
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation and synthesis of cryptosanguinolentine (isocryptolepine), a naturally-occurring bioactive indoloquinoline alkaloid
Elida N. Thobokholt
 ,
Enrique L. Larghi
,
Andrea B. J. Bracca
* and
Teodoro S. Kaufman
*
,
Enrique L. Larghi
,
Andrea B. J. Bracca
* and
Teodoro S. Kaufman
*
Instituto de Química Rosario (IQUIR, CONICET-UNR), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, S2002LRK Rosario, Argentina. E-mail: kaufman@iquir-conicet.gov.ar; bracca@iquir-conicet.gov.ar; Fax: +54-341-4370477; Tel: +54-341-4370477
First published on 19th May 2020
Abstract
Cryptosanguinolentine (isocryptolepine) is one of the minor naturally-occurring monomeric indoloquinoline alkaloids, isolated from the West African climbing shrub Cryptolepis sanguinolenta. The natural product displays such a simple and unique skeleton, which chemists became interested in well before it was found in Nature. Because of its structure and biological activity, the natural product has been targeted for synthesis on numerous occasions, employing a wide range of different strategies. Hence, discussed here are aspects related to the isolation of isocryptolepine, as well as the various approaches toward its total synthesis.
 Elida N. Thobokholt | Elida N. Thobokholt was born in Rosario (Santa Fe, Argentina) and studied at the School of Biochemical and Pharmaceutical Sciences at the National University of Rosario, where she earned her BS degree in Food Science and Technology in 2018. Shortly after, she joined the group of Dr Kaufman as a Doctoral student at IQUIR. Currently, she is developing novel approaches to the synthesis of naturally occurring indoloquinoline alkaloids, under the supervision of Drs Bracca and Larghi. |
 Enrique L. Larghi | Enrique L. Larghi was born in Rosario (Santa Fe, Argentina). He received his B.S. from the UNR (Argentina) in 1997 and immediately after he moved to the UFSM (RS-Brazil) where he obtained MSc and Ph. D. degrees under the advice of Profs. Silveira (1997) and Morel (1999). After a short experience in the Argentine pharmaceutical industry, he returned to IQUIR, joining Prof. Kaufman's group as a postdoctoral fellow first, and then as a staff member of CONICET. Currently, he is an Independent Researcher and Adjunct Professor at the UNR. His current interests cover the synthesis of heterocyclic natural products and their derivatives. |
 Andrea B. J. Bracca | Andrea B. J. Bracca was born in Rosario (Santa Fe, Argentina) and graduated in 2001 with a BS in Biotechnology from the National University of Rosario. She received her Ph. D. in 2009 under the guidance of Prof. Kaufman. After a two year period of postdoctoral training, she returned to work in Dr Kaufman's group as an Assistant Research Scientist at the Argentine National Research Council (CONICET) at the Institute of Chemistry of Rosario (IQUIR). Currently, Dr Bracca is an Adjunct Researcher and develops research work in the area of the total synthesis of heterocyclic natural products and their most relevant analogues. |
 Teodoro S. Kaufman | Teodoro S. Kaufman was born near Moisés Ville (Santa Fe, Argentina). He graduated in Biochemistry (1982) and Pharmacy (1985) at the National University of Rosario (UNR), and received his Ph. D. (1987) under the guidance of Prof. Edmundo A. Rúveda. After two years of postdoctoral training at The University of Mississippi (USA), he returned to Rosario. Currently, he is a full Professor of the UNR, Research Head of the Argentine National Research Council (CONICET) and Head of the Institute of Chemistry of Rosario (IQUIR). His research interests include heterocyclic chemistry and the synthesis and evaluation of bioactive natural products and analogues. |
1. Introduction
It is now common knowledge that after millions of years of co-existing under different external pressures (territory, food, predators, etc.), the communities of organisms sharing the same ecosystem underwent co-evolution as a survival strategy. As a result, some of their secondary metabolic routes suffered changes over time, gaining the ability to become resistant to their environment by producing certain low molecular weight metabolites. These small molecules are collectively known as natural products.It has been found that the natural products may act as small ligands for certain macromolecular targets found within the living organisms or, more often, within other living organisms of the same ecosystem.1 In this way, the natural products assist the producing organism to sustain the hostilities of the environmental pressures and survive. Interestingly, many human proteins contain structural domains that are very similar to some of the macromolecular targets affected by natural products; therefore, it was not unexpected to find out that the small molecule natural products originally produced with a different aim (reproduction, signaling, communication, defense), can interact with human proteins and modulate different responses.2
As a result of the natural selection process, natural products display wide chemical diversity and chemical specificity, which enhances their ability to interact with diverse biorelevant macromolecules, turning them into a unique and rich source of compounds for new drug development.
Empirically, humankind took advantage of the general properties of these natural products, and medicinal plants have been used in virtually all cultures as a source of medicines since the beginning of time.3 Furthermore, statistical analysis performed on the new chemical entities reported between 1981 and 2018 confirmed this situation, revealing that 40% of them were derived from natural products or were natural products themselves. This proportion increases to 64% among anticancer drugs and to 75% when only antibiotics are considered.4
Since the abundance of natural products in extracts is invariably low and therefore the former are often isolated in tiny amounts, when they show signs of being useful, it is usually found that their natural supply is not enough to satisfy the demand at a reasonable scale, such as that needed for biological testing. At this point, the development of efficient synthetic pathways to guarantee convenient access to these compounds becomes a requirement.
Furthermore, natural products are currently seen not only as potential sources of inspiration for the development of new medicines through structural diversification; they are also highly regarded as potentially suitable probes to explore the interactions within and between cells, as a logical means to understand the inner-workings of the complex molecular machinery that sustains life.5 However, since Nature has produced such wonderfully complex molecules that no synthetic chemist could ever dream of, natural products are still a relevant challenge to organic chemists, who advance the frontiers of knowledge by devising new strategies and reagents for their synthesis.
2. Known naturally-occurring indoloquinoline alkaloids
Naturally-occurring indoloquinolines are a small family of alkaloids, which have been isolated mainly from Cryptolepis sanguinolenta (Lind.) Schlechter, family Asclepiadaceae.6 However, other plants such as Justicia betonica L. (Acanthaceae),7 J. secunda8 and Sida rhombifolia L. (Malvaceae)9 are also natural sources of alkaloids that exhibit the indoloquinoline motif. These unique natural heterocycles bear both indole and quinoline rings, fused through their pyrrole and pyridine rings.Considering this limiting characteristic, only four isomeric ring systems are possible (Fig. 1), namely indolo[3,2-c] quinoline (1), indolo[3,2-b]quinoline (quinindoline, 2), indolo [2,3-b]quinoline (3) and indolo[2,3-c]quinoline (4). The cores of natural indoloquinolines correspond to isomers 1–3, and despite the skeleton of 4 being synthesized, there are no natural examples of compounds of class 4.10
 | ||
| Fig. 1 Some isomers of the indoloquinoline skeleton. | ||
Their main source of indoloquinolines in Nature, Cryptolepis sanguinolenta, is a tropical scrambling and twinning shrub, indigenous to West and Central Africa.11 The study of the plant began in the early 1950s and cryptolepine was the first alkaloid to be isolated.12 To date, over a dozen naturally occurring indoloquinolines have been discovered, including quinindoline (3),7 quindoline (2),13 quindolinone (5),14 neocryptolepine (6),15 cryptosanguinolentine (isocryptolepine, 7),16 cryptolepine (8),17 11-isopropylcryptolepine (9)18 and cryptolepinone (hydroxycryptolepine, 10).19 These are monomeric members of this family (Fig. 2), however, several dimeric natural indoloquinolines have also been reported.19–21
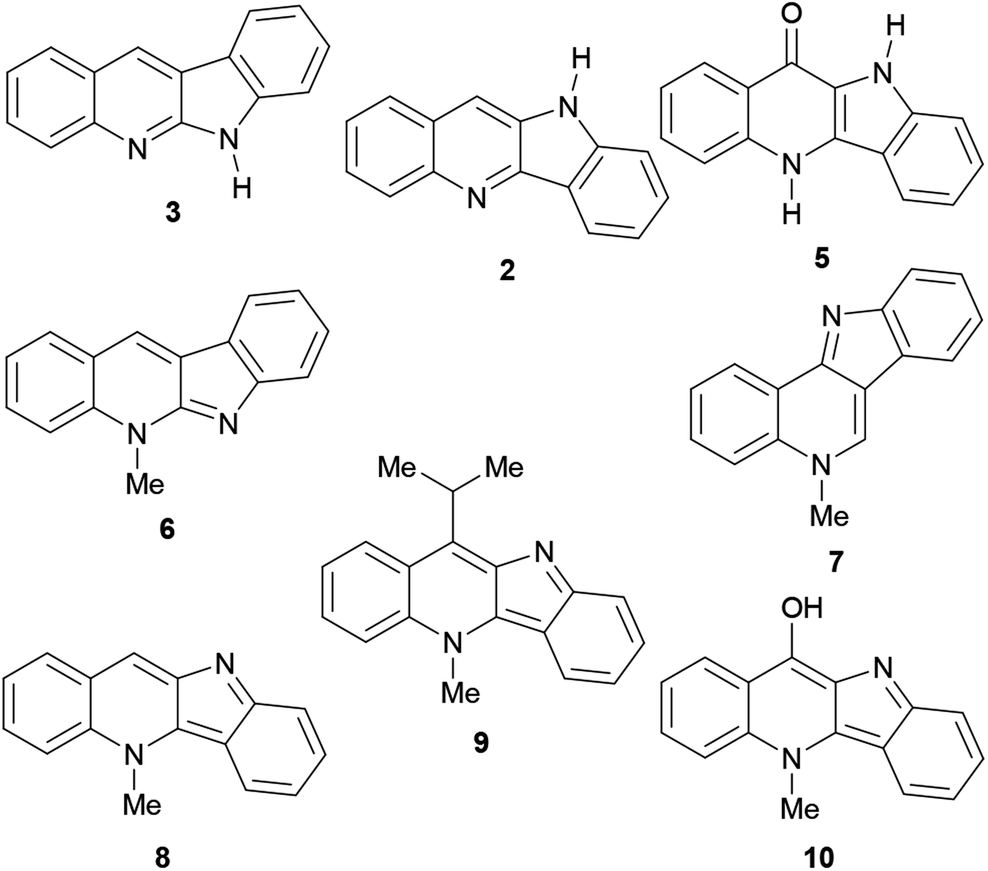 | ||
| Fig. 2 Monomeric naturally-occurring indoloquinolines. | ||
The plant has long been employed in the dyeing of textiles and leather.22 In addition, a decoction of its roots is frequently used in traditional folk medicine, to treat fevers, malaria,23 upper respiratory infections and venereal diseases.24 The root has been used in the Congo as a bitter stomachic,24c and in Nigeria for the therapy of colics, rheumatism, and urogenital infections.25 The plant has been demonstrated to possess antibacterial activity and to exert vasodilation.21b The pharmacological activities of different indoloquinoline alkaloids, their analogs and derivatives, have been reported in several articles. The most relevant ones are their antitumoral and antiparasitic activities.26
3. Isocryptolepine: its isolation and properties
Isocryptolepine was first obtained as a degradation product during investigations on the constitution of indigo-yellow.27 However, the heterocycle was first isolated from a natural source (Cryptolepis sanguinolenta) in 1995 by the Bodo group as a non-crystalline substance, along with cryptolepine and quindoline. Only 5 mg were obtained from 60 g of air-dried roots ground to a fine powder (0.08% w/w). Its chemical structure was determined after exhaustive analysis of its infrared (IR) and nuclear magnetic resonance (NMR) spectroscopic and mass spectrometric (MS) and data.16 Interestingly, on the bases of spectral grounds, it has also been conjectured that the previously isolated and partially characterized alkaloid CSA-3 may be identical to isocryptolepine.13Shortly after, the same alkaloid was isolated again by the Tackie group, from the same source in 1996, under the name cryptosanguinolentine.28 The natural product, obtained as a yellowish residue (1.7 mg from 3.14 kg of plant roots; 0.5 × 10−4% w/w), was isolated along with cryptotackieine (neocryptolepine); due to the scarcity of the material, preparative reverse-phase high-performance liquid chromatography (HPLC) and micro NMR techniques were required to confirm its structure. At a later date, the same group reported a more detailed account of their isolation, with the obtaining of another 15 alkaloids.21a
The 1H, 13C and 15N NMR spectra of isocryptolepine and its protonated form were assigned as part of a systematic NMR study of four isomeric indoloquinoline alkaloids.29 All of the NMR signals were assigned using 2D correlation techniques. Interestingly, in the 15N NMR [a gradient selected single-quantum multiple-bond correlation (GSQMBC) experiment], the quinoline-N (N-5) was observed in the usual range at δ 135.6 ppm, whereas the indole-N (N-11), which has is imine-like in nature, was found to be significantly deshielded (δ 248.1 ppm).
Density functional theory (DFT) calculations of the chemical shielding constants were also performed, allowing a detailed investigation of the effects of protonation and solvation. The calculations were able to reproduce the main experimental trends observed upon protonation, and it was observed that the inclusion of solvent effects in the computations improved the agreement with the experimental 13C NMR data.
1H NMR data was also recorded in order to determine that the pKa of the natural product is 9.8. This result was obtained through a graphical estimation of the inflection point and also by the use of the Henderson–Hasselbalch equation [eqn (1)],30 which showed a linear dependence of the chemical shift on the pH of the sample:31
| pH = pKa + log[δmax − δ]/[δ − δmin] | (1) |
The effect of cyclodextrin (CD) encapsulation on the photophysics of isocryptolepine, was studied using steady state and time-resolved fluorescence spectroscopy.32 In the excited state, it was observed that the natural product exists mainly in its zwitterionic form, exhibiting appreciable emission from the π–π* state upon excitation at a specific wavelength.
Due to the presence of the hydrophobic nanocavities of CDs, the existence of zwitterions in the excited state allows a mutual interaction to form dimers, triggered through coulombic interactions. This was evidenced by treating the fluorophores with CDs with different cavity spaces and employing steady state fluorescence measurements. Moreover, the photophysical behavior of the heterocycle was found to be modulated by the nature of the cyclodextrin. In addition, trapped water molecules inside the bigger cavity of γ-CD seemed to quench the fluorescence of the zwitterions.
Isocryptolepine has been demonstrated to show antimicrobial,33 anti-trypanosomal,34 antifungal,35 anti-inflammatory, antimalarial (including β-haematin inhibition),36 antitrypanosomal, antileishmanial and cytotoxic activities,34,36c,37 as well as the ability to interact with DNA,38 promote low density lipoprotein uptake in HepG2 cells,39 and be useful as a treatment for intestinal disorders. Most of these properties are common to other natural indoloquinolines, which differing in potency and selectivity.
As a result of its profile, during the last 20 years, this natural product became a recurrent target for total synthesis as well as a useful scaffold for the preparation of analogs40 and derivatives,41 with the aim of improving its activity profile (mainly its potency and selectivity). Furthermore, its attractive structure turned its synthesis into a fertile field for testing the scope and efficiency of new and imaginative C–C and C–N bond forming reactions. Detailed below are the different approaches that have been designed to synthesize isocryptolepine.
4. Total syntheses of isocryptolepine
Isocryptolepine is an angularly fused indoloquinoline. As observed in the case of other indoloquinolines, there has been some relevant synthetic work that focused on targeting isocryptolepine before it was found in nature, in 1996. However, most of the synthetic efforts toward the alkaloid have been carried out over the last 20 years, where the natural product has been totally synthesized over 25 times, by different groups around the world; furthermore, some of them can be credited with more than one synthesis.We have previously classified the synthetic approaches toward the indoloquinoline alkaloids neocryptolepine (6) and quindoline (2) into three main groups, based on the chemical structure of the starting materials involved in the sequence, including: (a) benzenoids, (b) quinolines and (c) indoles.42 Accordingly, except for the advances recorded before the isolation of isocryptolepine from C. sanguinolenta, the total syntheses of the natural product are classified here to fit into these three categories. In addition, for the sake of clarity, the syntheses are ordered chronologically.
4a. Early synthetic studies before the isolation of isocryptolepine from a natural source
The first reported total synthesis of the structure of isocryptolepine was disclosed in 1950 by Kermack and Storey,43 who synthesized derivatives of the tetracycle 1 for biological testing, in an attempt to find new antimalarials. These authors employed a modification of the Graebe–Ullmann carbazole synthesis for the preparation of the tetracyclic framework of their prospective compounds and also synthesized isocryptolepine via the N-methylation of 1.Thus, 4-chloroquinoline (11) was condensed with ortho-phenylenediamine (12) at 140 °C at 20–30 mmHg for 10–20 min, resulting in 4-(2-aminoanilino)quinoline (13), in 76% yield (Scheme 1). After this, 13 was submitted to a Graebe–Ullmann protocol by diazotization with HNO2 to afford the benzotriazole derivative 14 in 70% yield. Further heating of 14 with syrupy phosphoric acid, gave 1 in 77% yield, after the loss of nitrogen and rearrangement.
 | ||
| Scheme 1 Reagents and conditions: (a) 140 °C, 20–30 mm Hg, 10–20 min (76%); (b) NaNO2, 1 N HCl, H2O, 5 °C (70%); (c) H3PO4, heat (77%); (d) MeI, PhNO2, 2 h, 100 °C (72%); (e) (1) 15, MW (160 W); (2) pyrophosphoric acid, 160 W, 4–6 min (60% overall). | ||
The product was obtained as a violet fluorescent compound, which exhibited increased fluorescence when dissolved in concentrated sulfuric acid. Finally, the tetracycle was heated in a steam bath with excess methyl iodide in nitrobenzene, furnishing the methiodide of 7 in 72% yield. The base of the latter could be freed by treatment with aqueous ammonia, followed by drying of the resulting isocryptolepine monohydrate.
In the reaction mechanism (Scheme 2), the primary amino group of 13 is diazotized with HNO2 to give the diazonium salt intermediate A, which undergoes an intramolecular cyclization to the protonated benzotriazole intermediate B. After deprotonation, benzotriazole 14 loses nitrogen to afford the diradical species C, which in its conformation D undergoes a radical rearrangement to the carbene intermediate E. Then, cyclization takes place to afford intermediate F, which further aromatizes to 1, via a [1,3] proton shift.
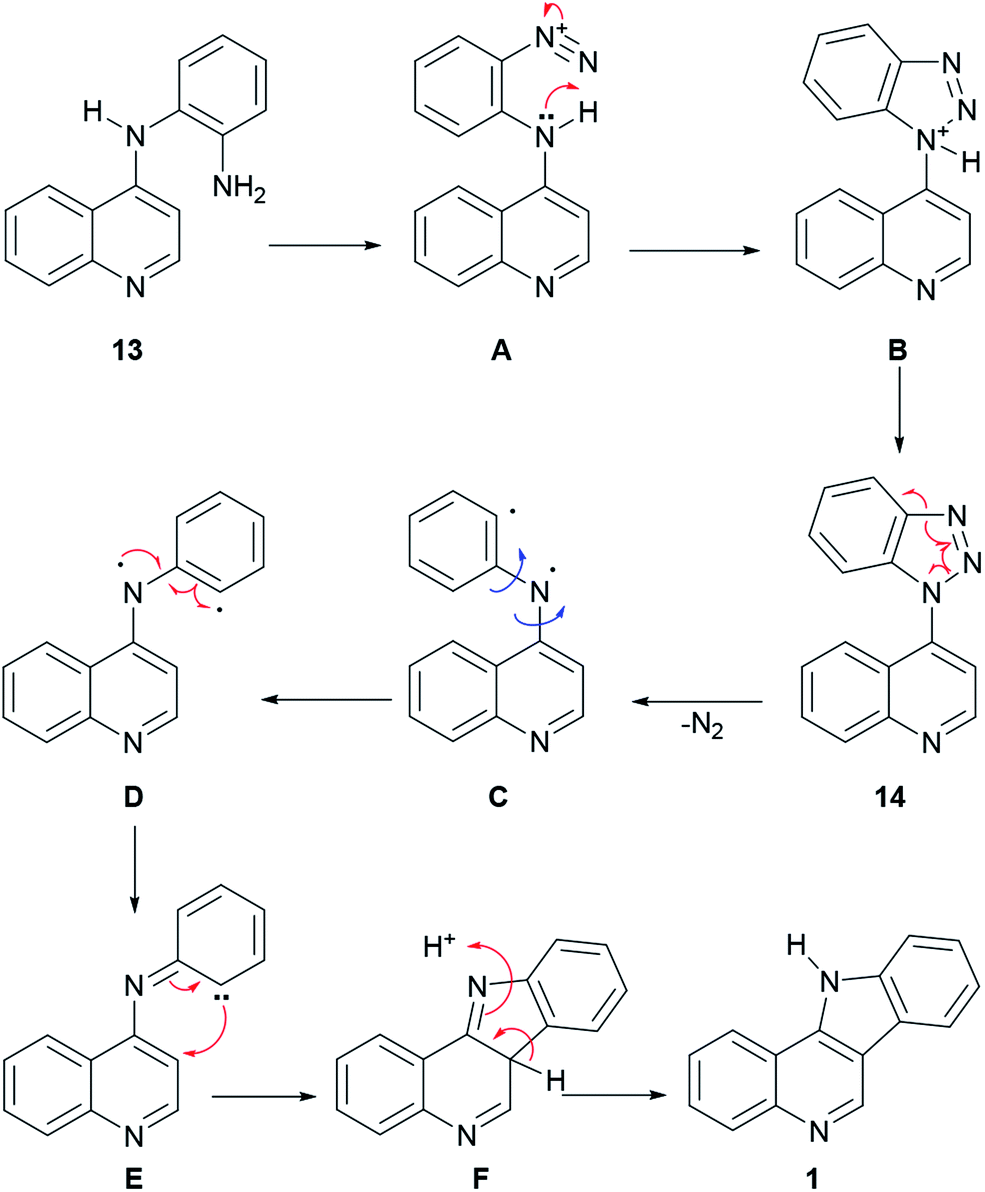 | ||
| Scheme 2 Proposed mechanism of the Graebe–Ullmann synthesis of isocryptolepine. | ||
This contribution complemented the earlier findings by Clemo and Perkin,44 which can be regarded as a synthesis of an advanced intermediate of the natural product. These authors demonstrated that upon heating with aqueous sulfuric acid, the phenylhydrazone of 1,2,3,4-tetrahydroquinolin-4-one underwent a one-pot Fischer indolization and subsequent dehydrogenation to give 1. Employing the same approach but starting with the phenylhydrazone of N-methyl-1,2,3,4-tetrahydroquinolin-4-one (obtained from N-methylaniline in 15% yield) Braunholtz and Mann also synthesized isocryptolepine in 1955 in an unspecified yield.45
In 1965, Roussel et al. reported an alternative approach toward 1 (Scheme 3) by way of the 1,2,3,4-tetrahydroquinolin-4-one 16.46 The ketone was best prepared in 68% yield by direct cyclization of β-N-phenylalanine (15, obtained in turn from aniline and methyl acrylate) with PPA at 130 °C for 20 min. However, 16 could also be accessed in three steps and in substantially the same yield via the aza-Michael addition of aniline to methyl acrylate, followed by ester hydrolysis and cyclization.
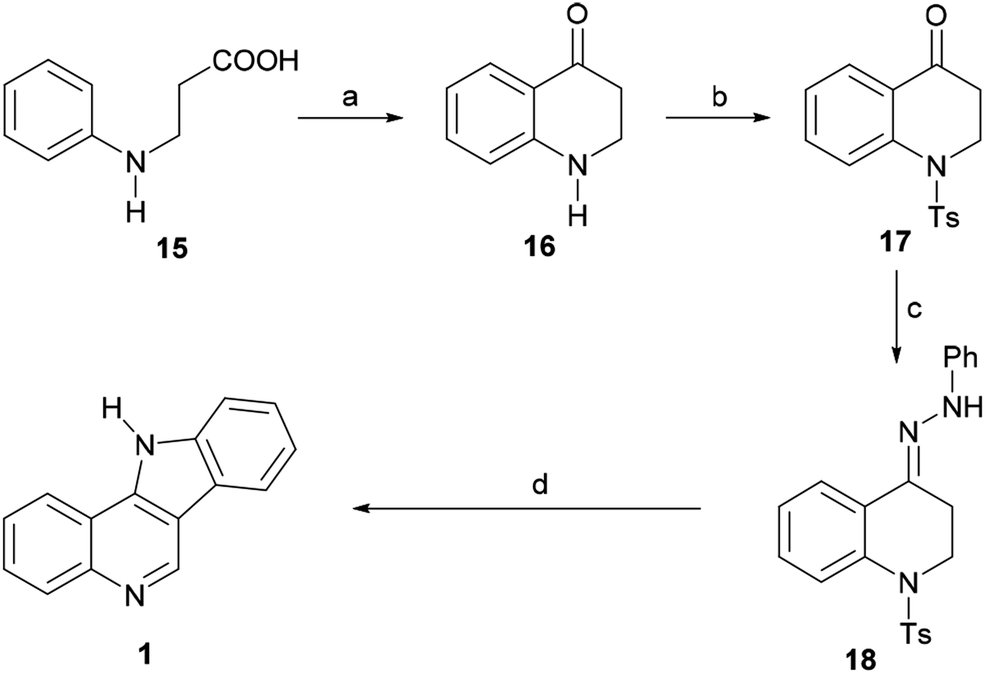 | ||
| Scheme 3 Reagents and conditions: (a) PPA, 130 °C, 20 min (68%); (b) TsCl, Et3N, CH2Cl2; (c) PhNHNH2, EtOH, AcOH (cat), 80 °C, 90 min, 74%; (d) AcOH, H2SO4, 10 min, Δ, 50%. | ||
The latter was converted into the tosylamide 17 and further indolized in 50% overall yield by first heating it for 90 min at 80 °C with phenylhydrazine in EtOH, to give the hydrazone 18, and then heating it for 10 min in AcOH, under H2SO4 promotion. The cyclization stage was accompanied by both detosylation and dehydrogenation toward 1.
In 1968, García et al. disclosed an additional alternative process of synthesizing [3,2-c]indoloquinoline (1).47 These authors prepared 3-(2-fluorobenzoyl)indole (19) via the acylation of indolylmagnesium bromide with o-fluorobenzoyl chloride (Scheme 4) and found that the subsequent treatment of the latter with ethylenediamine in refluxing pyridine gave the substituted aminobenzoylindole imine derivative 20 in 54% yield via the nucleophilic exchange of fluorine for an amine.48 Upon acid treatment, 20 underwent rearrangement to afford a mixture of the quinolone 21 and the indoloquinoline 1. However, heating the quinolone in diphenyl ether under reflux gave the fused heterocycle 1 as the sole product, apparently in very low yield.
 | ||
| Scheme 4 Reagents and conditions: (a) H2NCH2CH2NH2, pyridine, reflux, 20 h (54%); (b) 2 N H2SO4, EtOH, reflux, 72 h (70%); (c) Ph2O, reflux (low yield). | ||
In 1993, the Alvarez-Builla group improved and simplified the original sequence of Kermack and Storey,38a,49 by reporting a one-pot Graebe–Ullmann approach to the tetracyclic precursor 1 of the natural product, as a surrogate for the traditional Fischer indolization approach (Scheme 1). The process, which was later employed by Murray et al. for the preparation of analogs for biological evaluation as antimalarials,36d could be carried out in an open vessel using a domestic microwave oven.
The strategy involved two irradiation stages; first, a solventless mixture of 11 and 15 was irradiated at 160 W to obtain the benzotriazole derivative 14 in 90% yield; then, without purification of the benzotriazole, a second irradiation process was performed at 160 W in the presence of pyrophosphoric acid to produce the tetracycle 1 in 60% yield after 4–6 min. Interestingly, it was observed that the attained yield under these conditions was similar to that obtained by applying conventional heating. It was also concluded that irradiation of a solventless mixture of the pure reactants was more efficient than using their supported forms, over silica gel or montmorillonite.
4b. Syntheses from benzenoids
 | ||
| Scheme 5 Reagents and conditions: (a) ClCH2CO2H, NaOH (69%); (b) PPA, 130 °C, 10 min (96%); (c) PhNHNH2, AcOH (cat.), EtOH, 80 °C, 1.5 h (87%); (d) H2SO4, AcOH, 100 °C, 10 min (56%); (e) MeI, K2CO3, Me2CO, reflux, 3 h (82%); (f) MeI, PhMe, ∼20 °C (64%); (g) K2CO3, H2O, ∼20 °C (93%); (f) I2, TBHP, CHCl3, rt, 14 h (75%). | ||
The final product was obtained from N-tosylaniline (22) via a six-step sequence in an overall yield of 19.2%. To that end, the authors performed the N-alkylation of 22 with β-chloropropionic acid51 to obtain the sulfonamide 23 in 69% yield and cyclized the latter by heating at 130 °C with polyphosphoric acid,52 to access the required intermediate carbonyl derivative 17 in 96% yield.
In turn, the 1,2,3,4-tetrahydroquinolin-4-one 17 was exposed to phenylhydrazine under AcOH catalysis, furnishing 87% yield of the intermediate hydrazone 18. Subsequently, this compound was subjected to indolization with H2SO4 in acetic acid at 100 °C, to give 1 in 56% yield. Exposure of the tetracycle 1 to MeI afforded the mono-methiodide 24 in 64% yield. Final basic treatment to free the base gave isocryptolepine (7) in 93% yield. Interestingly, in the presence of K2CO3, the bis-N-methylated isocryptolepinium derivative 26 was obtained in 82% yield. More recently, 26 was accessed by Volvoikar and Tilve in 75% yield from 25, employing a cross-dehydrogenative coupling reaction with TBHP and I2.53
The mechanism of the transformation of the hydrazone intermediate 18 into the tetracycle 1 is depicted in Scheme 6. It is proposed that the protonation of 18 to afford A would give B after tautomerization. In turn, the latter could become protonated (C) and rearrange to D followed by rearomatization to give the anilino-iminium ion E. Nucleophilic attack of the aniline of the iminium moiety would afford intermediate F, which could then provide 1 after losing ammonia and then toluenesulfinic acid, driven by the possibility of aromatization.
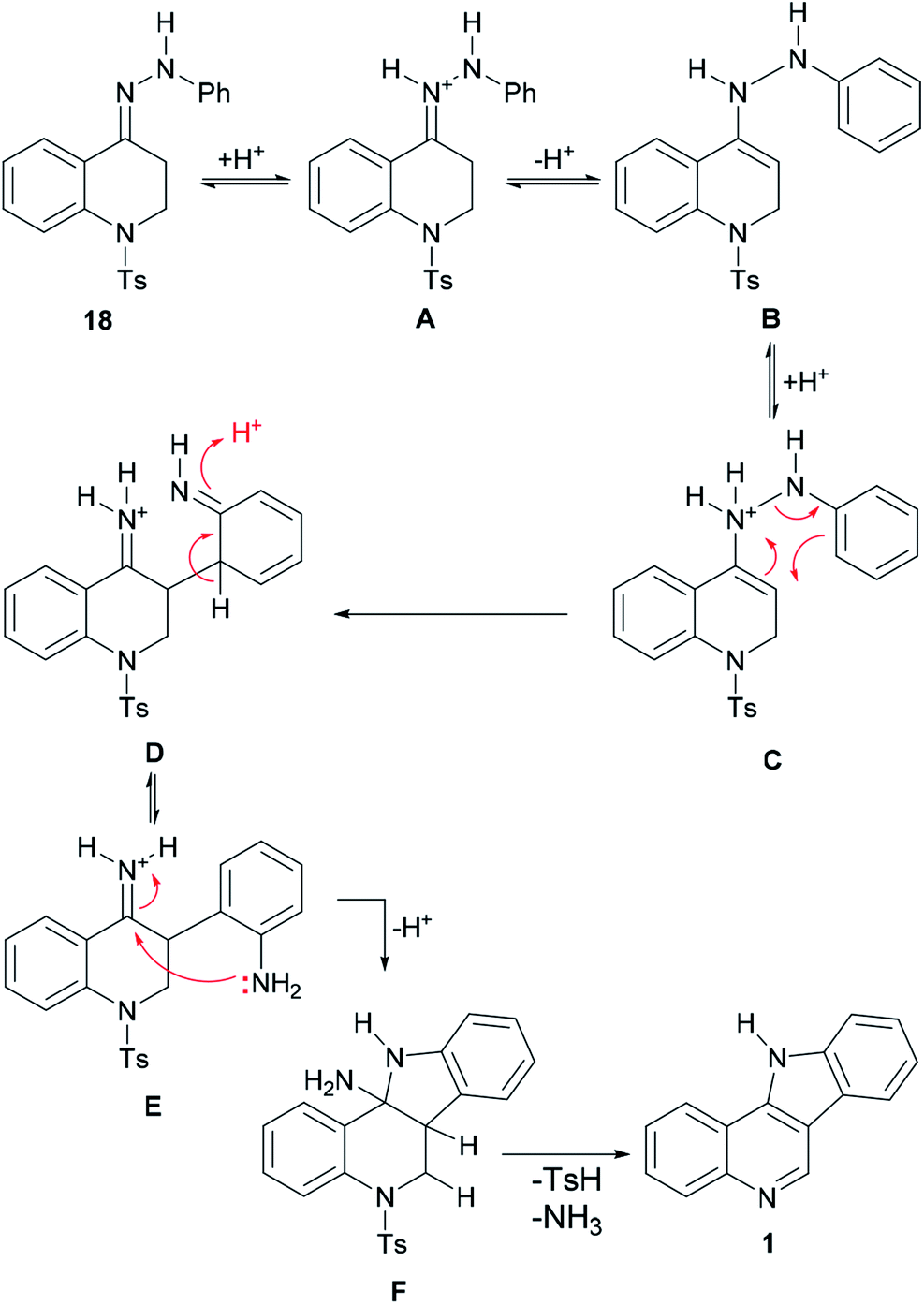 | ||
| Scheme 6 Mechanism of the indolization of hydrazone 18 toward indoloquinoline 1. | ||
In their rather long synthetic sequence, the phosphonium salt 27 was condensed with the azidobenzaldehyde 28 employing K2CO3 and dibenzo-18-crown-6, to give the stilbene derivative 29 in 85% yield as a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Z:E isomers (Scheme 7). Next, the Staudinger reaction of the azide 29 with n-Bu3P provided the iminophosphorane 30 which was hydrolyzed to the aniline 31 in 84% overall yield, as a mixture of isomers (Z:E = 7:1). Subsequently, the catalytic isomerization of the double bond with the thiophenol/AIBN reagent system afforded E-aminostilbene 31 in 92% yield. This compound contains the nitrogen atoms required to install both heterocyclic rings.
:1 mixture of Z:E isomers (Scheme 7). Next, the Staudinger reaction of the azide 29 with n-Bu3P provided the iminophosphorane 30 which was hydrolyzed to the aniline 31 in 84% overall yield, as a mixture of isomers (Z:E = 7:1). Subsequently, the catalytic isomerization of the double bond with the thiophenol/AIBN reagent system afforded E-aminostilbene 31 in 92% yield. This compound contains the nitrogen atoms required to install both heterocyclic rings.
 | ||
| Scheme 7 Reagents and conditions: (a) K2CO3, dibenzo-18-crown-6, CH2Cl2, rt (85%, Z/E = 4:1); (b) n-Bu3P, CH2Cl2, rt; (c) THF–H2O, rt (84%, Z/E = 7:1); (d) PhSH, AIBN, PhH, reflux (92%); (e) triphosgene, CH2Cl2, Et3N, 0 °C → rt, 1 h; (f) PhNO2, MW, 150 °C, 12 min (80% overall); (g) CH3I, DMF, 60 °C (82%); (h) H2, Pd/C, EtOH, rt (91%); (i) (1) NaNO2, H2SO4; (2) NaN3, H2O (85%); (j) (1) Me3P, PhNO2, rt, 45 min; (2) MW, 180 °C, 30 min (40% overall); (k) o-xylene, 150 °C (82%); (l) Red-Al, PhMe, reflux, 32 h (90%). | ||
Treatment of E-31 with triphosgene and further microwave-promoted cyclization of the resulting isocyanate 32 furnished the quinolin-2-one derivative 33 in 80% overall yield.55 The latter was next transformed into the common intermediate 36 by means of a three-step sequence involving N-methylation to give 34 (82% yield) followed by Pd/C-mediated catalytic hydrogenation to afford 35 in 91% yield and final one-pot treatment of the amine with HNO2, and subsequent reaction of the resulting diazo intermediate with NaN3 to provide the azide derivative 36 in 85% yield.
The indolization of the azide 36 toward the framework of isocryptolepine was performed by refluxing the azide in ortho-xylene, which afforded 37 (isocryptolepinone) in 82% yield. Interestingly, 37 was previously prepared from N-methyl-4-azido-3-phenylquinolin-2-one, by refluxing in dimethylformamide (DMF) for 2 h (72% yield) through quite a similar nitrene-based reaction, and in 61% yield via irradiation with a low pressure mercury lamp at room temperature for 4 h.56 More recently, the Sagar group reported the synthesis of the lactam 37 in 68% yield via the reductive cyclization of 34 under MoO2Cl2(DMF)2 catalysis.57
Final reduction of the carbonyl moiety with Red-Al in toluene at reflux gave isocryptolepine (7) in 90% yield. Interestingly, treatment of 36 with the highly reactive Me3P gave an intermediate iminophosphorane, which upon microwave irradiation in nitrobenzene at 150–180 °C for 30 min furnished cryptotackieine (6) in 40% yield via an aza-Wittig type reaction.58
Unlike most of the previous strategies, which relied predominantly on the formation of the indole by ring closure to culminate the synthesis, their approach was characterized by the formation of the quinoline ring in the final step.59
The sequence was based on Kundu's modified version of the Pictet–Spengler cyclization.60 To find the correct conditions, they first put in place a Fischer indole synthesis between phenylhydrazine (38) and 2-methylaminoacetophenone (39)61 to afford the intermediate 40 (Scheme 8).62
 | ||
| Scheme 8 Reagents and conditions: (a) (1) AcOH (5 mol%), EtOH, 6 h; (2) PPA, 100 °C, 10 min; (b) (CH2O)n, TFA, MeCN, 80 °C (sealed tube), 2 h; (c) air [O] (86% overall); (d) MeI, PhMe, reflux, 2 h; (e) Tf2O, Ph3PO, r.t., 15 min (75%), (f) (1) HCOH, CuI, L-proline, NH3·H2O, K2CO3, DMSO 100 °C, 12 h; (2) HCl, air, 120 °C, 8 h (32%). | ||
The latter was then subjected to a Pictet–Spengler reaction with paraformaldehyde in the presence of TFA, to directly furnish the indoloquinoline 44. Presumably 44 was formed through the intermediacy of imine 43. In turn, 44 underwent spontaneous aerobic oxidation to afford 1 in 86% yield.63 Then, the tetracycle was subjected to selective N-methylation, by treatment with MeI in refluxing toluene to furnish isocryptolepine.
Interestingly, when exposed to Hendrickson's reagent (Tf2O-PPh3O), the N-formyl derivative 41 gave 1 in 75% yield, through a regioselective 6-endo-cyclization.64 The precursor 41 was elaborated efficiently via a gold(III)-catalyzed 5-endo-dig cyclization.65 On the other hand, a regioselective copper-catalyzed one-pot cascade reaction of bromoarene 42 with formaldehyde and aqueous ammonia in DMSO has been developed, which affords 1 in 32% yield.66
Thus, the required imine 47 was synthesized by a reaction between 45 and 46 under AcOH catalysis, employing microwave irradiation at 80 °C on a methanolic solution of the reactants (Scheme 9). The reaction of the as-formed nitro-imine 47 with KtBuO in THF at 0 °C gave the indole 48 in 72% overall yield, presumably through the anionic species A, which may lose PPh3 after undergoing cyclization to the intermediate B, which in turn undergoes a final H-shift and aromatization.
 | ||
| Scheme 9 Reagents and conditions: (a) (1) AcOH, MeOH, MW (80 °C), 10 min (72%); (b) KtBuO, THF, 25 °C, 1 h; (c) Fe0, HCl, EtOH (90%); (d) (CH2O)n, TFA, MeCN, 80 °C (81%); (e) MeI, PhMe (91%). | ||
Reduction of the nitro group with elemental iron (Fe0) and HCl in EtOH to aniline 40 and further formylation of the indole moiety with paraformaldehyde and TFA with subsequent in situ cyclization completed the tetracycle 1 in 82% yield. Finally, according to Agarwal et al.,59 the methylation of 1 with MeI in toluene gave 7 in 91% yield.
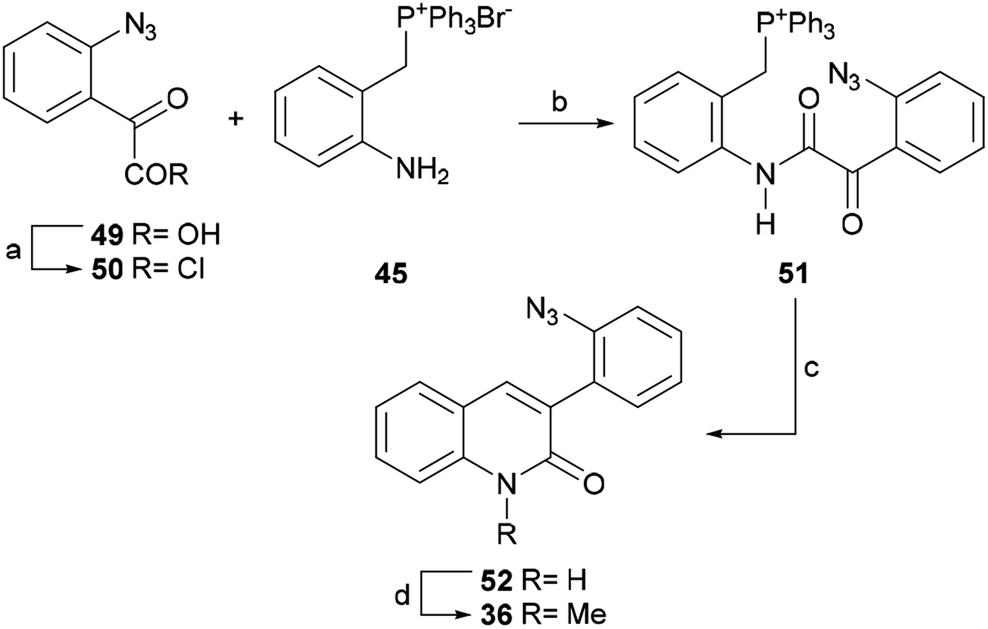 | ||
| Scheme 10 Reagents and conditions: (a) SOCl2, PhH, reflux, 1 h or (COCl)2, CH2Cl2, rt, 3 h; (b) CH2Cl2, rt, 12 h; (c) KtBuO, THF, rt, 5 h (62% overall); (d) MeI, K2CO3, DMF, 60 °C, 8 h (98%). | ||
The product was coupled with the phosphonium bromide 45 and the resulting amide 51 was treated with potassium tert-butoxide at room temperature, where it underwent an intramolecular Wittig reaction to afford lactam 52 in 62% overall yield from 49.
The N-methylation of 52 with methyl iodide in the presence of K2CO3 in DMF gave 36 in 98% yield. This lactam is a key intermediate in the total synthesis of isocryptolepine by Molina.54b The latter group invested in nine steps to arrive at the intermediate 36, in 22% overall yield, whereas this shortcut advantageously accomplished the same purpose in 60% yield over 4 steps in only two steps.
 | ||
| Scheme 11 Reagents and conditions: (a) (1) HCl, 30 min, 80 °C; (2) NaNO2, 0 °C, 40 min; (3) furfural, CuCl2, H2O (53%); (4) NaBH4, AlCl3, THF, 0 °C, 20 min; then reflux, 2 h (67%); (5) RANEY® nickel, NH2NH2, EtOH, reflux, 2 h (76%); (b) 2-NO2C6H4CHO, HCl, AcOH, 30–35 °C, 1.5 h (58%); (c) Fe0, AcOH, reflux, 5 min (86%); (d) (1) MeI, PhNO2; (2) NH4OH, H2O (86%). | ||
The reaction of 55 with 2-nitrobenzaldehyde afforded the expected indole derivative 56, but in comparative lower yield (58%) than for other aldehydes. This was explained as being the result of a combination of unfavorable steric and electronic effects of the ortho-nitro moiety, which affected the iminium ion attack on the furan ring.
A possible reaction mechanism includes a first step, where the Schiff base A is formed by the condensation of 55 with the aldehyde. Then, protonation of this intermediate (B) should drive the ring opening of the furan motif and afford the oxaaza-spirocyclic intermediate C, which after undergoing ring opening of the dihydrofuran moiety triggered by aromatization of the nitrogen ring, affords the 2-substituted indole 56.
In the next stage, the reduction of the nitro group, various reagent systems were studied (Zn/AcOH, Zn/NaOH, RANEY® nickel, Fe/AcOH), with the finding that Fe in AcOH directly affords the indoloquinoline 1 in 86% yield. The formation of 1 could result from the reduction of the nitro group to the corresponding aniline 57, followed by an intramolecular Michael addition to give intermediate D,74 and aromatization of the pyridine ring of the latter via the rather unusual elimination of acetone.75 For the N-methylation stage, the best result (86% yield) was obtained using the Kermack–Storey procedure, employing MeI in nitrobenzene.43
Their contribution included the preparation of the tetracycle 1, which constitutes a formal total synthesis of isocryptolepine.76 For this purpose, the authors specifically prepared the 3,4-dihaloquinoline 63 (Scheme 12).
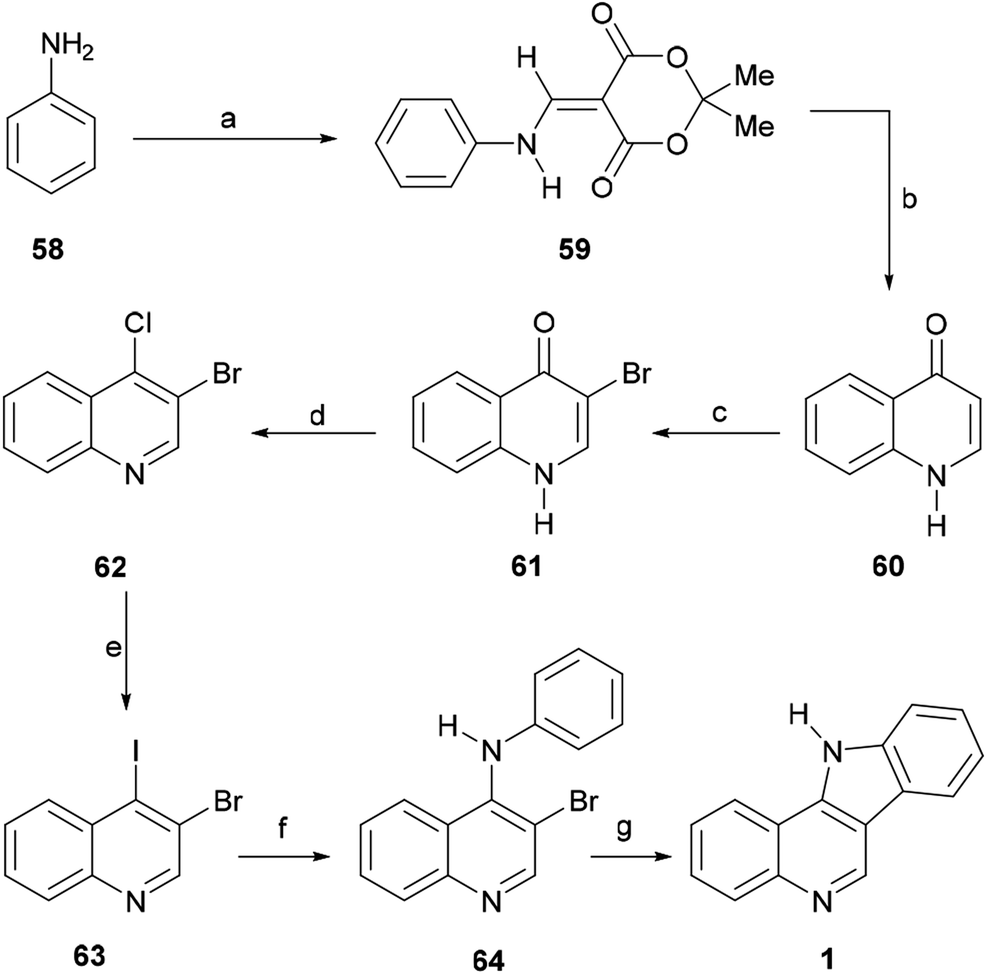 | ||
| Scheme 12 Reagents and conditions: (a) Meldrum's acid, HC(OMe)3, reflux (77%); (b) Ph2O, 250 °C (92%); (c) Br2, AcOH, reflux (99%); (d) POCl3, MeCN, reflux (93%); (e) NaI, MeCN, reflux (55%); (f) PhNH2, Pd(OAc)2, Xantphos, Cs2CO3, PhMe, MW (120 °C), 1.5 h (95%); (g) PdCl2(PPh3)2, NaOAc, DMA, MW (150 °C), 1 h (64%). | ||
The key 1H-quinolin-4-one intermediate 60 was obtained in 77% yield, via the condensation of the aniline 58 with Meldrum's acid in the presence of trimethyl orthoformate to afford 59, followed by cyclative thermolysis of the latter in diphenyl ether.77 The direct halogenation of 60 with bromine in AcOH78 gave the expected 3-bromo-1H-quinolin-4-one 61 in 99% yield, whilst treatment of the latter with POCl3 afforded 3-bromo-4-chloroquinoline (62) in93% yield. Final installation of an iodide motif was accomplished via a Finkelstein-like displacement of the chloride with NaI in MeCN, affording the desired heterocycle 63 in 55% yield.79
Next, the dihaloquinoline 63 was coupled with aniline in toluene, employing palladium catalysis, with Xantphos as a ligand and Cs2CO3 as a base, under microwave irradiation at 120 °C for 90 min, to selectively afford 64 in 95% yield. Final cyclization of 64 to the indoloquinoline motif 1 was performed in 64% yield, through an intramolecular Heck-type reaction in DMA, under microwave irradiation (1 h at 150 °C), using PdCl2(PPh3)2 as a catalyst and NaOAc as a base.80
The Maes group prepared analogous intermediates through a different synthetic sequence and developed several syntheses of various indoloquinolines. An interesting advantage of the Kámán synthesis over the previous reports by Maes et al. on the tandem palladium-catalyzed approaches to indoloquinolines is that in the latter cases, small amounts of the undesired regioisomer were detected after the ring closure.10a,80
Their approach was applied in 2016 to the synthesis of highly functionalized indoles, including isocryptolepine, via C–H/N–H functionalization of (hetero)-arylamines with tethered alkynes.85 The synthesis (Scheme 13) commenced with a Sonogashira cross-coupling between N-methyl-2-iodoaniline (65) and the acetylenic acetal 66 in Et3N–THF at room temperature. The subsequent reaction of the resulting intermediate, obtained in 81% yield, with phenylisocyanate in toluene, gave the urea 67 in 78% yield.
 | ||
| Scheme 13 Reagents and conditions: (a) (1) Pd(Ph3P)2Cl2, CuI, Et3N:THF (1:1, v/v), rt, 12 h (81%); (2) PhNCO, Et3N, PhMe, rt, 48 h (78%); (b) Cp2Fe, MeOH:THF (1:5, v/v), Na2CO3, nBu4NBF4, reflux, 20 h; electrolysis at constant current (5 mA) with a reticulated vitreous carbon anode, Pt cathode; (c) 0.2 N HCl, rt, 30 min (63% overall); (d) NaOH, EtOH, reflux, 2 h (91%). | ||
Compound 67 became a suitable substrate for the electrolysis, which was carried out at a constant current of 10 mA on the gram-scale, in the presence of ferrocene as a redox relay reagent. The electrolyte was a mixture of sodium carbonate and tetrabutylammonium tetrafluorborate in THF–MeOH. A platinum plate was used as a cathode and reticulated vitreous carbon served as an anode.
This process afforded the tetracyclic acetalic compound 68, which after its subsequent acid-promoted hydrolysis resulted in the formation of the formyl-substituted indole 69 in 63% overall yield. Compound 69 was readily transformed into isocryptolepine (7) in one step and 91% yield, via the base-promoted hydrolysis of the urea motif, which freed the N-methylaniline moiety, enabling its spontaneous intramolecular condensation with the free formyl group. Among the advantages of this approach are the facts that it proceeds through the liberation of H2 and does not require the use of noble-metal catalysts and external oxidants for the cyclization stages.
A plausible mechanism for the electrochemical formation of the product was proposed based on previous studies.84 Accordingly, the anodic oxidation of [Cp2Fe] to [Cp2Fe]+ takes place with the concomitant cathodic reduction of MeOH to form the methoxide anion and release H2. Then, the base deprotonates the amino group of the substrate 67 to furnish the anion A, which is a much better electron donor than its neutral precursor.
Therefore, single-electron transfer (SET) takes place between intermediate A and [Cp2Fe]+ to afford the electron-deficient nitrogen-centered radical B86 and regenerate Cp2Fe.87 It was suggested that the SET is so efficient, that it competes well with the cathodic reduction of [Cp2Fe]+, enabling the electrolysis to be executed in an undivided cell. Next, the intermediate B participates in a rare 6-exo-dig cyclization83 to give the vinyl radical C, which then undergoes a second cyclization with the aryl ring, to afford the delocalized tetracyclic radical D. Finally, the rearomatization of the latter by electron and proton eliminations generates the product 68.
In that direction, in 2017 Aksenov et al. conjectured that PPA could promote a one pot synthesis of the intermediate hydrazone 70 required89 for Fischer indolization (Scheme 14), as well as drive to completion the formation of the indole moiety (71), and also participate in the subsequent intramolecular Vilsmeier reaction between an aromatic amine and a carboxylic acid, for the final cyclization leading to isocryptolepine (7).90
 | ||
| Scheme 14 Reagents and conditions: (a) HCO2H (cat.), EtOH, reflux, 30 min, or neat, 120 °C, 20 min; (b) 80% PPA, 80 °C, 2 h; 130 °C, 90 min (83%). | ||
However, PPA failed to promote the synthesis of the hydrazone between ortho-aminoacetophenone (39) and phenylhydrazine (38), favoring self-condensation of the ketone, and this step had to be performed in EtOH under formic acid catalysis91 or even better, under solventless conditions, for 10 min at 120 °C. Furthermore, despite PPA was able to promote the Vilsmeier cyclization with various carboxylic acids, but formic acid proved to be unstable to the reaction conditions, decomposing into CO and CO2 and furnishing 7 in only 25% yield.92
Therefore, a formic acid surrogate was sought and triazine was proposed to fulfill this place. The authors found that once hydrazone 70 was formed, it could be treated with PPA to afford indole 71, and shortly after with triazine (72) to produce isocryptolepine as the sole reaction product in an optimized 82% yield, using a convenient metal-free process.
The proposed reaction mechanism of the transformation involving triazine as a formic acid equivalent comprises the addition of the triazine (72) to the Fischer indolization product 71 to produce species A. Next, ring opening of the dihydrotriazine and subsequent intramolecular electrophilic attack of the resulting linear species B at C3 of the indole furnishes the six-membered heterocyclic ring (C). Finally, proton transfer, followed by an elimination (D) anchimerically-assisted by the quinoline nitrogen, affords isocryptolepine. An alternate deprotonation/elimination sequence leading to 7 has also been proposed. In addition, the same group devised a second generation PPA-mediated one-pot three-component synthesis of the indoloquinoline scaffold employing nitroalkanes. The use of MeNO2 in place of the triazine 72 furnished 7.93
Interestingly, the use of dimethyl sulfoxide (DMSO) as a one-carbon atom synthon in a similar transformation leading to the unsubstituted indoloquinoline skeleton in 69% yield was recently reported.94 In this case, it was proposed that two mechanistic pathways are possible. In the first case, at high temperature, DMSO may generate methyl radicals (Me˙), which can react with O2 to produce a peroxy radical and then CH2O.95 In turn, this could result in imine formation and a subsequent electrophilic aromatic substitution to produce the observed product. It is also possible that DMSO could decompose at high temperature, and produce CH2O directly.96
4c. Syntheses from quinolines
The total syntheses of isocryptolepine, which employs quinolines as starting materials, comprise synthetic efforts where haloquinolines or 1,2,3,4-tetrahydroquinolin-4-one derivatives were employed. They complement other accomplishments, such as that of Bogányi and Kámán,76 where quinolines were prepared as intermediates from structurally simple benzenoids. | ||
| Scheme 15 Reagents and conditions: (a) Pd(0), DME, H2O, NaHCO3, reflux, 4 h (90%); (b) 20% H2SO4, reflux, 24 h (93%); (c) (1) NaNO2, HCl (conc.), 0 °C, 1 h; (2) NaN3, 0 °C, 1 h (80%); (d) 1,2-Cl2C6H4, 180 °C, 5 h (75%); (e) (1) Me2SO4, CH3CN, reflux, 5 h; (2) K2CO3, H2O (93%); (f) PhMgBr, THF, 0 °C, 15 min (46%). | ||
The thermal cyclization of 77 was executed in refluxing dichlorobenzene, and proceeded in 70% yield to exclusively furnish the required tetracycle 1 in 75% yield,101 by way of a singlet nitrene intermediate that inserts into the C4–H bond. The reaction of 1 with Me2SO4 in MeCN resulted in the regioselective alkylation of the pyridine nitrogen, affording isocryptolepine (7) in 93% yield after freeing the base by treatment with potassium carbonate.
An alternate pathway for accessing 1 in one pot and 46% yield from 78 was recently disclosed by Ess, Kürti and coworkers, through the electrophilic amination of the quinoline C4–H bond with PhMgBr.102 On the other hand, the Sydnes group disclosed the preparation of tetracycle 1 from the aniline derivative 76 in 73% yield, employing a palladium-catalyzed C–H activation/C–N bond formation strategy.103
 | ||
| Scheme 16 Reagents and conditions: (a) Pd(OAc)2, BINAP, K2CO3, dioxane, reflux, overnight (60%), or Pd2(dba)3, Xantphos, Cs2CO3, dioxane, reflux, overnight (81%); (b) Pd2(dba)3, P(t-Bu)3, K3PO4, dioxane, 120 °C (closed vessel), 3 h (95%); (c) (1) CH3I, DMF, 80 °C, 1 h; (2) overnight, r.t.; (3) aq. Na2CO3 (57%); (d) Pd2Cl2(dba)3 (5 mol%), P(Bu)3 (20 mol%), K3PO4 (20 mol%), dioxane, 125 °C closed vessel, 24 h (82%, from 79, R = Cl) or PdCl2(PPh3)2 (1 mol%), NaOAc·3H2O (1.47 mol%), DMA, MW, 200 °C, 10 min (79%, from 80, R = Br). | ||
However, the use of Xantphos as a ligand improved the yield to 81% when Cs2CO3 was employed as a base.108 In order to execute the required direct arylation of 81, a nucleophilic palladium catalyst system better than Pd/BINAP was deemed necessary.109 Therefore, trialkylphosphine ligands, which are more electron rich than their triaryl counterparts, were examined, with the finding that the cyclization could be carried out to afford a 95% yield of 1 when tributylphosphine was used,110 in the presence of potassium phosphate.
The direct arylation leading to 1 was considered a notable example of the intramolecular Pd-catalyzed arylation of an electron-deficient heteroaromatic. Examples of these reactions are scarcer in the literature compared to those of electron-rich heteroarenes.111 Final, methylation of 1 with MeI in DMF, followed by treatment with Na2CO3 to release the base, afforded isocryptolepine (7) in 57% yield.
The synthetic strategy was further improved by these authors in 2008,112 with the development of an auto-tandem palladium-catalyzed consecutive C–N and C–C bond forming process, which enabled the coupling of 4-chloroquinoline (11) and 2-chloroaniline (79), somehow reminiscent of the auto-tandem syntheses of other carbazole systems.106c,113 Employing the catalytic system previously used for the direct arylation [Pd2Cl2(dba)3, P(tBu)3], with K3PO4 as a mild base for improved compatibility, the tetracyclic compound 1 was obtained in 82% yield. It was also observed that despite K3PO4 not dissolving completely in the reaction medium, the addition of an excess of reagent can speed-up the reaction.
 | ||
| Scheme 17 Reagents and conditions: (a) AcOHgl/12 N HCl (4:1), 135 °C, 5 h (65%); (b) POCl3, 100 °C, 8 h (62%); (c) H2, 10% Pd/C (70%). | ||
To that end, 82 was exposed to phenylhydrazine hydrochloride (38), in a 4:1 mixture of glacial AcOH and concentrated HCl, to give the indoloquinolinone 37 in 65% yield. The latter existed predominantly in the hydroxy form (83); therefore, the required aromatizative deoxygenation of 83 was performed as a two-step process, by reaction with POCl3 (ref. 115) to easily form the intermediate chloro-derivative 84 (62% yield), which was hydrogenolyzed over 10% Pd/C to give isocryptolepine (7) in 70% yield.
The authors observed that the yield of product for the photocyclization in protic solvents was significantly higher than that in benzene alone; hence, the reactions were carried out in a benzene:methanol:sulfuric acid (60:30:1, v/v/v) solution, to afford the tetracycle 1 in 78% yield.10b The final N-methylation of 1 was performed with methyl sulfate in acetonitrile, furnishing isocryptolepine (7) in 83% yield after freeing the base with K2CO3. Furthermore, a similar approach culminating with the photoannulation of a 3-iodoquinoline derivative was employed by this group for the synthesis of a 6-methyl derivative of isocryptolepine.116
In 2006, the same group reported an efficient three-step heteroatom directed photoannulation approach toward the natural product (Scheme 18).10b In their synthetic sequence, 4-chloroquinoline (1) was thermally aminated at 200 °C in 74% yield with 2-chloroaniline (79),117 and the resulting 4-anilinoquinoline derivative 81 (also prepared by Maes et al.) was submitted to an oxidative photoannulation in various solvents and in the presence of iodine, as a mild oxidant.
 | ||
| Scheme 18 Reagents and conditions: (a) 200 °C, 5 h (74%); (b) hν, PhH:MeOH:H2SO4 (60:30:1, v/v/v), I2, rt (78%); (c) Me2SO4, MeCN, reflux, 6 h, K2CO3 (83%). | ||
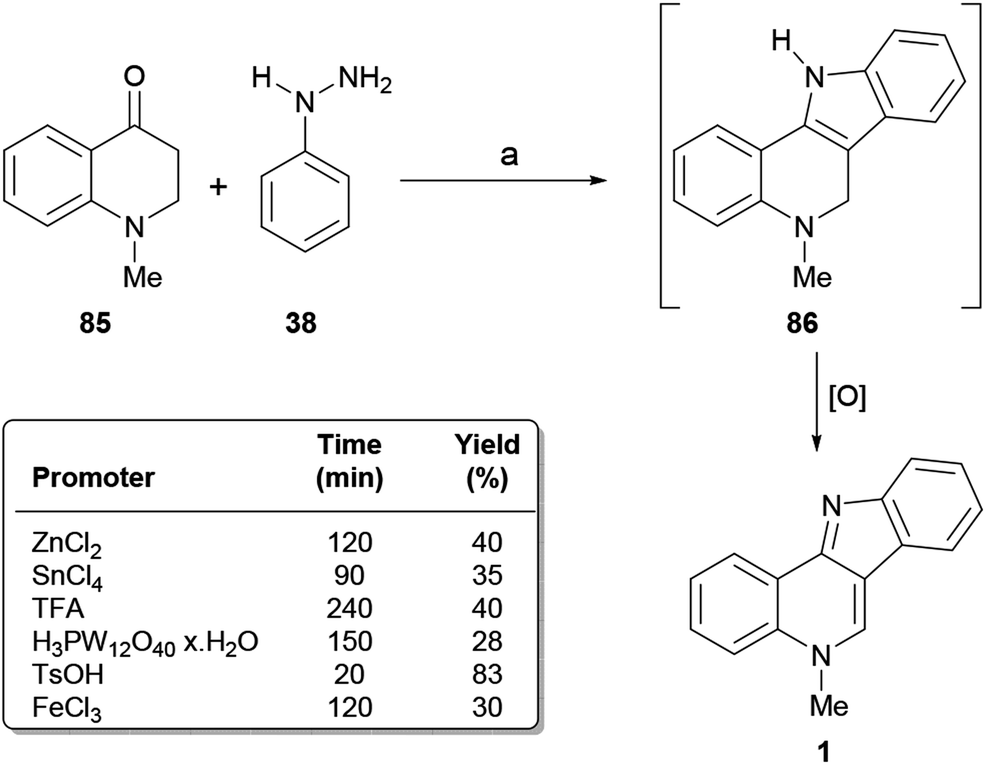 | ||
| Scheme 19 Reagents and conditions: (a) Promoter (1.1 equiv.), neat, air, 100 °C. Reaction times and overall yields are shown in the table. | ||
The transformation involves the cyclocondensation toward the tetracycle 86 and its further dehydrogenation in situ10b,89a,121 to afford the final product. The authors tested different alternative acidic promoters, discovering that heating with a 1.1 equiv. TsOH at 100 °C gave the best result (83% yield).
4d. Syntheses from indoles
The approaches to the total synthesis of isocryptolepine from indoles are characterized by the use of N-protected and/or 3-substituted indoles as starting materials. Although certain syntheses have used an unprotected indole derivative and others have started with 2-substituted indoles, they are fewer in number. Electron withdrawing groups (benzenesulfonyl, Boc) do not always meet C–C or C–N bond formation requirements; in such cases protective groups such as SEM, MOM and benzyl ether have been used.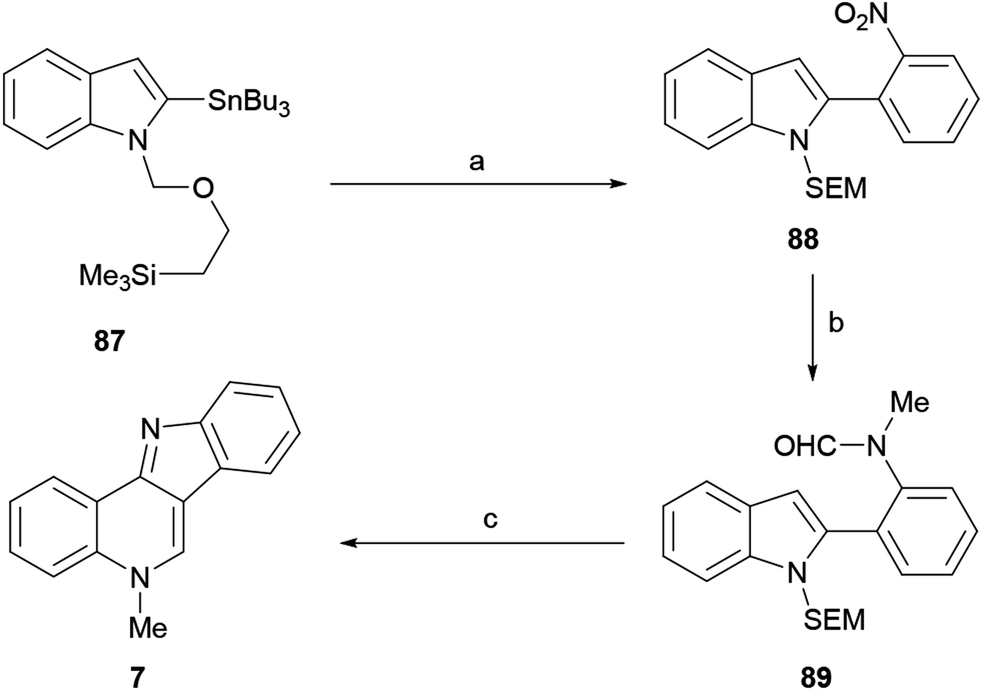 | ||
| Scheme 20 Reagents and conditions: (a) Pd(PPh3)4, 2-IC6H4NO2, THF, reflux (98%); (b) (1) H2, Pd/C, EtOH, rt (98%); (2) AFA, THF, −20 °C; (3) NaH, THF, rt; (4) MeI (95% overall); (c) EtOH, 10% H2SO4, reflux (50%). | ||
Therefore, the 2-tributylstannyl indole 87 was N-protected as the SEM derivative and coupled with 2-iodonitrobenzene under palladium catalysis, to give the 2-(ortho-nitrophenyl)indole 88 in 98% yield. The catalytic hydrogenation of the nitro group (98% yield), followed by N-formylation with acetic formic anhydride (AFA)123 and subsequent N-methylation of the resulting amine with methyl iodide, gave 89 in 95% overall yield.
The latter was subjected to an intramolecular Vilsmeier ring closure with sulfuric acid in ethanol, to afford 7 in 50% yield. Interestingly, protection of the starting indole as the N-phenylsulfonyl derivative produced complex mixtures, presumably due to the deactivating effect of the sulfonyl moiety toward the final electrophilic attack.
 | ||
| Scheme 21 Reactions and conditions: (a) AcOHgl, reflux, 3 h (85%); (b) (1) hν (254 nm), PhH–MeOH, rt, 2:1; (2) I2, 48 h (67% overall); (c) Me2SO4, MeCN, reflux, 6 h, K2CO3 (83%). | ||
Mechanistically, 91 first underwent photo-isomerization of the E-isomer to its Z-congener; then, the excited S1 state of the Z-isomer underwent an orbital symmetry-controlled conrotatory ring closure to the tetracyclic intermediate A.
These types of dihydro intermediates are sensitive to oxygen;121 therefore, A underwent aromatization by way of the loss of the allylic hydrogens to form 1 in 67% yield, in the presence of iodine which acted as a mild oxidizing agent.
The final regioselective methylation of 1 with Me2SO4 in MeCN, in the presence of K2CO3, gave isocryptolepine in 83% yield. This total synthesis was complemented in 2006 with a three step photoannulation approach, which started from a quinoline derivative.10b
 | ||
| Scheme 22 Reagents and conditions: (a) MeCN, rt, 72 h (77%); (b) Pd(TFA)2, Ag2CO3, 5% DMSO, DMF, 50 °C, 48 h (50%); (c) TBAF, THF, rt, 6 h (80%); (d) LiAlH4, dioxane, 1 h (98%). | ||
Next, a projected decarboxylative Heck-type cyclization was executed following the protocol of Myers, by treatment of 94 with 20 mol% Pd(TFA)2 and Ag2CO3 in a mixture of 5% DMSO and DMF at 50 °C for 48 h,130 to give lactam 95 in 50% yield. Interestingly, a similar Heck cyclization was reported earlier by the Mérour group.131 In that process, lactam 95 was desulfonylated in 80% yield, by treatment with TBAF in THF at room temperature for 6 h,54 and the resulting tetracycle 37 was further reductively deoxygenated to isocryptolepine in 98% yield, by treatment with LiAlH4 in hot dioxane.
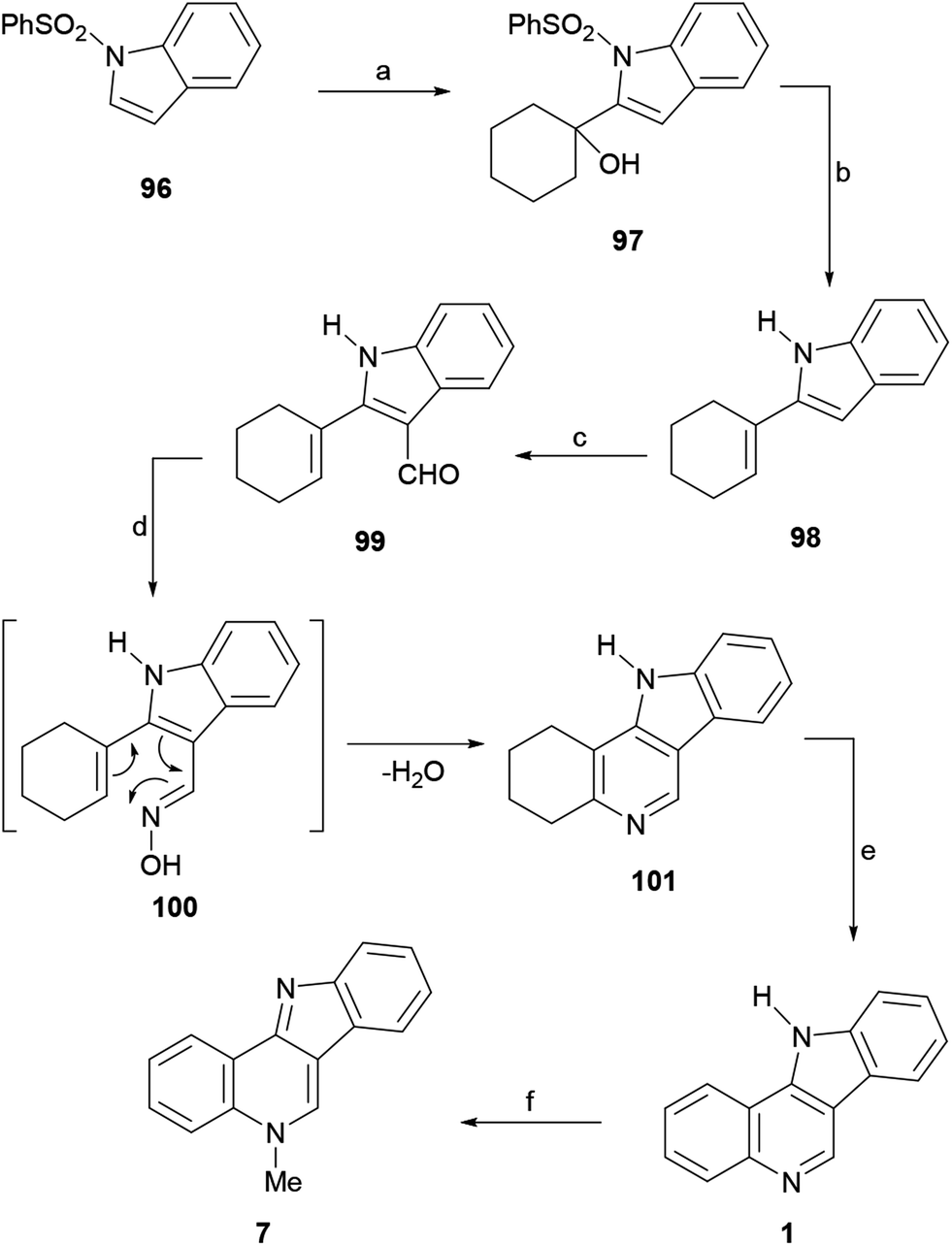 | ||
| Scheme 23 Reagents and conditions: (a) LDA, THF, −78 °C, cyclohexanone, 4 h (86%); (b) TBAF, THF, reflux, 2 h (84%); (c) POCl3, DMF, rt, 1 h (88%); (d) NH2OH·HCl, NaAcO, dioxane, reflux, 24 h (78%); (e) Pd/C, 1,2-Cl2C6H4, reflux, 20 h (88%); (f) (1) MeI, DMF, 80 °C, 1 h; (2) Na2CO3 (85%). | ||
The subsequent Vilsmeier–Haack formylation of 98 gave 99 in 88% yield, while attempted oximation of the latter with hydroxylamine in refluxing dioxane surprisingly furnished the tetracycle 101, presumably as a result of the initial formation of oxime 100 followed by a thermal intramolecular 6π-electrocyclization/elimination process. Therefore, the sequence continued with the dehydrogenation of 101 using Pd/C in ortho-dichlorobenzene, which afforded 1 in good yield. Finally, treatment of 1 with methyl iodide to afford the methiodide and sodium carbonate to free the base, gave 7 in 37% overall yield.
 | ||
| Scheme 24 Reagents and conditions: (a) Pd(PPh3)4, Na2CO3, DME, reflux, 12 h (99%); (b) TFA, CH2Cl2, rt, 12 h (94%); (c) MOMCl, NaH, DMF, rt, 12 h (91%); (d) 1 M LiOH, THF, 60 °C, 12 h (98%); (e) DPPA, Et3N, PhMe, MW, 100 °C, 10 min (99%); (f) Tf2O, Py, CH2Cl2, rt, 30 min (83%); (g) Et3SiH, Pd(OAc)2, dppp, DMF, 60 °C, 1 h (91%); (h) MeOH, HC(OMe)3, CF3SO3H, MeNO2, 100 °C, 30 min (96%); (i) (1) MeI, PhMe, reflux, 2 h; (2) 28% NH4OH (90%). | ||
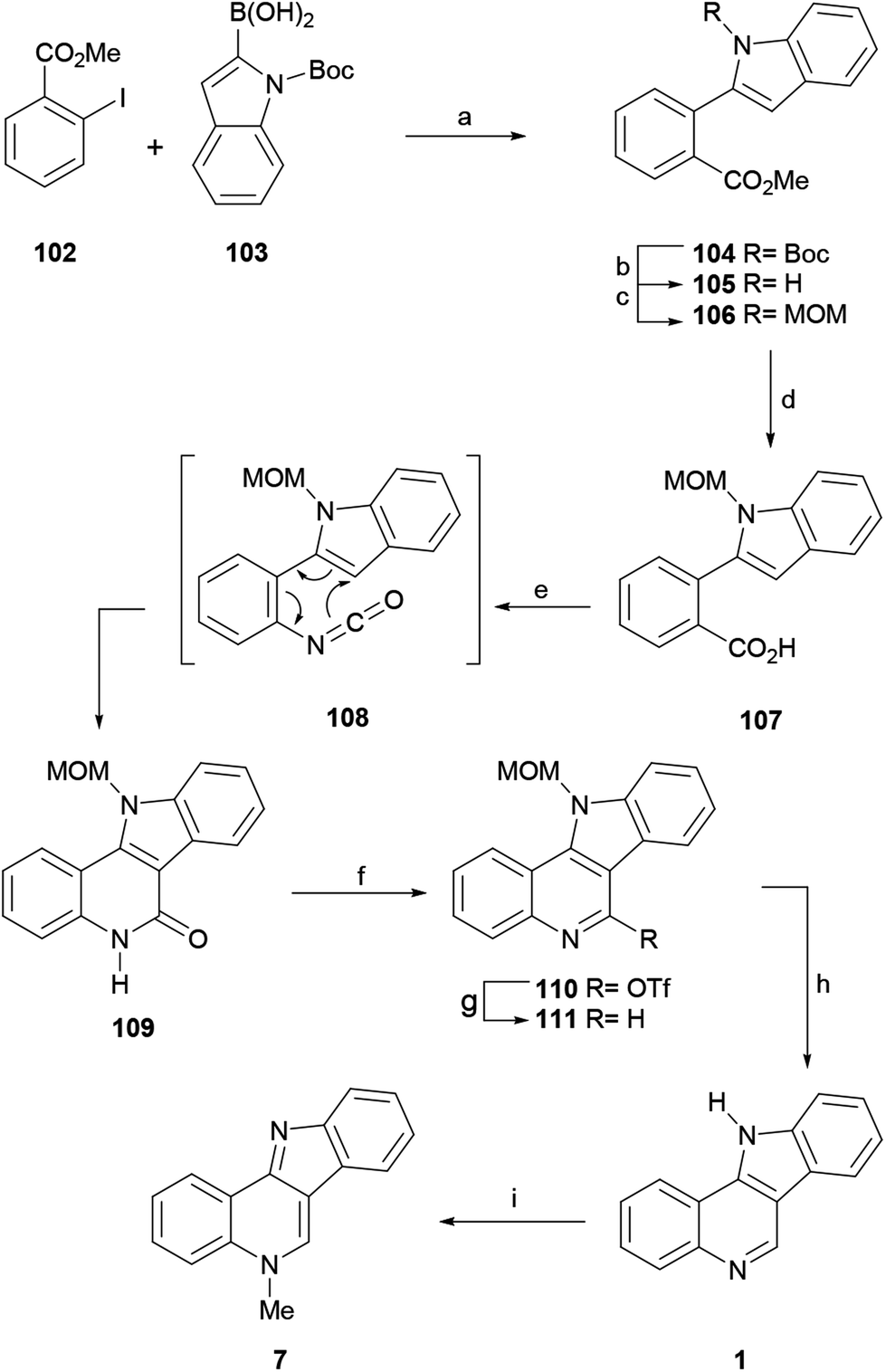
Following the procedure of Beaumont, these authors subjected the 2-iodobenzoate 102 and the indole-2-boronic acid 103 to a Suzuki–Miyaura cross-coupling reaction and obtained 104 in 99% yield. Since Boc proved to be an unsuitable protecting group for the advanced stages of the synthesis, 104 was exposed to TFA, resulting in the deprotection of the N-Boc group in 94% yield (105).135 After trying different protecting groups, the authors selected the MOM group, which has minimum steric demand, to pursue the synthesis.
Hence, treatment of 105 with MOMCl in the presence of NaH gave the N-MOM-indolylbenzoate 106 in 91% yield and subsequent basic hydrolysis of the ester moiety with 1 M LiOH furnished the carboxylic acid 107 in 98% yield. The microwave-assisted Curtius rearrangement of the latter with DPPA in toluene at 100 °C quickly provided tetracycle 109, resulting from a subsequent one-pot thermal 6π-azaelectrocyclic reaction, in an optimized 99% yield, without the isolation of the isocyanate intermediate 108.
The deoxygenation of the lactam, required to complete the total synthesis, was achieved by treatment of 109 with triflic anhydride and pyridine, which afforded triflate 110 in 83% yield. Next, the subsequent reductive cleavage of the latter with Et3SiH under Pd catalysis, according to the procedure of Katsuki,136 provided the N-MOM-indoloquinoline 111 (91% yield), which was deprotected with triflic acid and trimethyl orthoformate in MeOH and nitromethane,137 to give indolo[3,2-c]quinoline 1 in 96% yield. The final methylation of 1 with MeI in toluene59 furnished isocryptolepine in 90% yield after release of the base.
Speculating that the iminium ion could react with an aromatic nucleophile through an electrophilic aromatic substitution to afford a N-arylmethyl arene, in 2012 the Tummatorn group devised the four-step total synthesis of isocryptolepine (Scheme 25).139
 | ||
| Scheme 25 Reagents and conditions: (a) TfOH, CH2Cl2, rt, 14 h (94%); (b) DDQ, CH2Cl2, rt (94%); (c) 2 M NaOH, MeOH, reflux (95%); (d) (1) Me2SO4, MeCN; (2) K2CO3 (78%). | ||
In their synthetic sequence, benzyl azide (112) and N-phenylsulfonyl indole (113) were exposed to TfOH in CH2Cl2 to afford the tetracyclic compound 114 in 94% yield. Presumably, this reaction involved two sequential steps, namely the nucleophilic addition to the iminium ion followed by the intramolecular annulation of the resulting intermediate, resulting in a formal aza-[4 + 2]-cycloaddition.
Dehydrogenation of 114 with DDQ afforded 115 in 94% yield, which was desulfonylated in 95% yield with 2 M NaOH in refluxing MeOH to give the related indoloquinoline 1. The regioselective methylation of the latter with methyl sulfate finally furnished isocryptolepine in 78% yield. Functionalization of the precursor 114 has also been reported.140 On the other hand, the basic synthetic sequence was employed as a key strategy toward the development of analogs of the natural product.141
The Lin group studied the palladium-catalyzed coupling of N-methoxycarboxamides with iodobenzene (117).144 They found that the reaction is capable of forming both C–C and C–N bonds in the presence of a suitable ligand, an oxidant and an indispensable silver source. PPh3 proved to be one of the best ligands to activate the metal,145 whereas silver carbonate proved to be the most appropriate source of silver, also acting as an oxidant.
These authors applied the developed method to a new total synthesis of isocryptolepine (Scheme 26). Thus, the methoxyamide 116 was coupled with iodobenzene (117) under the optimized conditions to afford the tetracycle 118 in 67% yield. A mixed AcOH–DMA solvent was required in order to avoid decomposition of the starting material due to the considerable acidity of the solvent at the required high temperature.146
 | ||
| Scheme 26 Reagents and conditions: (a) Pd(OAc)2 (10 mol%), PPh3 (20 mol%), Ag2CO3 (2 equiv.), AcOH, DMA (3:1, v/v), 100 °C, 6 h (67%); (b) (1) NaH, DMF, 120 °C, 30 min; (2) MeI, rt, 5 h; (3) NaH, 140 °C, 6 h (77%); (c) (1) NaBH4, BF3·Et2O THF, rt, 30 min; (2) reflux, 12 h (84%). | ||
Next, the latter was exposed to MeI and an excess of NaH at elevated temperature, in order to achieve simultaneous debenzylation and selective N-methylation with concomitant demethoxylation and afford the key intermediate 37 in 77% yield.147 The final deoxygenation of the lactam in 37 was accomplished by partial reduction with NaBH4 in THF under the assistance of BF3·Et2O, followed by the in situ dehydration of the resulting lactol to afford 7 in 84% yield.
The reaction mechanism for the tandem C–C/C–N bond formation reaction (Scheme 27) involves the reaction of the starting material 116 with Pd(OAc)2 to form a five-membered palladacycle A, which undergoes oxidative addition of iodobenzene (117) to form the PdIV species B. Next, B suffers a reductive elimination to give the arylated intermediate C, regenerating the catalyst.
 | ||
| Scheme 27 Proposed reaction mechanism for the tandem C–C/C–N bond formation reaction. | ||
Subsequent deprotonation of the amine in C and C–H activation affords the seven-membered palladacycle D,148 which undergoes a reductive elimination to furnish 118 and a Pd0 species. The latter is finally oxidized by Ag2CO3 to regenerate the active PdII species for the next catalytic cycle.
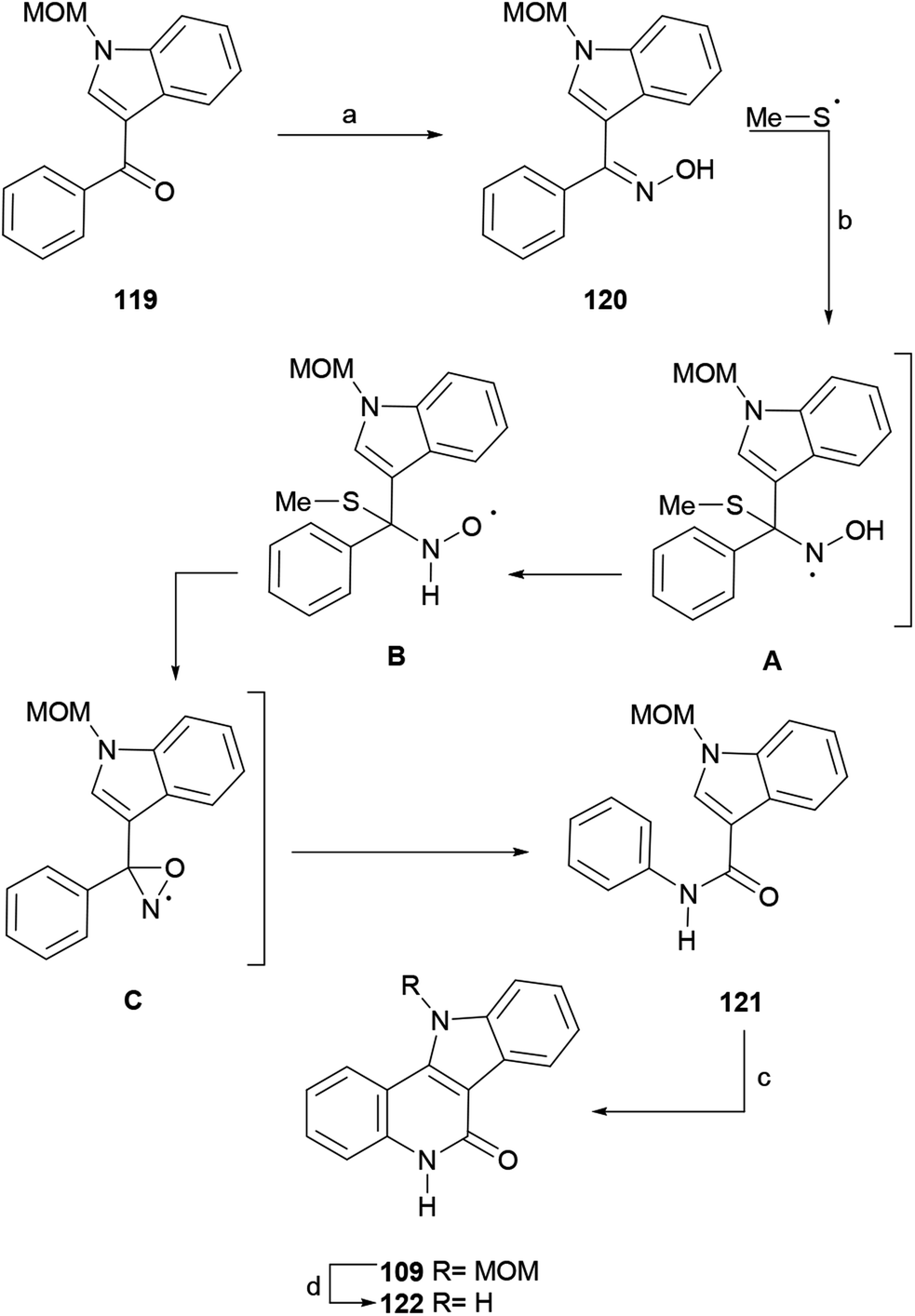 | ||
| Scheme 28 Reagents and conditions: (a) NH2OH·HCl, NaOAc, MeOH:H2O (10:3, v/v), reflux, 24 h (88%); (b) (NH4)2S2O8, DMSO, dioxane, 6 h (51%); (c) Pd(OAc)2, Cu(OAc)2, AcOH, 18 h (66%); (d) 4 N HCl, dioxane, 4 h (95%). | ||
The synthetic sequence commenced with 3-benzoylindole,151 which in the successful synthetic sequence was protected with a MOM group to afford 119, since electron withdrawing protecting groups (benzoyl, tosyl) failed during more advanced stages. In turn, 119 was converted to oxime 120 in 88% yield under conventional conditions, and the latter was subjected to the standard oxidation protocol to furnish the radical Beckmann rearrangement product, the amide 121, in moderate yield (51%).
A mechanism for this rearrangement was proposed. It was suggested that the (NH4)2S2O8-DMSO system is a source of methylsulfanyl radicals (MeS˙), which can attack the oxime moiety of 120 and afford the radical intermediate A. The latter can undergo a rearrangement to B and be further converted into the oxaziridine C,152 with the loss of MeSH.
In turn, the latter intermediate finally undergoes a rearrangement to afford amide 121. Radical intermediates have also been observed in the photochemically driven Beckmann rearrangement.153
The as-formed intermediate 121 was exposed to Pd(OAc)2 and Cu(OAc)2 in AcOH at 120 °C, which promoted an intramolecular double C–H activation, affording 109 in 66% yield. A similar C–C bond formation under UV light irradiation (254 nm) was reported earlier.154 Compound 109 is a key intermediate in Hibino's synthesis of isocryptolepine;134 therefore, this sequence constitutes a formal total synthesis of isocryptolepine. Additionally, compound 109 was deprotected with HCl in dioxane to obtain indoloquinolinone 122, a well-known bioactive scaffold, in 95% yield.147a,155
The general transformation was optimized to use 0.35 equiv. of Cu(OAc)2 and 1.2 equiv. CsF as a base, in the presence of 0.5 equiv. of TBAI, which enhances the generation of benzyne at 80 °C in DMF:MeCN (1:1, v/v). The reaction took place under oxygen as a final oxidant, for 12 h. The synthetic sequence leading to the isocryptolepine precursor involved the use of 5-methoxy-8-aminoquinoline as a removable sacrificial N,N-ligand158 and the Kobayashi benzyne precursor 124.159
In the synthetic sequence, 124 was induced to react with the indole-3-carboxamide derivative 123 to afford 125 in 62% yield (Scheme 29), carrying a removable MOM group.
 | ||
| Scheme 29 Reagents and conditions: (a) 124, Cu(OAc)2, CsF, TBAI, DMF:MeCN (1:1, v/v), O2, 80 °C, 12 h (62%); (b) (1) BBr3, CH2Cl2 (67%); (2) K2Cr2O7, AcOH:H2O (1:1, v/v) (65%); (c) see ref. 134. | ||
Then, the quinoline auxiliary was removed via a two-step protocol, including the selective O-demethylation of the quinolyl methyl ether with BBr3 to free the phenol group, which was further oxidized with K2Cr2O7 in AcOH:H2O (1:1, v/v), to afford lactam 109 in 65% yield, Hibino's intermediate toward isocryptolepine.134
This reaction system avoids the use of sensitive and expensive noble metals and oxidants, and complements previous metal-based strategies.
A plausible mechanism for this C–H/N–H cascade annulation was presented, where a Cu(III)-mediated organometallic C–H activation takes place (Scheme 30).160 Thus, 123 coordinates with Cu(OAc)2 to generate an anionic complex A ligated by a N,N-bidentate directing group.161 Then, the complex undergoes acetate-assisted intramolecular C–H concerted metalation/deprotonation to afford the five-membered ring complex B, which then undergoes carbocupration with the aryne generated from precursor 124 to furnish the intermediate C. The final reductive elimination of CuOAc C provides the desired annulation product (125), with the catalyst being regenerated for the next cycle in the presence of oxygen.
 | ||
| Scheme 30 Proposed reaction mechanism for the copper(II)-mediated selective cascade C–H/N–H annulation strategy. | ||
4e. Summary of the synthetic efforts toward isocryptolepine
In general, the reported synthetic routes toward isocryptolepine required multistep linear sequences with the corresponding isolation and purification stages of the intermediate products. A comparison of them (Table 1), in terms of complexity and overall yield, is difficult and not straightforward, due to the variable nature and structural complexity of the starting materials. In addition, many syntheses of isocryptolepine are actually formal total syntheses, reaching only a previously known advanced intermediate toward the natural product.| Year | Group | Classa | No. of steps | Main reaction(s) in the strategy | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a Classification according to the structure of the starting material. B = benzenoid; Q = quinoline; I = indole.b Formal total synthesis. Data correspond to the synthesis of the unsubstituted tetracycle 1.c Formal total synthesis. Data correspond to the synthesis of the azide 36, an intermediate in the synthesis of Molina et al.d Formal total synthesis. Data correspond to the synthesis of 109, the tetracyclic intermediate of Hibino.e Formal total synthesis. Data correspond to the synthesis of 109, the tetracyclic intermediate of Hibino. | ||||||
| 1950 | Kermack | Q | 4 | Graebe–Ullmann of 4-benzotriazolylquinoline | 29.5 | 43 |
| 1955 | Mann | Q | 3 | Fischer indolization of N-methyl tetrahydroquinolin-4-one | <15 | 45a |
| 1993 | Alvarez-Builla | Q | 2b | Graebe–Ullmann of 4-benzotriazolylquinoline | 54 | 49 |
| 1996 | Novikov | B | 6 | Fischer indolization of N-tosyl tetrahydroquinolin-4-one | 15.5 | 50 |
| 1997 | Tímari | Q | 5 | Suzuki cross-coupling; nitrene cyclization | 46.7 | 97 |
| 1998 | Joule | I | 3 | Suzuki coupling; Vilsmeier closure | 45.6 | 122 |
| 1999 | Molina | B | 11 | Cyanate and nitrene cyclizations | 24.6 | 54a |
| 2002 | Mohan | I | 3 | Photochemical ring closure | 47.3 | 89a |
| 2003 | Maes | Q | 3 | Buchwald–Hartwig; intramolecular direct arylation | 43.9 | 104 |
| 2005 | Mohan | Q | 3 | Fisher indolization of N-methyl tetrahydroquinolin-2,4-dione | 45.5 | 114 |
| 2006 | Mohan | Q | 3 | Heteroatom directed photoannulation | 64.7 | 10b |
| 2007 | Miki | I | 4 | Decarboxylative Heck-cyclization | 30.2 | 129 |
| 2008 | Maes | Q | 2 | Auto-tandem Pd-catalyzed consecutive C–N and C–C bond formation | 46.7 | 112 |
| 2009 | Kundu | B | 2b | Modified Pictet–Spengler | 86 | 59 |
| 2009 | Kumar | Q | 1 | Fisher indolization; oxidation of N-methyl tetrahydroquinolin-4-one | 83 | 120 |
| 2010 | Kraus | B | 5 | Electrocyclization of the anion of a Schiff base | 47.8 | 68 |
| 2010 | Kraus | B | 4c | Intramolecular Wittig reaction | 60.8 | 69 |
| 2011 | Kusurkar | I | 6 | 6π-Electrocyclization | 37.1 | 132 |
| 2012 | Butin | B | 4 | Furan to indole recyclization; Michael addition | 11.6 | 73 |
| 2012 | Hibino | I | 9 | Curtius rearrangement; 6π-electrocyclization | 53.6 | 134 |
| 2012 | Tummatorn | I | 4 | Aza-[4 + 2]-cycloaddition | 65.5 | 139 |
| 2013 | Bogányi | B | 7b | Buchwald–Hartwig amination; Heck-type biaryl coupling | 21.8 | 76 |
| 2014 | Lin | I | 3 | Pd(II)-Catalyzed direct dehydrogenative annulation | 43.3 | 144 |
| 2016 | Lu & Xu | B | 4 | Electrosynthesis. Cyclization of acetylenic acetal via amidyl radicals | 36.2 | 85 |
| 2016 | Mhaske | I | 4d | Radical Beckmann rearrangement; Pd-catalyzed double C–H activation | 29.6 | 150 |
| 2017 | Aksenov & Rubin | B | 2 | Fischer indolization; Vilsmeier reaction with triazine | 83 | 90 |
| 2018 | Zhang | I | 2e | Selective cascade C–H/N–H annulations | 27 | 156 |
However, it is worth noting that several very short approaches, consisting of just one to three steps using commercially available materials, have been reported, and various approaches have allowed access to the natural product in more than 60% overall yield.
5. Conclusions and perspective
The indoloquinoline heterocyclic system is strongly associated with the West African plant Cryptolepis sanguinolenta, although other natural sources are known. Cryptosanguinolentine (isocryptolepine) has been isolated twice from this plant, widely used in traditional medicine.Its unique framework and interesting pharmacological properties have meant that over the last 20 years isocryptolepine has become a test ground for new synthetic approaches toward C–C and C–N bond formation, with or without the use of precious metal catalysts. The natural product has been the target of over 25 synthetic efforts, where the tetracycle was built from benzenoids, quinolines and/or indoles via the stepwise construction of the heterocyclic moieties or through the successive assembly of the remaining heterocyclic rings.
The natural product has also gained recognition as an important and privileged scaffold in drug discovery due to the broad spectrum of its biological activities. Hence, many of the developed strategies have proved to be useful for the synthesis of more complex analogs and derivatives of the natural product, embodied with more potent and/or selective activity, mainly as cytotoxic and antimalarial agents. Therefore, it can be foreseen that the virtuous cycle, which involves new chemistries and novel structures for improved bioactive compounds, will keep going in the future, meaning that novel total syntheses of isocryptolepine will keep being disclosed.
Conflicts of interest
There are no conflicts to declareAcknowledgements
This work was financially supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Grant PUE IQUIR 2016) and the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, Grant PICT 2017-0149). E. N. T. thanks ANPCyT for her Doctoral Fellowship. The authors also gratefully acknowledge the Agencia Santafesina de Ciencia, Tecnología e Innovación Productiva (ASaCTeI) for the institutional grant AC 2015-0005 to IQUIR, which enabled the acquisition of a 400 MHz NMR spectrometer.Notes and references
- R. D. Firn and C. G. Jones, Nat. Prod. Rep., 2003, 20, 382–391 RSC.
- R. S. Bon and H. Waldmann, Acc. Chem. Res., 2010, 43, 1103–1114 CrossRef CAS PubMed.
- (a) L. Hoareau and E. J. Da Silva, Electron. J. Biotechnol., 1999, 2, 56–70 Search PubMed; (b) G. Samuelsson, Drugs of Natural Origin: A Textbook of Pharmacognosy, Swedish Pharmaceutical Press, Stockholm, Sweden, 5th edn, 2004 Search PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- J. Hong, Curr. Opin. Chem. Biol., 2011, 15, 350–354 CrossRef CAS PubMed.
- P. T. Parvatkar and P. S. Parameswaran, Curr. Org. Synth., 2016, 13, 58–72 CrossRef CAS.
- G. V. Subbaraju, J. Kavitha, D. Rajasekhar and J. I. Jimenez, J. Nat. Prod., 2004, 67, 461–462 CrossRef CAS PubMed.
- A. I. Calderón, A. Hodel, J.-L. Wolfender, M. P. Gupta, M. Correa and K. Hostettmann, Nat. Prod. Res., 2013, 27, 1335–1342 CrossRef PubMed.
- O. S. Chaves, Y. C. F. Teles, M. M. de Oliveira Monteiro, L. G. Mendes Jr, M. de Fátima Agra, V. de Andrade Braga, T. M. S. Silva and M. F. V. de Souza, Molecules, 2017, 22, 94 CrossRef PubMed.
- (a) S. Hostyn, B. U. W. Maes, L. Pieters, G. L. F. Lemière, P. Mátyus, G. Hajós and R. Dommisse, Tetrahedron, 2005, 61, 1571–1577 CrossRef CAS; (b) T. Dhanabal, R. Sangeetha and P. S. Mohan, Tetrahedron, 2006, 62, 6258–6263 CrossRef CAS; (c) M. O. Sydnes, Stud. Nat. Prod. Chem., 2020, 64, 59–84 Search PubMed; (d) M. O. Sydnes, Targets Heterocycl. Syst., 2019, 23, 201–219 Search PubMed.
- J. Hutchinson and J. M. Dalziel, Flora of Tropical West Africa, The Whitefrairs Press Ltd., London, UK, 1963, p. 102 Search PubMed.
- E. Gellert, C. R. Raymond-Hamet and E. Schlittler, Helv. Chim. Acta, 1951, 34, 642–651 CrossRef CAS.
- (a) D. Dwuma-Badu, J. S. K. Ayim, N. Y. Y. Fiagbe, J. E. Knapp, P. L. Schiff Jr and D. J. Slatkin, J. Pharm. Sci., 1978, 67, 433–434 CrossRef CAS PubMed; (b) M. V. Méndez, S. O. Simonetti, T. S. Kaufman and A. B. J. Bracca, New J. Chem., 2019, 43, 10803–10813 RSC.
- R. C. Crouch, A. O. Davis, T. D. Spitzer, G. E. Martin, M. H. M. Sharaf, P. L. Schiff Jr, C. H. Phoebe and A. N. Tackie, J. Heterocycl. Chem., 1995, 32, 1077–1080 CrossRef CAS.
- (a) M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr and G. E. Martin, J. Heterocycl. Chem., 1996, 33, 239–243 CrossRef CAS; (b) M. V. Méndez, D. A. Heredia, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, RSC Adv., 2017, 7, 28298–28307 RSC.
- J.-L. Pousset, M.-T. Martin, A. Jossang and B. Bodo, Phytochemistry, 1995, 39, 735–736 CrossRef CAS.
- (a) S. Y. Ablordeppey, C. D. Hufford, R. F. Borne and D. Dwama-Badu, Planta Med., 1990, 56, 416–417 CrossRef CAS PubMed; (b) A. N. Tackie, M. H. M. Sharaf, P. L. Schiff Jr, G. L. Boye, R. C. Crouch and G. E. Martin, J. Heterocycl. Chem., 1991, 28, 1429–1435 CrossRef CAS.
- (a) C. E. Hadden, M. H. M. Sharaf, J. E. Guido, R. H. Robins, A. N. Tackie, C. H. Phoebe Jr, P. L. Schiff Jr and G. E. Martin, J. Nat. Prod., 1999, 62, 238–240 CrossRef CAS PubMed; (b) D. M. Fort, J. Litvak, J. L. Chen, Q. Lu, P. W. Phuan, R. Cooper and D. E. Bierer, J. Nat. Prod., 1998, 61, 1528–1530 CrossRef CAS PubMed.
- A. Paulo, E. T. Gomes and P. J. Houghton, J. Nat. Prod., 1995, 58, 1485–1491 CrossRef CAS.
- (a) M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr, L. Howard, C. Myers, C. E. Hadden, S. K. Wrenn, A. O. Davis, C. W. Andrews, D. Minick, R. L. Johnson, J. P. Shockcor, R. C. Crouch and G. E. Martin, Magn. Reson. Chem., 1995, 33, 767–778 CrossRef CAS; (b) K. Cimanga, T. de Bruyne, L. Pieters, M. Claeys and A. Vlietnick, Tetrahedron Lett., 1996, 37, 1703–1706 CrossRef CAS; (c) K. Blinov, M. Elyashberg, R. Martirosian, S. G. Molodtsov, A. J. Williams, A. N. Tackie, M. H. M. Sharaf, P. L. Schiff Jr, R. C. Crouch, G. E. Martin, C. E. Hadden, J. E. Guido and K. A. Mills, Magn. Reson. Chem., 2003, 41, 577–584 CrossRef CAS.
- (a) M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr, R. L. Johnson, D. Minick, C. W. Andrews, R. C. Crouch and G. E. Martin, J. Heterocycl. Chem., 1996, 33, 789–797 CrossRef CAS; (b) A. N. Tackie, G. L. Boye, M. H. M. Sharaf, P. L. Schiff Jr, R. C. Crouch, T. D. Spitzer, R. L. Johnson, J. Dunn, D. Minick and G. E. Martín, J. Nat. Prod., 1993, 56, 653–670 CrossRef CAS.
- J. E. Saxton, in The Alkaloids, Chemistry and Physiology, ed. R. H. F. Manske and H. L. Hoimes, Academic Press, New York, USA, 1965, vol. 8, p. 19 Search PubMed.
- G. L. Boye and O. Ampofo, Proceedings at the First International Symposium on Cryptolepine, University of Science and Technology, Kumasi, Ghana, 1983 Search PubMed.
- (a) M. Iwu, Handbook of African Medicinal Plants, CRC Press, Boca Raton, USA, 1993 Search PubMed; (b) J. Kerharo, La Pharmacopée Sénégalaise Traditionnelle, Plantes Médicinales et Toxiques. Editions Vigot Frères, Paris, France, 1974 Search PubMed; (c) J. M. Watt and M. G. Breyer-Brandwijk, The Medicinal and Poisonous Plants of Southern and Eastern Africa. E. & S. Livingstone Ltd, London, UK, 1962, pp. 128–129 Search PubMed; (d) K. Cimanga, T. de Bruyne, A. Lasure, B. Van Poel, L. Pieters, M. Claeys, D. Vanden Berghe, K. Kambu, L. Tona and A. J. Vlietinck, Planta Med., 1996, 62, 22–27 CrossRef CAS PubMed.
- B. Oliver-Bever, Medicinal Plants in Tropical West Africa, Cambridge University Press, Cambridge, UK, 1986, p. 41 Search PubMed.
- (a) E. L. Larghi, A. B. J. Bracca, A. A. Arroyo Aguilar, D. A. Heredia, J. L. Pergomet, S. O. Simonetti and T. S. Kaufman, Curr. Top. Med. Chem., 2015, 17, 1683–1707 CrossRef PubMed; (b) P. T. Parvatkar and S. G. Tilve, Bioactivities and Synthesis of Indoloquinoline Alkaloids: Cryptolepine, Isocryptolepine and Neocryptolepine in: Bioactive Heterocycles: Synthesis and Biological Evaluation, ed. K. L. Ameta, R. P. Pawar and A. J. Domb, Nova Science Publishers, Inc., New York, USA, 2013, ch. 10, pp. 217−234 Search PubMed; (c) E. V. K. S. Kumar, J. R. Etukala and S. Y. Ablordeppey, Mini-Rev. Med. Chem., 2008, 8, 538–554 CrossRef CAS PubMed; (d) C. W. Wright, J. Pharm. Pharmacol., 2007, 59, 899–904 CrossRef CAS PubMed.
- H. de Diesbach, E. de Bie and F. Rubli, Helv. Chim. Acta, 1934, 17, 113–128 CrossRef CAS.
- A. N. Tackie, C. H. Phoebe Jr and G. E. Martin, J. Heterocycl. Chem., 1996, 33, 239–243 CrossRef.
- J. Tousek, S. VanMiert, L. Pieters, G. Van Baelen, S. Hostyn, B. U. W. Maes, G. Lemiere, R. Dommisse and R. Marek, Magn. Reson. Chem., 2008, 46, 42–51 CrossRef CAS PubMed.
- E. Breitmaier and K. H. Spohn, Tetrahedron, 1973, 29, 1145–1152 CrossRef CAS.
- L. Grycova, R. Dommisse, L. Pieters and R. Marek, Magn. Reson. Chem., 2009, 47, 977–981 CrossRef CAS PubMed.
- P. Ghosh, S. S. Jaffer and P. Purkayastha, J. Phys. Chem. B, 2011, 115, 2046–2054 CrossRef CAS PubMed.
- W. Peczynska-Czoch, F. Pognan, L. Kaczmarek and J. Boratyriski, J. Med. Chem., 1994, 37, 3503–3510 CrossRef CAS PubMed.
- G. Van Baelen, S. Hostyn, L. Dhooghe, P. Tapolcsányi, P. Mátyus, G. Lemière, R. Dommisse, M. Kaiser, R. Brun, P. Cos, L. Maes, G. Hajós, Z. Riedl, I. Nagy, B. U. W. Maes and L. Pieters, Bioorg. Med. Chem., 2009, 17, 7209–7217 CrossRef CAS PubMed.
- J.-C. Li, R.-X. Wang, Y. Sun, J.-K. Zhu, G.-F. Hu, Y.-L. Wang, R. Zhou, Z.-M. Zhao, Y.-Q. Liu, J.-W. Peng, Y.-F. Yan and X.-F. Shang, Bioorg. Chem., 2019, 92, 103266 CrossRef CAS PubMed.
- (a) P. Grellier, L. Ramiaramanana, V. Millerioux, E. Deharo, J. Schrével, F. Frappier, F. Ikigalo, B. Bodo and J. L. Pousset, Phytother. Res., 1996, 10, 317–321 CrossRef CAS; (b) S. Van Miert, S. Hostyn, B. U. W. Maes, K. Cimanga, R. Brun, M. Kaiser, P. Mátyus, R. Dommisse, G. Lemière, A. Vlietinck and L. Pieters, J. Nat. Prod., 2005, 68, 674–677 CrossRef CAS PubMed; (c) N. Wang, K. J. Wicht, K. Imai, M.-Q. Wang, T. A. Ngoc, R. Kiguchi, M. Kaiser, T. J. Egan and T. Inokuchi, Bioorg. Med. Chem., 2014, 22, 2629–2642 CrossRef CAS PubMed; (d) L. R. Whittell, K. T. Batty, R. P. M. Wong, E. M. Bolitho, S. A. Fox, T. M. E. Davis and P. E. Murray, Bioorg. Med. Chem., 2011, 19, 7519–7525 CrossRef CAS PubMed; (e) K. Rujimongkon, M. Mungthin, J. Tummatorn, S. Ampawong, P. Adisakwattana, U. Boonyuen and O. Reamtong, PLoS One, 2019, 14, e0220871 CrossRef CAS PubMed.
- (a) G. Van Baelen, C. Meyers, G. L. F. Lemière, S. Hostyn, R. Dommisse, L. Maes, K. Augustyns, A. Haemers, L. Pieters and B. U. W. Maes, Tetrahedron, 2008, 64, 11802–11809 CrossRef CAS; (b) N. Wang, M. Switalska, L. Wang, E. Shaban, M. I. Hossain, I. E. T. El Sayed, J. Wietrzyk and T. Inokuchi, Molecules, 2019, 24, 2121 CrossRef CAS PubMed.
- (a) A. Molina, J. J. Vaquero, J. L. García-Navio, J. Alvarez-Builla, B. Pascual-Teresa, F. Gago, M. M. Rodrigo and M. Ballesteros, J. Org. Chem., 1996, 61, 5587–5599 CAS; (b) P. Ghosh and P. Purkayastha, RSC Adv., 2014, 4, 22442–22448 RSC.
- M. E. Aggrey, H. H. Li, W.-Q. Wang, W. Song, Y. Wang and L.-J. Xuan, Rev. Bras. Farmacogn., 2017, 28, 654–657 Search PubMed.
- (a) P. Schmitt, C. H. Nguyen, J.-S. Sun, D. S. Grierson, E. Bisagni, T. Garestier and C. Hélène, Chem. Commun., 2000, 763–764 CAS; (b) J. Lavrado, S. A. Ohnmacht, I. Correia, C. Leitão, S. Pisco, M. Gunaratnam, R. Moreira, S. Neidle, D. J. V. A. dos Santos and A. Paulo, ChemMedChem, 2015, 10, 836–849 CrossRef CAS PubMed; (c) J. Lavrado, H. Brito, P. M. Borralho, S. A. Ohnmacht, N.-S. Kim, C. Leitão, S. Pisco, M. Gunaratnam, C. M. P. Rodrigues, R. Moreira, S. Neidle and A. Paulo, Sci. Rep., 2015, 5, 9696 CrossRef PubMed; (d) H. Falke, A. Chaikuad, A. Becker, N. Loaëc, O. Lozach, S. A. Jhaisha, W. Becker, P. G. Jones, L. Preu, K. Baumann, S. Knapp, L. Meijer and C. Kunick, J. Med. Chem., 2015, 58, 3131–3143 CrossRef CAS PubMed.
- (a) N. Wang, K. J. Wicht, E. Shaban, T. A. Ngoc, M.-Q. Wang, I. Hayashi, M. I. Hossain, Y. Takemasa, M. Kaiser, I. E. T. El Sayed, T. J. Egan and T. Inokuchi, Med. Chem. Commun., 2014, 5, 927–931 RSC; (b) L. K. Filak, S. Göschl, P. Heffeter, K. G. Samper, A. E. Egger, M. A. Jakupec, B. K. Keppler, W. Berger and V. B. Arion, Organometallics, 2013, 32, 903–914 CrossRef CAS PubMed; (c) C.-M. Lu, Y.-L. Chen, H.-L. Chen, C.-A. Chen, P.-J. Lu, C.-N. Yang and C.-C. Tzeng, Bioorg. Med. Chem., 2010, 18, 1948–1957 CrossRef CAS PubMed.
- (a) A. B. J. Bracca, D. A. Heredia, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2014, 7979–8003 CrossRef CAS; (b) M. V. Méndez, A. B. J. Bracca and T. S. Kaufman, Synthesis, 2018, 50, 1417–1429 CrossRef; (c) O. N. Nadein, D. A. Aksenov, G. M. Abakarov, N. A. Aksenov, L. G. Voskressensky and A. V. Aksenov, Chem. Heterocycl. Compd., 2019, 55, 905–932 CAS.
- W. O. Kermack and N. E. Storey, J. Chem. Soc., Chem. Commun., 1950, 607–612 CAS.
- G. R. Clemo and W. H. Perkin Jr, J. Chem. Soc., 1924, 125, 1608–1622 RSC.
- (a) J. T. Braunholtz and F. G. Mann, J. Chem. Soc., 1955, 381–392 RSC; (b) J. A. C. Allison, J. T. Braunholtz and F. G. Mann, J. Chem. Soc., 1955, 403–408 Search PubMed; (c) J. T. Braunholtz and F. G. Mann, J. Chem. Soc., 1958, 3368–3377 RSC.
- O. Roussel, P. Buu-Hop and P. Jacquignon, J. Chem. Soc., 1965, 5458–5464 RSC.
- E. E. García, J. G. Riley and R. I. Fryer, J. Org. Chem., 1968, 33, 2868–2874 CrossRef.
- R. I. Fryer, J. V. Earley and L. H. Sternbach, J. Chem. Soc., 1963, 4979–5004 RSC.
- A. Molina, J. J. Vaquero, J. L. García-Navío and J. Alvarez-Builla, Tetrahedron Lett., 1993, 34, 2673–2676 CrossRef CAS.
- S. V. Dubovitskii, O. S. Radchenko and V. L. Novikov, Russ. Chem. Bull., 1996, 45, 2656–2657 Search PubMed.
- W. Hückel and L. Hagedorn, Chem. Ber., 1957, 90, 752–754 CrossRef.
- W. S. Johnson and E. L. Woroch, J. Am. Chem. Soc., 1949, 71, 1901–1905 CrossRef CAS.
- P. S. Volvoikar and S. G. Tilve, Org. Lett., 2016, 18, 892–895 CrossRef CAS PubMed.
- (a) P. M. Fresneda, P. Molina and S. Delgado, Tetrahedron Lett., 1999, 40, 7275–7278 CrossRef CAS; (b) P. M. Fresneda, P. Molina and S. Delgado, Tetrahedron, 2001, 57, 6197–6202 CAS.
- P. Molina, M. Alajarin and A. Vidal, Tetrahedron, 1990, 46, 1063–1078 CrossRef CAS.
- W. Stadlbauer, A.-S. Karem and T. Kappe, Monatsh. Chem., 1987, 118, 81–89 CrossRef CAS.
- C. Narayana, P. Kumari and R. Sagar, Org. Lett., 2018, 20, 4240–4244 CrossRef CAS PubMed.
- P. Molina and M. J. Vilaplana, Synthesis, 1994, 1197–1218 CAS.
- P. K. Agarwal, D. Sawant, S. Sharma and B. Kundu, Eur. J. Org. Chem., 2009, 292–303 CrossRef CAS.
- (a) B. Kundu, D. Sawant and R. Chhabra, J. Comb. Chem., 2005, 7, 317–321 CrossRef CAS PubMed; (b) B. Kundu, D. Sawant, P. Partani and A. P. Kesarwani, J. Org. Chem., 2005, 70, 4889–4892 CrossRef CAS PubMed; (c) S. Duggineni, D. Sawant, B. Saha and B. Kundu, Tetrahedron, 2006, 62, 3228–3241 CrossRef CAS; (d) S. Sharma, B. Saha, D. Sawant and B. Kundu, J. Comb. Chem., 2007, 9, 783–792 CrossRef CAS PubMed; (e) S. Sharma, B. Saha, D. Sawant and B. Kundu, Tetrahedron, 2008, 64, 8676–8684 CrossRef.
- (a) A. K. Kiang, A. F. Mann, A. F. Prior and A. Topham, J. Chem. Soc., 1956, 1319–1331 RSC; (b) R. L. Duncan, G. C. Helsey and R. F. Boswell, J. Heterocycl. Chem., 1973, 10, 65–70 CrossRef CAS.
- R. S. Stabler, Synth. Commun., 1994, 24, 123–129 CrossRef.
- (a) M. A. J. Duncton, L. M. Smith, S. Burdzovic-Wizeman, A. Burns, H. Liu, Y. Mao, W. C. Wong and A. S. Kiselyov, J. Org. Chem., 2005, 70, 9629–9631 CrossRef CAS PubMed; (b) L. Zheng, J. Xiang, Q. D. Daang and X. Bai, J. Comb. Chem., 2005, 7, 813–815 CrossRef CAS PubMed; (c) E. David, P.-S. Rostaing and M. Lemaire, Tetrahedron, 2007, 63, 8999–9006 CrossRef CAS; (d) X. L. Wang, X. F. Zheng, R. H. Liu, J. Reiner and J. B. Chang, Tetrahedron, 2007, 63, 3389–3394 CrossRef CAS; (e) S. Gracia, J. Schulz, S. Pellet-Rostaing and M. Lemaire, Synlett, 2008, 1852–1856 CAS; (f) X. Che, L. Zheng, Q. Dang and X. Bai, J. Org. Chem., 2008, 73, 1147–1149 CrossRef CAS PubMed.
- M. Xu, Q. Hou, S. Wang, H. Wang and Z.-J. Yao, Synthesis, 2011, 626–634 CAS.
- (a) M. Alfonsi, A. Acradi, M. Aschi, G. Bianchi and F. Marinelli, J. Org. Chem., 2005, 70, 2265–2273 CrossRef CAS PubMed; (b) A. Arcadi, G. Bianchi and F. Marinelli, Synthesis, 2004, 610–618 CAS.
- S. Guo, L. Tao, W. Zhang, X. Zhang and X. Fan, J. Org. Chem., 2015, 80, 10955–10964 CrossRef CAS PubMed.
- G. A. Kraus and H. T. Guo, Org. Lett., 2008, 10, 3061–3063 CrossRef CAS PubMed.
- G. A. Kraus, H. Guo, G. Kumar, G. Pollock III, H. Carruthers, D. Chaudhary and J. Beasley, Synthesis, 2010, 1386–1393 CrossRef CAS.
- G. A. Kraus and H. Guo, Tetrahedron Lett., 2010, 51, 4137–4139 CrossRef CAS.
- J. Yang, H. Song, X. Xiao, J. Wang and Y. Qin, Org. Lett., 2006, 8, 2187–2190 CrossRef CAS PubMed.
- A. V. Butin, F. A. Tsiunchik, V. T. Abaev and V. E. Zavodnik, Synlett, 2008, 1145–1148 CrossRef CAS.
- A. V. Butin, M. G. Uchuskin, A. S. Pilipenko, F. A. Tsiunchik, D. A. Cheshkov and I. V. Trushkov, Eur. J. Org. Chem., 2010, 920–926 CrossRef CAS.
- M. G. Uchuskin, A. S. Pilipenko, O. V. Serdyuk, I. V. Trushkov and A. V. Butin, Org. Biomol. Chem., 2012, 10, 7262–7265 RSC.
- (a) S. Cacchi, G. Fabrizi, P. Pace and F. Marinelli, Synlett, 1999, 620–622 CrossRef CAS; (b) L. M. Werbel, S. J. Kesten and W. R. Turner, Eur. J. Med. Chem., 1993, 28, 837–852 CrossRef CAS.
- (a) K. D. Park, S. J. Cho, J. S. Moon and S. U. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 6551–6554 CrossRef CAS PubMed; (b) G. E. Lee, H.-S. Lee, S. D. Lee, J.-H. Kim, W.-K. Kim and Y.-C. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 954–958 CrossRef CAS PubMed; (c) O. Tsuge, S. Kanemasa and S. Takenaka, Bull. Chem. Soc. Jpn., 1987, 60, 1489–1495 CrossRef CAS; (d) A. Katritzky, N. E. Grzeskowiak, J. Alvarez-Bulla and A. Tarraga-Tomas, J. Prakt. Chem., 1983, 325, 177–187 CrossRef CAS.
- B. Bogányi and J. Kámán, Tetrahedron, 2013, 69, 9512–9519 CrossRef.
- R. Cassis, R. Tapia and J. A. Valderrama, Synth. Commun., 1985, 15, 125–133 CrossRef CAS.
- N. Boudet, J. R. Lachs and P. Knochel, Org. Lett., 2007, 9, 5525–5528 CrossRef CAS PubMed.
- (a) B. J. Margolis, K. A. Long, D. L. T. Laird, J. C. Ruble and S. R. Pulley, J. Org. Chem., 2007, 72, 2232–2235 CrossRef CAS PubMed; (b) C. Wolf, G. E. Tumambac and C. N. Villalobos, Synlett, 2003, 1801–1804 CrossRef CAS.
- S. Hostyn, B. U. W. Maes, V. G. Baelen, A. Gulevskaya, C. Meyers and K. Sits, Tetrahedron, 2006, 62, 4676–4684 CrossRef CAS.
- (a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (b) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC; (c) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed; (d) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC; (e) K. A. Ogawa and A. J. Boydston, Chem. Lett., 2015, 44, 10–16 CrossRef CAS; (f) R. Francke, Beilstein J. Org. Chem., 2014, 10, 2858–2873 CrossRef PubMed; (g) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef CAS PubMed; (h) S. R. Waldvogel and S. Möhle, Angew. Chem., Int. Ed., 2015, 54, 6398–6399 CrossRef CAS PubMed.
- (a) T. Morofuji, A. Shimizu and J. Yoshida, J. Am. Chem. Soc., 2014, 136, 4496–4499 CrossRef CAS PubMed; (b) T. Morofuji, A. Shimizu and J. Yoshida, J. Am. Chem. Soc., 2015, 137, 9816–9819 CrossRef CAS PubMed; (c) W. J. Gao, W. C. Li, C. C. Zeng, H. Y. Tian, L. M. Hu and R. D. Little, J. Org. Chem., 2014, 79, 9613–9618 CrossRef CAS PubMed; (d) S. J. Yoo, L. J. Li, C. C. Zeng and R. D. Little, Angew. Chem., Int. Ed., 2015, 54, 3744–3747 CrossRef CAS PubMed; (e) B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2014, 53, 5210–5213 CrossRef CAS; (f) B. Elsler, A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Chem.–Eur. J., 2015, 21, 12321–12325 CrossRef CAS PubMed.
- N. Fuentes, W. Kong, L. Fernandez-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964–973 CrossRef CAS PubMed.
- L. Zhu, P. Xiong, Z. Y. Mao, Y. H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef CAS PubMed.
- Z.-W. Hou, Z.-Y. Mao, H.-B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef CAS PubMed.
- (a) Y. F. Wang, H. Chen, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 11980–11983 CrossRef CAS PubMed; (b) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640–4643 CrossRef CAS PubMed; (c) X. Q. Hu, J. R. Chen, Q. Wei, F. L. Liu, Q. H. Deng, A. M. Beauchemin and W. J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 12163–12167 CrossRef CAS PubMed; (d) G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226–9229 CrossRef CAS PubMed; (e) D. C. Miller, G. J. Choi, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 13492–13495 CAS.
- (a) U. Jahn and P. Hartmann, Chem. Commun., 1998, 209–210 RSC; (b) F. Kafka, M. Holan, D. Hidasova, R. Pohl, I. Cisarova, B. Klepetarova and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 9944–9948 CrossRef CAS PubMed.
- (a) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, L. V. Frolova, A. Kornienko, I. V. Magedov and M. Rubin, Chem. Commun., 2013, 49, 9305–9307 RSC; (b) A. V. Aksenov, A. N. Smirnov, I. V. Magedov, M. R. Reisenauer, N. A. Aksenov, I. V. Aksenova, A. L. Pendleton, G. Nguyen, R. K. Johnston, M. Rubin, A. Carvalho, R. De Kiss, V. Mathieu, F. Lefranc, J. Correa, D. A. Cavazos, A. J. Brenner, B. A. Bryan, S. Rogelj, A. Kornienko and L. V. Frolova, J. Med. Chem., 2015, 58, 2206–2220 CrossRef CAS PubMed; (c) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, A. S. Bijieva and M. Rubin, Org. Biomol. Chem., 2014, 12, 9786–9788 RSC; (d) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, J. P. Matheny and M. Rubin, RSC Adv., 2015, 5, 8647–8656 RSC.
- (a) R. N. Kumar, T. Suresh and P. S. Mohan, Tetrahedron Lett., 2002, 43, 3327–3328 CrossRef CAS; (b) A. V. Aksenov, D. A. Aksenov, G. D. Griaznov, N. A. Aksenov, L. G. Voskressensky and M. Rubin, Org. Biomol. Chem., 2018, 16, 4325–4332 RSC.
- A. V. Aksenov, D. A. Aksenov, N. A. Orazova, N. A. Aksenov, G. D. Griaznov, A. De Carvalho, R. Kiss, V. Mathieu, A. Kornienko and M. Rubin, J. Org. Chem., 2017, 82, 3011–3018 CrossRef CAS PubMed.
- H. Z. Alkhathlan, M. A. Al-Saad, H. M. Al-Hazimi, K. A. Al-Farhan and A. A. Mousa, J. Chem. Res., 2002, 587–588 CrossRef CAS.
- (a) A. V. Aksenov, N. A. Aksenov, Z. V. Dzhandigova, D. A. Aksenov and M. Rubin, RSC Adv., 2015, 5, 106492–106497 RSC; (b) N. A. Aksenov, A. Z. Gasanova, G. M. Abakarov, I. V. Aksenova and A. V. Aksenova, Russ. Chem. Bull., 2019, 68, 836–840 CrossRef CAS.
- N. A. Aksenov, A. V. Aksenov, A. Kornienko, A. de Carvalho, V. Mathieu, D. A. Aksenov, S. N. Ovcharov, G. D. Griaznove and M. Rubin, RSC Adv., 2018, 8, 36980–36986 RSC.
- C. Xie, Z. Zhang, D. Li, J. Gong, X. Han, X. Liu and C. Ma, J. Org. Chem., 2017, 82, 3491–3499 CrossRef CAS PubMed.
- (a) A. R. Katritzky, X. Lan, O. Yang and O. V. Denisko, Chem. Rev., 1998, 98, 409–584 CrossRef CAS PubMed; (b) J. Chen, B. Liu, D. Liu, S. Liu and J. Cheng, Adv. Synth. Catal., 2012, 354, 2438–2442 CrossRef CAS; (c) H. Fei, J. Yu, Y. Jiang, H. Guo and J. Cheng, Org. Biomol. Chem., 2013, 11, 7092–7095 RSC; (d) Z. Zhang, Q. Tian, J. Qian, Q. Liu, T. Liu, L. Shi and G. Zhang, J. Org. Chem., 2014, 79, 8182–8188 CrossRef CAS PubMed; (e) J. Qian, Z. Zhang, Q. Liu, T. Liu and G. Zhang, Adv. Synth. Catal., 2014, 356, 3119–3124 CrossRef CAS.
- (a) V. Traynelis and W. Hergenrother, J. Org. Chem., 1964, 29, 221–222 CrossRef CAS; (b) K. Tasneem and K. Z. Khan, Asian J. Chem., 2008, 20, 929–933 CAS.
- G. Tímari, T. Soos and G. Hajos, Synlett, 1997, 1067–1068 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- W. Fuhrer and H. W. Gschwend, J. Org. Chem., 1979, 44, 1133–1136 CrossRef CAS.
- P. Molina, A. Lorenzo and E. Aller, Tetrahedron, 1992, 48, 4601–4616 CrossRef CAS.
- F. Trecourt, F. Mongin, M. Mallet and G. Queguiner, Synth. Commun., 1995, 25, 4011–4024 CrossRef CAS.
- H. Gao, Q.-L. Xu, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2014, 53, 2701–2705 CrossRef CAS PubMed.
- K. S. Håheim, I. T. U. Helgeland, E. Lindbäck and M. O. Sydnes, Tetrahedron, 2019, 75, 2949–2957 CrossRef.
- T. H. M. Jonckers, B. U. W. Maes, G. L. F. Lemière, G. Rombouts, L. Pieters, A. Haemers and R. A. Dommisse, Synlett, 2003, 615–618 CAS.
- (a) D. Barañano, G. Mann and J. F. Hartwig, Curr. Org. Chem., 1997, 1, 287–305 CrossRef; (b) C. G. Frost and P. Mendonça, J. Chem. Soc., Perkin Trans. 1, 1998, 2615–2623 RSC; (c) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS; (d) A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131–201 CrossRef CAS.
- (a) D. E. Ames and D. Bull, Tetrahedron, 1982, 38, 383–387 CrossRef CAS; (b) T. Iwaki, A. Yasuhara and T. Sakamoto, J. Chem. Soc., Perkin Trans. 1, 1999, 1505–1510 RSC; (c) R. B. Bedford and C. S. Cazin, Chem. Commun., 2002, 2310–2311 RSC.
- C. Wolf and R. Lerebours, J. Org. Chem., 2003, 68, 7077–7084 CrossRef CAS PubMed.
- J. Yin, M. M. Zhao, M. A. Huffman and J. M. McNamara, Org. Lett., 2002, 4, 3481–3484 CrossRef CAS PubMed.
- P. Fitton and E. A. Rick, J. Organomet. Chem., 1971, 28, 287–291 CrossRef CAS.
- (a) A. F. Littke and G. C. Fu, J. Org. Chem., 1999, 64, 10–11 CrossRef CAS PubMed; (b) A. Z. Ehrentraut and M. Beller, Synlett, 2000, 1589–1592 CAS.
- I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC.
- C. Meyers, G. Rombouts, K. T. . J. Loones, A. Coelho and B. U. W. Maes, Adv. Synth. Catal., 2008, 350, 465–470 CrossRef CAS.
- (a) L. Ackermann and A. Althammer, Angew. Chem., Int. Ed., 2007, 46, 1627–1629 CrossRef CAS PubMed; (b) R. B. Bedford and M. Betham, J. Org. Chem., 2006, 71, 9403–9410 CrossRef CAS PubMed.
- T. Dhanabal, R. Sangeetha and P. S. Mohan, Tetrahedron Lett., 2005, 46, 4509–4510 CrossRef CAS.
- S. Sunder and N. P. Peet, J. Heterocycl. Chem., 1978, 15, 1379–1382 CrossRef CAS.
- P. Pitchai, P. S. Mohan and R. M. Gengan, Indian J. Chem., Sect. B, 2009, 48, 692–696 Search PubMed.
- R. Saari, J. C. Toermae and T. Nevalainen, Bioorg. Med. Chem., 2011, 19, 939–950 CrossRef CAS PubMed.
- (a) K. M. Kasiotis, N. Fokialakis and S. A. Haroutounian, Synthesis, 2006, 1791–1802 CAS; (b) S. Chandrasekhar, K. Vijeender and C. Sridhar, Tetrahedron Lett., 2007, 48, 4935–4937 CrossRef CAS.
- K. Matsumoto, A. Tanaka, I. Yukio, N. Hayashi, M. Toda and R. A. Bulman, Heterocycl. Commun., 2003, 9, 9–12 CAS.
- D. Kumar, M. Kumar and V. S. Rao, Chem. Lett., 2009, 38, 156–157 CrossRef CAS.
- K. A. Muszkat and E. Fischer, J. Chem. Soc. B, 1967, 662–678 RSC.
- P. E. Murray, K. Mills and J. A. Joule, J. Chem. Res., Synop., 1998, 377 (J. Chem. Res., Miniprint, 1998, 1435–1447) RSC.
- P. Strazzolini, A. G. Giumanini and S. Cauci, Tetrahedron, 1990, 46, 1081–1118 CrossRef CAS.
- (a) S. T. Selvi and P. S. Z. Mohan, Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 1337–1341 Search PubMed; (b) S. T. Selvi and P. S. Mohan, Heterocycl. Commun., 1999, 6, 533–535 Search PubMed.
- S. T. Selvi and P. S. Mohan, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1999, 38, 1118–1120 Search PubMed.
- (a) L. Jayabalan and P. Shanmugam, Synthesis, 1991, 217–220 CrossRef CAS; (b) E. Schendera, L.-N. Unkel, P. P. Huyen Quyen, G. Salkewitz, F. Hoffmann, A. Villinger and M. Brasholz, Chem.–Eur. J., 2020, 26, 269–274 CrossRef CAS PubMed.
- E. R. El-Sawy, A. B. Abdelwahab and G. Kirsch, Synthesis, 2018, 50, 4525–4538 CrossRef CAS.
- (a) Y. Miki, Y. Tsuzaki, H. Hibino and Y. Aoki, Synlett, 2004, 2206–2208 CrossRef CAS; (b) Y. Miki, Y. Aoki, H. Miyatake, T. Minematsu and H. Hibino, Tetrahedron Lett., 2006, 47, 5215–5218 CrossRef CAS.
- Y. Miki, M. Kuromatsu, H. Miyatake and H. Hamamoto, Tetrahedron Lett., 2007, 48, 9093–9095 CrossRef CAS.
- (a) D. Tanaka, S. P. Romeril and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 10323–10333 CrossRef CAS PubMed; (b) D. Tanaka and G. Myers, Org. Lett., 2004, 6, 433–436 CAS; (c) A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250–11251 CrossRef CAS PubMed.
- A. Mouladdib, B. Joseph, A. Hasnaoui and J. Mérour, Synthesis, 2000, 549–556 CrossRef.
- D. G. Hingane and R. S. Kusurkar, Tetrahedron Lett., 2011, 52, 3686–3688 CrossRef CAS.
- A. Yasuhara and T. Sakamoto, Tetrahedron Lett., 1998, 39, 595–596 CrossRef CAS.
- (a) K. Hayashi, T. Choshi, K. Chikaraishi, A. Oda, R. Yoshinaga, N. Hatae, M. Ishikura and S. Hibino, Tetrahedron, 2012, 68, 4274–4279 CrossRef CAS; (b) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Nat. Prod. Rep., 2019, 36, 354–401 RSC.
- S. Beaumont, P. Retailleau, P. Dauban and R. H. Dodd, Eur. J. Org. Chem., 2008, 5162–5175 CrossRef CAS.
- H. Katsuki, P. K. Datta, H. Hayakawa and H. Suenaga, Synthesis, 1995, 1348–1350 CrossRef.
- T. Kuwada, M. Fukui, M. Hirayama, J. Nobuhiro, T. Choshi and S. Hibino, Heterocycles, 2002, 58, 326–332 Search PubMed.
- W. H. Pearson and W. K. Fang, Isr. J. Chem., 1997, 37–39 Search PubMed.
- J. Tummatorn, C. Thongsornkleeb and S. Ruchirawat, Tetrahedron, 2012, 68, 4732–4739 CrossRef CAS.
- T. Maibunkaew, C. Thongsornkleeb, J. Tummatorn, A. Bunrit and S. Ruchirawat, Synlett, 2014, 25, 1769–1775 CrossRef.
- P. Aroonkit, C. Thongsornkleeb, J. Tummatorn, S. Krajangsri, M. Mungthin and S. Ruchirawat, Eur. J. Med. Chem., 2015, 94, 56–62 CrossRef CAS PubMed.
- (a) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS PubMed; (b) S. Ding, Z. Shi and N. Jiao, Org. Lett., 2010, 12, 1540–1543 CrossRef CAS PubMed; (c) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed; (d) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (e) B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed; (f) C. Zhu, R. Wang and J. R. Falck, Chem.–Asian J., 2012, 7, 1502–1514 CrossRef CAS PubMed; (g) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS.
- (a) Z. Shi, Y. Cui and N. Jiao, Org. Lett., 2010, 12, 2908–2911 CrossRef CAS PubMed; (b) J. Zheng, Y. Zhang and S. Cui, Org. Lett., 2014, 16, 3560–3563 CrossRef CAS PubMed.
- X. Chen, P. Sun, J. Xu, X. Wu, L. Kong, H. Yao and A. Lin, Tetrahedron Lett., 2014, 55, 7114–7117 CrossRef CAS.
- E. T. Nadres, A. Lazareva and O. Daugulis, J. Org. Chem., 2011, 76, 471–483 CrossRef CAS PubMed.
- J. Karthikeyan and C. H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880–9883 CrossRef CAS PubMed.
- (a) X. Zhang, D. Zhang-Negrerie, J. Deng, Y. Du and K. Zhao, J. Org. Chem., 2013, 78, 12750–12759 CrossRef CAS PubMed; (b) H. Zhong, D. Yang, S. Wang and J. Huang, Chem. Commun., 2012, 3236–3238 RSC.
- (a) O. Daugulis and V. G. Zaitsev, Angew. Chem., Int. Ed., 2005, 44, 4046–4048 CrossRef CAS PubMed; (b) A. Lazareva and O. Daugulis, Org. Lett., 2006, 8, 5211–5213 CrossRef CAS PubMed.
- (a) P. S. Mahajan, S. D. Tanpure, N. A. More, J. M. Gajbhiye and S. B. Mhaske, RSC Adv., 2015, 5, 101641–101646 RSC; (b) D. N. Garad, S. D. Tanpure and S. B. Mhaske, Beilstein J. Org. Chem., 2015, 11, 1008–1016 CrossRef CAS PubMed.
- P. S. Mahajan, V. T. Humne, S. D. Tanpure and S. B. Mhaske, Org. Lett., 2016, 18, 3450–3453 CrossRef CAS PubMed.
- A. M. Cook and C. Wolf, Chem. Commun., 2014, 50, 3151–3154 RSC.
- (a) A. Lattes, E. Oliveros, M. Riviere, C. Belzeck, D. Mostowicz, W. Abramskj, C. Piccinni-Leopardi, G. Germain and M. Van Meersschel, J. Am. Chem. Soc., 1982, 104, 3929–3934 CrossRef CAS; (b) T. Oine and T. Mukai, Tetrahedron Lett., 1969, 10, 157–160 CrossRef.
- (a) O. E. Hutt, T. L. Doan and G. I. Georg, Org. Lett., 2013, 15, 1602–1605 CrossRef CAS PubMed; (b) B. T. Smith, J. A. Wendt and J. Aube, Org. Lett., 2002, 4, 2577–2584 CrossRef CAS PubMed; (c) Y. Ogata, K. Takagi and K. Mizuno, J. Org. Chem., 1982, 47, 3684–3687 CrossRef CAS; (d) T. Sasaki, S. Eguchi and T. Toru, J. Chem. Soc. D, 1970, 1239–1240 RSC.
- V. Dave and E. W. Warnhoff, Tetrahedron, 1975, 31, 1255–1258 CrossRef CAS.
- (a) Y. L. Chen, C. H. Chung, I. L. Chen, P. H. Chen and H. Y. Jeng, Bioorg. Med. Chem., 2002, 10, 2705–2712 CrossRef CAS PubMed; (b) Z. L. Xiao, N. C. Waters, C. L. Woodard, Z. Y. Li and P. K. Li, Bioorg. Med. Chem. Lett., 2001, 11, 2875–2878 CrossRef CAS PubMed; (c) A. Walser, G. Silverman, T. Flynn and R. I. Fryer, J. Heterocycl. Chem., 1975, 12, 351–358 CrossRef CAS.
- T. Y. Zhang, C. Liu, C. Chen, J. X. Liu, H. Y. Xiang, W. Jiang, T. M. Ding and S. Y. Zhang, Org. Lett., 2018, 20, 220–223 CrossRef CAS PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed; (b) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS PubMed; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (d) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (e) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635–645 CrossRef CAS PubMed.
- (a) G. He, S. Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124–11128 CrossRef CAS PubMed; (b) C. Yamamoto, K. Takamatsu, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 7675–7684 CrossRef CAS PubMed.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211–1214 CrossRef.
- (a) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068–12073 CrossRef CAS PubMed; (b) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef CAS PubMed.
- (a) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed; (b) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797–9804 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |