
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomo-condensation of acetophenones toward imidazothiones†
Phuc Hoang Pham‡
 ab,
Phuc Thai Thien Nguyen‡ab,
Thuy Thu Buiab,
Hien Nhat Traab,
Tung Thanh Nguyen*ab and
Nam Thanh Son Phan*ab
ab,
Phuc Thai Thien Nguyen‡ab,
Thuy Thu Buiab,
Hien Nhat Traab,
Tung Thanh Nguyen*ab and
Nam Thanh Son Phan*ab
aFaculty of Chemical Engineering, Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam. E-mail: tungtn@hcmut.edu.vn; ptsnam@hcmut.edu.vn
bVietnam National University Ho Chi Minh City, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam
First published on 4th November 2020
Abstract
Direct synthesis of imidazothiones from simple, commercial substrates is not known. We report a method for the condensation of acetophenones, elemental sulfur, and ammonium acetate as a nitrogen source to obtain the hitherto challenging five-membered heterocycles. Functionalities such as halogen, trifluoromethyl, cyano, methylthio, and heteroaryl groups were well tolerated.
Amino-2H-imidazoles such as commercial lanabecestat or a pyrimidine derivative, A (Scheme 1) are potent inhibitors against BACE-1 (β-site APP cleaving enzyme), and are thus useful in studies for treating Alzheimer's disease.1 Until now, available methods for the synthesis of amino-2H-imidazoles often proceed through substitution of imidazothiones with ammonia.1d It should be noted that this approach suffers from the use of toxic, flammable hydrogen sulfide gas.2 Development of continuous-flow conditions has recently increased the synthetic practicality.2b,c In 2012, Gravenfors and co-workers reported a rare study of substrate scope with respect to synthesis of 2,5-diaryl imidazothiones from acetophenones, α-oxoacetates, and ammonia, followed by sulfur exchange of the amide intermediates.1d Arguably, methods for directly assembling simple, commercial substrates to obtain such five-membered heterocycles are still deficient.
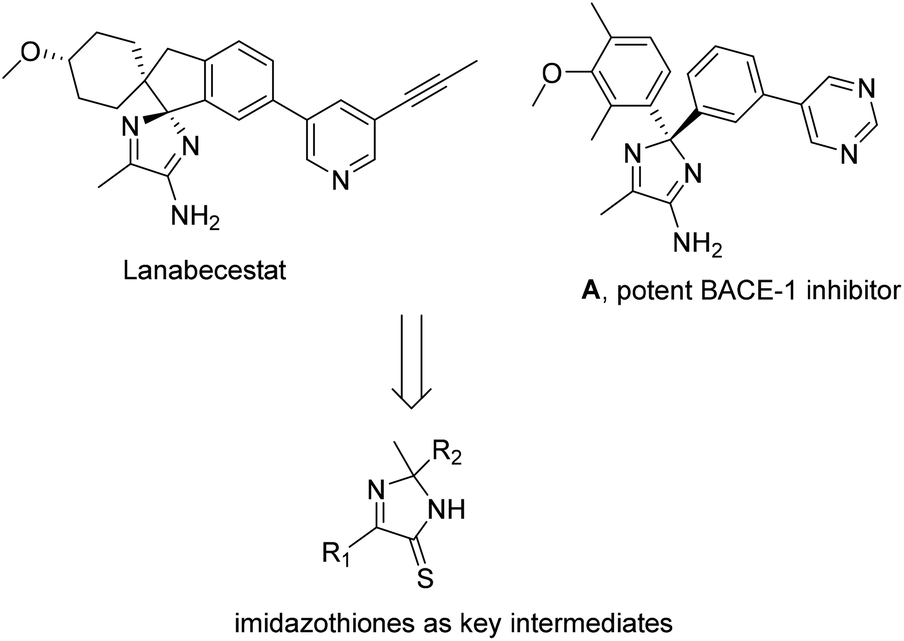 | ||
| Scheme 1 Importance of imidazothiones. | ||
Studies of using elemental sulfur in organic synthesis has been extensively investigated over the last few years, probably because it is cheap, abundant, and easy to handle. The element has replaced the need of heavy phosphorus–sulfur complexes such as Lawesson's reagent and P2S5 in synthesis of thioamides.3 Many methods for incorporation of small molecules into polycyclic S-heterocycles have been reported.4 In such transformations, C–H bonds in acetophenones or pseudo oxime esters have been considered as the major targets.
Oxime-assisted annulation of elemental sulfur and activated C–H bonds α to carbonyl group often afford synthetically challenging polyheterocycles.5 A few examples of furnishing benzothienothiazoles via functionalization of C–H bonds in acetophenone oximes have been reported.5b,c Nevertheless, the available methods have required a two-step prefunctionalization of aryl ketones. Arguably it would be more advantageous if C–H bonds in acetophenones could be directly functionalized, thus avoiding the unnecessary preparation of oxime substrates. Only a couple of reports, to our best knowledge, have described attempts to convert methyl C–H bonds of acetophenones without the need of N-oxime esters. Nguyen reported a synthesis of 2,4-diarylthiophenes via homo-annulation of acetophenones and elemental sulfur.5e The substrate scope was limited to simple functional groups such as alkyls, alkoxys, and halogens. Our group has recently presented an annulation of acetophenones, elemental sulfur, and urea as a nitrogen source.5f The reactions afforded benzothienothiazoles that were compatible with many functionalities such as methylthio, protected alcohol, and heterocycle groups. As a continuation of our interests in functionalization of C–H bonds in absence of transition metals, herein a method for synthesis of imidazothiones from simple acetophenones, elemental sulfur, and ammonium acetate has been developed. It should be noted that such an one-pot formation of quaternary carbon from simple reagents is relatively rare in the literature.
While optimizing the reaction of acetophenone 1a with elemental sulfur and a nitrogen source such as NH4OAc to obtain benzothienothiazole 2a (eqn (1), Scheme 2),5f we early observed the formation of imidazothione 3a, especially at the lower temperature (eqn (2), Scheme 2). Our study then commenced on obtaining the simplest diphenyl imidazothione 3a from homocoupling of acetophenone 1a. The reaction was optimized with respect to temperature, nitrogen source, and solvent. The results are presented in Table 1. A trace amount of the desired product was detected when running the reaction at room temperature (entry 1). Increasing the temperature up to 60 °C dramatically increased the yield of 3a (entry 2). However, above 60 °C some by-products could be detected, thus leading to drops in yield (entry 3). Unlike our previous report,5f urea could not be used to incorporate nitrogen atoms into the product (entry 4). Since NH3 did not give the product, the presence of acetate anion was pivotal to the formation of 3a (entry 5). No solvents rather than DMSO were effective in this transformation (entries 6 and 7). Lastly, the reaction could reach to the equilibrium only after 6 h (entry 8).
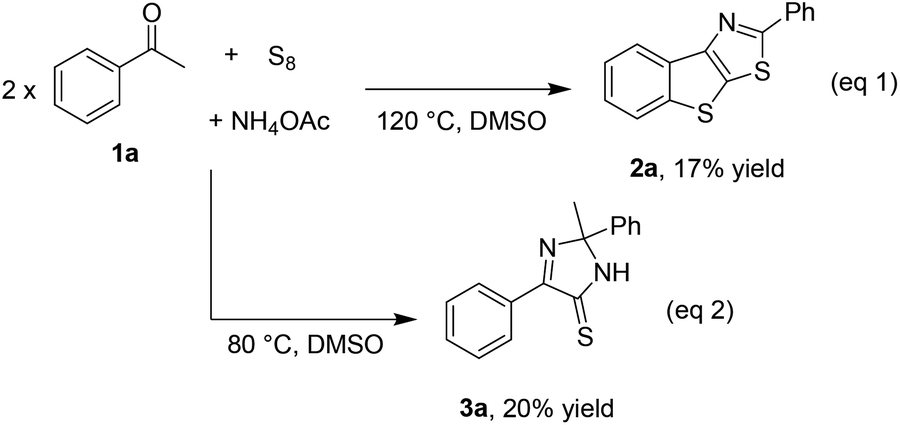 | ||
| Scheme 2 An early observation of imidazothione. | ||
|
||||
|---|---|---|---|---|
| Entry | Temperature (°C) | Nitrogen source | Solvent | Yield of 3a (%) |
| a Acetophenone (0.2 mmol), elemental sulfur (0.3 mmol, 32 g mol−1), nitrogen source (0.5 mmol, based on nitrogen), solvent (1 mL, [1a] = 0.2 M), for 24 h. Yields of 3a are GC yields using diphenyl ether internal standard.b NH3 as a 28% aqueous solution.c 6 h. Abbreviations: r.t. = room temperature, n.d. = not determined. | ||||
| 1 | r.t. | NH4OAc | DMSO | Trace |
| 2 | 60 | NH4OAc | DMSO | 84 |
| 3 | 70 | NH4OAc | DMSO | 71 |
| 4 | 60 | Urea | DMSO | n.d. |
| 5b | 60 | NH3 | DMSO | n.d. |
| 6 | 60 | NH4OAc | Dioxane | n.d. |
| 7 | 60 | NH4OAc | DMF | n.d. |
| 8c | 60 | NH4OAc | DMSO | 84 |

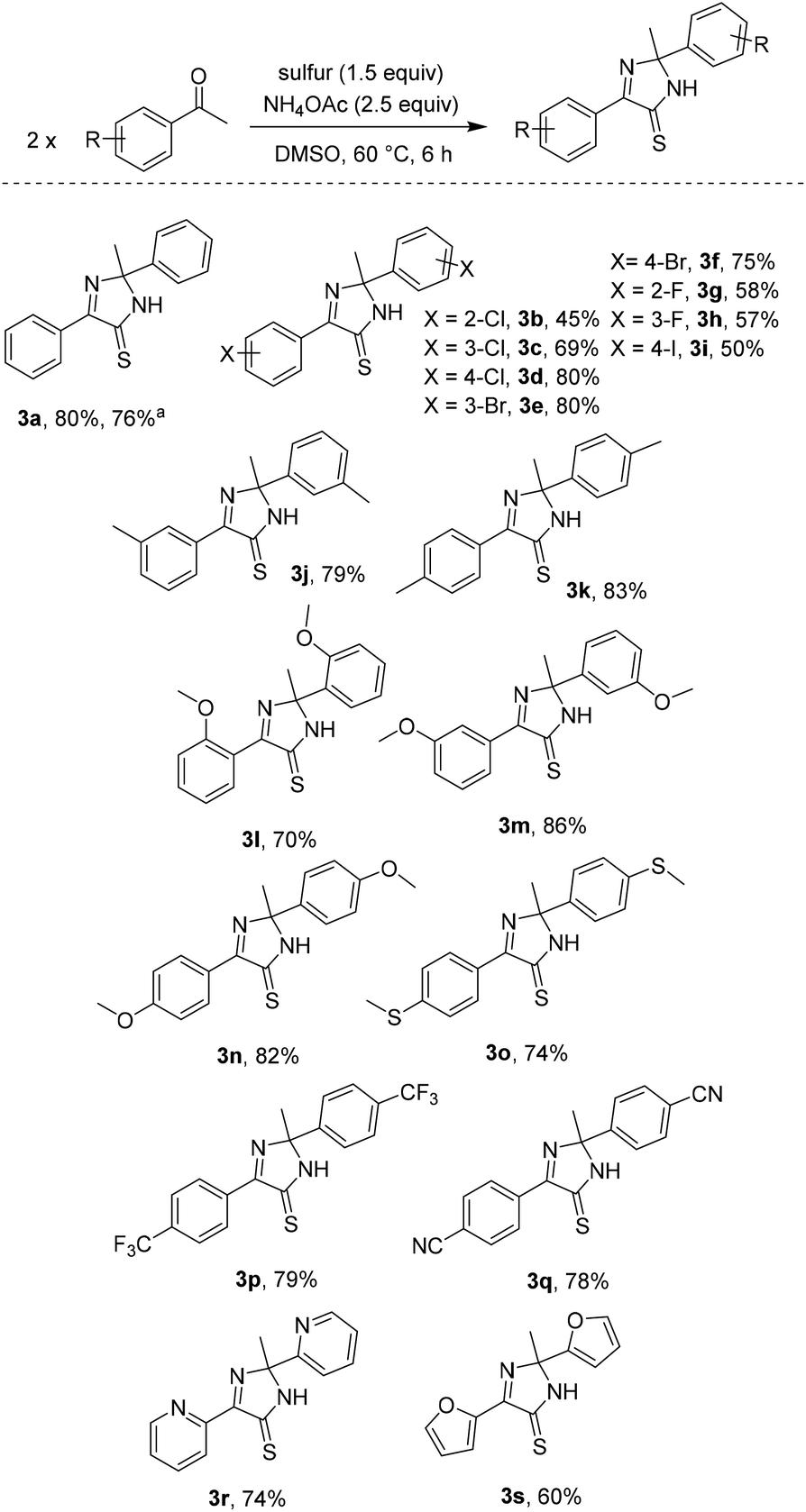 | ||
| Scheme 3 Reaction scope with respect to acetophenones. Reagents and conditions: acetophenones (0.2 mmol), sulfur (0.3 mmol, 32 g mol−1), NH4OAc (0.5 mmol), DMSO (1 mL, [acetophenones] = 0.2 M), 60 °C, 6 h. Yields are isolated yields. Please see the ESI† for more details. a2 mmol scale. | ||
Substrate scope with respect to acetophenones was next studied (Scheme 3). Structures of some imidazothiones were confirmed by the results of HMQC and HMBC spectra.6 The simple diphenyl imidazothione 3a was isolated in good yield, even in a 2 mmol scale. Halo-substituted acetophenones afforded the products regardless of positions of the substituents (3b–3i). Since synthesis of bromo- (3e, 3f) and iodo- (3i) containing imidazothiones was viable, late stage functionalization of such molecules could be possible.1d Methyl (3j, 3k), methoxy (3l–3n), and methylthio (3o) functionalities were all compatible with reaction conditions. Uses of acetophenones containing electron-withdrawing groups were also attempted. Trifluoromethyl- (3p) and cyano- (3q) substituted imidazothiones could be obtained in good yields. However, running the reaction of 4-nitroacetophenone did not give the desired product. Furthermore, our conditions were applied for synthesis of polyheterocyclic imidazothiones (3r, 3s). It should be noted that 2-acetylheteroarenes sometimes were not competent substrates.
Although most of possible intermediates were unstable and difficult to independently prepare, some mechanistic studies still have been investigated (Scheme 4). Similar to previous studies,5f,g compounds afforded by oxidative sulfuration of α C–H bonds were detected by GC-MS if the coupling of 1a and 2a was stopped after 1 h (eqn (1)). The annulation was somewhat affected by the presence of radical quenchers such as TEMPO or 1,1-diphenylethylene, especially if excess amount of these compounds was used (eqn (2)). Since DMSO was much effective more than other solvents, we envisaged that it could play roles in oxidation steps. Indeed, 53% and 28% of product 3a were obtained if 3 equivalents of DMSO were used in solvents 1,4-dioxane and CH3CN, respectively (eqn (3)). Analysis of the crude mixture obtained from the reaction of 4′-chloroacetophenone and 4′-methoxyacetophenone disclosed a 0.8/0.3/1 ratio of three coupling products, as the major was the heterocoupling imidazothione (eqn (4)). Given that only one cross-coupled product was obtained, it was likely that electronic properties of substituents have played a somewhat important role during reaction course. Ongoing experiments will include the isolation of synthetically challenging cross-coupled imidazothiones.
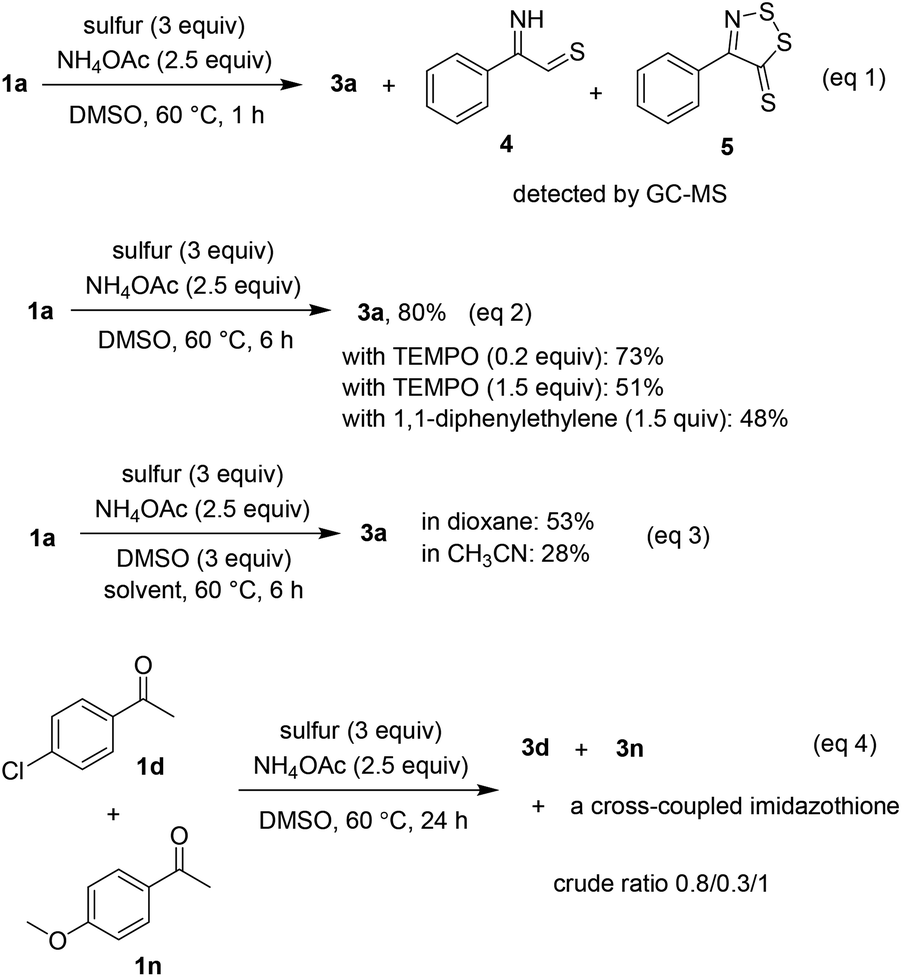 | ||
| Scheme 4 Mechanistic considerations. | ||
With the results in hand, we proposed a possible mechanism for the annulation (Scheme 5). An imine 6 was obtained when acetophenone 1a reacted with NH4OAc, followed by a Willgerodt–Kindler sulfuration to afford the intermediate 7. Oxidation, presumably promoted by DMSO, would furnish an iminothial 4. This intermediate was unstable, either generating a dithiazolethione 5 or reacting with 6 following a [3 + 2] cycloaddition to afford 8. After isomerization (8 → 9) and oxidation of C–S bond, the imidazothione 3a was obtained.
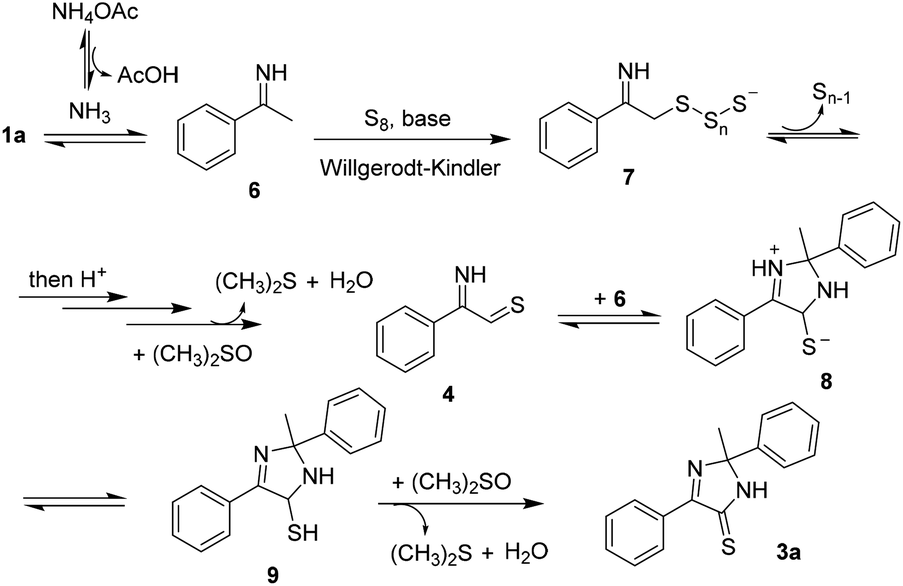 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
In conclusion, we have developed a simple method for synthesis of imidazothiones from annulation of methyl C–H bonds in acetophenones with elemental sulfur and NH4OAc. DMSO was used as a solvent as well as oxidant in these reactions. A wide range of functionalities including halogen, methylthio, cyano groups were compatible with reaction condition. Heteroaryl methyl ketones were also competent substrates. The method would offer a rapid route to assemble complex polycyclic structures from simple, commercial substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Vietnam National Foundation for Science and Technology Development (NAFOSTED) is acknowleged for supporting this research under project code 104.01-2019.340.Notes and references
- For reviews: (a) K. Lao, N. Ji, X. Zhang, W. Qiao, Z. Tang and X. Gou, J. Drug Targeting, 2019, 27, 164 CrossRef CAS; (b) A. K. Ghosh and H. L. Osswald, Chem. Soc. Rev., 2014, 43, 6765 RSC . Selected examples:; (c) S. Eketjäll, J. Janson, K. Kaspersson, A. Bogstedt, F. Jeppsson, J. Fälting, S. B. Haeberlein, A. R. Kugler, R. C. Alexander and G. Cebers, J. Alzheimer's Dis., 2016, 50, 1109 Search PubMed; (d) Y. Gravenfors, J. Viklund, J. Blid, T. Ginman, S. Karlström, J. Kihlström, K. Kolmodin, J. Lindström, S. von Berg, F. von Kieseritzky, C. Silvo, B.-M. Swahn, L.-L. Olsson, P. Johansson, S. Eketjäll, J. Fälting, F. Jeppsson, K. Strömberg, J. Janson and F. Rahm, J. Med. Chem., 2012, 55, 9297 CrossRef CAS; (e) E. G. Vega-Hissi, R. Tosso, R. D. Enriz and L. J. Gutierrez, Int. J. Quantum Chem., 2015, 115, 389 CrossRef CAS; (f) F. Asinger, H. Offermanns, D. Neuray and F. A. Dagga, Monatsh. Chem., 1970, 101, 500 CrossRef CAS.
- (a) F. Asinger, W. Schäfer and F. Haaf, Justus Liebigs Ann. Chem., 1964, 672, 134 CrossRef CAS; (b) D. Cantillo, P. A. Inglesby, A. Boyd and C. O. Kappe, J. Flow Chem., 2017, 7, 29 CrossRef CAS; (c) D. Znidar, D. Cantillo, P. Inglesby, A. Boyd and C. O. Kappe, Org. Process Res. Dev., 2018, 22, 633 CrossRef CAS.
- (a) T. Guntreddi, R. Vanjari and K. N. Singh, Org. Lett., 2014, 16, 3624 CrossRef CAS; (b) P. Zhang, W. Chen, M. Liu and H. Wu, J. Org. Chem., 2018, 83, 14269 CrossRef CAS.
- For review: (a) T. B. Nguyen, Adv. Synth. Catal., 2017, 359, 1066 CrossRef CAS . Selected examples:; (b) B. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 10496 CrossRef CAS; (c) T. B. Nguyen, L. Ermolenko, P. Retailleau and A. Al-Mourabit, Angew. Chem., Int. Ed., 2014, 53, 13808 CrossRef CAS; (d) L. Meng, T. Fujikawa, M. Kuwayama, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 10351 CrossRef CAS; (e) J.-R. Guo, J.-F. Gong and M.-P. Song, Org. Biomol. Chem., 2019, 17, 5029 RSC; (f) Y. Xie, X. Chen, Z. Wang, H. Huang, B. Yi and G.-J. Deng, Green Chem., 2017, 19, 4294 RSC.
- (a) X. Wang, X. Qiu, J. Wei, J. Liu, S. Song, W. Wang and N. Jiao, Org. Lett., 2018, 20, 2632 CrossRef CAS; (b) H. Huang, Z. Xu, X. Ji, B. Li and G.-J. Deng, Org. Lett., 2018, 20, 4917 CrossRef CAS; (c) H. Huang, Q. Wang, Z. Xu and G.-J. Deng, Adv. Synth. Catal., 2019, 361, 591 CrossRef CAS; (d) Y. Liao, Y. Peng, H. Qi, G.-J. Deng, H. Gong and C.-J. Li, Chem. Commun., 2015, 51, 1031 RSC; (e) T. B. Nguyen and P. Retailleau, Green Chem., 2018, 20, 387 RSC; (f) P. H. Pham, K. X. Nguyen, H. T. B. Pham, T. T. Nguyen and N. T. S. Phan, Org. Lett., 2019, 21, 8795 CrossRef CAS; (g) P. Zhou, Y. Huang, W. Wu, J. Zhou, W. Yu and H. Jiang, Org. Lett., 2019, 21, 9976 CrossRef CAS.
- Please see the ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of optimization, experimental procedures, and characterization of unknown compounds. See DOI: 10.1039/d0ra03047c |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |