
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free oxidative coupling of arylmethylamines with indoles: a simple, environmentally benign approach for the synthesis of 3,3′-bis(indolyl)methanes†
Vikas D. Kadu *a,
Sankala Naga Chandrudub,
Mahesh G. Hublikara,
Dattatraya G. Rauta and
Raghunath B. Bhosalea
*a,
Sankala Naga Chandrudub,
Mahesh G. Hublikara,
Dattatraya G. Rauta and
Raghunath B. Bhosalea
aSchool of Chemical Sciences, Punyashlok Ahilyadevi Holkar Solapur University, Solapur-413255, Maharashtra, India. E-mail: vikaskadu1@gmail.com
bDepartment of Chemistry, College of Engineering, Rayalseema University, Kurnool-518002, Andhra Pradesh, India
First published on 17th June 2020
Abstract
The efficient metal-free oxidative coupling of arylmethylamines with indoles has been developed using molecular oxygen as a green oxidant. The present reaction provides a novel route towards the synthesis of 3,3′-bis(indolyl)methanes in excellent yields of up to 95% via C–C and C–N bond formation. This attractive and environmentally friendly one-pot protocol is a simple procedure that features inexpensive acetic acid as the catalyst and molecular oxygen as the sole oxidant, and it supports a wide substrate scope with the good tolerance of functional groups.
Introduction
The indole scaffold is the most ubiquitous nitrogen heterocycle and is present in various natural products, alkaloids, hormones, neurotransmitters, pharmaceuticals and agrochemicals.1 This useful structural scaffold has been established as an essential component of heterocyclic compounds in pharmacology and medicinal chemistry.2 Typically, along with various indole derivatives, bis(indolyl)methanes have been extensively recognized as a privileged class due to their presence in natural products2 and their wide-ranging applications in pharmaceuticals.1,3 In the last few years, a large number of bis(indolyl)methanes (BIMs) have been referred to as extremely privileged structures due to their presence in various terrestrial and marine natural sources, where they exhibit a wide range of biological applications.4–7Biologically active bis(indolyl)methanes are depicted in Fig. 1. Simple 3,3′-bis(indolyl)methanes, such as arundine, have been used in cancer chemotherapy, while vibrindole A displays excellent antibacterial activity8 and arsindoline A shows excellent antitumor activity towards various cancer cell lines.9 Bis(indolyl)methanes are also efficacious at preventing cancer owing to their capability to modify established cancer-causing estrogen metabolites.10 Recently, the M. Shiri group have reviewed the cancer chemotherapy effectiveness and various biological activities of BIMs.11 Also, these compounds may normalize the anomalous cell growth that is related to cervical dysplasia.12,13 Therefore, their preparation has generated additional interest from synthetic organic chemists and biologists.
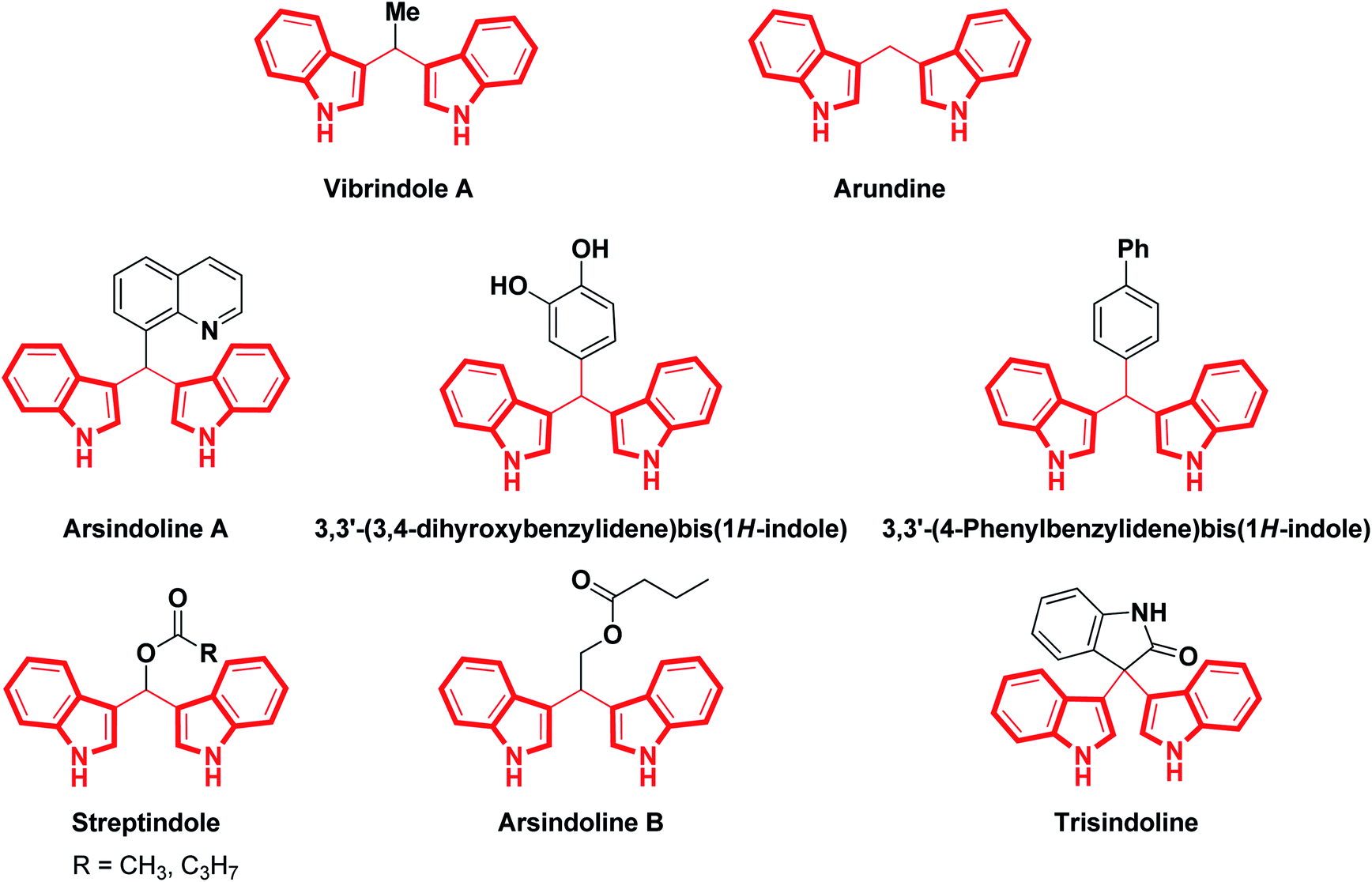 | ||
| Fig. 1 Some biologically active bis(indolyl)methanes. | ||
Traditionally, 3,3′-bis(indolyl)methanes have been synthesized via the condensation of aldehydes with indoles in the presence of diverse catalysts and strong acids.14–16 Alternatively, 3,3′-bis(indolyl)methanes can be synthesized from alkynes and indoles using ruthenium catalysts.17 Also, a primary alcohol can undergo transfer hydrogenation in the presence of precious metal complexes, followed by condensation with indoles to furnish BIMs.18–20 Then, Xiang et al. demonstrated the decarboxylative deamination of amino acids using an I2/PTA catalyst, and the Zhang research group described the utility of a tetramethylethylenediamine (TMEDA) carbon source in the presence of CuCl2 for BIM synthesis with indoles.21,22 Previously, the Sankala research group reported the synthesis of 3,3′-benzylidenebis(1H-indole), using an iron(II) triflate catalyst, from benzylamine with indoles.23 Furthermore, the platinum-catalyzed bisindolylation reaction of indoles and allenes has also led to the construction of 3,3′-bis(indolyl)methanes (Scheme 1).24
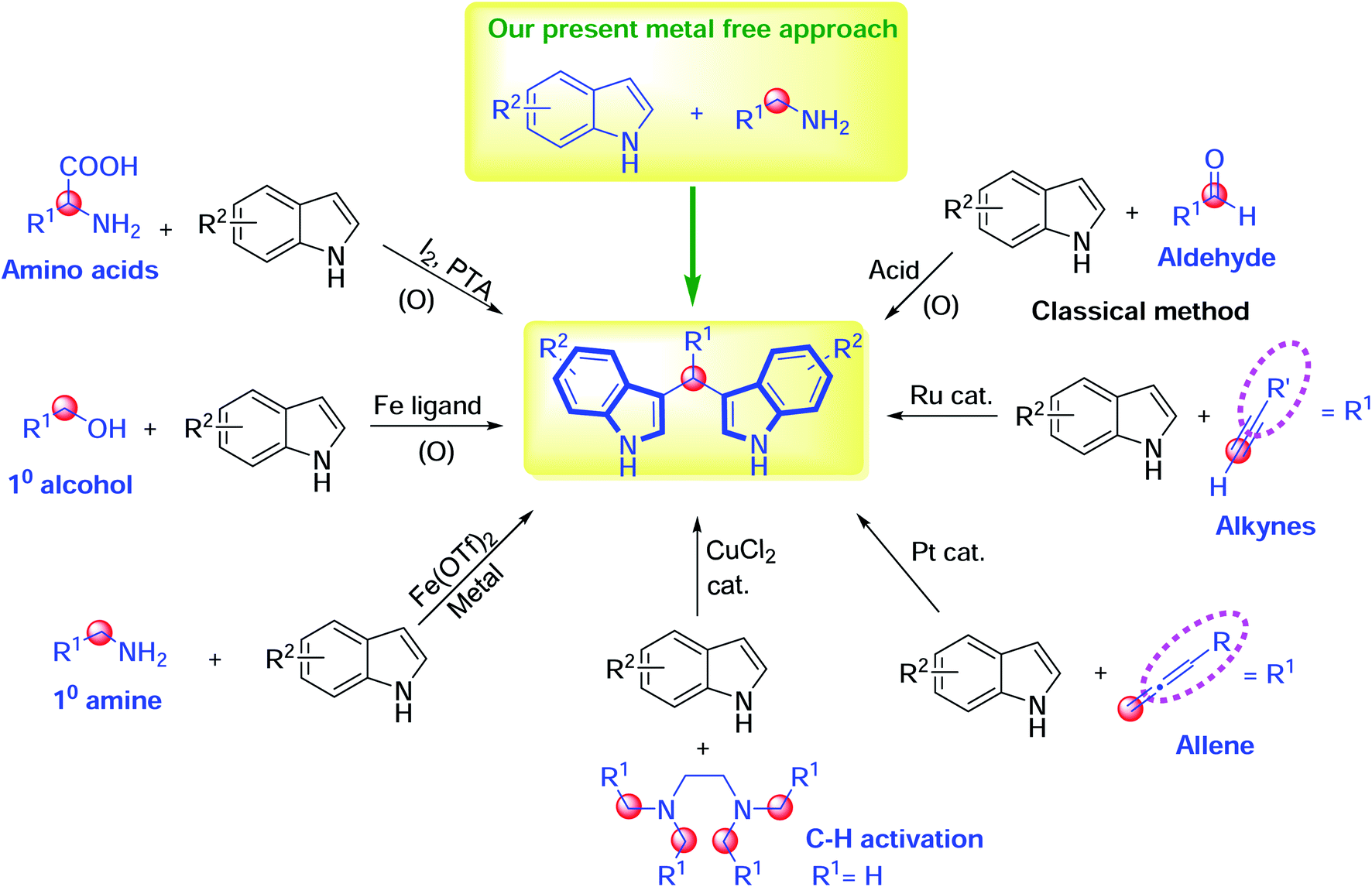 | ||
| Scheme 1 Approaches for the synthesis of 3,3′-bis(indolyl)methanes. | ||
Although these methods are effective for 3,3′-bis(indolyl)methane synthesis, they all have at least one shortcoming, such as the use of toxic or precious metals, high catalyst loadings, expensive ligands, volatile solvents, large amounts of promoters, and/or hydrophobic oxidants; long reaction times; lower yields; tedious work-up procedures; and the formation of byproducts under microwave and ultrasound conditions. Moreover, removing metal impurities from the desired products is a highly challenging task for chemists and biologists. In this context, the versatile applications of BIMs have attracted great demand for the development of a new modified method. Herein, the present protocol provides a simple, attractive and environmentally friendly one-pot approach for the synthesis of functionalized bis(indolyl)methanes with the good tolerance of a wide range of functional groups.
Results and discussion
We commenced our investigation with benzylamine 1a (1.1 mmol) and indole 2a (2 mmol) as model substrates to optimize the reaction parameters, as depicted in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (1.1 mmol), 2a (2.0 mmol), catalyst (30 mol%), solvent (1 mL), at 120 °C, under an O2 balloon unless otherwise mentioned.b Isolated yield.c 10 mol% of catalyst was used.d 5 mol% catalyst was used.e Under atmospheric air.f Under N2.g Reaction was carried out at 80 °C. | ||||
| 1 | TFA | Toluene | 12 | 62 |
| 2 | MsOH | Toluene | 12 | 50 |
| 3 | TfOH | Toluene | 12 | 35 |
| 4 | AcOH | Toluene | 12 | 71 |
| 5 | PivOH | Toluene | 12 | 65 |
| 6 | AcOH | Ethylene glycol | 12 | 25 |
| 7 | AcOH | DMF | 12 | 31 |
| 8 | AcOH | Chlorobenzene | 9 | 92 |
| 9 | AcOH | — | 9 | 75 |
| 10c | AcOH | Chlorobenzene | 9 | 92 |
| 11d | AcOH | Chlorobenzene | 9 | 79 |
| 12c,e | AcOH | Chlorobenzene | 9 | 81 |
| 13c,f | AcOH | Chlorobenzene | 12 | 35 |
| 14c,g | AcOH | Chlorobenzene | 12 | 76 |
Initially, a series of toluene-mediated acid systems was tested. For this, benzylamine 1a (1.1 mmol) and indole 2a (2 mmol) were studied in the presence of trifluoroacetic acid (TFA) (30 mol%) and toluene (1 mL) at 120 °C for 12 h under an O2 atmosphere; this gratifyingly afforded the 3,3′-bis(indolyl)methane 3aa in 62% yield (Table 1, entry 1). Then, reaction optimization was carried out with different acids, such as MsOH, TfOH, AcOH and PivOH, under similar reaction conditions; the desired product 3aa was obtained and yields are reported in Table 1 (entries 2–5). All the toluene/acid systems promoted 3,3′-bis(indolyl)methane 3aa formation, and the AcOH/toluene combination was observed to be most effective (yield of 3aa: 71%; entry 4). For further yield improvements, the reaction conditions were optimized using the polar solvents ethylene glycol and DMF in AcOH (30 mol%) at 120 °C, but 3,3′-bis(indolyl)methane 3aa was procured in low yields of 25% and 31%, respectively (Table 1, entries 6 and 7). Then, the transformation was accelerated in the non-polar solvent chlorobenzene and, significantly, the anticipated product 3,3′-bis(indolyl)methane 3aa was produced in 91% yield (Table 1, entry 8). After that, neat reaction conditions were tested, and the yield of product 3aa shifted from 91% to 75% (Table 1, entry 9). Subsequently, we attempted various catalyst loading levels from 30 mol% to 10 mol% to further optimize the reaction conditions (Table 1, entries 10–11). A lesser amount of AcOH catalyst (10 mol%) secured the same yield (92%) of product 3aa. However, further lowering the amount of AcOH catalyst (5 mol%) reduced the yield of product 3aa from 92% to 79%. Also, the reaction was executed under atmospheric air, and this furnished 3aa in 81% yield (Table 1, entry 12). Next, the reaction was performed under a nitrogen atmosphere, and this afforded a 35% yield of the desired product 3aa (Table 1, entry 13). After that, a decrease in the reaction temperature from 120 °C to 80 °C was also studied, and this resulted in a lower yield of 3aa (Table 1, entry 14). Overall, the optimal conditions were identified as: 1a: 1.1 mmol; 2a: 2.0 mmol; AcOH: 10 mol%; toluene: 1 mL; 120 °C; and under an O2 atmosphere.
With the optimized conditions in hand, we investigated the scope of amines as coupling partners (Table 2). To our delight, a wide range of amines (1a–1r), with electron-donating and electron-withdrawing groups, furnished the desired products (3aa–3ra) without any difficulties under the optimized reaction conditions. Moreover, a variety of functional groups, such as methoxy, methyl, halogen, trifluoromethyl, naphthyl and thiophene, could be significantly maintained under the optimized reaction conditions. Electronic factors slightly influenced the yields of the desired products. Benzylamines with electron-donating groups at the para position (1b–1e) furnished the corresponding 3,3′-BIMs in slightly higher yields than benzylamines with electron-withdrawing groups (1f–1g). Furthermore, benzylamines with halogen substituents also afforded the desired products in good to moderate yields. Sterically hindered benzylamines (1i–1n), such as those with 2-chloro, 2-methoxy, 4-chloro-2-fluoro, 2-chloro-4-fluoro, and 2,3-dichloro substituents on the benzene rings, also imparted the corresponding 3,3′-BIMs with up to 85–75% yields (3ia–3ma,Table 2).

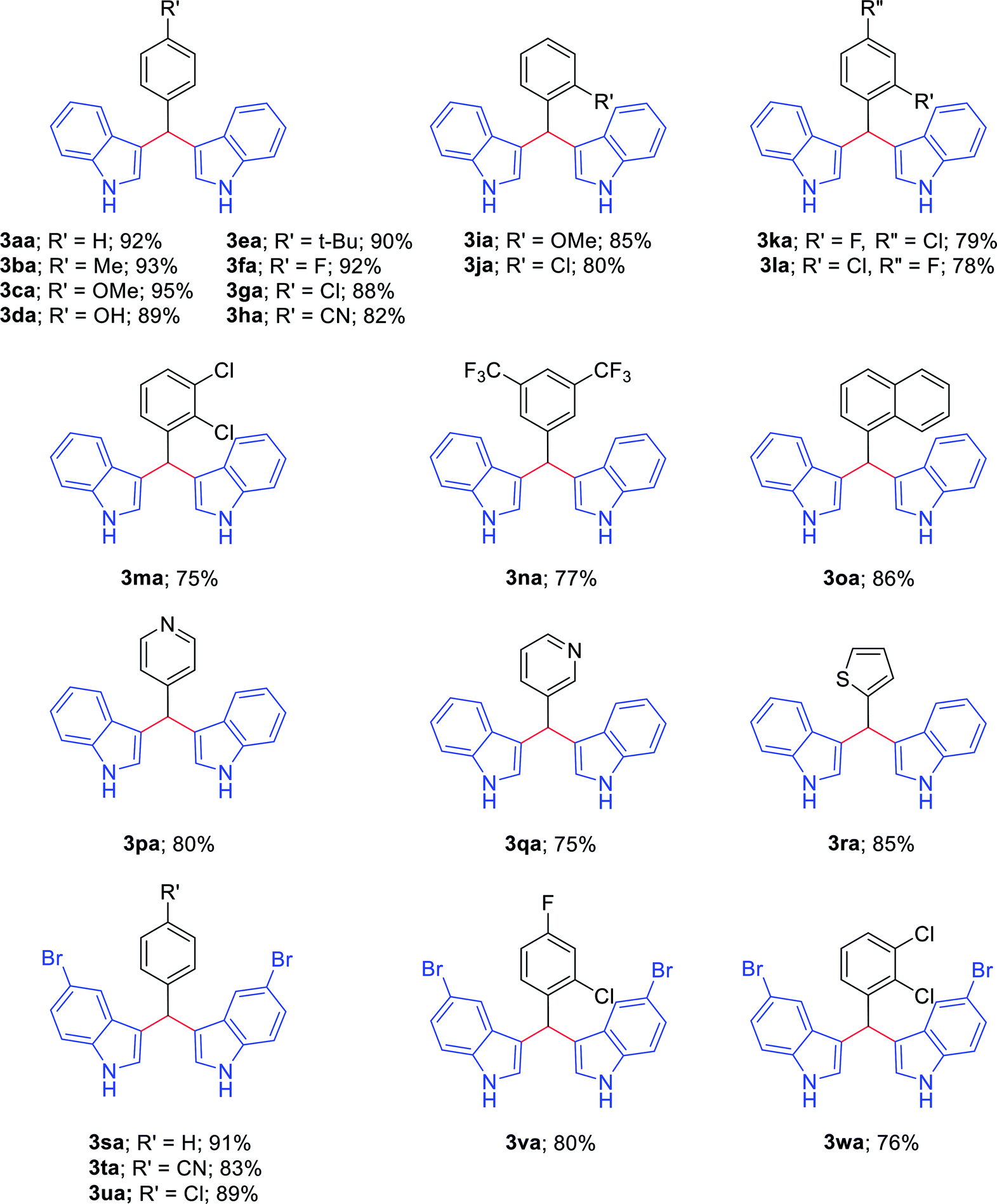
Excitingly, having a strong electron-withdrawing group at the meta position of 3,5-bis(trifluoromethyl)benzylamine (1n) also resulted in 3,3′-[3,5-bis(trifluoromethyl)-benzylidene]bis(1H-indole) (3na) in good yield.
To expand the scope of the reaction, heteroaryl methylamines, such as 4-picolylamine, 3-picolylamine and 2-thiophene-methylamine, were further investigated, and the desired products (3pa–3ra) could be obtained with notable yields of 80%, 75%, and 85%, respectively. In addition, 5-bromo indole (2b) tolerated various substituted arylmethylamines well and furnished 3,3′-BIMs (3sa–3wa) in good to high yields of 91%, 83%, 89%, 80%, and 76%, respectively.
To gain more insight into the reaction mechanism, we conducted some control experiments (Scheme 2). Initially, N-4-methyl-N-(4-methylbenzylidene)benzylamine was prepared from 4-methylbenzylamine at 120 °C in chlorobenzene under a molecular oxygen atmosphere using acetic acid (10 mol%) (I). Afterwards, this intermediate was treated with two equivalents of indole, furnishing a bis(indolyl)methane (II) (Scheme 2); this was also achieved using 1.5 mmol of benzylamine and 2.0 mmol of indole. From previous literature reports and the above control experiments, we propose a tentative reaction mechanism for the synthesis of a bis(indolyl)methane from benzylamine 1a and indole 2a under metal-free conditions (Scheme 3). At first, an oxidative process involving benzylamine 1a results in the formation of the corresponding aldimine 8. Then, this aldimine 8 undergoes further transamination with another molecule of amine to furnish the corresponding homo-coupled product N-benzylidinebenzylamine 5. In the next step, indole 2a undergoes a Mannich reaction with N-benzylidinebenzylamine 5 to afford the C-3 alkylated intermediate 9, which undergoes further oxidation resulting in the formation of the azafulvene intermediate 10. In the final step, azafulvene 10 reacts with another molecule of indole 2a, resulting in the formation of the desired product 3,3′-bis(indolyl)methane 3aa. For testing the practicability of the reaction on a large scale, the reaction of benzylamine (1a, 19.5 mmol) and indole (2a, 30 mmol) was investigated, and it afforded 4.59 g of 3aa in 91% yield, showing no major loss of efficiency. As a result, this protocol has utility as a practical process for preparing bis(indolyl)methanes.
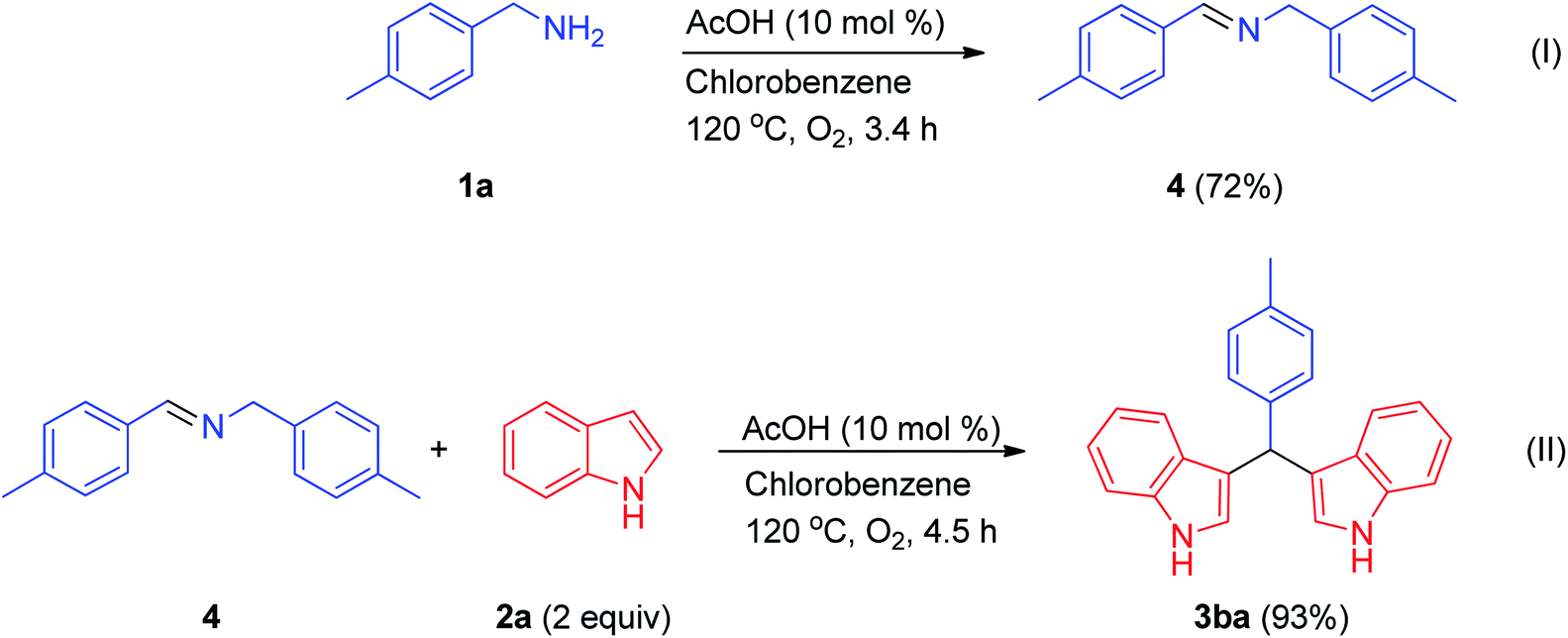 | ||
| Scheme 2 Control experiments. | ||
 | ||
| Scheme 3 The proposed reaction mechanism. | ||
Additionally, we studied the ability of our methodology to synthesize some biologically active bis(indolyl)methanes, such as 3,3′-(4-methoxybenzylidene)bis(1H-indole) (3ca), 3,3′-(4-hydroxybenzylidene)bis(1H-indole) (3da), and 3,3′-(4-tert-butylbenzylidene)bis(1H-indole) (3ea) (Table 2). The compounds 3ca and 3da induce nuclear NR4A1-dependent apoptosis and inhibit the growth of colon, bladder, lung and pancreatic cancer cells25–27 through the activation or deactivation of receptors. Compound 3ea inhibits the proliferation of the invasive estrogen receptor-negative MDA-MB-231 and MDA-MB-453 human breast cancer cell lines.28
Conclusions
In summary, we have developed a simple AcOH-catalyzed novel metal-free oxidative coupling reaction between benzylamines and indoles to synthesize a diverse range of functionalized bis(indolyl)methanes. The synthesis of bis(indolyl)methanes involves aerobic oxidative addition, cleavage, and sequential coupling in a one-pot procedure using green molecular oxygen as the oxidant, making the transformation very economical and environmentally benign.Experimental section
General information
All reactions were carried out in oven-dried glassware using dry solvents under a molecular oxygen atmosphere, unless stated otherwise. Iron salts were purchased from Sigma-Aldrich and were used as received. All other chemicals were used as received from commercial sources. Reactions were monitored via TLC on 0.25 mm Merck silica gel plates (60 F254) using UV light for visualization. Column chromatography purification was performed using silica gel (100–200 mesh). Melting points were measured using Büchi melting point apparatus and are uncorrected. IR spectra were recorded using a Spectrum FT-IR spectrophotometer. NMR spectra were recorded using a Bruker 400 MHz spectrometer (1H: 400 MHz, 13C: 100 MHz), using DMSO-d6 or CDCl3 as the solvent with TMS as the internal standard at room temperature. Mass spectra were recorded using a 6530 Accurate-Mass Q-TOF LC/MS (Agilent Technologies).General procedure for the synthesis of bis(indolyl)methanes (3)
Benzylamine 1 (1.1 mmol), indole 2 (2.0 mmol), AcOH (10 mol%), and dry chlorobenzene (2 mL) were added to a 25 mL round bottom flask. The round bottom flask was equipped with an O2 balloon, and the reaction mixture was stirred at 110 °C until the complete consumption of indole 2 occurred, as monitored via TLC. After the reaction was finished, the reaction mixture was cooled to room temperature, diluted with CH2Cl2 (10 mL), and washed with water (2 × 10 mL). The organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified via silica gel column chromatography using a hexane/ethyl acetate mixture to afford the corresponding bis(indolyl)methane products 3.Characterization data from the pure bis(indolyl)methanes products
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture as the eluent to afford 4-methyl-N-(4-methylbenzylidene)benzylamine (4) (232 mg, 72% yield) as a pale yellow solid.
:1) mixture as the eluent to afford 4-methyl-N-(4-methylbenzylidene)benzylamine (4) (232 mg, 72% yield) as a pale yellow solid.IR (cm−1): 3022, 2923, 2853, 1646, 1514, 1174, 1021, 811, 711; 1H NMR (400 MHz, CDCl3): δ 8.24 (s, 1H), 7.07–7.26 (m, 8H, ArH), 4.5 (d, J = 5.6 Hz, 2H), 2.35 (s, 3H), 2.34 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 160.9, 137.5, 134.5, 129.6, 129.5, 129.3, 129.2, 127.8, 126.9, 41.97, 21.1 ppm; 13C NMR DEPT-135 (100 MHz, CDCl3): 160.9 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, up), 129.6, 129.5, 127.8, 126.9, 41.9 (CH2, down), 21.1 (2 × CH3, up) ppm.
CH, up), 129.6, 129.5, 127.8, 126.9, 41.9 (CH2, down), 21.1 (2 × CH3, up) ppm.
CH, sp2), 40.2 (up, CH, sp3) ppm.Conflicts of interest
There are no conflicts to declare.References
- R. Tjalkens and S. Safe, US Pat., 8580843, 2013.
- S. A. Morris and R. J. Andersen, Tetrahedron, 1990, 46, 715–720 CrossRef CAS.
- A. Ramirez, S. Garcia-Rubio, A. Ramírez and S. García-Rubio, Curr. Med. Chem., 2003, 10, 1891–1915 CrossRef CAS PubMed.
- J. K. Porter, C. W. Bacon, J. D. Robbins, D. S. Himmelsbach and H. C. Higman, J. Agric. Food Chem., 1977, 25, 88–93 CrossRef CAS.
- T. R. Garbe, M. Kobayashi, N. Shimizu, N. Takesue, M. Ozawa and H. Yukawa, J. Nat. Prod., 2000, 63, 596–598 CrossRef CAS PubMed.
- K.-B. Oh, W. Mar, S. Kim, J.-Y. Kim, T.-H. Lee, J.-G. Kim, D. Shin, C. J. Sim and J. Shin, Biol. Pharm. Bull., 2006, 29, 570–573 CrossRef CAS PubMed.
- E. Fahy, B. C. M. M. Potts, D. J. Faulkner and K. Smith, J. Nat. Prod., 1991, 54, 564–569 CrossRef CAS.
- R. Bell, S. Carmeli and N. Sar, J. Nat. Prod., 1994, 57, 1587–1590 CrossRef CAS PubMed.
- S. X. Cai, D. H. Li, T. J. Zhu, F. P. Wang, X. Xiao and Q. Q. Gu, Helv. Chim. Acta, 2010, 93, 791–795 CrossRef CAS.
- M. A. Zelgis, J. Med. Food, 1998, 1, 67–82 CrossRef.
- M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed.
- C. Thomas, US Pat., 4500689, 1985.
- M. C. Bell, P. Crowley-Nowick, H. L. Bradlow, D. W. Sepkovic, D. Schmidt-Grimminger, P. Howell, E. J. Mayeaux, A. Tucker, E. A. Turbat-Herrera and J. M. Mathis, Gynecol. Oncol., 2000, 78, 123–129 CrossRef CAS PubMed.
- V. D. Kadu, D. N. Nadimetla, M. G. Hublikar, D. G. Raut and R. B. Bhosale, Lett. Org. Chem., 2020, 17, 61–67 CrossRef CAS.
- R. M. N. Kalla, J. V. John, H. Park and I. Kim, Catal. Commun., 2014, 57, 55–59 CrossRef CAS.
- S. A. R. Mulla, A. Sudalai, M. Y. Pathan, S. A. Siddique, S. M. Inamdar, S. S. Chavan and R. S. Reddy, RSC Adv., 2012, 2, 3525 RSC.
- D. Xia, Y. Wang, Z. Du, Q. Y. Zheng and C. Wang, Org. Lett., 2012, 14, 588–591 CrossRef CAS PubMed.
- S. Zhang, W. Fan, H. Qu, C. Xiao, N. Wang, L. Shu, Q. Hu and L. Liu, Curr. Org. Chem., 2012, 16, 942–948 CrossRef CAS.
- A. E. Putra, K. Takigawa, H. Tanaka, Y. Ito, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2013, 6344–6354 CrossRef CAS.
- T. Nobuta, A. Fujiya, N. Tada, T. Miura and A. Itoh, Synlett, 2012, 23, 2975–2979 CrossRef CAS.
- J. Xiang, J. Wang, M. Wang, X. Meng and A. Wu, Org. Biomol. Chem., 2015, 13, 4240–4247 RSC.
- L. Zhang, C. Peng, D. Zhao, Y. Wang, H. J. Fu, Q. Shen and J. X. Li, Chem. Commun., 2012, 48, 5928–5930 RSC.
- K. Gopalaiah, S. N. Chandrudu and A. Devi, Synthesis, 2015, 47, 1766–1774 CrossRef CAS.
- M. P. Muñoz, M. C. De La Torre and M. A. Sierra, Chem.–Eur. J., 2012, 18, 4499–4504 CrossRef PubMed.
- D. C. Sung, K. Yoon, S. Chintharlapalli, M. Abdelrahim, P. Lei, S. Hamilton, S. Khan, S. K. Ramaiah and S. Safe, Cancer Res., 2007, 67, 674–683 CrossRef PubMed.
- S. Dae Cho, S.-O. Lee, S. Chintharlapalli, M. Abdelrahim, S. Khan, K. Yoon, A. M. Kamat and S. Safe, Mol. Pharmacol., 2010, 77, 396–404 CrossRef PubMed.
- S.-O. Lee, X. Li, E. Hedrick, U.-H. Jin, R. B. Tjalkens, D. S. Backos, L. Li, Y. Zhang, Q. Wu and S. Safe, Mol. Endocrinol., 2014, 28, 1729–1739 CrossRef PubMed.
- K. Vanderlaag, Y. Su, A. E. Frankel, R. C. Burghardt, R. Barhoumi, G. Chadalapaka, I. Jutooru and S. Safe, BMC Cancer, 2010, 10, 669 CrossRef CAS PubMed.
- B. P. Bandgar, A. V. Patil and V. T. Kamble, ARKIVOC, 2007, 252–259 Search PubMed.
- B. S. Liao, J. T. Chen and S. T. Liu, Synthesis, 2007, 2007, 3125–3128 CrossRef.
- R. Ghorbani-Vaghei and S. M. Malaekehpoor, Org. Prep. Proced. Int., 2010, 42, 175–182 CrossRef CAS.
- A. Zare, A. Parhami, A. R. Moosavi-Zare, A. Hasaninejad, A. Khalafi-Nezhad and M. H. Beyzavi, Can. J. Chem., 2009, 87, 416–421 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03221b |
| This journal is © The Royal Society of Chemistry 2020 |