
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of micro–mesoporous ZSM-5 zeolite with microcrystalline cellulose as co-template and catalytic cracking of polyolefin plastics
Hongchang Yua,
Fuwei Lia,
Wen Hea,
Caifeng Songa,
Yansong Zhanga,
Zhixia Li *a and
Hongfei Linb
*a and
Hongfei Linb
aSchool of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China. E-mail: zhixiali@hotmail.com
bGuangxi Bossco Environmental Protection Technology Co., Ltd., Nanning 530007, China
First published on 9th June 2020
Abstract
Micro–mesoporous ZSM-5 with different Si/Al ratios (MZ-X, X = 27, 80, 150) were synthesized by adding microcrystalline cellulose (MCC) as co-template into the hydrothermal synthesis process of zeolites. The resultant ZSM-5 were used for catalytic cracking of high density polyethylene (HPDE) and polypropylene. It was found that introduction of MCC significantly enhanced the formation of mesopores and strong acid sites. MZ-27 achieved the highest oil yield: 21.5% for HPDE and 32.1% for polypropylene, and the light aromatics (BTEX) selectivity therein were 87.6% and 79.7%, respectively. HZ-150 (MCC-free ZSM-5, Si/Al = 150) achieved the highest gas yield: 85.4% for HDPE and 76.7% for polypropylene, and the light olefins (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 2–4) selectivity therein were 44% and 48.3%, respectively. The dense acidic sites and mesoporous structure of MZ-27 were responsible for its better activity for producing aromatic products. The moderate acidity and microporous structure of HZ-150 were helpful for producing light olefins from catalytic cracking of polyolefin plastics.
2–4) selectivity therein were 44% and 48.3%, respectively. The dense acidic sites and mesoporous structure of MZ-27 were responsible for its better activity for producing aromatic products. The moderate acidity and microporous structure of HZ-150 were helpful for producing light olefins from catalytic cracking of polyolefin plastics.
1. Introduction
Light olefins such as ethylene and propylene, and light aromatics such as benzene, toluene, ethylbenzene and xylene (BTEX), are important raw materials for numerous chemical industrial processes.1 Currently, commercial production of light olefins and aromatics mainly depends on petroleum-based routes: the former involves steam cracking of naphtha and light diesel, and the latter mainly utilizes naphtha catalytic reformate. Along with the oil resource's gradual depletion, comes the huge challenge of developing non-petroleum routes for production of these fundamental raw materials.Polyolefin plastics are extensively used in industrial production and daily life. Global production of resins and fibers increased from 2 Mt in 1950 to 380 Mt in 2015, a compound annual growth rate of 8.4% was reported during that period.2 Meanwhile, the discharge of waste plastics has caused serious environmental problem and waste of the resources.3–6 The main treatment solutions of waste plastics are landfills, incineration, mechanical recycling and thermal degradation.7,8 Among them, thermal degradation is the most promising technology from resource recycling point of view.9 Several papers reviewed research results on thermal and catalytic pyrolysis of plastics.10–12 The thermal pyrolysis was generally carried out at high temperature, and achieved a wide product distribution including gases, liquid oils, waxes and solid residue. The catalytic pyrolysis could convert plastics into valuable products e.g. waxes, BTX and light olefins with high selectivity at lower temperature and reaction time. Product selectivity strongly depended on the natures of catalysts (porosity and acidity). A microporous catalyst with high acidity e.g. HZSM-5 produced more gases, while macroporous catalysts with less acidity e.g. MCM-41 enhanced the formation of liquid oils. The spent FCC catalyst was used for the cracking of plastics because of its cheapness and high activity, which obtained a high liquid yield, mainly including gasoline fractions.11
In order to minimize heat transfer limitations and physical problems (sticky nature) in the pyrolysis of plastics, different reactors have been proposed, e.g. fixed bed, fluidized bed, spouted bed and screw kiln.10 The benefits for the tubular fixed bed reactors is that the gas residence time in these reactors could be regulated by adjusting the flow rate of carrier gas. Fluidized bed reactors are characterized by their high heat and mass transfer rates. However, bed defluidization due to agglomeration of particles coated with fussed plastic would occur under severe operating conditions. Spouted beds are an alternative to fluidized beds, and are characterized by its vigorous gas–solid contact. Accordingly, bed defluidization could be avoided. Besides, screw kilns were used for pyrolysis of plastics, which make polymer pyrolysis handy and smooth. The plastics alone or the mixture of plastics with catalysts was feed into reactor, and polymer residence time was controlled by varying the rotation speed of the screw.
In situ catalytic cracking is the primary way for all kinds of reactors. However, it has to face two main drawbacks: impurities in waste plastics would cause inactivation of catalysts, and there is serious diffusional restriction for bulky polymeric molecules accessing the active sites inside the porous catalysts. In order to improve the efficiency of the catalytic step, ex situ catalytic cracking technology was developed. Ex situ cracking can be arranged by a fixed bed reactor (or fluid bed/spouted bed reactor) connected in-line with a tubular reactor, with the aim of the first reactor being pyrolysis to yield oil and waxes. In the second reactor, the volatiles formed in the first reactor are subsequently pyrolyzed at high temperatures and short residence times. A high gases yield is easily obtained by ex situ cracking. Another advantage by ex situ cracking is that two steps (pyrolysis and catalytic cracking) can be operated under different conditions.10
So far, a lot of researches have been carried out on catalytic cracking of waste plastics for production of chemicals. For instance, plastic film wastes were cracked over nanocrystalline HZSM-5 zeolite for selective production of light olefins.9 Pyrolysis-catalysis of high density polyethylene (HPDE) was carried out in a fixed bed, two stage reactor for the production of aromatics, and found that loading of 1 wt% Ga, Fe or Ru on the Y-zeolite catalysts led to a higher production of aromatic hydrocarbons in the product oil (aromatic content >90%).13 Nishino et al.14 used a Ga-ZSM-5 catalyst for the catalytic pyrolysis of industrial plastic waste, a liquid product yield >50% consisting of >80% aromatic compounds were obtained. These results indicate that the zeolites especially HZSM-5, are the most effective catalyst for catalytic conversion of waste plastics into aromatics. Aguado et al.15 studied the catalytic cracking of PP over HMCM-41, SiO2–Al2O3 and HZSM-5, and reported 99.2% conversion for HMCM-41 and only 11.3% for HZSM-5. The higher activities of HMCM-41 was deemed to be associated with the presence of highly accessible acid sites in the mesoporous catalysts. A mesoporous structure of catalysts would be helpful for the catalytic cracking of bulky polymers.
Cellulose is a chain macromolecule, and represents one kind of major renewable and cheap resource. In this work, three micro–mesoporous ZSM-5 with different Si/Al ratio (27, 80, 150) were hydrothermally synthesized using tetrapropyl ammonium bromide as main template, and microcrystalline cellulose (MCC) as co-template, with the aim of reducing the production cost of mesoporous catalysts which are often produced by using expensive organic structure directing agents.16–18 The effect of MCC addition on acidity and pore structure of zeolites were investigated. The obtained ZSM-5 were used for catalytic cracking of HDPE and PP to produce high added-value chemicals e.g. light olefins and BTEX.
2. Experimental
2.1 Materials
Colloidal silica (Ludox, 40%, Guangzhou Suize Co. Ltd), ammonium chloride (NH4Cl, 99.5%, Sinopharm Chemical Reagent Co. Ltd), sodium aluminate (NaAlO2, Al2O3 wt% = 50 Sinopharm), sodium hydroxide (NaOH, 96%, Aladdin Reagents), tetrapropyl ammonium bromide (TPABr, 98%, Aladdin), microcrystalline cellulose (MCC, Beijing Solarbio Science & Technology Co. Ltd), and sulfuric acid (H2SO4, 98%) were used as received. PP (d = 910 kg m−3) and HDPE (d = 951 kg m−3) were cylindrical pellets with particle size about 4 mm, provided by China National Petroleum Corporation.2.2 Synthesis of the catalysts
ZSM-5 samples with different Si/Al ratio (27, 80, 150) were synthesized according to a hydrothermal method.19,20 0.8 g MCC was soaked in 10 g H2O for 24 h to obtain the MCC suspension, denoted as MCS. The molar compositions of three ZSM-5 precursors was 200SiO2–xAl2O3–35NaOH–10TPABr–15H2SO4–2445H2O. Precursors A, B and C were obtained by setting x at 7.4, 2.5 and 1.33, respectively. A typical synthesis process was as follows: first, a certain amount of NaOH and NaAlO2 were dissolved in H2O to form solution I. A certain amount of Ludox and template were dispersed in H2O to form solution II. Solution I was added into solution II drop by drop with agitation to form gel mixture. A certain amount of moderate concentrated H2SO4 was added to the mixture to adjust gel pH to 9 to obtain the precursors. Then, MCS was added into the precursor gel, and continuously stirred for 4 h. Finally, the resultant gel was transferred into a stainless-steel Teflon-lined autoclave and crystallized at 180 °C for 48 h. The obtained solid product was filtered, washed with distilled water until the pH of the filtrate was 7, and then dried at 105 °C overnight. The resultant product was afterwards calcined at 550 °C for 6 h to remove the organic templates. The obtained sample was defined as NaZSM-5 zeolite. HZSM-5 zeolite was obtained by ion-exchanging NaZSM-5 zeolite two times with 1 mol L−1 NH4Cl solution (liquid/solid = 10 mL g−1) at 80 °C for 4 h. The solid sample was filtered, dried at 105 °C overnight, and calcined at 550 °C for 6 h. The obtained HZSM-5 was tableted, granulated and sieved to obtain 20–30 mesh catalyst. The three catalyst samples derived from three precursors A, B and C were correspondingly denoted as MZ-X (X = 27, 80, 150) according to the different Si/Al ratio. As reference, the MCC-free zeolites (denoted as HZ-X, X = 27, 80, 150) were synthesized according to the above procedure but no addition of MCS.2.3 Characterizations of catalysts
The crystalline structure of samples was determined by X-ray powder diffraction (XRD) using a Rigaku SmartLab3 instrument in the range of 5–50 °C at a scanning rate of 8° min−1. The surface morphology of the catalysts was observed on Hitachi SU8220 scanning electron microscopy (SEM). N2 adsorption–desorption isotherm was performed at −196 °C, employing the Quantachrome NOVA 2200e instruments. The pore size distribution was obtained using the non-local density functional theory (NLDFT) model.The ammonia temperature-programmed desorption (NH3-TPD) was carried out by AutoChemII2920 to characterize the acidity of the catalysts. Prior to adsorption experiments, 300 mg of catalyst was pretreated at 200 °C for 1 h in a He flow. Upon cooling to 50 °C, the samples were saturated with a NH3 flow for 1 h, and then the physically absorbed NH3 was removed through purging with He gas for 1 h. The samples were then heated to 800 °C at a heating rate of 10 °C min−1 in He flow. Desorbed ammonia was recorded by a thermal conductivity detector (TCD). The total acidity amount was calculated from the peak area compared to a standard peak area of 1 mL of NH3 based on NH3-TPD curve. The distribution of weak acid, medium acid, and strong acid was calculated from the peak area from Gaussian fitting of the NH3-TPD profiles.
Infrared spectroscopic (IR) analysis was carried out on a Shimadzu spectrometer (IRTracer-100) with scanning range from 4000 cm−1 to 400 cm−1 using the KBr pellet method. The thermal stability of coke was analysed by a thermogravimetric analyzer (TGA, 209F3, Tarsus) in a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 (v/v) O2/N2 mixture.
:70 (v/v) O2/N2 mixture.
2.4 Catalytic performance tests
HDPE and PP were dried at 80 °C for 2 h before use. The catalytic cracking experiments were performed in a fixed-bed quartz tube reactor. For each run, the catalyst (0.5 g) was loaded into reactor, and heated to 550 °C in N2 stream (the flow rate: 40 mL min−1). The plastic sample (1.0 g) was fed in the reactor in three equal installments at a 5 min interval, and reacted for another 11 min. After flowing out from the reactor, the products were cooled at about −10 °C. The obtained gas was collected with a gas collecting bag, and its volume was recorded. Liquid products were weighed and analysed.2.5 Product analysis
The gas products were analysed using gas chromatography (GC, FULI-9790II, China) equipped with a flame ionization detector (FID) and a capillary column of HP-PLOT/Q. The following temperature program was used: hold at 25 °C for 4 min, heated to 240 °C at 15 °C min−1, and hold at 240 °C for 15 min. The qualitative and quantitative analyses were performed using the standard gases.The liquid products were analysed by GC-MS (GC7820A, MS 5977E, Agilent) equipped with a HP-5MS fused silica capillary column (30 m × 250 μm × 0.25 μm). The GC had been calibrated by typical polymer pyrolytic oil components using standard chemicals such as benzene, toluene, p-xylene and m-xylene. The oven temperature program was set as follows: hold at 60 °C for 5 min, heated to 290 °C at 10 °C min−1, and hold at this temperature for 5 min; the injector and interface temperature were maintained at 290 °C.
The total gas production (TGP), the liquid yield (YL), the coke yield (YC), the gas yield (YG), the selectivity to the light olefin (SLO), the selectivity to BTEX aromatics (SBTEX), were calculated by the following equations, respectively:
| TGP (mL g−1) = VTGP/M0 × 100% | (1) |
| YL (%) = M1/M0 × 100% | (2) |
| YC (%) = (Mc0 − Mc1)/M0 × 100% | (3) |
| YG (%) = 1 − YL − YC | (4) |
| SLO (%) = x(C2H4) + x(C3H6) + x(C4H8) | (5) |
| SBTEX (%) = y(benzene) + y(toluene) + y(ethylbenzene) + y(xylene) | (6) |
2.6 Stability experiments of catalysts
The stability of the catalysts was determined by continuous catalytic cracking of HDPE and PP over MZ-27 at 550 °C in N2 stream for 600 min. For each test, 0.3 g plastic sample was fed in the reactor every 5 min until the end of reaction. Liquid products were collected for 1 min every 30 min. Liquid yield and BTEX selectivity were analysed.3. Result and discussion
3.1 Characterization of the catalysts
The XRD patterns of all samples are shown in Fig. 1. The XRD were similar, and consisted of sharp peaks near 8°, 8.8°, 23.1° and 23.3°, matched well with the characteristic peaks of ZSM-5.21 Lattice parameters of catalysts were calculated and depicted in Table 1. The lattice constants were close to the value of ZSM-5 reported in a previous study.22 The relative crystallinity (calculated based on the sum of peak area between 22.5–25° in XRD pattern of the ZSM-5 samples compared to that of HZ-150) is 79.6% for HZ-27, 70.1% for MZ-27, 99.2% for HZ-80, 72.5% for MZ-80 and 90.2% for MZ-150. The crystallinity of MCC-adding catalysts decreased compared to the MCC-free catalysts. MCC could depolymerize to cellulose monomers, and even degrade to other micromolecules like monosaccharide and carboxylic acids during the process of gel crystallization.23,24 These degraded components could hinder crystallization reaction of silica species and aluminium species, consequently leading to the decrease in the crystallinity of MZ-X catalysts. The pH value of the reaction solution after crystallization reduced to 8 (initial pH was 9), indicating that hydrolysis reaction of MCC occurred and produced acidic compounds.25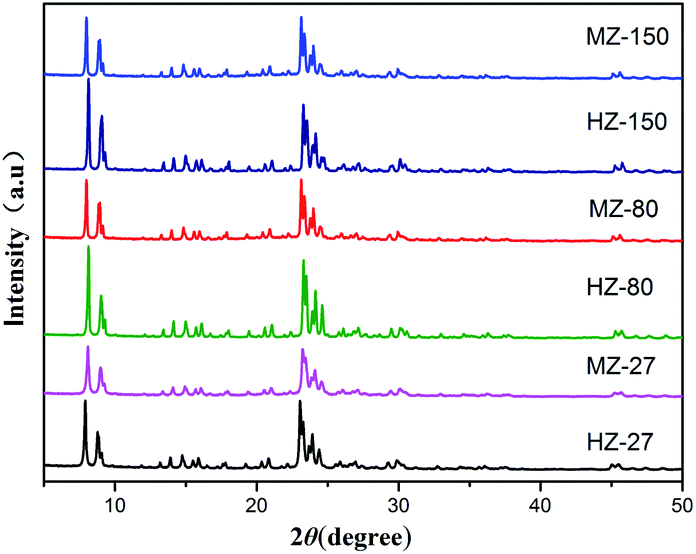 | ||
| Fig. 1 XRD patterns of the HZ-X and MZ-X samples. | ||
| Catalysts | Relative crystallinity (%) | Lattice constants (nm) | Vcell (nm3) | ||
|---|---|---|---|---|---|
| a | b | c | |||
| HZ-27 | 79.6 | 1.9992 | 1.9919 | 1.3347 | 5.315 |
| MZ-27 | 70.1 | 2.0102 | 1.9897 | 1.3413 | 5.365 |
| HZ-80 | 99.2 | 2.0088 | 1.9802 | 1.3386 | 5.325 |
| MZ-80 | 72.5 | 2.0133 | 1.9828 | 1.3326 | 5.320 |
| HZ-150 | 100 | 1.9987 | 1.9902 | 1.3401 | 5.331 |
| MZ-150 | 90.2 | 2.0087 | 1.9815 | 1.3371 | 5.322 |
Fig. 2 shows the IR spectra of all samples. There was strong bands at 796 cm−1, which is owing to the corresponding T–O–T symmetric stretching mode, 546 cm−1 resulted from the framework vibration at pentacyl ring which is characteristic of the MFI-type zeolite structure, and 450 cm−1 caused from vibration of the T–O–T bending bond, where T is Si or Al atoms.26 These results indicated that all samples contained ZSM-5 structure.
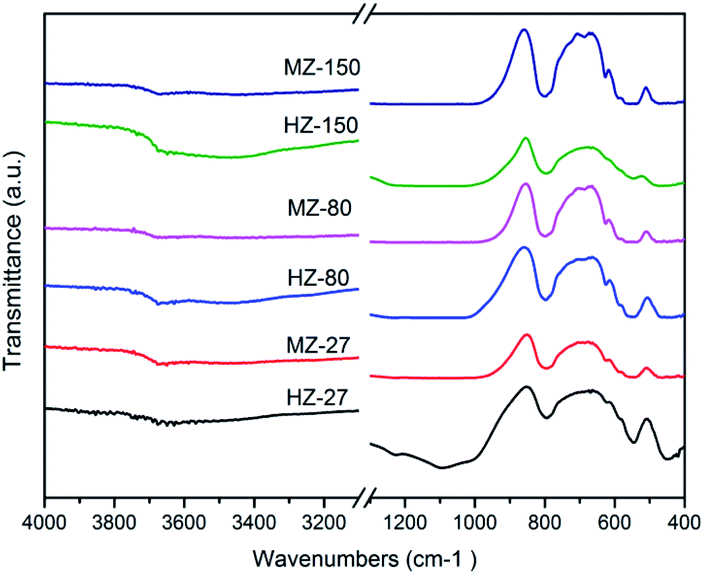 | ||
| Fig. 2 IR spectra of the HZ-X and MZ-X catalysts. | ||
The N2 adsorption–desorption isotherms and the pore size distribution of samples are illustrated in Fig. 3. As can be seen in Fig. 3(a), HZ-27, MZ-27 and MZ-80 showed an approximately IV-type isotherm with a hysteresis loop at a high relative pressure (P/P0 = 0.4–0.9), indicating the presence of mesopores. As shown in Fig. 3(b), the major pore diameter of all samples was around 3 nm. More large pores with diameter 3–14 nm were present in MZ-27 and MZ-80. All samples had a high specific surface area (>370 m2 g−1) and an average pore diameter about 3–4 nm (Table 2). The Sext and Vmeso of MZ-X samples were obviously larger than the corresponding HZ-X samples with the same Si/Al ratio. These results indicated that introduction of MCC into the hydrothermal synthesis process of zeolites significantly enhanced the formation of mesopores. The presence of mesopores in ZSM-5 is useful for accelerating the diffusion of bulkier molecules and accessing the active sites on catalysts.27
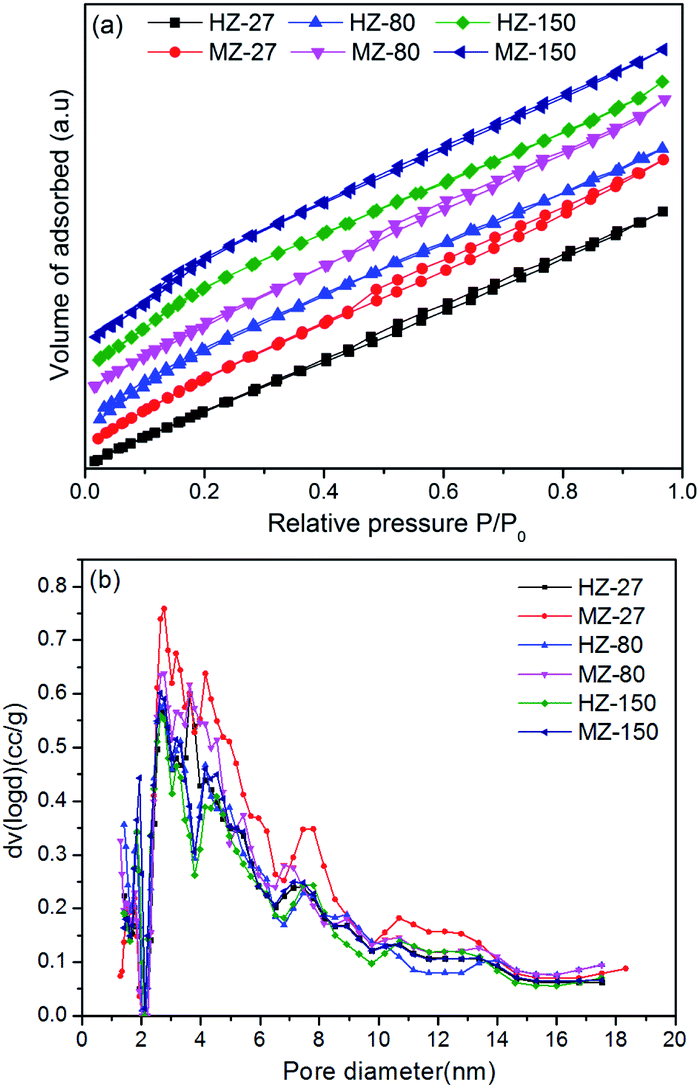 | ||
| Fig. 3 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of HZ-X and MZ-X catalysts. | ||
| Catalyst | SBETa (m2 g−1) | Sextb (m2 g−1) | Daverc (nm) | Vtotald (cm3 g−1) | Vmicrob (cm3 g−1) | Vmesoe (cm3 g−1) | Total acid (cm3 g−1) | Percentage of acid sites in total acid (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Weak acid | Medium acid | Strong acid | ||||||||
| a SBET (BET surface area) was obtained from the adsorption isotherm.b Sext (external surface areas) and Vmicro (micropore volumes) were calculated using t-plot method.c Daver (average pore size) was obtained from average pore size.d Vtotal (total pore volumes) was obtained at the relative pressure (P/P0) = 0.99.e Vmeso = Vtotal − Vmicro. | ||||||||||
| HZ-27 | 412.9 | 253.1 | 3.67 | 0.378 | 0.067 | 0.311 | 4.68 | 62.05 | 24.65 | 13.30 |
| MZ-27 | 371.6 | 282.7 | 4.28 | 0.447 | 0.054 | 0.393 | 4.49 | 50.39 | 35.57 | 14.04 |
| HZ-80 | 461.3 | 276.9 | 3.27 | 0.388 | 0.092 | 0.296 | 3.11 | 57.94 | 33.87 | 8.19 |
| MZ-80 | 453.3 | 341.3 | 3.73 | 0.442 | 0.078 | 0.364 | 2.83 | 55.57 | 21.95 | 18.46 |
| HZ-150 | 524.3 | 257.3 | 3.22 | 0.394 | 0.111 | 0.283 | 1.09 | 48.02 | 38.11 | 13.86 |
| MZ-150 | 485.6 | 303.2 | 3.25 | 0.421 | 0.101 | 0.320 | 0.98 | 44.69 | 36.40 | 18.91 |
The acid amount and relative acid strength in ZSM-5 samples were determined by NH3-TPD. The results are shown in Fig. 4 and Table 2. The weak acid, medium acid and strong acid were defined in the temperature region 100–230 °C, 230–350 °C, and 350–550 °C, respectively, in NH3-TPD curves. It was found that peak area and total acid amount decreased with increasing Si/Al ratio. This was consistent with the findings reported in previous research.28 The acid amount is related to the density of acidic sites, which are generated by Al species in the catalysts consisting of silica and alumina.29 In addition, it can be seen in Table 2, more strong acid sites were formed in MCC-adding samples compared to the MCC-free samples with same Si/Al ratio. This could be due to MCC-derived degraded compounds inducing the formation of new Brønsted acid sites on catalysts during crystallization process.
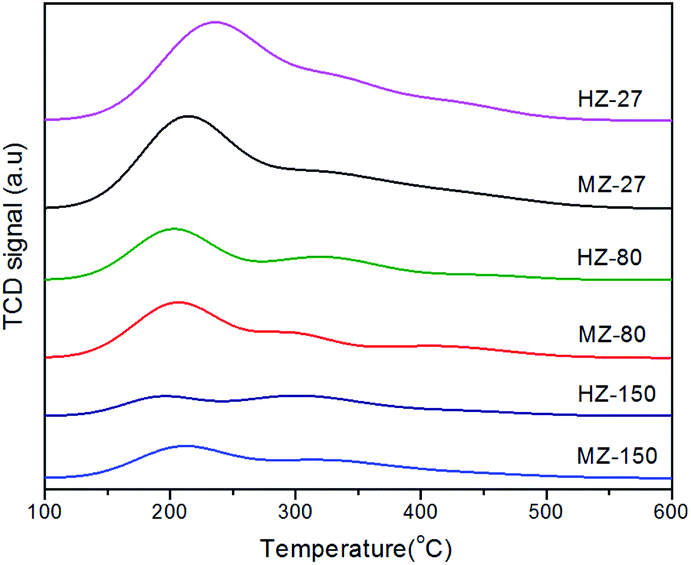 | ||
| Fig. 4 NH3-TPD profiles of the MZ-X and HZ-X samples. | ||
SEM images of the prepared ZSM-5 samples is shown in Fig. 5. As can be seen, HZ-27 and MZ-27 existed as spherical particles, and were actually aggregates of small needle-like crystals. HZ-80, MZ-80, HZ-150 and MZ-150 showed a typical “coffin” morphology.30 The difference would arise from the different crystallization reaction mechanisms. In the case of preparation of HZ-27 and MZ-27, zeolites was likely produced via a solid hydrogel transformation mechanism, and the high content of Na+ in the gel (added with introduction of NaAlO2) could play important structure directing role. The presence of Na+ helped to form the primary aluminosilicate species of five-membered structures in the initial gel, and subsequently facilitate the condensation or rearrangement of the primary structure building units.31,32 Compared with HZ-27, the particle size of the aggregates of MZ-27 were larger, and the needle-like crystals in the aggregates were obviously larger and more dispersive. This was probably ascribed to a synergistic structure directing effect of MCC. There is a lot of –OH groups on the surface of MCC, the considerable interaction between –OH and silicon and aluminum species (e.g. hydrogen bond) probably induces the growth of zeolite crystals in different direction; besides, the negative-charged degraded products from basic hydrolysis of MCC (e.g. glucose, straight-chain and branch-chain hydroxy-monocarboxylic and dicarboxylic acids) would produce steric effect,25 and hamper the cross-linking reaction of silicon and aluminum species in horizontal direction, consequently producing one-way growing needle-like crystals. Therefore, we speculated that there will be a synergy effect between TPABr, Na+ and MCC.
 | ||
| Fig. 5 SEM images of the prepared ZSM-5 samples: (a) HZ-27, (b) MZ-27, (c) HZ-80, (d) MZ-80, (e) HZ-150, (f) MZ-150. | ||
In the case of preparation of zeolites with higher Si/Al ratio e.g. HZ-80, MZ-80, HZ-150 and MZ-150, zeolites could be produced by following a solution-mediated transport mechanism. Owing to lack of Na+ in gel, there were little primary structure building units of five-membered structures formed in the solid-phase of the gel, and the nucleation of gel should occur in the liquid phase.31 The production of large ZSM-5 crystals indicated the lack of nuclei in the liquid phase of gel. The interaction between MCC and silicon and aluminum species could restrain the diffusion of silicon and aluminum species in the solid-phase of the gel into the liquid phase, and thus influence the formation of nuclei as well as growth of crystals, consequently leading to the formation of a larger particle of MZ-80 compared to HZ-80.
3.2 Catalyst performance
Catalytic cracking reactions of HDPE and PP were carried out at 550 °C in N2 stream over all prepared ZSM-5 samples. Gas yield, liquid yield, and product distribution are summarized in Tables 3 and 4.| Catalysts | ||||||
|---|---|---|---|---|---|---|
| HZ-27 | MZ-27 | HZ-80 | MZ-80 | HZ-150 | MZ-150 | |
| YTGP (mL g−1) | 433 | 402 | 446 | 432 | 468 | 444 |
| YL (%) | 20.8 | 21.5 | 17.5 | 18.8 | 13.9 | 14.3 |
| YC (%) | 1.6 | 1.2 | 1.2 | 0.8 | 0.7 | 0.5 |
| YG (%) | 77.6 | 77.3 | 81.3 | 80.4 | 85.4 | 85.2 |
|
||||||
| Gas product (%) | ||||||
| C2H4 | 13.8 | 13.5 | 13.1 | 12.9 | 13.9 | 14.1 |
| C3H6 | 17.3 | 13.8 | 18.5 | 17.3 | 24.8 | 19.8 |
| n-C4H8 | 5.5 | 4.0 | 5.3 | 6.1 | 5.3 | 8.9 |
| CH4 | 4.4 | 6.6 | 4.9 | 3.0 | 2.2 | 2.3 |
| C2H6 | 2.9 | 3.7 | 1.3 | 2.9 | 1.6 | 2.4 |
| C3H8 | 16.9 | 20.3 | 18.3 | 18.9 | 10.3 | 13.9 |
| n-C4H10 | 4.7 | 4.6 | 4.9 | 5.8 | 6.7 | 5.2 |
| C2–4 |
36.6 | 31.3 | 36.9 | 36.3 | 44.0 | 42.8 |
| C1–4 | 28.9 | 35.2 | 29.4 | 30.6 | 20.8 | 23.8 |
| Others | 34.5 | 33.5 | 33.7 | 33.1 | 35.2 | 33.4 |
|
||||||
| Liquid product (%) | ||||||
| Benzene | 5.9 | 8.7 | 4.9 | 6.4 | 4.5 | 4.6 |
| Toluene | 23.4 | 28.3 | 21.3 | 25.7 | 19.6 | 23.2 |
| Ethylbenzene | 7.3 | 5.7 | 5.3 | 6.5 | 6.3 | 6.7 |
| Xylene | 41.3 | 44.9 | 42.5 | 46.2 | 35.8 | 38.4 |
| BTEX | 77.9 | 87.6 | 74 | 84.5 | 66.2 | 72.9 |
| Aliphatic hydrocarbons | 7.8 | 1.8 | 8.1 | 3 | 19.2 | 16.9 |
| Others | 14.3 | 10.6 | 17.9 | 12.2 | 14.6 | 10.2 |
| Catalysts | ||||||
|---|---|---|---|---|---|---|
| HZ-27 | MZ-27 | HZ-80 | MZ-80 | HZ-150 | MZ-150 | |
| YTGP (mL g−1) | 353 | 331 | 367 | 348 | 412 | 381 |
| YL (%) | 29.3 | 32.1 | 24.1 | 27.2 | 22.8 | 25.3 |
| YC (%) | 1.2 | 1.0 | 0.8 | 0.6 | 0.5 | 0.2 |
| YG (%) | 69.5 | 66.9 | 75.1 | 72.2 | 76.7 | 74.5 |
|
||||||
| Gas product (%) | ||||||
| C2H4 | 15.0 | 12.8 | 15.5 | 14.3 | 15.1 | 13.1 |
| C3H6 | 17.1 | 14.5 | 20.1 | 17.3 | 25.6 | 22.4 |
| n-C4H8 | 4.9 | 4.5 | 6.3 | 5.4 | 7.6 | 9.1 |
| CH4 | 8.1 | 6.5 | 6.9 | 6.1 | 3.7 | 3.1 |
| C2H6 | 5.9 | 4.7 | 5.4 | 5.9 | 3.5 | 4.6 |
| C3H8 | 17.4 | 23.1 | 20.1 | 22.8 | 10.8 | 13.9 |
| n-C4H10 | 4.5 | 3.8 | 4.6 | 4.6 | 3.4 | 4.0 |
| C2–4 |
37.0 | 31.8 | 41.9 | 37.0 | 48.3 | 44.6 |
| C1–4 | 35.9 | 38.1 | 37.0 | 39.4 | 21.4 | 25.6 |
| Others | 27.1 | 30.1 | 21.1 | 23.6 | 30.3 | 29.8 |
|
||||||
| Liquid product (%) | ||||||
| Benzene | 7.5 | 8.1 | 6.2 | 7.9 | 5.3 | 4.8 |
| Toluene | 23.3 | 24.9 | 22.1 | 23.5 | 18 | 22.6 |
| Ethylbenzene | 5.4 | 5.6 | 5 | 5.1 | 5.1 | 4.9 |
| Xylene | 38.2 | 41.1 | 37.8 | 38.5 | 32.4 | 31.9 |
| BTEX | 74.4 | 79.7 | 71.1 | 75 | 60.8 | 64.2 |
| Aliphatic hydrocarbons | 12.2 | 11.5 | 17.8 | 15.8 | 30.7 | 30.3 |
| Others | 13.4 | 8.8 | 11.1 | 9.2 | 8.5 | 5.5 |
2–4) and alkanes (C1–4) were the main products from catalytic cracking of HDPE. It was also found that YG increased while YL and YC decreased with increasing Si/Al ratio of the catalysts. Besides, MZ-X achieved a higher liquid yield, but a lower gas yield than the corresponding HZ-X catalysts with same Si/Al ratio. YC on MZ-X were lower than on the corresponding HZ-X. The maximum YTGP (468 mL g−1) and the highest C2–4 selectivity (44%) were achieved over the HZ-150. The highest YL (21.5%) and BTEX selectivity (87.6%) were achieved over MZ-27.Combing the analysis on acidity and pore structure of catalysts, we deduced that the amount of acidic sites was mainly responsible for the formation of liquid products and coke, while a mesoporous structure facilitated the formation of liquid products. Acidic active sites were necessary for improving the decomposition of polymers via carbonium ion and β-scission reaction mechanisms. Meanwhile, acidic active sites were also necessary for the formation of aromatic products via cyclization and dehydrogenation reaction of cracking fragments. However, the over-crowded acidic sites could cause excessive coupling reaction of aryl groups through a carbonium ion reaction mechanism, leading to the formation of cokes (e.g. HZ-27). Therefore, optimization and modification of the acidic property of zeolites are important to improve product selectivity. Addition of MCC in synthesis process of zeolites enhanced the formation of mesopores, and thus improved the mass transfer and diffusion rate of reactants and intermediate products, consequently restraining the secondary reactions e.g. coking and accelerating the formation of liquid products.33 The presence of a substantial proportion of benzene, toluene and xylene in liquid product (Table 3), indicates that catalytic cracking of HDPE over MZ-27 is a potential technique to selectively produce BTEX via a non-petroleum route.
For the catalysts with high Si/Al ratio e.g. HZ-150 and MZ-150, the moderate acid sites on zeolites (Table 2) could be adequate for catalytic scission of polymers into micromolecule fragments, but inadequate for the formation of aromatics-rich liquid products via multi-position adsorption and activation of these micromolecule fragments.34 Therefore, a lot of gas products were produced on these catalysts. The presence of more aliphatic hydrocarbons (16.9–19.2%) in HZ-150 and MZ-150 derived liquid products, indicates an incomplete aromatization reactions occurring on these catalysts.
If considering the importance of both light olefins and BTEX in industrial applications, HZ-150 and MZ-27 exhibited their own great application potential.
Artetxe et al. carried out a similar research on cracking HDPE on HZSM-5 with different Si/Al ratios in a thermal-catalytic two-step unit (spouted bed connected by fixed bed).35 The maximum light olefin yield was 58 wt%, obtained at 500 °C using the most acidic catalyst (Si/Al = 30). This disagrees with our results. The maximum light olefin yield (37.6%) in our study was achieved over the weakest acidic catalyst (HZ-150). The smaller pore could inhibit polymer oligomers accessing the active sites inside the pore of zeolite; the high temperature (550 °C) could accelerate secondary reactions e.g. aromatization and coking; the fixed bed reactor used in our study could cause heat transfer limitation. These factors led to a lower light olefin yield in our study. Other studies on cracking plastics over HZSM-5 were found to be about selective production of light olefins. Plastics were commonly cracked in fluidized-bed or spouted bed reactors at a temperature of 400–500 °C. A gas yield of 60–88% with light olefins selectivity of 40–75% were achieved.36,37 Besides, the light aromatics selectivity in MZ-27 derived oil is 87.6%, which is similar to that reported in previous study, obtained from catalytic pyrolysis of industrial plastic waste on Ga-ZSM-5 (aromatic content >80%).15 In order to improve light aromatics yield, an increase in residence time should be considered.
It was found that regardless of PP and HDPE, propylene selectivity increased whereas C1–4 selectivity decreased with increasing Si/Al ratio of catalysts. This would be ascribed to the acidity of catalysts. The dense acid sites present on HZ-27 and MZ-27 helped to improve cracking of polymers, and also subsequent secondary reactions of cracking fragments, consequently leading to formation of more stable gas (C1–4) and liquid products (e.g. aromatics). The catalysts with high Si/Al ratio e.g. HZ-150 and MZ-150, had less acid sites, and thus a relatively weaker secondary reaction of cracking fragments could occur on its surfaces, therefore, more propylene was present in gas products. Isopentane, pentylene, isobutylene and butadiene were probably produced during the cracking process but could not be identified with the present standard gas. Accordingly, they were classified in other gas products.
The TGA and DTG profiles of the spent catalysts are shown in Fig. 6. The weight loss below 200 °C mainly resulted from the removal of physically adsorbed water, the weight loss between 200–500 °C could be attributed to the oxidation of light coke, while weight loss between 500–800 °C was probably due to the oxidation of heavy coke such as stable graphite.41 The order of weight loss between 200–800 °C (coke amount) for different catalysts was: HZ-27 > MZ-27 > MZ-80 > MZ-150. The trend of coke amount from TGA agrees with the results obtained from calcination method. These results indicate that a mild acidity and a larger pore structure would be helpful for restraining the formation of coke and improving the lifetime of catalysts.
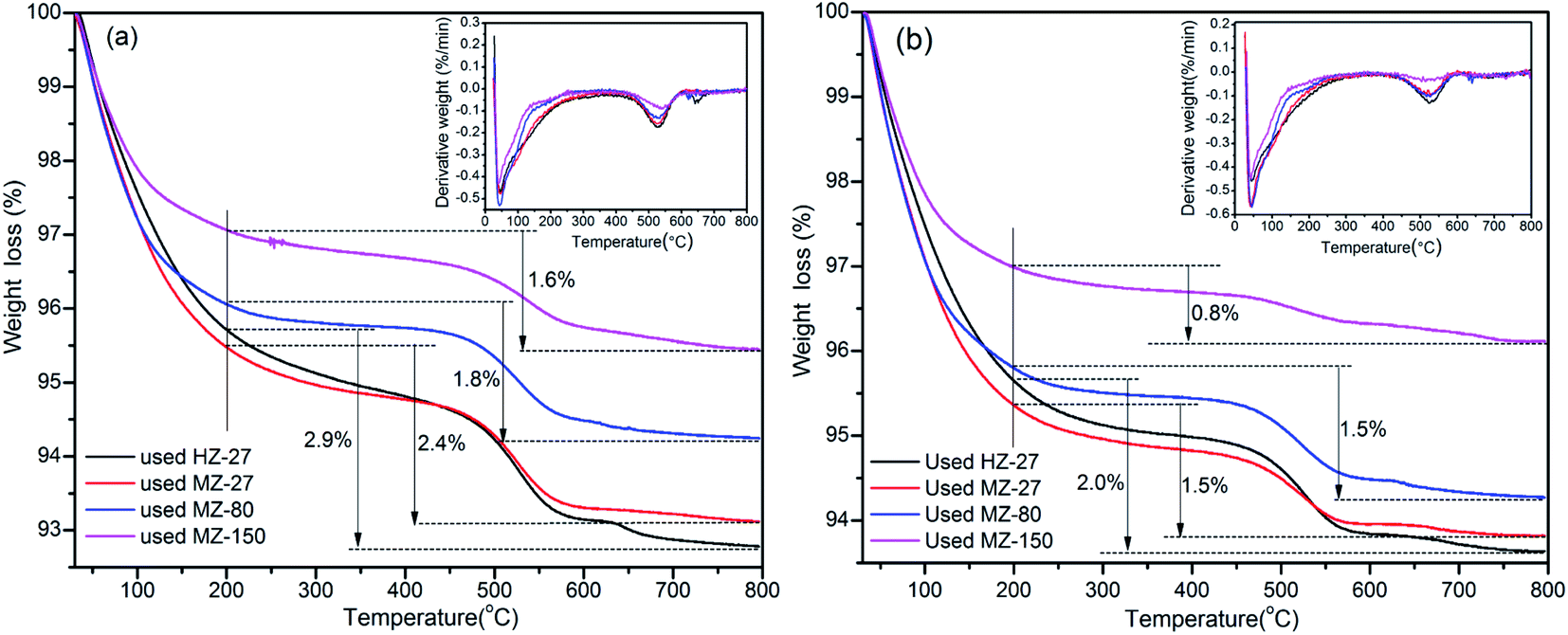 | ||
| Fig. 6 TGA and DTG profiles of the used catalysts from cracking HDPE (a) and PP (b). | ||
| No. | RT (min) | Compounds | Molecular formula | Catalysts and feedstocks area (%) | |||
|---|---|---|---|---|---|---|---|
| HZ-27 (HDPE) | MZ-27 (HDPE) | HZ-27 (PP) | MZ-27 (PP) | ||||
| a —: not detected. | |||||||
| 1 | 1.71 | Cyclopentane, methyl- | C6H12 | 1.0 | 0.5 | 1.5 | 0.8 |
| 2 | 1.81 | 1,4-Pentadiene, 2-methyl- | C6H10 | 0.9 | 0.3 | 0.8 | 0.7 |
| 3 | 1.87 | Benzene | C6H6 | 5.9 | 8.8 | 7.5 | 8.1 |
| 4 | 1.97 | Cyclopentane, 1,3-dimethyl-, trans- | C7H14 | 0.5 | — | 0.5 | 0.4 |
| 5 | 2.04 | Heptane | C7H16 | 0.5 | 0.3 | 0.6 | — |
| 6 | 2.17 | Cyclopentene, 1,5-dimethyl- | C7H12 | 0.3 | 0.2 | 0.5 | 0.3 |
| 7 | 2.25 | Cyclohexane, methyl- | C7H14 | 0.5 | 0.4 | 0.2 | — |
| 8 | 2.34 | Cyclopentane, ethyl- | C7H14 | 0.4 | — | 0.1 | 0.1 |
| 9 | 2.40 | Cyclohexene, 4-methyl- | C7H12 | 0.5 | — | 0.4 | 0.3 |
| 10 | 2.52 | Cyclobutane, (1-methylethylidene)- | C7H12 | 1.0 | 0.5 | 0.8 | 0.8 |
| 11 | 2.64 | Heptane, 4-methyl- | C8H14 | — | — | — | 0.9 |
| 12 | 2.70 | Toluene | C7H8 | 23.4 | 28.3 | 23.4 | 24.9 |
| 13 | 3.05 | Octane | C8H18 | 0.8 | — | 0.6 | 0.2 |
| 14 | 3.24 | 1,3-Dimethyl-1-cyclohexene | C8H14 | 0.4 | — | 1.2 | 1.3 |
| 15 | 3.41 | Heptane, 2,4-dimethyl- | C9H20 | — | — | 0.5 | 0.5 |
| 16 | 3.63 | Cyclohexane, 1,3,5-trimethyl- | C9H18 | — | — | 1.6 | 1.6 |
| 17 | 4.01 | 1,2,4,4-Tetramethylcyclopentene | C9H16 | — | — | 0.6 | 0.3 |
| 18 | 4.15 | Cyclohexane, 1,3,5-trimethyl-, (1-alpha, 3-alpha, 5-beta)- | C9H18 | — | — | 2.0 | 2.3 |
| 19 | 4.31 | Ethylbenzene | C8H10 | 7.4 | 5.7 | 5.4 | 5.6 |
| 20 | 4.57 | p-Xylene | C8H10 | 36.3 | 36.6 | 31.5 | 32.8 |
| 21 | 5.18 | o-Xylene | C8H10 | 5.1 | 8.3 | 6.8 | 8.4 |
| 22 | 5.30 | Nonane | C9H20 | 0.5 | 0.3 | — | — |
| 23 | 6.85 | Benzene, propyl- | C9H12 | 0.6 | 5.3 | 0.4 | 0.3 |
| 24 | 7.05 | Benzene, 1-ethyl-3-methyl- | C9H12 | 6.9 | 0.2 | 2.8 | 1.2 |
| 25 | 7.51 | Benzene, 1-ethyl-2-methyl- | C9H12 | — | 1.2 | 2.5 | 1.8 |
| 26 | 7.84 | Benzene, 1,2,3-trimethyl- | C9H12 | 0.8 | — | 1.3 | 0.8 |
| 27 | 8.19 | Nonane, 2,6-dimethyl- | C11H14 | — | 0.6 | 0.7 | 1.3 |
| 28 | 8.79 | Indane | C9H10 | 0.6 | 0.3 | 0.6 | 0.4 |
| 29 | 9.14 | Benzene, 1-methyl-3-propyl- | C10H14 | 0.5 | — | 0.5 | 0.4 |
| 30 | 9.22 | Benzene, 1-methyl-4-propyl- | C10H14 | 0.8 | — | 0.6 | 0.3 |
| 31 | 9.76 | Benzene, 1-methyl-2-(2-propenyl)- | C10H12 | — | — | 0.2 | 0.2 |
| 32 | 9.86 | Benzene, 1-methyl-4-(2-propenyl)- | C10H12 | — | — | 0.2 | 0.4 |
| 33 | 10.08 | Undecane | C11H24 | 0.3 | 0.4 | — | — |
| 34 | 10.87 | 1H-Indene, 2,3-dihydro-4-methyl- | C10H12 | 0.3 | 0.2 | 0.5 | 0.3 |
| 35 | 11.06 | Benzene, 1-methyl-1,2-propadienyl- | C10H10 | 0.2 | 1 | — | — |
| 36 | 11.67 | Naphthalene | C10H8 | 1.6 | — | 1.1 | 0.8 |
| 37 | 11.85 | Dodecane | C12H26 | 0.6 | — | — | — |
| 38 | 12.44 | Dodecane, 4,6-dimethyl- | C14H30 | — | 0.8 | 0.5 | 0.2 |
| 39 | 13.44 | Naphthalene, 1-methyl- | C11H10 | 1.1 | — | 1.5 | 0.9 |
| 40 | 15.51 | 2,3-Dimethyldodecane | C14H30 | — | — | 0.5 | 0.2 |
| 41 | 15.87 | Hexadecane, 3-methyl- | C14H30 | — | — | 0.5 | 0.1 |
| 42 | 17.29 | Cetene | C16H32 | 0.4 | — | — | — |
Combined with the results of current reports,40,42 the possible reaction pathway of HDPE and PP decomposition were concluded as follows. Catalytic cracking of polyolefins includes pyrolysis and catalytic cracking reactions. The polyolefin is firstly cracked into volatile alkyl radicals via random scission of the long chains of the polymers at high temperature according to free radical mechanism.34 The volatile components subsequently undergo a series of further secondary reactions including cracking, Diels–Alder reaction, dehydrogenation, hydrogen transfer and aromatization upon passing through catalyst bed according to a carbenium ion mechanism, consequently leading to the formation of gas products e.g. ethylene and propylene, and liquid products including cyclic hydrocarbons (1,3-dimethyl-cyclopentane, methyl-cyclohexane, 1,5-dimethyl-cyclopentene, 4-methyl-cyclohexene) and aromatic products.42 Compared to HDPE, more C9 hydrocarbons (propylene oligomer) present in PP-derived oil, which would result from breakage of PP chain at tertiary carbons.
3.3 Stability of the catalysts
Fig. 7 shows the results for the continuous cracking of HDPE and PP in 300 min. The collected liquid oil during 1 min ranged from 0.02–0.03 g for HDPE, and 0.03–0.04 g for PP. BTEX selectivity decreased from 87.6% to 79.3% for HDPE, and from 80% to 65.7% for PP after 300 min. Xylene and toluene selectivity decreased slightly with increasing reaction time. Carbon deposition on catalysts could result in the decrease in catalytic activity. The presence of the pendant methyl groups increases the effective cross-section of the PP oligomers, indicates that PP molecules are more difficult to diffuse into the pore of the zeolite compared to HDPE. This inhibits the aromatization reaction of PP. The stability of the synthesized MZ-27 catalyst should be further improved in the successive research.38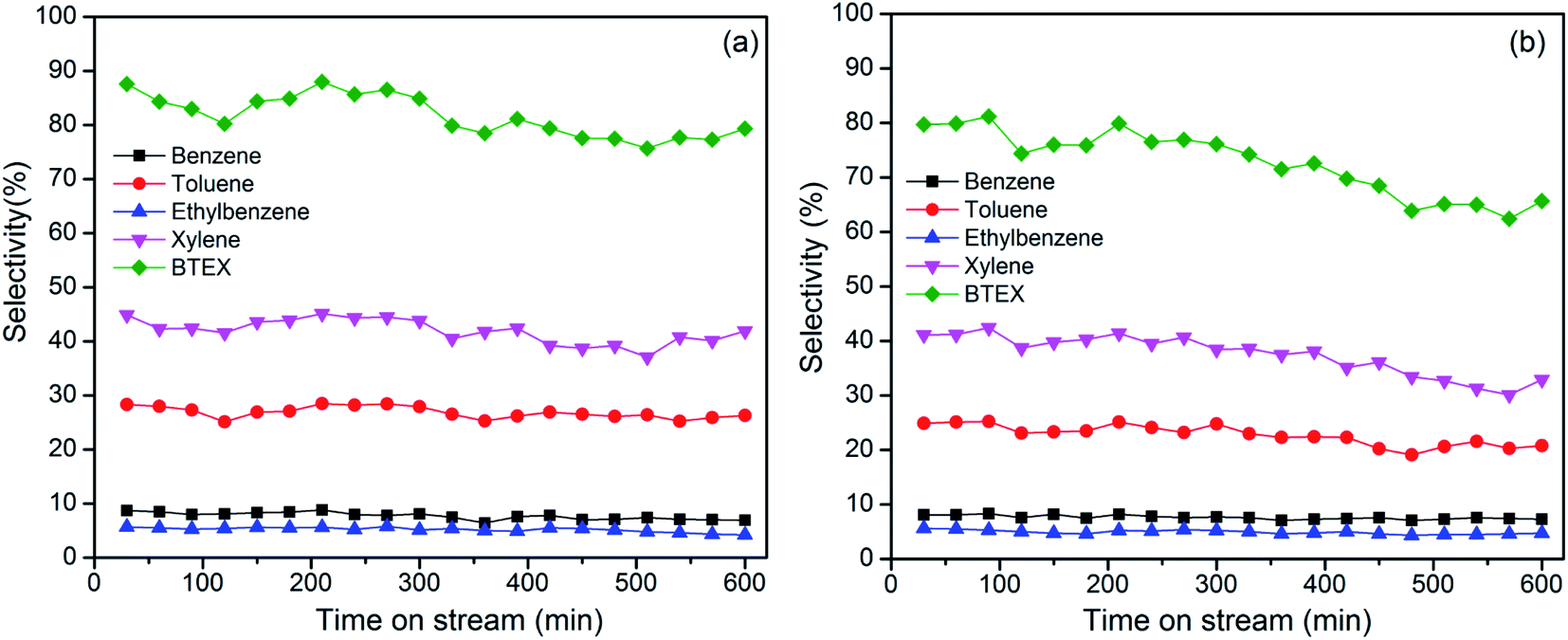 | ||
| Fig. 7 BTEX selectivity with time on stream from cracking of HDPE (a) and PP (b) over MZ-27 at 550 °C. | ||
4. Conclusions
ZSM-5 zeolites with micro–mesopore structure were successfully prepared by introducing MCC as co-template into hydrothermal synthesis process. The higher Si/Al ratio led to the formation of zeolites with less acid sites. MCC-adding ZSM-5 (MZ-X) exhibited higher mesopore volume and more strong acid sites compared to the MCC-free HZ-X zeolites. The resultant ZSM-5 were used for catalytic cracking of HDPE and PP. MZ-27 which possessed more acid sites and mesopores achieved the highest oil yields: 21.5% for HDPE and 32.1% for PP, and the BTEX selectivity therein were 87.6% and 79.7%, respectively. HZ-150 which possessed more micropores and less acid sites obtained the highest gas yields: 85.4% for HDPE and 76.7% for PP, and the C2–4 selectivity therein were 44% and 48.3%, respectively. It was concluded that the acidity and porous structure of catalysts significantly affected the conversion and product distribution. MCC as an economical and renewable resource showed potential application prospects to be used as template to modulate pore structure and acidity of zeolites.
Conflicts of interest
Authors declare no competing interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21566004), and the Scientific Research Foundation of Guangxi University (XGZ120081).Notes and references
- C. D. Zhang, G. Kwaket, Y. J. Lee, K. W. Jun, R. X. Gao, H. G. Park, S. Kim, J. E. Min, S. C. Kang and G. F. Guan, Microporous Mesoporous Mater., 2019, 284, 316–326 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. D. A. Sharuddin, F. Abnisa, W. A. M. W. Daud and M. K. Aroua, Energy Convers. Manage., 2016, 115, 308–326 CrossRef.
- A. K. Panda, R. K. Singh and D. K. Mishra, Renewable Sustainable Energy Rev., 2010, 14, 233–248 CrossRef CAS.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- G. H. Song, H. Zhang, H. B. Duan and M. Xu, Resour., Conserv. Recycl., 2018, 130, 226–227 CrossRef.
- H. A. Arafat, K. Jijakli and A. Ahsan, J. Cleaner Prod., 2015, 105, 233–240 CrossRef.
- W. K. Kamal and P. Vijayabalan, Waste Manag., 2016, 51, 91–96 CrossRef PubMed.
- D. P. Serrano, J. Aguado, J. M. Escola, E. Garagorri, J. M. Rodrgueza, L. Morselli, G. Palazzi and R. Orsic, Appl. Catal., B, 2004, 49, 257–265 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2017, 73, 346–368 CrossRef CAS.
- R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan and A. S. Nizami, Process Saf. Environ. Prot., 2016, 102, 822–838 CrossRef CAS.
- B. Kunwar, H. N. Cheng, S. R. Chandrashekaran and B. K. Sharma, Renewable Sustainable Energy Rev., 2016, 54, 421–428 CrossRef CAS.
- K. Akubo, M. A. Nahil and P. T. Williams, J. Energy Inst., 2019, 92, 195–202 CrossRef CAS.
- J. Nishino, M. Itoh, H. Fujiyoshi and Y. Uemichi, Fuel, 2008, 87, 3681–3686 CrossRef CAS.
- J. Aguado, J. L. Sotelo, D. P. Serrano, J. A. Calles and J. M. Escola, Energy Fuels, 1997, 11, 1225–1231 CrossRef CAS.
- Z. H. Zhang, H. Cheng, H. Chen, K. Q. Chen, X. Y. Lu, P. K. Ouyang and J. Fu, Bioresour. Technol., 2018, 256, 241–246 CrossRef CAS PubMed.
- D. P. Gamliel, H. J. Cho, W. Fan and J. A. Valla, Appl. Catal., A, 2016, 522, 109–119 CrossRef CAS.
- H. Chen, X. Shi, J. F. Liu, K. C. Jie, Z. H. Zhang, X. B. Hu, Y. M. Zhu, X. Y. Lu, J. Fu, H. Huang and S. Dai, J. Mater. Chem. A, 2018, 6, 21178–21185 RSC.
- Q. Y. Wang, S. T. Xu, J. R. Chen, Y. X. Wei, J. Z. Li, D. Fan, Z. X. Yu, Y. Qi, Y. L. He, S. L. Xu, C. Y. Yuan, Y. Zhou, J. B. Wang, M. Z. Zhang, B. L. Su and Z. M. Liu, RSC Adv., 2014, 4, 21479–21491 RSC.
- Q. Zhang, S. Hu, L. L. Zhang, Z. J. Wu, Y. J. Gong and T. Dou, Green Chem., 2013, 1, 77–81 Search PubMed.
- S. Vichaphund, D. Aht-ong, V. Sricharoenchaikul and D. Atong, Renewable Energy, 2015, 79, 28–37 CrossRef CAS.
- X. Li, C. Gu and B. Zhong, J. Mol. Catal., 1992, 6, 104–112 CAS.
- K. Niemelä and E. Sjöström, Biomass, 1986, 11, 215–221 CrossRef.
- B. Y. Yang and R. Montgomery, Carbohydr. Res., 1996, 280, 27–45 CrossRef CAS.
- G. Nazeri, S. B. Liaw, Y. Yu and H. W. Wu, Fuel, 2018, 218, 174–178 CrossRef CAS.
- Y. Sang, Q. Z. Jiao, H. S. Li, Q. Wu, Y. Zhao and K. N. Sun, J. Nanopart. Res., 2014, 16, 2755 CrossRef.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodriguez and A. Peral, J. Catal., 2010, 276, 152–160 CrossRef CAS.
- C. Engtrakul, C. Mukarakate, A. K. Starace, K. A. Magrini, A. K. Rogers and M. M. Yung, Catal. Today, 2016, 269, 175–181 CrossRef CAS.
- L. Rodríguez-González, F. Hermes, M. Bertmer, E. Rodríguez-Castellón, A. Jiménez-López and U. Simon, Appl. Catal., A, 2007, 328, 174–182 CrossRef.
- S. Sang, F. Chang, Z. Liu, C. He, Y. He and L. Xu, Catal. Today, 2004, 93–95, 729–734 CrossRef CAS.
- C. Y. Liu, W. Y. Gu, D. J. Kong and H. C. Guo, Microporous Mesoporous Mater., 2014, 183, 30–36 CrossRef CAS.
- N. Y. Kang, B. S. Song, C. W. Lee, W. C. Choi, K. B. Yoon and Y. K. Park, Microporous Mesoporous Mater., 2009, 118, 361–372 CrossRef CAS.
- Y. H. Kim, K. H. Lee, C. M. Nam and J. S. Lee, ChemCatChem, 2012, 4, 1143–1153 CrossRef CAS.
- K. Sun, Q. X. Huang, Y. Chi and J. H. Yan, Waste Manag., 2018, 81, 128–137 CrossRef CAS PubMed.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2013, 52, 10637–10645 CrossRef CAS.
- M. D. R. Hernandez, A. N. Garcia, A. Gomez, J. Agullo and A. Marcilla, Ind. Eng. Chem. Res., 2006, 45, 8770–8778 CrossRef CAS.
- J. F. Mastral, C. Berrueco, M. Gea and J. Ceamanos, Polym. Degrad. Stab., 2006, 91, 3330–3338 CrossRef CAS.
- Q. Zhou, L. Zheng, Y. Z. Wang, G. M. Zhao and B. Wang, Polym. Degrad. Stab., 2004, 84, 493–497 CrossRef CAS.
- G. Elordi, M. Olazar, M. Artetxe, P. Castano and J. Bilbao, Appl. Catal., A, 2012, 415–416, 89–95 CrossRef CAS.
- Y. M. Ni, A. M. Sun, X. L. Wu, G. L. Hai, J. L. Hu, T. Li and G. X. Li, J. Colloid Interface Sci., 2011, 361, 521–526 CrossRef CAS PubMed.
- P. Castaño, G. Elordi, M. Olazar, A. T. Aguayo, B. Pawelec and J. Bilbaoa, Appl. Catal., B, 2011, 104, 91–100 CrossRef.
- K. Sun, Q. Huang, M. Ali, Y. Chi and J. Yan, Energy Fuels, 2018, 32, 5471–5479 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |