
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structure, and antitumor activity of 2,9-disubstituted perhydro 2,3a,7b,9,10a,14b-hexaazadibenzotetracenes†
Elena B. Rakhimova *,
Victor Yu. Kirsanov,
Elena V. Tret'yakova,
Leonard M. Khalilov,
Askhat G. Ibragimov,
Lilya U. Dzhemileva*,
Vladimir A. D'yakonov and
Usein M. Dzhemilev
*,
Victor Yu. Kirsanov,
Elena V. Tret'yakova,
Leonard M. Khalilov,
Askhat G. Ibragimov,
Lilya U. Dzhemileva*,
Vladimir A. D'yakonov and
Usein M. Dzhemilev
Institute of Petrochemistry and Catalysis, Russian Academy of Sciences, 141 Prospekt Oktyabrya, 450075 Ufa, Russian Federation. E-mail: rakhimovaelena@mail.ru; dzhemilev@mail.ru
First published on 4th June 2020
Abstract
Catalytic methods for the synthesis of previously unknown 2,9-disubstituted 3bR*,7aR*,10bR*,14aR*-cis-14c,14d-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzotetracenes have been developed. The structures were established by 1D (1H, 13C) and 2D (COSY, HSQC, HMBC) NMR spectroscopy, MALDI TOF/TOF mass spectrometry, and X-ray diffraction analysis. Primary screening of the synthesized perhydro hexaazadibenzotetracenes for antitumor activity was carried out.
Introduction
World literature describes methods for the synthesis of N,N′-disubstituted trans-1,2-diaminocyclohexane derivatives1–4 and their cyclic adducts,5–7 including poly-8 and macroheterocycles.9,10 Compounds containing the trans-1,2-diaminocyclohexane moiety in the molecule often exhibit antitumor activities11–14 and antiproliferative properties15 and are also of interest as N-containing chiral ligands,16 bifunctional organocatalysts,17 and catalyst systems for enantioselective transformations.18,19Recently,20 we demonstrated the possibility of one-pot synthesis of (3bS*,7aR*,10bR*,14aS*)-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes involving the catalytic reaction of cis-1,6,7,12-tetraazaperhydrotetracene with cycloaminomethylating reagents. As further development of research in the field of synthesis of annulated polyazapolycycles20–23 and to design an efficient synthetic route to compounds with potential biological activities, we attempted to prepare perhydro 2,3a,7b,9,10a,14b-hexaazadibenzotetracenes from trans-1,6,7,12-tetraazaperhydrotetracene.
Results and discussion
As the starting substrate, we chose trans-1,6,7,12-tetraazaperhydrotetracene, which was obtained in situ from (±)-trans-1,2-diaminocyclohexane. Unlike acyclic aliphatic diamines, diaminocyclohexanes have fairly rigid configurations. In trans-1,2-diaminocyclohexane, both amino groups are located in equatorial positions and the H2N–C–C–NH2 dihedral angle is close to 60°; therefore, trans-1,2-diaminocyclohexane behaves as a nearly planar building block.10 Preliminary experiments demonstrated that, without a catalyst, N,N-bis(methoxymethyl)-N-propylamine, obtained in situ,24 reacts with trans-1,6,7,12-tetraazaperhydrotetracene (1) at 60 °C to give 2,9-dipropyl-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracene (2) in ∼20% yield. In order to increase the yield of the target heterocycle 2, the reaction of bis(methoxymethyl)propylamine with 1,6,7,12-tetraazaperhydrotetracene was conducted in the presence of 5 mol% SmCl3·6H2O, which was successfully used in our previous studies for heterocyclization reactions.20,22 The heterocyclization of trans-1,6,7,12-tetraazaperhydrotetracene with N,N-bis(methoxymethyl)-N-propylamine in the presence of this catalyst furnished (3bR*,7aR*,10bR*,14aR*)-2,9-dipropyl-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracene (2) in 72% yield. Similar results were obtained when the n-propyl residue at the nitrogen atom was replaced by other alkyl groups (iso-Pr, n-Bu, tert-Bu). The intermolecular heterocyclization of trans-1,6,7,12-tetraazaperhydrotetracene with N,N-bis(methoxymethyl)-N-alkylamines under the chosen conditions [5 mol% SmCl3·6H2O, MeOH, 20 °C, 3 h] resulted in the selective formation of (3bR*,7aR*,10bR*,14aR*)-2,9-dialkyl-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes (3–5) in 68–75% yields (Scheme 1). | ||
| Scheme 1 Intermolecular heterocyclization of N,N-bis(methoxymethyl)-N-alkylamines with trans-1,6,7,12-tetraazaperhydrotetracene. | ||
The 1H NMR spectra of compounds 2–5 show four doublets for the AB spin system at 2.31–4.39 ppm with geminal spin–spin coupling constants of 8.5 and 12.0 Hz, corresponding to the methylene protons at carbons in positions H-1,8 and H-3,10, respectively. In the 13C NMR spectra, nine cage signals exhibit pair resonances, since compounds 2–5 are centrosymmetric structures. It is of interest that, because of the steric compression of the C-3,4 and C-10,11 carbon atoms, the signals of the C3,10 and C4,11 atoms in the 13C NMR spectra are located in a higher field, at δ 64–69 ppm and δ 24 ppm, respectively. Meanwhile, the loosely positioned C-7,8 and C-1,14 atoms are characterized by downfield shifts of the signals, δ C1,8 = 69–75 ppm and δ C7,14 = 28 ppm. Similarly, the carbon signals δ C3b,10b = 55 ppm are located in a higher field than the carbon signals δ C7a,14a = 64 ppm. Note that (3bS*,7aR*,10bR*,14aS*)-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes earlier obtained from (±)-cis-1,2-diaminocyclohexane20 do not exhibit spectral features of this type. The signals in the 1H and 13C NMR spectra of (3bR*,7aR*,10bR*,14aR*)-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes were assigned using 2D homo- (COSY) and heteronuclear (HSQC, HMBC) NMR experiments. The structures proposed for compounds 2–5 are confirmed by molecular ion peaks present in positive ion MALDI TOF/TOF mass spectra.
In recent years, 1,3,5-trisubstituted 1,3,5-triazinanes have been actively used as effective reagents in both aminomethylation25–27 and cycloaddition28–32 reactions. Therefore, in our subsequent experiments, we attempted to perform selective synthesis of 2,9-dicycloalkyl-substituted perhydro 2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes by the catalytic reaction between trans-1,6,7,12-tetraazaperhydrotetracene (1) and 1,3,5-tricycloalkyl-1,3,5-triazinanes. It was found that under the optimal conditions [5 mol% NiCl2·6H2O, MeOH, 20 °C, 3 h], 1,3,5-tricycloalkyl-1,3,5-triazinanes selectively react with trans-1,6,7,12-tetraazaperhydrotetracene 1 giving (3bR*,7aR*,10bR*,14aR*)-2,9-dicycloalkyl-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes (6–11) in 66–85% yields (Scheme 2). Note that, owing to the presence of two chiral centers at C-1′ and C-1′′, the 1H and 13C NMR spectra of 11 show the presence of a diastereomeric pair and diastereomeric splitting33,34 of signals of both the norbornane moieties and the hexaazaperhydrodibenzotetracene cage.
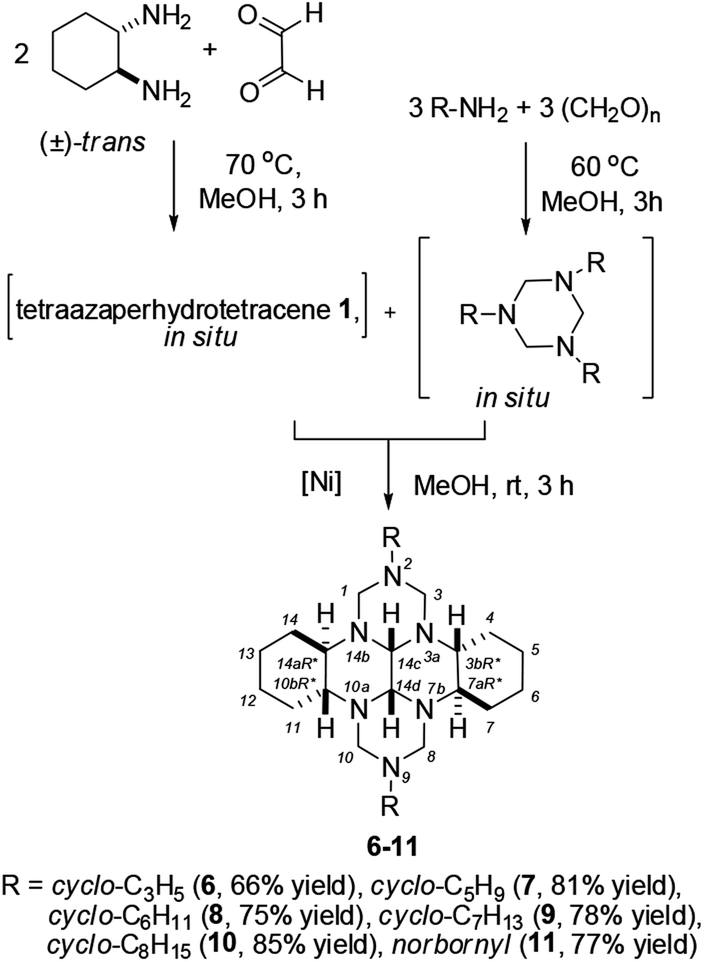 | ||
| Scheme 2 Recyclization of 1,3,5-tricycloalkyl-1,3,5-triazinanes with trans-1,6,7,12-tetraazaperhydrotetracene. | ||
Recrystallization of compound 9 from chloroform yielded transparent lamellar crystals. The crystals have a triclinic crystal lattice (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The independent part of the unit cell includes a molecule of 9 and two chloroform molecules (Fig. 1).
). The independent part of the unit cell includes a molecule of 9 and two chloroform molecules (Fig. 1).
 | ||
| Fig. 1 The independent part of the unit cell of compound 9. | ||
Non-hydrogen atoms are represented by thermal vibration ellipsoids (p = 30%).
The molecule of 9 is a fused polyazapolycyclic system, with cycloheptane substituents being attached to the N2 and N9 ring nitrogen atoms. The cycloheptane rings have the chair conformation and occupy equatorial positions relative to the hexaazaperhydrodibenzotetracene cage. The six-membered carbo and aza rings in the hexaazaperhydrodibenzotetracene moiety of 9 also take the chair conformation, but have a different type of ring coupling as compared with the previously studied20 compound containing a similar polyazapolycyclic cage. Indeed, in compound 9, the piperazine rings are cis-coupled, which is confirmed by the size of the H14c–C14c–C14d–H14d torsion angle of 54.2(5)°. The piperazine and cyclohexane rings are trans-coupled, with the H3b–C3b–C7a–H7a and H10b–C10b–C14a–H14a torsion angles being 178.8(4)° and 176.0(3)°, respectively. The conformational structures of the polyazapolycyclic moiety of compound 9 established by X-ray diffraction experiments and by proton–proton correlation 2D NMR spectra completely coincide.
The broad range of biological activities of heterocyclic molecules with the adamantane cage35 attracts attention of researchers and stimulates development of new efficient synthetic routes to cage compounds containing various heterocyclic moieties. Therefore, our subsequent experiments were directed towards development of a selective synthesis of 2,9-diadamantyl-substituted perhydro 2,3a,7b,9,10a,14b-hexaazadibenzotetracenes. As previously,22 because of the bulky adamantyl substituent, we were unable to prepare N,N-bis(methoxymethyl)-N-adamantylamines or 1,3,5-triadamantyl-1,3,5-triazinanes as the starting aminomethylating reagents. Therefore, we attempted to conduct a one-pot catalytic cyclocondensation of adamantylamines with formaldehyde and trans-1,6,7,12-tetraazaperhydrotetracene (1) to prepare previously undescribed 2,9-diadamantyl-substituted perhydro 2,3a,7b,9,10a,14b-hexaazadibenzotetracenes. The reaction was catalyzed by granulated highly crystalline zeolite Y36 with high phase purity in the H form, which is known to be most active in reactions of this type.22 In the presence of 10 wt% zeolite Ymmm as a catalyst, the one-pot multicomponent condensation of adamantylamines (adamantyl-1-amine, adamantyl-2-amine, 1-hydroxyadamantyl-3-amine) with formaldehyde and trans-1,6,7,12-tetraazaperhydrotetracene (1) under the chosen conditions [MeOH, 20 °C, 3 h] resulted in the selective formation of (3bR*,7aR*,10bR*,14aR*)-2,9-diadamantyl-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes (12–14) in 51–78% yields (Scheme 3).
 | ||
| Scheme 3 Cyclocondensation reaction of adamantylamines, formaldehyde, and trans-1,6,7,12-tetraazaperhydrotetracene. | ||
Relying on the above Experimental results, we put forward the assumption that 2,9-disubstituted perhydro 2,3a,7b,9,10a,14b-hexaazadibenzotetracenes containing natural metabolites with pronounced biological activity as substituents could be obtained by a one-pot reaction protocol. An accessible compound easily isolated from pine pitch is maleopimaric acid, a diene adduct of levopimaric acid and maleic anhydride.37 Levopimaric diene adducts and their derivatives, are known to possess a wide range of biological activities including antitumor, antiulcer, anti-inflammatory, antimicrobial, antiviral, and other properties.38–43 Chemical modifications of these diene adducts result in a broad spectrum of cytotoxic activity in vitro and in vivo,38 as well as the ability of some of them to be effective inducers of tumor cell apoptosis.44 Maleopimaric acid and its derivatives show a broad range of biological activities, including anti-inflammatory, antiulcer, cytotoxic, and bactericidal ones.45,46 Our research was directed towards amino derivatives of methyl maleopimarate.47 It was found that the hydrazide and imido-amine of methyl maleopimarate reacts with formaldehyde and trans-tetraazaperhydrotetracene 1 under conditions we developed [10 wt% zeolite Ymmm, MeOH, 20 °C, 3 h] to give dimeric (3bR*,7aR*,10bR*,14aR*)-perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes (15–16) in 17–20% yields (Scheme 4).
 | ||
| Scheme 4 Multicomponent condensation between amino derivatives of maleopimaric acid methyl ester, formaldehyde, and trans-1,6,7,12-tetraazaperhydrotetracene. | ||
A cytotoxic effect of perhydro hexaazadibenzotetracenes synthesized hereby was determined based on IC50 for six tumor cell cultures (Jurkat, K562, U937, A549, A2780 and T74D) and the same for normal fibroblasts (Fibroblats). The compound 14 was demonstrated to possess a cytostatic activity (a proliferation-restrictive activity) with regard to cells in all studied lines. The observed effect was the most prominent in the cell line U937. The average IC50 value for the compound 14 constituted 0.1 μM ± 0.3, whilst in fibroblasts the IC50 value was 0.3 μM ± 0.1, respectively (p < 0.05). In order to elucidate a mechanism underlying the proliferation-restrictive effect exerted by the tested compound 14 in studied tumor cultures, we have studied the apoptosis and have demonstrated the influence of said compound on cell cycle phases in the U937 cell culture. Thus, upon addition of the above mentioned compound in different concentrations to tumor cells, the apoptosis-related processes have been observed that manifested in dose-dependent accumulation of cells in early- and late-stage apoptosis with a maximum value of 90% achieved at a concentration of the tested compound of 0.2 μM.
Additionally, we have studied the influence of a leader compound on a pattern of cell distribution in cell cycle phases in the U937 tumor culture. In various concentrations, the compound 14 caused changes of the same type in the pattern of cell distribution over the cell cycle phases. Thus, at concentrations of 0.05 μM and 0.1 μM, a dose-dependent increase in pre-G0 cell fraction was observed (9.02% at a concentration of the tested compound of 0.1 μM and 7.44% at a concentration of 0.05 μM), which is indicative of activation of apoptotic processes in conducted experiments. Moreover, the compound 14 arrested U937 cells in the phases G1 and G2 (Fig. 2). Upon increasing concentration of the tested compound in cell cultures, the value associated with G1 and G2 phases was reduced (G1 – 39.13% and G2 – 5.14%, as compared to a control: G1 – 61.23% and G2 – 12.10%). Removal of the compound 14 from a culture medium did not restore progression of U937 cells between the cell cycle phases. It is highly probable that the leader compound tested hereby acts as a cytotoxic agent and exerts irreversible effect on tumor cells (Fig. 3).
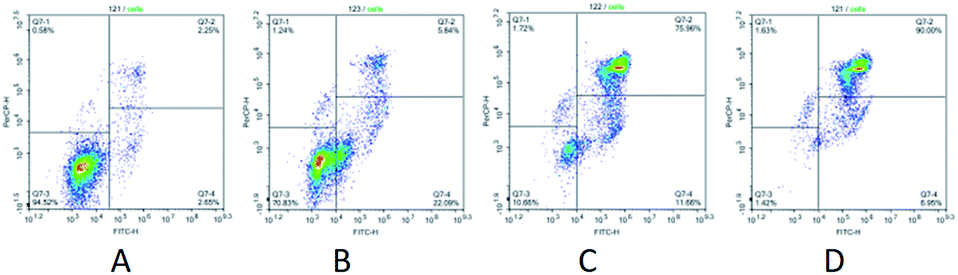 | ||
| Fig. 2 U937 tumor cells treated with the test compound 14 in different concentrations and stained with annexin V/7AAD. Flow cytometry data: (A) control; (B) 14, 0.05 μM; (C) 14, 0.1 μM; (D) 14, 0.2 μM. The histogram shows the apoptosis rate (%); *p < 0.05 when compared with the control group (n = 4). | ||
 | ||
| Fig. 3 Cell cycle phases of U937 cells treated with the test compound 14: (A) control; (B) 14, 0.1 μM; (C) 14, 0.05 μM. Incubation time of U937 cells with 14 is 48 h. The DNA histograms of U937 cells' cell cycle were analyzed by flow cytometry for PI staining. The G0/G1%, S%, and G2 + M% phases of the cells were analyzed using flow cytometry. Flow cytometric histograms are representative of three separate experiments. Data are presented with the means ± SD and mean values of three independent experiments. *p < 0.05 and **p < 0.01, compared with the control group. | ||
Conclusions
Thus, we developed efficient one-pot methods for the synthesis of previously undescribed 2,9-disubstituted perhydro-2,3a,7b,9,10a,14b-hexaazadibenzo[fg,op]tetracenes with pronounced antitumor activity based on intermolecular heterocyclization of N,N-bis(methoxymethyl)-N-alkylamines or ring transformation of 1,3,5-tricycloalkyl-1,3,5-triazinanes with trans-tetraazaperhydrotetracene catalyzed by d- and f-element salts, and multicomponent condensation of adamantylamines or amino derivatives of methyl maleopimarate with formaldehyde and trans-tetraazaperhydrotetracene catalyzed by metal silicates. The structural feature of the synthesized perhydro hexazadibenzotetracenes is the presence of the R*,R*,R*,R*-relative configuration of chiral centers at the carbon atoms C3b,7a,10b,14a and a cis-junction of the cycles along the C14c–C14d bond.Experimental section
The NMR spectra, including two-dimensional homo- (COSY, NOESY) and heteronuclear (HSQC, HMBC) spectra, were recorded on a Bruker Avance 500 spectrometer at 500.17 MHz for 1H and 125.78 MHz for 13C according to standard Bruker procedures. CDCl3 was used as the solvent, and tetramethylsilane, as the internal standard. The MALDI TOF/TOF mass spectra (positive ion detection, 2,5-dihydroxybenzoic acid matrix) were obtained on a Bruker AutoflexTM III Smartbeam mass spectrometer. Samples were prepared by the dried drop technique. Solutions of a matrix and analyte were mixed at a ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 100:1, and a drop of the resulting mixture was applied to a target and dried in a stream of warm air. The sample was transferred from the target to the gas phase by laser pulses (200 pulses at a frequency of 100 Hz) using a solid state UV laser (λ 355 nm). The elemental analyses were obtained on a Carlo Erba 1106 analyzer. The melting points were determined on a PHMK 80/2617 melting point apparatus. The reaction progress was monitored by TLC using Sorbfil plates (PTSH-AF-V), visualization with iodine vapor. Column chromatography was performed on KSK silica gel (100–200 μm).
:1 to 100:1, and a drop of the resulting mixture was applied to a target and dried in a stream of warm air. The sample was transferred from the target to the gas phase by laser pulses (200 pulses at a frequency of 100 Hz) using a solid state UV laser (λ 355 nm). The elemental analyses were obtained on a Carlo Erba 1106 analyzer. The melting points were determined on a PHMK 80/2617 melting point apparatus. The reaction progress was monitored by TLC using Sorbfil plates (PTSH-AF-V), visualization with iodine vapor. Column chromatography was performed on KSK silica gel (100–200 μm).
Heterocyclization of N,N-bis(methoxymethyl)-N-alkylamines with trans-1,6,7,12-tetraazaperhydrotetracene (1) (general method)
A round bottom flask equipped with a magnetic stir bar was charged with MeOH (5 mL), SmCl3·6H2O (0.018 g, 0.05 mmol), and the appropriate N,N-bis(methoxymethyl)alkylamine (2.00 mmol), prepared in situ by the reported procedure,24 and the mixture was stirred at r.t. for 30 min. Next, trans-1,6,7,12-tetraazaperhydrotetracene (1; 1.00 mmol) in MeOH (5 mL), prepared in situ by the reaction of (±)-trans-cyclohexane-1,2-diamine (0.23 g, 2.00 mmol) with 40 wt% aq glyoxal (0.14 g, 1.00 mmol), was added and the resultant mixture was stirred at 20 °C for 3 h. The mixture was then concentrated, and the residue was purified by column chromatography (silica gel, MeOH). Compounds 2–5 were obtained as white powdery crystals (recrystallized from CHCI3). The final products 2–5 were identified by spectroscopic methods.Recyclization of 1,3,5-tricycloalkyl-1,3,5-triazinanes with trans-1,6,7,12-tetraazaperhydrotetracene (1) (general method)
A round-bottomed flask equipped with a magnetic stirrer bar was charged with MeOH (5 mL), NiCl2·6H2O (0.012 g, 0.05 mmol), and the appropriate 1,3,5-tricycloalkyl-1,3,5-triazinane (2.00 mmol), prepared in situ by the reported procedure,21 and the mixture was stirred at r.t. for 30 min. trans-1,6,7,12-Tetraazaperhydrotetracene (1; 1.00 mmol) in MeOH (5 mL), prepared in situ by the reaction of (±)-trans-cyclohexane-1,2-diamine (0.23 g, 2.00 mmol) with 40 wt% aq glyoxal (0.14 g, 1.00 mmol) was added, and the mixture was stirred at 20 °C for 3 h, then concentrated. The white precipitate was filtered and washed twice with MeOH (5 mL). Compounds 6–11 were obtained as white powdery crystals (recrystallized from CHCl3). The final products 6–11 were identified by spectroscopic methods., with a = 10.1228(10), b = 10.5481(11), c = 19.5768(19) Å, α = 78.370(9), β = 85.218(8) and γ = 72.376(9)°, V = 1950.8(4) Å3, Z = 2, Dcalc = 1.300 g cm−3, μ = 0.473 cm−1. The X-ray diffraction measurements for compound 9 was performed on an Agilent Xcalibur (Eos, Gemini) automated four-circle diffractometer (graphite monochromator, MoKα radiation, λ = 0.71073 Å, ω-scan mode, 2θmax = 62°) at ambient temperature (293–298 K). Collected data were processed using the program CrysAlisPro.48 Structures determinations were carried out with the OLEX2 program.49 The structures were solved by direct methods and refined by the full-matrix least-squares method in the anisotropic approximation for non-hydrogen atoms. All hydrogen atoms are generated using the proper HFIX command and refined isotropically using the riding model. The calculations were performed using the SHELX program package.50,51 The molecular plots were drawn using Mercury.52 Refinement was converged with R1 = 0.0751, wR2 = 0.1721 [2594 reflections with I > 2s(I)] and R1 = 0.2600, wR2 = 0.2723 for all data (8968 reflections). Atomic coordinates, bond lengths, bond angles and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre as a CIF deposition with file number CCDC 1973770.Multicomponent condensation reaction of adamantylamines or amino derivatives of methyl maleopimarate with formaldehyde and trans-1,6,7,12-tetraazaperhydrotetracene (1) (general method)
A round bottom flask equipped with a magnetic stir bar was charged with MeOH (5 mL), zeolite Y36 (10 wt%), the appropriate adamantylamine (2.00 mmol) or amino derivative of methyl maleopimarate (2.00 mmol), and formaldehyde (0.45 mL, 4.00 mmol). Next, trans-1,6,7,12-tetraazaperhydrotetracene (1; 1.00 mmol) in MeOH (5 mL), prepared in situ by the reaction of (±)-trans-cyclohexane-1,2-diamine (0.23 g, 2.00 mmol) with 40 wt% aq glyoxal (0.14 g, 1.00 mmol),was added and the resultant mixture was stirred at 20 °C for 3 h, then concentrated. The white precipitate was collected by filtration and washed twice with MeOH (5 mL). Compounds 12–16 were obtained as powdery crystals (recrystallized from CHCl3). The final products 12–16 were identified by spectroscopic methods.Biological assay data
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research has been financially supported by the Russian Foundation for Basic Research (grant 18-29-09068), Scholarships of the President of the Russian Federation for young scientists and graduate students (SP-197.2019.4) and the President of Russian Federation for Government support of Leading Scientific Schools (Grant SS-5240.2018.3). The work has been carried out in accordance with the approved plans for research projects at the Institute of Petrochemistry and Catalysis of the Russian Academy of Sciences (RAS) on the topic titled “Metal complex and heterogeneous catalysis in the design of macroheterocycles and heteroatomic compounds”. State Registration No. AAAA-A19-119022290010-9 (2019–2021). The structural studies of synthesized compounds (2–16) were carried out at the “Agidel” Center for Collective Use at the Ufa Federal Research Center of the Russian Academy of Sciences with the financial support of the Russian Ministry of Education and Science (grant 2019-05-595-000-058).References
- L. Alakonda and M. Periasamy, J. Organomet. Chem., 2009, 694, 3859 CrossRef CAS.
- M. Dalai and M. Periasamy, Tetrahedron: Asymmetry, 2009, 20, 1247 CrossRef CAS.
- Á. L. Fuentes de Arriba, D. G. Seisdedos, L. Simón, V. Alcázar, C. Raposo and J. R. Morán, J. Org. Chem., 2010, 75, 8303 CrossRef PubMed.
- P. J. Boratyński, A. E. Nowak and J. Skarżewski, Synthesis, 2015, 47, 3797 CrossRef.
- M. Periasamy, N. Sanjeevakumar and P. O. Reddy, Synthesis, 2012, 44, 3185 CrossRef CAS.
- E. Wojaczyńska, J. Bąkowicz, M. Dorsz and J. Skarżewski, J. Org. Chem., 2013, 78, 2808 CrossRef PubMed.
- B. Zaleska, R. Socha, M. Karelus, E. Szneler, J. Grochowski and P. Serda, J. Org. Chem., 2003, 68, 2334 CrossRef CAS PubMed.
- A. Rivera, D. Quiroga, L. Jimenez-Cruz, K. Fejfarova and M. Dušek, Tetrahedron Lett., 2012, 53, 345 CrossRef CAS.
- M. Padmaja and M. Periasamy, Tetrahedron: Asymmetry, 2004, 15, 2437 CrossRef CAS.
- N. E. Borisova, M. D. Reshetova and Yu. A. Ustynyuk, Russ. Chem. Rev., 2007, 76, 785 CrossRef CAS.
- M. Dragoun, T. Günther, C. Frias, A. Berkessel and A. Prokop, J. Cancer Res. Clin. Oncol., 2018, 144, 685 CrossRef CAS PubMed.
- A. R. Khokhar, S. Al-Baker, S. Shamsuddin and Z. H. Siddik, J. Med. Chem., 1997, 40, 112 CrossRef CAS PubMed.
- F. Morales, A. Ramírez, C. Morata-Tarifa, S. A. Navarro, J. A. Marchal, J. M. Campos and A. Conejo-García, Future Med. Chem., 2017, 9, 293 CrossRef CAS PubMed.
- K. H. Omer, A. A. Seliman, M. Altaf, N. Casagrande, D. Aldinucci, S. Altuwaijri and A. A. Isab, Polyhedron, 2015, 102, 773 CrossRef CAS.
- J. Iwanejko, E. Wojaczyńska, J. Trynda and J. Wojaczyński, Tetrahedron, 2017, 73, 2276 CrossRef CAS.
- I. A. Dvornikova, E. V. Buravlev, L. L. Frolova, Yu. V. Nelyubina, I. Yu. Chukicheva and A. V. Kuchin, Russ. J. Org. Chem., 2011, 47, 1130 CrossRef CAS.
- S. Eröksüz, J. M. Neudörfl and A. Berkessel, Synlett, 2017, 28, 1278 CrossRef.
- D. A. Evans and D. Seidel, J. Am. Chem. Soc., 2005, 127, 9958 CrossRef CAS PubMed.
- D. A. Evans, S. Mito and D. Seidel, J. Am. Chem. Soc., 2007, 129, 11583 CrossRef CAS PubMed.
- E. B. Rakhimova, V. Yu. Kirsanov, E. S. Mescheryakova, L. M. Khalilov, A. G. Ibragimov and U. M. Dzhemilev, Synlett, 2018, 29, 1861 CrossRef CAS.
- E. B. Rakhimova, V. Yu. Kirsanov, R. A. Zainullin, A. G. Ibragimov and U. M. Dzhemilev, J. Chem., 2016, 2016, 8406172 Search PubMed.
- E. B. Rakhimova, V. Yu. Kirsanov, E. S. Meshcheryakova, L. M. Khalilov, B. I. Kutepov, A. G. Ibragimov and U. M. Dzhemilev, Tetrahedron, 2017, 73, 6880 CrossRef CAS.
- E. B. Rakhimova, V. Yu. Kirsanov, E. S. Mescheryakova, L. M. Khalilov, A. G. Ibragimov, L. U. Dzhemileva, V. A. D'yakonov and U. M. Dzhemilev, ACS Med. Chem. Lett., 2019, 10, 378 CrossRef CAS PubMed.
- E. B. Rakhimova, R. A. Ismagilov, E. S. Meshcheryakova, L. M. Khalilov, A. G. Ibragimov and U. M. Dzhemilev, Tetrahedron Lett., 2014, 55, 6367 CrossRef CAS.
- S. Oda, B. Sam and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 8525 CrossRef CAS PubMed.
- X. Lian, L. Lin, K. Fu, B. Ma, X. Liu and X. Feng, Chem. Sci., 2017, 8, 1238 RSC.
- J. Gong, S.-W. Li, S. Qurban and Q. Kang, Eur. J. Org. Chem., 2017, 25, 3584 CrossRef.
- D. Ji and J. Sun, Org. Lett., 2018, 20, 2745 CrossRef CAS PubMed.
- C. Zhu, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 11867 CrossRef CAS PubMed.
- S. Peng, S. Cao and J. Sun, Org. Lett., 2017, 19, 524 CrossRef CAS PubMed.
- L. K. B. Garve, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2017, 56, 9226 CrossRef CAS PubMed.
- L. K. B. Garve, A. Kreft, P. G. Jones and D. B. Werz, J. Org. Chem., 2017, 82, 9235 CrossRef CAS PubMed.
- L. M. Khalilov, A. Yu. Spivak, E. V. Vasil'eva, A. A. Fatykhov, N. A. Prokhorova and G. A. Tolstikov, Chem. Nat. Compd., 1991, 27, 318 CrossRef.
- L. M. Khalilov, A. A. Panasenko, R. R. Muslukhov and G. A. Tolstikov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1988, 37, 1569 CrossRef.
- S. A. Kon'kov, I. K. Moiseev, M. N. Zemtsova and K. M. Bormasheva, Russ. Chem. Rev., 2014, 83, 377 CrossRef.
- B. I. Kutepov, O. S. Travkina, I. N. Pavlova, A. N. Khazinova, N. G. Grigor'eva and M. L. Pavlov, Russ. J. Appl. Chem., 2015, 88, 65 CrossRef CAS.
- L. H. Zalkov, R. A. Ford and J. P. Kutney, J. Org. Chem., 1962, 27, 3535 CrossRef.
- E. V. Tretyakova, I. E. Smirnova, O. B. Kazakova, G. A. Tolstikov, N. P. Yavorskaya, I. S. Golubeva, R. B. Pugacheva, G. N. Apryshko and V. V. Poroikov, Bioorg. Med. Chem., 2014, 22, 6481 CrossRef CAS PubMed.
- O. B. Flekhter, E. V. Tret'yakova, N. S. Makara, S. F. Gabdrakhmanova, N. Zh. Baschenko, F. Z. Galin, F. S. Zarudii and G. A. Tolstikov, Russ. Pharm. Chem. J., 2003, 37, 142 CrossRef CAS.
- O. B. Kazakova, E. V. Tret'yakova, I. E. Smirnova, L. V. Spirikhin, G. A. Tolstikov, I. V. Chudov, G. V. Bazekin and A. F. Ismagilova, Russ. J. Bioorg. Chem., 2010, 36, 257 CrossRef CAS PubMed.
- E. V. Tretyakova, I. E. Smirnova, E. V. Salimova, T. M. Pashkova, O. L. Kartashova, V. N. Odinokov and L. V. Parfenova, Russ. J. Bioorg. Chem., 2017, 43, 317 CrossRef CAS.
- E. V. Tret'yakova, G. F. Zakirova, E. V. Salimova, O. S. Kukovinets, V. N. Odinokov and L. V. Parfenova, Med. Chem. Res., 2018, 27, 2199 CrossRef.
- E. V. Tretyakova, I. E. Smirnova, E. V. Salimova and V. N. Odinokov, Bioorg. Med. Chem., 2015, 23, 6543 CrossRef CAS PubMed.
- E. V. Tretyakova, E. V. Salimova, L. V. Parfenova, M. M. Yunusbaeva, L. U. Dzhemileva, V. V. D'yakonov and U. M. Dzhemilev, Anti-Cancer Agents Med. Chem., 2019, 19, 1172 CrossRef CAS PubMed.
- O. B. Kazakova, E. V. Tret'yakova, O. S. Kukovinets, G. A. Tolstikov, T. I. Nazyrov, I. V. Chudov and A. F. Ismagilova, Russ. J. Bioorg. Chem., 2010, 36, 762 CrossRef CAS.
- J. Wang, Y. P. Chen, K. Yao, P. A. Wilbon, W. Zhang, L. Ren, J. Zhou, M. Nagarkatti, Ch. Wang, F. Chu, X. He, A. W. Decho and Ch. Tang, Chem. Commun., 2012, 48, 916 RSC.
- E. V. Tretyakova, E. V. Salimova and L. V. Parfenova, Russ. J. Bioorg. Chem., 2018, 44, 547 CrossRef CAS.
- CrysAlis PRO/2012, Agilent Ltd, Yarnton, Oxfordshire, England, 2012 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for products, X-ray data. CCDC 1973770. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03209c |
| This journal is © The Royal Society of Chemistry 2020 |