 °C for 30 min. Furthermore, the devices using 33 were stable even after 50 thermal cycles from −45 °C to 85 °C. The authors speculated that the strong interaction between the perovskite layer and the HTM layer led to the good performance of the devices. GI-XRD revealed that 33 was stacked on the perovskite layer in a face-on orientation. In addition, the physical adhesion of the perovskite layer and the HTL were confirmed by the tape test.
Guo et al. synthesized three Pc derivatives (34, 35, and 36) with different types of substituents and different central metals.77 The PCEs of the fabricated devices were 12.5% for 34, 13.7% for 35, and 15.7% for 36. SEM images of the compounds showed that the film of 36 had few pinholes. The film of 36 also exhibited the highest hole mobility. Because hole mobility was related to the hysteresis of the devices, few hysteresis was found in the device for 36. They also investigated device stability under light irradiation at 60 °C. The spiro-OMeTAD based device fabricated under the same conditions showed an efficiency drop of more than 90% after 500 h of irradiation. Interestingly, the performance decreased less than 30% for the devices with 36. This difference of stability was derived from the hydrophilicity of the HTL. The water contact angle on the film of 36 was 92.27°, whereas that of the spiro-OMeTAD film was 58.22°.
Hu et al. reported the mixture HTL composed form Pc derivatives and another organic compound.78 They employed a low-cost octamethyl Pc derivative (37) and poly(3-hexylthiophene-2,5-diyl) (P3HT). The optimal ratio of compound 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P3HT was 1:1, which gave a PCE of 16.6%. This value was superior to that of the spiro-OMeTAD device (16.1%) fabricated under the same conditions. The device using 37 with P3HT showed better hole mobility than that of a device with only P3HT as HTL. AFM measurements confirmed that the film containing both 37 and P3HT was smoother. The contact angle of the 37/P3HT composite was larger than that of P3HT alone, and the efficiency of the device with the 37/P3HT composite maintained 90% of its device efficiency even after 800 h at 75% humidity.
:P3HT was 1:1, which gave a PCE of 16.6%. This value was superior to that of the spiro-OMeTAD device (16.1%) fabricated under the same conditions. The device using 37 with P3HT showed better hole mobility than that of a device with only P3HT as HTL. AFM measurements confirmed that the film containing both 37 and P3HT was smoother. The contact angle of the 37/P3HT composite was larger than that of P3HT alone, and the efficiency of the device with the 37/P3HT composite maintained 90% of its device efficiency even after 800 h at 75% humidity.
2.4 Porphyrins and phthalocyanines at other places in perovskite solar cells
There are a few examples of using Pcs as additives in the HTLs of perovskite solar cells. Zhang et al. reported perovskite solar cells using the water-soluble Pc 38 (Fig. 2) as an additive in a poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) HTL of a perovskite solar cell.79 A PCE of 18.90% was obtained when 10 wt% 38 was added to PEDOT:PSS (Table 2). Wang et al. also reported the use of 38 with F4-TCNQ as a dopant for high device performance.80 Due to the strong electron affinity of F4-TCNQ, 38 was effectively p-doped, reducing the series resistance of the devices. As a result, the device employing 38:F4-TCNQ as the HTM exhibited a PCE of 16.14% in a p–i–n structure and 20.16% in an n–i–p structure, respectively. Interfacial engineering has also been reported in which porphyrin derivatives were used as a cathode buffer layer in perovskite solar cells. Liu et al. synthesized a porphyrin 39 that was soluble in alcohol through introduction of pyridinium acetylene at the meso position.81 By making the compound soluble in alcohol, it could be used as a substitute for bathocuproine (BCP), which has been widely used in solar devices. Compound 39 was used as a cathode buffer because it has a LUMO level equivalent to that of BCP and a HOMO level higher than that of PCBM according to electrochemical measurements. When the device was fabricated, VOC, JSC, and FF improved compared with those using BCP, and PCE increased from 15.6% to 17.5%.
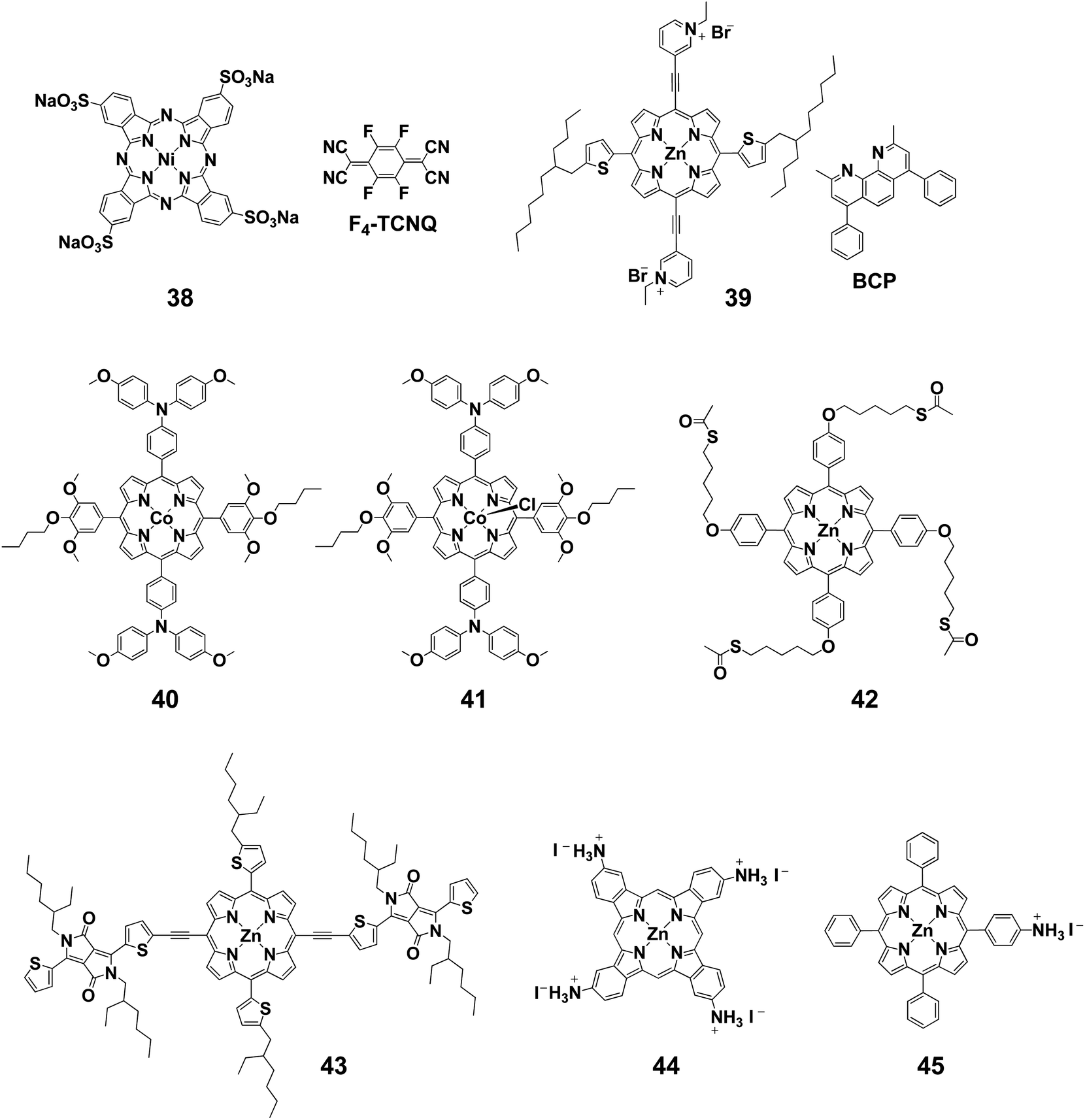 | ||
| Fig. 2 Structures of porphyrins and phthalocyanines for interface engineering. | ||
| Type | HTM, ETM hybrid, or active layer | VOC [V] | JSC [mA cm−2] | FF [%] | PCE [%] | Ref. |
|---|---|---|---|---|---|---|
| HTL | PEDOT:PSS + 38 (10 wt%) | 1.08 | 23.01 | 77.0 | 18.90 | 79 |
| HTL | 38 + F4-TCNQ (2.5 wt%) | 0.96 | 21.71 | 77.0 | 16.14 | 80 |
| HTL | Spiro-OMeTAD/38 + F4-TCNQ (2.5 wt%) | 1.12 | 24.32 | 74.0 | 20.16 | 80 |
| ETL | 39 | 1.07 | 21.15 | 77.75 | 17.5 | 81 |
| ETL | BCP | 1.02 | 19.98 | 76.0 | 15.6 | 81 |
| HTL | 40 + 41 (ETM, TiO2) | 1.12 | 23.61 | 74.45 | 19.61 | 82 |
| HTL | 40 + 41 (ETM, TiO2:sinapoyl malate) | 1.13 | 23.62 | 76.66 | 20.47 | 82 |
| HTL | PEDOT:PSS/42 | 0.93 | 21.90 | 69.0 | 14.05 | 83 |
| Hybrid | 43 | 0.93 | 18.19 | 56.3 | 9.52 | 84 |
| Hybrid | 43:PC61BM | 1.04 | 23.32 | 78.4 | 19.00 | 84 |
| Active layer | 33 | 1.02 | 22.60 | 75.0 | 17.30 | 85 |
| Active layer | 440.5 MAn−1PbnI3n+1 | 1.11 | 23.55 | 77.28 | 20.26 | 86 |
| Active layer | 45 + MAI + PdI2 | 1.09 | 22.64 | 73.66 | 18.26 | 87 |
Cao et al. synthesized Co(II)/(III) porphyrins (40 and 41) that have lower HOMO and LUMO levels than those of spiro-OMeTAD.82 They anticipated smooth charge carrier transfer by energy level alignment with the perovskite layer. When Co(II)/(III) porphyrin was used as an HTM instead of spiro-OMeTAD, the PCEs of 30 devices were slightly improved from 19.1 ± 1.0% to 19.7 ± 1.1%. The device using Co(II)/(III) porphyrin was thermally stable, and PCE was almost entirely maintained after annealing at 85 °C for 1000 h in a nitrogen atmosphere, whereas the PCE of the device using spiro-OMeTAD dropped to 20% of its initial value. After thermal annealing, cross-sectional SEM measurements showed that the crystal structure of perovskite was disrupted in the spiro-OMeTAD-based devices, but was maintained in the Co(II)/(III) porphyrin-based device.
The Huang group reported the use of porphyrin 42 between the PEDOT:PSS and perovskite layers in inverted perovskite solar cells.83 The HOMO levels of 42 are located between those of PEDOT:PSS and perovskite, so smooth hole transport can be expected. In fact, it contributed to the elimination of hysteresis and improvement of VOC, JSC, and FF, and PCE improved from 11.35% to 14.05%.
The group of Peng and Jen succeeded in reducing intermolecular recombination by using porphyrin derivative 43 and PC61BM as a bulk heterojunction (BHJ) layer on the perovskite layer in perovskite/BHJ hybrid solar cells.84 This BHJ layer works as an HTL in this hybrid system, enabling the absorption of long-wavelength light. The device performance of this system was higher than that of the porphyrin only system and the PCE remarkably improved from 9.38% to 18.3%, surpassing the spiro-OMeTAD-based device (17.1%). This was because JSC improved from 21.37 to 23.32 mA cm−2.
An example of using tetrakis(tert-butyl) compound 33 as an additive in the perovskite active layer has been reported by Wu et al.85 They showed that PCE increased from 15.3% to 17.3% by doping a small amount of 33 (4.4 × 10−3 mM) into the perovskite photoactive layer. SEM images of the perovskite layer showed pinholes in the films without the additive, which disappeared upon addition of 33. Pinholes are known to cause charge recombination by allowing contact between the perovskite and TiO2 layers respectively above and below the HTM later. Therefore, prevention of pinholes by the addition of 33 was effective in suppressing the charge recombination. It was also found that using 33 as an additive can reduce device hysteresis. Furthermore, the EQE and UV-Vis spectra showed that the efficiency of light absorption was also improved by the addition of 33. This was reflected by the JSC value (no addition: 20.0 mA cm−2; with addition: 22.6 mA cm−2).
Cao et al. used a Pc derivative bearing cationic ammonium groups (44) as a passivating layer to form a 2D perovskite.86 Coating this compound on the perovskite layer induced interaction between the perovskite grains to form a 2D structure. As a result, both PCE and stability improved. When combined with the above-mentioned Co(II)- and Co(III)-based porphyrin compounds,82 the PCE was initially higher than 20% and the initial efficiency remained at more than 90% of this value under 45% humidity at 85 °C for 1000 h. Furthermore, large-scale application was made possible by stabilizing the perovskite grains. A large-area device with mono-ammonium zinc porphyrin 45 with an effective area of 1.96 cm2 showed a PCE of 18.3%.87
3. Conclusion
This review summarized the application of porphyrin and Pcs in perovskite solar cells as HTMs, as additives in photoactive layers, and as interfacial layers for energy level engineering. For example, spiro-OMeTAD, a typical HTM can be replaced with porphyrin and Pc derivatives to improve the stability of perovskite solar cells. In some cases, the improved stability came from the hydrophobicity of the porphyrin and Pc layers even at high humidity and temperature. Advantages related to the face-on orientation of porphyrins and Pcs were realized by spin-coating on a perovskite layer. Some porphyrin and Pc derivatives have good solubility in organic solvents allowing solution processing on flexible substrates for flexible perovskite solar cells.In addition, porphyrins and Pcs have been used to construct hybrid organic and perovskite solar cells, where a BHJ containing porphyrins absorbs long-wavelength light and act as the HTL of the perovskite solar cells. Porphyrins and Pcs were also used to modify interfaces to obtain good morphology of the active layers and interfaces. For instance, porphyrins containing cationic parts can act as an interfacial material to form a 2D perovskite structure. We hope this review article is useful for understanding how porphyrins and phthalocyanines can be used in perovskite solar cells to improve device performance and stability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grants-in-Aid for Scientific Research (JSPS KAKENHI Grant Numbers JP19K22181 and 19K15669), MEXT, Japan. This work was also supported by JST SICORP Grant Number JPMJSC18H1, Japan.Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS.
- N. S. Lewis and G. Crabtree, Basic Research Needs for Solar Energy Utilization: report of the Basic Energy Sciences Workshop on Solar Energy Utilization, April 18–21, 2005, US Department of Energy, Office of Basic Energy Science, 2005 Search PubMed.
- D. J. Des Marais, Science, 2000, 289, 1703–1705 Search PubMed.
- H. Wang, B. Cobb, A. van Breemen, G. Gelinck and Z. Bao, Adv. Mater., 2014, 26, 4588–4593 CrossRef CAS.
- C. W. Tang and A. C. Albrecht, J. Chem. Phys., 1975, 62, 2139–2149 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Kay and M. Grätzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- A. Kay, R. Humphry-Baker and M. Grätzel, J. Phys. Chem., 1994, 98, 952–959 CrossRef CAS.
- X.-F. Wang and H. Tamiaki, Energy Environ. Sci., 2010, 3, 94–106 RSC.
- J.-J. Cid, M. García-Iglesias, J.-H. Yum, A. Forneli, J. Albero, E. Martínez-Ferrero, P. Vázquez, M. Grätzel, M. K. Nazeeruddin, E. Palomares and T. Torres, Chem.–Eur. J., 2009, 15, 5130–5137 CrossRef CAS.
- F. Silvestri, M. García-Iglesias, J.-H. Yum, P. Vázquez, M. Victoria Martínez-Díaz, M. Grätzel, M. K. Nazeeruddin and T. Torres, J. Porphyrins Phthalocyanines, 2009, 13, 369–375 CrossRef CAS.
- S. Eu, T. Katoh, T. Umeyama, Y. Matano and H. Imahori, Dalton Trans., 2008, 5476 RSC.
- D. Yang, R. Yang, K. Wang, C. Wu, X. Zhu, J. Feng, X. Ren, G. Fang, S. Priya and S. Liu, Nat. Commun., 2018, 9, 3239 CrossRef.
- M. Urbani, G. de la Torre, M. K. Nazeeruddin and T. Torres, Chem. Soc. Rev., 2019, 48, 2738–2766 RSC.
- G. Reddy, K. Devulapally, N. Islavath and L. Giribabu, Chem. Rec., 2019, 19, 2157–2177 CrossRef CAS.
- Z. Yu, A. Hagfeldt and L. Sun, Coord. Chem. Rev., 2020, 406, 213143 CrossRef CAS.
- J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 CrossRef.
- L. Bucher, N. Desbois, P. D. Harvey, G. D. Sharma and C. P. Gros, Sol. RRL, 2017, 1, 1700127 CrossRef.
- A. Mahmood, J.-Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, J. Mater. Chem. A, 2018, 6, 16769–16797 RSC.
- Y.-H. Chen, L.-Y. Lin, C.-W. Lu, F. Lin, Z.-Y. Huang, H.-W. Lin, P.-H. Wang, Y.-H. Liu, K.-T. Wong, J. Wen, D. J. Miller and S. B. Darling, J. Am. Chem. Soc., 2012, 134, 13616–13623 CrossRef CAS.
- S. Karpe, A. Cravino, P. Frère, M. Allain, G. Mabon and J. Roncali, Adv. Funct. Mater., 2007, 17, 1163–1171 CrossRef CAS.
- Y. Matsuo, Y. Sato, T. Niinomi, I. Soga, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 16048–16050 CrossRef CAS.
- C. Guo, L. Zhou and J. Lv, Polym. Polym. Compos., 2013, 21, 449–456 CAS.
- X. Huang, C. Zhu, S. Zhang, W. Li, Y. Guo, X. Zhan, Y. Liu and Z. Bo, Macromolecules, 2008, 41, 6895–6902 CrossRef CAS.
- J. Hatano, N. Obata, S. Yamaguchi, T. Yasuda and Y. Matsuo, J. Mater. Chem., 2012, 22, 19258 RSC; T. Yamamoto, J. Hatano, T. Nakagawa, S. Yamaguchi and Y. Matsuo, Appl. Phys. Lett., 2013, 102, 013305 CrossRef CAS; T. Nakagawa, J. Hatano and Y. Matsuo, J. Porphyrins Phthalocyanines, 2014, 18, 735–740 CrossRef; T. Sato, T. Nakagawa, H. Okada and Y. Matsuo, J. Porphyrins Phthalocyanines, 2015, 19, 451–458 CrossRef; T. Nakagawa, H. Wang, A. Zieleniewska, H. Okada, S. Aoyagi, D. M. Guldi and Y. Matsuo, Chem.–Asian J., 2018, 13, 3032–3039 CrossRef.
- L. Li, Y. Huang, J. Peng, Y. Cao and X. Peng, J. Mater. Chem. A, 2013, 1, 2144–2150 RSC.
- B. Walker, C. Kim and T.-Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS.
- S. Loser, H. Miyauchi, J. W. Hennek, J. Smith, C. Huang, A. Facchetti and T. J. Marks, Chem. Commun., 2012, 48, 8511 RSC.
- J. Huang, C. Zhan, X. Zhang, Y. Zhao, Z. Lu, H. Jia, B. Jiang, J. Ye, S. Zhang, A. Tang, Y. Liu, Q. Pei and J. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2033–2039 CrossRef CAS.
- Y. Lin, L. Ma, Y. Li, Y. Liu, D. Zhu and X. Zhan, Adv. Energy Mater., 2013, 3, 1166–1170 CrossRef CAS.
- T. Harschneck, N. Zhou, E. F. Manley, S. J. Lou, X. Yu, M. R. Butler, A. Timalsina, R. Turrisi, M. a. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Chem. Commun., 2014, 50, 4099 RSC.
- T. Bura, N. Leclerc, R. Bechara, P. Lévêque, T. Heiser and R. Ziessel, Adv. Energy Mater., 2013, 3, 1118–1124 CrossRef CAS.
- H. Gao, Y. Li, L. Wang, C. Ji, Y. Wang, W. Tian, X. Yang and L. Yin, Chem. Commun., 2014, 50, 10251 RSC.
- H. Qin, L. Li, F. Guo, S. Su, J. Peng, Y. Cao and X. Peng, Energy Environ. Sci., 2014, 7, 1397 RSC.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS.
- S. Arrechea, A. Aljarilla, P. de la Cruz, E. Palomares, G. D. Sharma and F. Langa, Nanoscale, 2016, 8, 17953–17962 RSC; G. Morán, S. Arrechea, P. de la Cruz, V. Cuesta, S. Biswas, E. Palomares, G. D. Sharma and F. Langa, J. Mater. Chem. A, 2016, 4, 11009–11022 RSC; M. Vartanian, R. Singhal, P. de la Cruz, G. D. Sharma and F. Langa, Chem. Commun., 2018, 54, 14144–14147 RSC.
- K. Ogumi, T. Nakagawa, H. Okada, R. Sakai, H. Wang and Y. Matsuo, J. Mater. Chem. A, 2017, 5, 23067–23077 RSC; K. Ogumi and Y. Matsuo, J. Porphyrins Phthalocyanines, 2019, 23, 1144–1148 CrossRef CAS.
- H. Wang, Q. Yue, T. Nakagawa, A. Zieleniewska, H. Okada, K. Ogumi, H. Ueno, D. M. Guldi, X. Zhu and Y. Matsuo, J. Mater. Chem. A, 2019, 7, 4072–4083 RSC.
- H. Wang, L. Xiao, L. Yan, S. Chen, X. Zhu, X. Peng, Xi. Wang, W.-K. Wong and W.-Y. Won, Chem. Sci., 2016, 7, 4301–4307 RSC; L. Xiao, S. Chen, K. Gao, X. Peng, F. Liu, Y. Cao, W.-Y. Wong, W.-K. Wong and X. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 30176–30183 CrossRef CAS.
- A. Zhang, C. Li, F. Yang, J. Zhang, Z. Wang, Z. Wei and W. Li, Angew. Chem., Int. Ed., 2017, 56, 2694–2698 CrossRef CAS.
- W. T. Hadmojo, D. Yim, H. Aqoma, D. Y. Ryu, T. J. Shin, H. W. Kim, E. Hwang, W.-D. Jang, I. H. Jung and S.-Y. Jang, Chem. Sci., 2017, 8, 5095–5100 RSC; Y. Guo, Y. Liu, Q. Zhu, C. Li, Y. Jin, Y. Puttisong, W. Chen, F. Liu, F. Zhang, W. Ma and W. Li, ACS Appl. Mater. Interfaces, 2018, 10, 32454–32461 CrossRef CAS.
- H.-H. Chou, Y.-H. Chiang, M.-H. Li, P.-S. Shen, H.-J. Wei, C.-L. Mai, P. Chen and C.-Y. Yeh, ACS Energy Lett., 2016, 1, 956–962 CrossRef CAS.
- Y.-H. Chiang, H.-H. Chou, W.-T. Cheng, Y.-R. Li, C.-Y. Yeh and P. Chen, ACS Energy Lett., 2018, 3, 1620–1626 CrossRef CAS.
- S. Chen, P. Liu, Y. Hua, Y. Li, L. Kloo, X. Wang, B. Ong, W.-K. Wong and X. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 13231–13239 CrossRef CAS.
- I. Jeon, H. Ueno, S. Seo, K. Aitola, R. Nishikubo, A. Saeki, H. Okada, G. Boschloo, S. Maruyama and Y. Matsuo, Angew. Chem., Int. Ed., 2018, 57, 4607–4611 CrossRef CAS.
- R. Azmi, U. H. Lee, F. T. A. Wibowo, S. H. Eom, S. C. Yoon, S. Y. Jang and I. H. Jung, ACS Appl. Mater. Interfaces, 2018, 10, 35404–35410 CrossRef CAS.
- U. H. Lee, R. Azmi, S. Sinaga, S. Hwang, S. H. Eom, T. W. Kim, S. C. Yoon, S. Y. Jang and I. H. Jung, ChemSusChem, 2017, 10, 3780–3787 CrossRef CAS.
- X. Lv, G. Xiao, X. Feng, J. Cao, X. Yao and J. Liu, Dyes Pigm., 2019, 160, 957–961 CrossRef CAS.
- G. Reddy, R. Katakam, K. Devulapally, L. A. Jones, E. Della Gaspera, H. M. Upadhyaya, N. Islavath and L. Giribabu, J. Mater. Chem. C, 2019, 7, 4702–4708 RSC.
- C. V. Kumar, G. Sfyri, D. Raptis, E. Stathatos and P. Lianos, RSC Adv., 2015, 5, 3786–3791 RSC.
- F. J. Ramos, M. Ince, M. Urbani, A. Abate, M. Grätzel, S. Ahmad, T. Torres and M. K. Nazeeruddin, Dalton Trans., 2015, 44, 10847–10851 RSC.
- G. Sfyri, N. Vamshikrishna, C. V. Kumar, L. Giribabu and P. Lianos, Sol. Energy, 2016, 140, 60–65 CrossRef CAS.
- E. Nouri, J. V. S. Krishna, C. V. Kumar, V. Dracopoulos, L. Giribabu, M. R. Mohammadi and P. Lianos, Electrochim. Acta, 2016, 222, 875–880 CrossRef CAS.
- P. Gao, K. T. Cho, A. Abate, G. Grancini, P. Y. Reddy, M. Srivasu, M. Adachi, A. Suzuki, K. Tsuchimoto, M. Grätzel and M. K. Nazeeruddin, Phys. Chem. Chem. Phys., 2016, 18, 27083–27089 RSC.
- K. T. Cho, K. Rakstys, M. Cavazzini, S. Orlandi, G. Pozzi and M. K. Nazeeruddin, Nano Energy, 2016, 30, 853–857 CrossRef CAS.
- S. Wu, Y. Zheng, Q. Liu, R. Li and T. Peng, RSC Adv., 2016, 6, 107723–107731 RSC.
- K. T. Cho, O. Trukhina, C. Roldan-Carmona, M. Ince, P. Gratia, G. Grancini, P. Gao, T. Marszalek, W. Pisula, P. Y. Reddy, T. Torres and M. K. Nazeeruddin, Adv. Energy Mater., 2017, 7, 1601733 CrossRef.
- X. Jiang, Z. Yu, H.-B. Li, Y. Zhao, J. Qu, J. Lai, W. Ma, D. Wang, X. Yang and L. Sun, J. Mater. Chem. A, 2017, 5, 17862–17866 RSC.
- X. Q. Jiang, Z. Yu, J. B. Lai, Y. C. Zhang, M. W. Hu, N. Lei, D. P. Wang, X. C. Yang and L. C. Sun, ChemSusChem, 2017, 10, 1838–1845 CrossRef CAS.
- I. Jeon, S. Seo, Y. Sato, C. Delacou, A. Anisimov, K. Suenaga, E. I. Kauppinen, S. Maruyama and Y. Matsuo, J. Phys. Chem. C, 2017, 121, 25743 CrossRef CAS.
- I. Jeon, R. Xiang, A. Shawky, Y. Matsuo and S. Maruyama, Adv. Energy Mater., 2019, 9, 1801312 CrossRef.
- L. Calio, J. Follana-Berna, S. Kazim, M. Madsen, H.-G. Rubahn, A. Sastre-Santos and S. Ahmad, Sustainable Energy Fuels, 2017, 1, 2071–2077 RSC.
- M. Cheng, Y. Li, M. Safdari, C. Chen, P. Liu, L. Kloo and L. Sun, Adv. Energy Mater., 2017, 7, 1602556 CrossRef.
- I. Jeon, J. Yoon, U. Kim, C. Lee, R. Xiang, A. Shawky, J. Xi, J. Byeon, H. M. Lee, M. Choi, S. Maruyama and Y. Matsuo, Adv. Energy Mater., 2019, 9, 1901204 CrossRef.
- G. Yang, Y.-L. Wang, J.-J. Xu, H.-W. Lei, C. Chen, H.-Q. Shan, X.-Y. Liu, Z.-X. Xu and G.-J. Fang, Nano Energy, 2017, 31, 322–330 CrossRef CAS.
- N. Ahn, I. Jeon, J. Yoon, E. I. Kauppinen, Y. Matsuo, S. Maruyama and M. Choi, J. Mater. Chem. A, 2018, 6, 1382–1389 RSC.
- I. Jeon, T. Chiba, C. Delacou, Y. Guo, A. Kaskela, O. Reynaud, E. I. Kauppinen, S. Maruyama and Y. Matsuo, Nano Lett., 2015, 15, 6665–6671 CrossRef CAS.
- X. Liu, Y. Wang, E. Rezaee, Q. Chen, Y. Feng, X. Sun, L. Dong, Q. Hu, C. Li and Z.-X. Xu, Sol. RRL, 2018, 2, 1800050 CrossRef.
- X. Zheng, Y. Wang, J. Hu, G. Yang, Z. Guo, J. Xia, Z. Xu and G. Fang, J. Mater. Chem. A, 2017, 5, 24416–24424 RSC.
- Q.-D. Dao, A. Fujii, R. Tsuji, Y. Takeoka and M. Ozaki, Org. Electron., 2017, 43, 156–161 CrossRef CAS.
- N. Torabi, A. Rahnamanic, H. Amrollahi, F. Mirjalili, M. A. Sadeghzade and A. Behjat, Org. Electron., 2017, 48, 211–216 CrossRef CAS.
- T. Lei, H. Dong, J. Xi, Y. Niu, J. Xu, F. Yuan, B. Jiao, W. Zhang, X. Hou and Z. Wu, Chem. Commun., 2018, 54, 6177–6180 RSC.
- T. Duong, J. Peng, D. Walter, J. Xiang, H. Shen, D. Chugh, M. Lockrey, D. Zhong, J. Li, K. Weber, T. P. White and K. R. Catchpole, ACS Energy Lett., 2018, 3, 2441–2448 CrossRef CAS.
- Y. C. Kim, T. Y. Yang, N. J. Jeon, J. Im, S. Jang, T. J. Shin, H. W. Shin, S. Kim, E. Lee, J. H. Noh, S. I. Seok and J. Seo, Energy Environ. Sci., 2017, 10, 2109–2116 RSC.
- J. Guo, X. Meng, H. Zhu, M. Sun, Y. Wang, W. Wang, M. Xing and F. Zhang, Org. Electron., 2019, 64, 71–78 CrossRef CAS.
- Q. Hu, E. Rezaee, Q. Dong, H. Shan, Q. Chen, L. Wang, B. Liu, J.-H. Pan and Z.-X. Xu, Sol. RRL, 2019, 3, 1800264 CrossRef.
- X.-F. Zhang, X. Zhou, L. Zhang and B. Xu, J. Mater. Chem. A, 2018, 6, 12515–12522 RSC.
- J.-M. Wang, Z.-K. Wang, M. Li, C.-C. Zhang, L.-L. Jiang, K.-H. Hu, Q.-Q. Ye and L.-S. Liao, Adv. Energy Mater., 2018, 8, 1701688 CrossRef.
- Y. Liu, J. Qi, X. Peng and Y. Cao, Org. Electron., 2018, 59, 414–418 CrossRef CAS.
- J. Cao, X. Lv, P. Zhang, T. T. Chuong, B. Wu, X. Feng, C. Shan, J. Liu and Y. Tang, Adv. Mater., 2018, 30, 1800568 CrossRef.
- B. Li, C. Zheng, H. Liu, J. Zhu, H. Zhang, D. Gao and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 27438–27443 CrossRef CAS.
- K. Gao, Z. Zhu, B. Xu, S. B. Jo, Y. Kan, X. Peng and A. K.-Y. Jen, Adv. Mater., 2017, 29, 1703980 CrossRef.
- S. Wu, Q. Liu, Y. Zheng, R. Li and T. Peng, J. Power Sources, 2017, 359, 303–310 CrossRef CAS.
- J. Cao, C. Li, X. Lv, X. Feng, R. Meng, Y. Wu and Y. Tang, J. Am. Chem. Soc., 2018, 140, 11577–11580 CrossRef CAS.
- C. Li, J. Yin, R. Chen, X. Lv, X. Feng, Y. Wu and J. Cao, J. Am. Chem. Soc., 2019, 141, 6345–6351 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |