
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural transformation and nature of defects in titanium carbide treated in different redox atmospheres
Maria Ivanovskayaa,
Evgeni Ovodok *a,
Dzmitry Kotsikaub,
Igor Azarkob,
Matej Micusikc,
Maria Omastovac and
Vyacheslav Golovanovd
*a,
Dzmitry Kotsikaub,
Igor Azarkob,
Matej Micusikc,
Maria Omastovac and
Vyacheslav Golovanovd
aResearch Institute for Physical and Chemical Problems of the Belarusian State University, 220006 Minsk, Belarus. E-mail: ovodok@bsu.by
bBelarusian State University, 220030 Minsk, Belarus
cPolymer Institute, Slovak Academy of Sciences, 845 41 Bratislava, Slovakia
dSouth-Ukrainian University, 65000 Odessa, Ukraine
First published on 6th July 2020
Abstract
The defect structure and phase formation processes occurring in the bulk and on the surface of titanium carbide (TiC) under thermal treatment in different ambient atmospheres (air, hydrogen, vacuum) were characterized using XRD, SEM, EPR, XPS and IR-spectroscopy. The oxidized states of both titanium (TiO2−x) and carbon, in the form of carbonate–carboxylate structures (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), were found on the surface of untreated TiC. Carbon vacancies were detected as paramagnetic defects in the crystalline lattice of TiC. The heat treatment of TiC in reducing conditions leads to an increase of the electrical conductivity associated with the formation of structural defects. Annealing of TiC in air causes its oxidation with the formation of an anatase-type TiO2 phase. Paramagnetic defects typical of both TiC and TiO2−x were revealed.
O), were found on the surface of untreated TiC. Carbon vacancies were detected as paramagnetic defects in the crystalline lattice of TiC. The heat treatment of TiC in reducing conditions leads to an increase of the electrical conductivity associated with the formation of structural defects. Annealing of TiC in air causes its oxidation with the formation of an anatase-type TiO2 phase. Paramagnetic defects typical of both TiC and TiO2−x were revealed.
1 Introduction
Carbides of group IV transition metals feature a unique set of physical, physical–chemical, magnetic and electric properties, which make them promising materials for practical use.1–3 Also, they appear to be convenient models for studying the character of chemical bonding and the effect of defects on functional features. A deeper understanding of these processes allows control of the structure and properties of materials and opens new opportunities for their application. Together with the chemical composition, the disordered crystal structure has been shown to contribute to many properties of carbides.1,2 The peculiarities that make TiC worthy of investigation are a wide homogeneity range from TiC0.47 to TiC0.95, mixed metallic and covalent chemical bonds and stability of the structure up to 2400 °C even at high defect concentration in the C and Ti sublattices. According to the most recent reports, carbon vacancies are the main defects in TiC.4–6The electric and magnetic properties of TiC are known to be very sensitive to the concentration of C-vacancies. However, there are contradictory points of view regarding their effect on the electric properties of TiC and on Ti–Ti interaction.1
Calculations of the energetic spectrum of TiC indicate a strong mixing of the C 2p and Ti 3d4s(4p) electronic states.1,2 The contributions of the 2s–2p states of carbon and 3d states of titanium to the hybridized d2sp3 band depend considerably on the lattice defectiveness. According to recent data, carbon vacancies are the most characteristic defects of TiC and induce disturbance in the binding and anti-binding bands.7 Various models of the TiC electronic structure have been proposed. Based on recent quantum-mechanical calculations and the electronic spectral data of TiC, a model of its electron state has been proposed.8,9 The model shows that the electron transfer is minor and occurs from titanium atoms to carbon: Tiδ+→Cδ− (δ ≪ 1).
There are no intrinsic paramagnetic centers in an ideal TiC crystal. Its paramagnetism is caused by defects occurring in the structure. The paramagnetic properties of titanium carbide are associated with the presence of structural defects both in the TiC crystal lattice and on its surface, where the formation of a non-stoichiometric TiO film is assumed. The substitution of carbon by oxygen in TiC may lead to a continuous transition from carbide to oxide since TiC and TiO are isostructural. A deviation from stoichiometry associated with the formation of both titanium and oxygen vacancies is typical of the TiO layer on the TiC surface (up to 15% of TiO lattice sites become vacant).3 A significant deficiency of oxygen leads to formation of titanium oxides in low oxidation states (Ti2O-type superstructure).10
Resonance of conductivity electrons and Ti3+ centers has been registered from TiC in the carbon matrix.4,5,11 It was shown that the distribution of TiC particles over the C matrix and the methods of preparation of both TiC and carbon might influence the properties of titanium carbide.
The growing interest in TiC structure in recent years has been caused by the fact that it is used to produce double carbides (MAX phases), precursors for the new layered 2D carbide materials called MXenes.12–14 The electronic states of titanium and carbon as well as the intrinsic defects in the TiC material are worth investigating, as there is a need to optimize the procedures of 2D-Ti3C2 preparation to control their composition and properties.15
The distinctive feature of TiC with a NaCl-type lattice is the layered structure of the crystals with interlacing close-packed Ti and C layers.1–3 Thermal treatment of TiC with additional sources of Al and Ti atoms leads to the formation of double carbide phases (MAX phases) in an inert atmosphere. The Ti3AlC2 MAX-phase has a hexagonal layered structure with P63/mmc symmetry.14 Atom layers consisting of titanium, carbon and aluminium are interlaced as Ti/C/Ti/Al/Ti/C/Ti. Nanosized Ti3C2 2D-carbide can be prepared by treating the Ti3AlC2 MAX-phase with various reactants (HF, LiF, and KOH) to result in Al removal.16–19 During this process, terminal (T) – (Ti3C2Tx) functional groups are attached to the surface of Ti3C2 particles. The resultant Ti3C2Tx thin structure consists of five layers with a chemically modified surface, which makes these materials promising for various applications due to their unusual properties.
The removal of Al from the Ti3AlC2 MAX-phase is often accompanied by partial corrosion of Ti3C2 to give TiOx. In addition, paramagnetic defects like Ti3+, oxygen vacancies, and O− centers are formed.13,19 It is important to control these processes, as they negatively influence the stability and properties of the materials.15 The TiO layer on the TiC surface might have a protective function, preventing further oxidation of the material. However, segregation of TiO on the TiC surface might have a negative effect when the material is used for preparation of a high quality MAX phase, as it is a source of oxygen.18
The aim of this paper is to study the structural transformations and nature of paramagnetic defects in titanium carbide with thermal treatment under the reducing and oxidizing atmospheres of air, hydrogen, and vacuum.
2 Experimental
Commercial samples of TiC (SIGMA-ALDRICH), graphene (A-12 GNP nanosize powder) and multiwall carbon nanotubes (MCNT) (NANOCYL® NC7000™) were studied.The titanium carbide was characterized in its initial state and after thermal treatment in air at 723 K for 1 h, in hydrogen at 473 K for 1 h, and under vacuum of 2 × 10−6 bar at 1573 K. Thermal treatment of the samples in hydrogen was performed in a tube furnace whose internal volume was filled with hydrogen. Heating of the samples in vacuum was carried out in quartz ampoules from which air had been evacuated. The particle size and morphology were characterized by scanning electron microscopy (SEM) using a LEO 1420 microscope. The X-ray diffraction measurements (XRD) were performed using a Philips X-ray PANalytical Empyrean diffractometer with CuKα radiation. The electron paramagnetic resonance (EPR) spectra of the powdered samples were recorded on a Varian E 112 spectrometer at 300 and 77 K. The measurements were carried out using a resonance frequency of 9.35 GHz (X-band), magnetic field modulation of 25 kHz, modulation amplitude of 1–4 G, and microwave power of 5 mW. The values of g-factors of EPR signals were determined in reference to Mn2+/MgO hyperfine structure lines. The half-width of the lines is denoted here as ΔB. The spin concentration was estimated in comparison to the standard of Mn2+ in MgO. The sample weight was selected experimentally in order to reduce the influence of skin effect and thus optimize the conditions of the EPR spectral recording. The change in electrical conductivity of the samples after thermal treatment was estimated indirectly by the variation of Q-factor of the spectrometer resonator.20 The Fourier transform infrared (FTIR) spectra were collected on an AVATAR-330 (Thermo Nicolet) spectrometer supplied with a diffuse reflectance accessory in the wavenumber range from 400 to 4000 cm−1. IR spectra were recorded from tablets using both transmission geometry and the Smart Diffuse Reflection accessory. Mixed powders of KBr and TiC were pressed into tablets under vacuum conditions. The layer thickness and amount of introduced TiC were selected empirically, achieving an optimal combination of layer transparency and sensitivity in recording vibrations. The X-ray photoelectron spectra (XPS) were measured from the surface of the original samples. XPS signals were recorded using a Thermo Scientific K-Alpha XPS system (Thermo Fisher Scientific Inc., UK) equipped with a microfocused, monochromatic Al Kα X-ray source (1486.68 eV). Thermo Scientific Avantage software, version 5.9917, was used for digital acquisition and data processing. Spectral calibration was achieved using the automated calibration routine and the internal Au, Ag, and Cu standards supplied with the K-Alpha system.
3 Results and discussion
Untreated TiC powder
SEM images revealed that the original titanium carbide powder consists of crystallites of heterogeneous sizes and with visible layered structure (Fig. 1a). The X-ray diffraction pattern shows the presence of single cubic phase TiC (Fig. 1b) with the unit cell parameter a = 4.326(3) Å.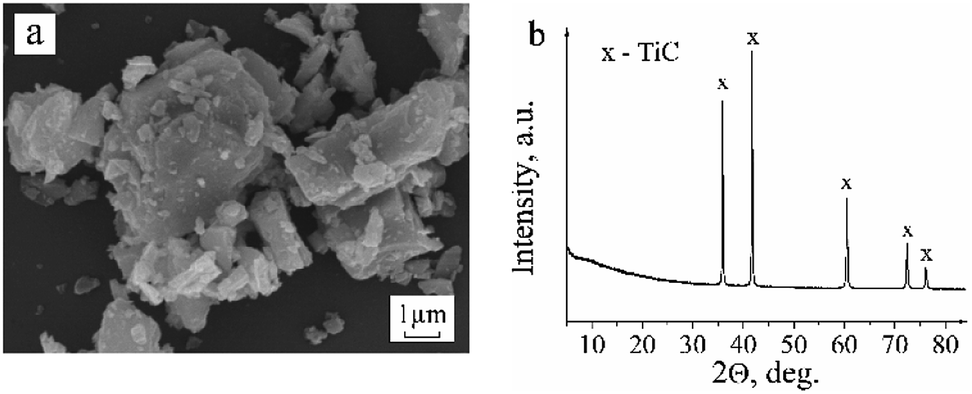 | ||
| Fig. 1 SEM image (a) and XRD pattern of the untreated commercial TiC powder (b). | ||
The EPR analysis results of the commercial TiC samples in the initial state and after heating in air and in hydrogen are summarized in Table 1 with the EPR parameters of the carbon materials (MCNT and graphene). The corresponding spectra are presented in Fig. 2.
| Sample | Wt., mg | The EPR signal at g = 2.000 ÷ 2.0027 | I, a.u. C, spin per g | Other EPR signals |
|---|---|---|---|---|
| Untreated TiC powder | 30–50 | 2.000; ΔB = 0.5 mT | 0.4–1.0 (2–4) × 1015 | — |
| Graphene powder A-12 GNP | 2 | 2.0027, ΔB = 1.5 mT ·C(H)– radical | 17, 6 × 1017 | — |
| MCNT NANOCYL® NC7000™ | 1 | 2.0027, ΔB = 2 mT ·C(H)– radical | 15–50, 3.2 × 1018 | — |
| TiC, annealed at 723 K in air, 1 h | 50 | 2.000, ΔB = 0.5 mT, a(13C) = 5.35 mT | 15, 6 × 1016 | g⊥ = 1.977; gII = 1.968, g⊥ = 1.943; gII = 1.934, g1=1.913; g2 = 1.906, g3 = 1.895 (Ti3+/TiO2−x) |
| TiC, annealed at 423 K in H2, 1 h | 2.3 | 2.001, ΔB = 0.6 mT | 8.7, 5.4 × 1016 | g⊥ = 2.060, ΔB = 10 mT gII = 2.037 (O−/TiC) or gII = 2.001, 1.95; 1.93 1.91 (Ti3+/TiO2–x) |
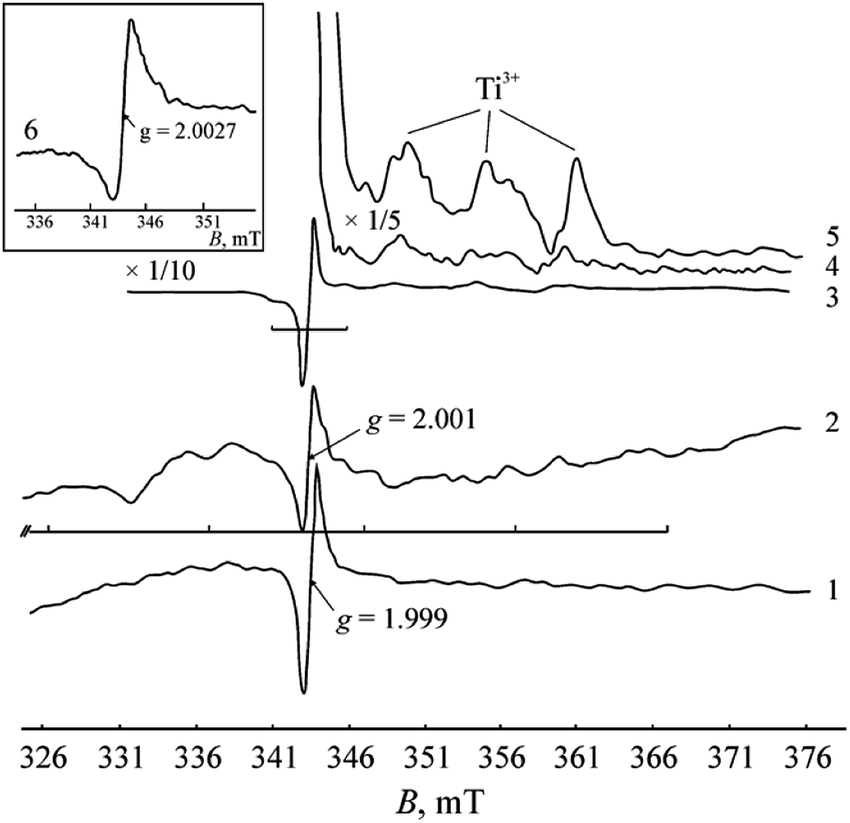 | ||
| Fig. 2 EPR spectra collected at 77 K: (1) untreated TiC powder, m = 50 mg; (2) TiC, annealed at 473 K in H2 atmosphere for 1 h, m = 2.3 mg; (3), (4), and (5) TiC, annealed at 723 K in air for 1 h, m = 50 mg, at different magnifications; (6) graphene powder (A-12 GNP). | ||
A single signal with g = 2.000 ± 0.001 is observed in the EPR spectrum of TiC. It could be related to the electronic defects of the titanium carbide lattice, but its nature needs to be clarified. The observed signal differs in both the shape and value of the g-factor from the signal typical of ·C radicals in the cyclic carbon structures of graphene and MCNT. Furthermore, its intensity in TiC is lower by about two orders of magnitude.
The broadened shape of the EPR signals in graphene and MCNT could result from the unresolved hyperfine structure of 1H atoms present in the carbon materials as impurities.
According to our IR spectroscopy data, graphene and MCNT contain CHx and OH functional groups with corresponding distinctive vibrations: ν(O–H) at 3665 and 3016 cm−1, ν(C–H) at 2915 cm−1, δ(O–H) at 1630 cm−1 and δ(Ti–O–H) at 1249 cm−1. The IR spectrum of MCNT differs from that of graphene in a large variety of vibrations, which shows the greater heterogeneity of the CHx and OH functional groups in MCNT.
From comparison of the EPR spectra of TiC and carbon materials, the appearance of the signal at g ≈ ge in the TiC spectrum cannot be caused by an admixture of carbon not included in the crystal lattice of carbide. Therefore, this signal can be assigned to the paramagnetic defects in the TiC crystal lattice. The main defects in TiC are both single and complex vacancies of carbon and titanium. There are no data on paramagnetic defects in TiC; however, as follows from numerous characterizations of SiC by EPR method, a resonance of both silicon and carbon vacancies can be observed. This resonance appears in spectra at g ≈ ge. As is noted in literature, a homogeneous MeIVC sample is very difficult to obtain and noticeable concentrations of vacancies are present in both sublattices.1 A large degree of disorder is found in the arrangement of vacancies, which can be present both in single form and as clusters. The most recent data indicate that the vacancies in the carbon sublattice are mainly formed in carbides of metals of group IV.18 Therefore, based on our redox experiments, we assign the observed EPR signal to the unpaired electron density localized in the carbon vacancy. The intensity of the observed signal substantially depends on the temperature. For this reason, it cannot be attributed to conduction electrons (Pauli paramagnetism), as shown by Augustyniak-Jabłokow et al.21
The presence of a TiO2/TiO phase on the TiC surface is confirmed by XPS. The corresponding results are presented in Table 2. The spectra of Ti 2p, O 1s and C 1s are shown in Fig. 3.
| Band | EBE, eV | At% | EBE, eV | aAt% | bAt% | State |
|---|---|---|---|---|---|---|
| a % from states of all Ti, O, C elements.b % from states of single element. | ||||||
| Ti 2p3/2 | 458.3 | 31.5 | 458.5 | 8.2 | 40.0 | Ti4+/TiO2 |
| 454.6 | 7.2 | 35.2 | Tiδ+/TiC | |||
| 455.6 | 3.0 | 14.6 | Ti2+/TiO | |||
| 457.6 | 2.1 | 10.2 | Ti3+/TiO2−x | |||
| O 1s | 530.5 | 19.8 | 529.9 | 19.6 | 60.5 | TiO2, TiO2−x |
| 531.5 | 10.5 | 32.4 | CO, O−/TiO2−x |
|||
| 533.0 | 2.3 | 7.1 | C–O | |||
| C 1s | 284.8 | 48.7 | 281.3 | 9.2 | 20.8 | Cδ−/TiC |
| 284.2; 284.8 | 8.1; 20.9 | 64.6 | C–C, C–H | |||
| 286.2 | 2.5 | 5.2 | C–O | |||
| 288.5 | 4.5 | 9.4 | O–CO |
|||
 | ||
| Fig. 3 XPS spectra of untreated TiC powder: (a) overall, (b) Ti 2p, (c) C 1s, (d) O 1s. | ||
The XPS data reveal the presence of various oxidized states of titanium and carbon on the TiC surface. In the Ti 2p signal, we have identified typical peaks for Ti carbides centered at ∼454.6 eV corresponding to the Ti–C bond, ∼455.6 eV corresponding to Ti2+ (C–Ti2+ or some TiO) and ∼457.6 eV corresponding to Ti3+ (C–Ti3+ or some Ti2O3) (Fig. 3).22 The Ti 2p signal at ∼458.5 eV corresponds to Ti4+ and is probably mainly TiO2, since it can be well correlated with the signal of O 1s centered at ∼529.9 eV corresponding to O2− (Table 2).
Signals from adventitious carbon coming from surface contamination are centered at ∼284.8 eV (sp3 carbon), ∼286.2 eV (C–O groups) and ∼288.5 eV (carboxyl OCO or carboxylate COO− groups). The signal at ∼284.2 eV (labelled “sp2” in Fig. 3c) could be assigned to the Ti-oxycarbides (Ti-OC) or some graphitic carbon.22 The typical signal for carbide at ∼281.3 eV (Fig. 3c) was detected. The signal at ∼282.6 eV (labelled “carbide II” in Fig. 3c) might be a satellite from TiC because of the conductive electrons available for shake-up-like events following core electron photoemission. At this position might also be some substoichiometric TiCx or C–Ti–O from the adsorption of OH groups on the surface of TiC.23 The C–Ti–O group is also detected in the O 1s signal centered at ∼531.5 eV, but this signal overlaps with organic contamination of the CO signal (Fig. 3d, Table 2).
The existence of carbonate–carboxylate structures on the TiC surface is confirmed by IR spectroscopy. The weak narrow lines at 412 and 531 cm−1 can be assigned to the vibration of the Ti–C bonds. The wide absorption lines with maxima at 838 cm−1, 1138–1374 cm−1 (with intermediate lines at 1235 and 1298 cm−1), 1506–1575 cm−1 and 1775 cm−1 characterize vibrations of the C–O bonds in carbonate–carboxyl groups. Taking into account the available data in the literature,24 it can be deduced that the observed vibrations refer to the following bonds: 838 cm−1 – δ(OCO); 1138 cm−1 – ν(C–C); 1235 cm−1 – ν(C–O) in bidentate carbonate; 1298 cm−1 – νas(COO) in bidentate carbonate; 1374 cm−1 – νs(COO−) (carboxylate group bonded to metal ion); 1506–1574 cm−1 – νas(COO−); 1775 cm−1 – ν(CO) (non-ionized carboxyl group). The bidentate and non-ionized COO-groups are weakly bound to the metal oxide surface. The presence of different carbonate–carboxylate surface structures demonstrates the heterogeneity of the sample surface, which is typical for defective layers of titanium dioxides.
TiC powder after thermal treatment in air
Annealing at 723 K in air causes oxidation of titanium carbide. The formation of anatase and rutile TiO2 phases is registered in the diffraction pattern (Fig. 4). As seen in the scanning electron microscopy images, the TiO2 occurs in the form of individual round-shaped particles that are evidently separated from TiC crystallites. IR spectroscopy also reflects the transformation of a portion of TiC into TiO2. A wide absorption band at 400–800 cm−1 with a maximum at 788 cm−1 assigned to the A2u LO Ti–O vibration in the nanosized rutile TiO2 powder appears in the IR spectrum.25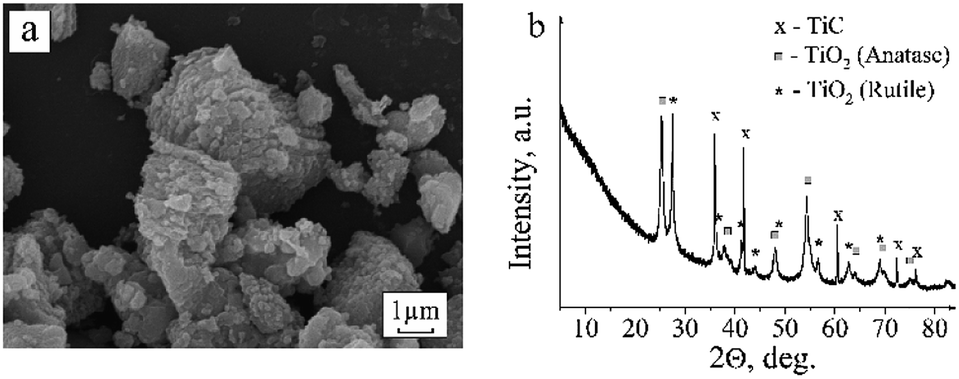 | ||
| Fig. 4 SEM picture (a) and XRD pattern (b) of the TiC powder annealed at 723 K in air for 1 h. | ||
In the EPR spectra of the samples calcined in air, a considerable increase in the signal intensity at g = 2.000 and the appearance of low-intensity signals with g-factors of 2.015, 1.983, 1.97, 1.94 and 1.91 are registered. The symmetrical arrangement of the lateral lines at 2.015 and 1.983 relative to the central line of g = 2.000 evidently corresponds to the hyperfine structure of the signal with a splitting value of 5.35 mT. The appearance of hyperfine structure can be evoked by the interaction of unpaired electron density localized in the carbon vacancy with nuclear magnetic moments of neighboring carbon 13C isotope atoms (nuclear spin I = ½). The ratio of the line intensities corresponds to the 13C hyperfine structure. The content of 13C isotope is known to be small (1.108%).26 Therefore, the hyperfine structure lines are only recorded in spectra when the main signal has a high intensity. Although chemical bonding between carbon atoms is absent in TiC, it has been noted in literature1,3 that the wave functions of atoms in TiC can extend over several unit cells, which explains the appearance of the hyperfine structure of 13C atoms.
The signals at g = 1.97, 1.94 and 1.91 undoubtedly originate from the Ti3+ centers (d1 electron state) in TiO2−x. It is known that the g-tensor of the Ti3+ centers (3d1 state) varies over a range of 1.99 ÷ 1.90 depending on the symmetry of the coordination environment.27 At high spectral intensities, it becomes possible to resolve the axial and triaxial shapes of the signals (see Fig. 2 and Table 1), which are typical of Ti3+ paramagnetic centers in titanium oxides. At 300 K, these signals are not observed in the spectrum, which corroborates their affiliation with Ti3+ centers.
The obtained results suggest that the partial oxidation of TiC during calcination in air is accompanied by the formation of a TiO2 phase. Such a process can generate defects in the TiC structure, as indicated by an increase in the signal intensity at g = 2.000.
The titanium dioxide phase formed at the rather low temperature of 723 K has a nonstoichiometric TiO2−x structure with stabilization of incompletely oxidized Ti3+ states. The presence of three Ti3+ signals with different parameters shows the heterogeneity of the titanium oxide structure being formed. The values of the g-factors differ considerably from the g-factors of the Ti3+ paramagnetic centers in the anatase and rutile crystalline phases. This difference may be caused by a surface arrangement of Ti3+ in the tetragonal coordination which is typical of highly defective TiO2−x oxides. The mechanism of the formation of Ti3+ centers and oxygen vacancies at the TiO2/Ti3C2 boundary has been considered by Yoon et al.19
TiC powder after thermal treatment in hydrogen
According to the XRD data, the new phases are not formed after exposure to hydrogen (Fig. 5b). Only the face-centered cubic phase of TiC with the same unit cell parameter as in the original sample was observed. As seen in the SEM images, there are no visible changes in the morphology of TiC crystallites, either (Fig. 5a). However, thermal treatment in H2 causes the electrical conductivity and the magnetic susceptibility of the sample to increase considerably. This conclusion was made based on the fact that the resonance of unpaired electrons becomes difficult to register. The difficulties in registration of EPR signals from samples with metallic conductivity are described in detail by Silveri et al.28 In materials with metallic conductivity, electrons interacting with a high-frequency external magnetic field can readily absorb microwave radiation and affect the intrinsic frequency of the resonator. Apparently, variation of the g-tensor in the range of 2.000 ÷ 2.001 can be evoked by the influence of electrical conductivity of the samples on the intrinsic frequency of the resonator. Dilution of the partially reduced sample by adding inert matrix (Al2O3) to the TiC powder caused neither elimination of the noise component nor improvement of the spectral recording conditions. To achieve acceptable conditions for registration of EPR spectra, the sample weight was considerably reduced (more than 20 times).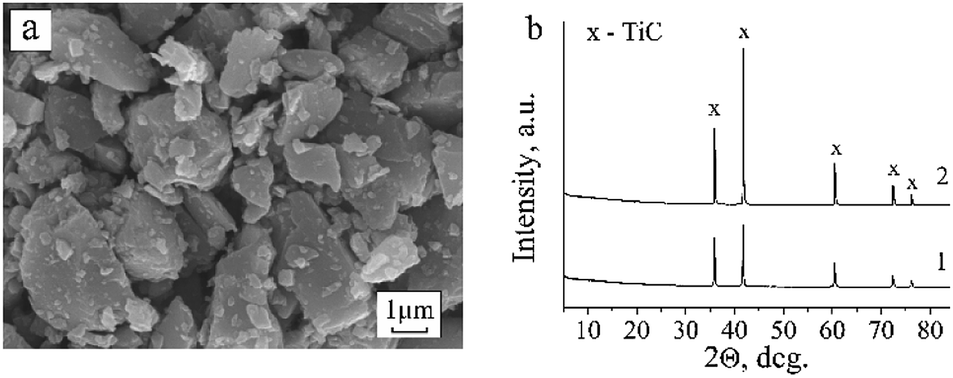 | ||
| Fig. 5 SEM image and XRD pattern of the TiC powder annealed at 723 K in H2 for 1 h (a and b(1)) and XRD pattern of TiC powder annealed at 1573 K in vacuum for 30 min (b(2)). | ||
Despite the significant differences in the recording conditions of the initial samples and the samples treated in H2, it can be concluded that the signal intensity at g = 2.001 increases after exposure to H2. Additionally, a wide signal with giso ≈ 2.06 and a series of low-intensity signals (2.037, 1.95, 1.93, 1.91) emerge against the noisy background (Fig. 2).
The increase in intensity of the g = 2.001 signal after annealing in hydrogen can be explained by the growing concentration of carbon vacancies at which unpaired electron density is localized. According to the calculations made by Khabibullin et al.,29 hydrogen adsorption on TiC is preferable on carbon atoms of the (001) and (111) planes. At elevated temperatures, formation of CHx fragments takes place. Their condensation and cyclization can generate highly defective aromatic hydrocarbons with stabilization of ˙C(H) radicals, which can also contribute to the signal at g = 2.001.
Annealing TiC in hydrogen leads to the appearance of IR absorption bands assigned to the following vibrations: ν(O–H) at 3790 cm−1, ν(O–H) (H2O) at 3467 cm−1, δ(O–H) at 1630 cm−1 and ν(C–H) at 2910 cm−1. These bands indicate the chemisorption of hydrogen on TiC and TiOx.
In the case of a high concentration of carbon vacancies, a transformation of the corresponding local energy levels into a band with a high density of electron states can be expected. Since magnetic susceptibility correlates with the concentration of such electron states, its observed increase after reducing TiC samples corroborates our assumption about the formation of carbon vacancies after treatment under hydrogen atmosphere.
Based on EPR measurements, we estimated the concentration of paramagnetic centers with g = 2.001 in the reduced sample as 5.4 × 1016 spins per g. At this concentration, only one vacancy per 100000 positions can be formed in the TiC crystal lattice. Such a low concentration of carbon vacancies cannot explain the strong change in the magnetic susceptibility and electric conductivity of TiC that takes place after exposure to hydrogen. The concentration of defects (vacancies) must be higher by 1–2 orders of magnitude and reach 1017 ÷ 1019 spins per g. Therefore, it can be assumed that not all vacancies present in the reduced sample were detected by EPR method. The observed single peak with g = 2.001 can only be related to isolated vacancies which create local energy levels. Under annealing in hydrogen, the increase in the concentration of vacancies can cause their arrangement into clusters of various dimensions and geometries.6 In the EPR measurements, such clusters of vacancies may not be detected for various reasons, including the absence of unpaired electron density. It is worth noting that the observed increase in electrical conductivity of the reduced sample could also be associated with its hydrogenation, which is typical of materials treated in hydrogen.
The appearance of a wide signal with giso ≈ 2.06 in the reduced sample suggests the formation of an O− center associated with an oxygen ion stabilized in the vicinity of a cation vacancy (Ti–O−–VTi). A similar center has been observed in the EPR spectra of many metal oxides. Its formation was explained by the rearrangement of a disordered oxide into a crystalline phase. A small peak at 2.037 could be an independent signal or the gII component of the signal with g⊥ = 2.06. A signal of similar axial shape but with different parameters (g⊥ = 2.048, gII = 2.035) was observed earlier in titanium dioxide doped with tin. It was assigned to the [VTi–O−] center. Similarly, signals with g⊥ = 2.0389, gII = 2.0032 and g⊥ = 2.0408, gII = 2.0032 were observed in MgO monocrystals and assigned to the [Vk–O−] and [O−–Vk–O−] conformations, respectively. Therefore, the resonance observed in the spectrum at g = 2.037 could be the g⊥ component of the independent signal, the gII component of which overlaps with the signal at g = 2.001.
Another model of the center with g⊥ = 2.06, gII = 2.002 includes an oxygen atom with a localized electron that links two neighboring titanium atoms through the oxygen vacancy. Such an O− signal should have a component gII = ge.30,31
The signal with g = 2.06 was observed in the EPR spectrum of TiO2 in the Q-band (λ = 8 mm), but was not registered within the X-band (λ = 3 cm).32 The authors relate this signal to exchange-coupled O−⋯O− centers with spin S = 1 and a strong magnetic interaction rather than to a single O− center. The resonance observed within the X-band has been attributed to the fine structure transitions of the triplet O−⋯O− center.
In the spectrum of the untreated TiC sample, the signal with g = 2.06 has been also registered at 300 K. After reducing the sample, the signal intensity increases and the line becomes isotropic and shifts toward higher g values (up to giso = 2.08). The described behavior of the signal indicates that it belongs to the O−⋯O− pairs with spin S = 1. The shift of the lines toward higher g values can be explained by the increase in magnetic interaction between the spins.
As follows from the aforesaid, the signal with giso ≈ 2.06 is undoubtedly related to the O− centers at the surface TiO2−x layer, but its structure requires refinement. The signal cannot be assigned to adsorbed anion-radical forms of oxygen because it still appears after treatment of the samples in vacuum. Although its intensity varies, these variations could be caused by other reasons, such as changes in the conditions of the spin–lattice relaxation due to the removal of adsorbed oxygen.
The signals with low intensity at 1.95, 1.93 and 1.91 registered against the background noise (Fig. 2) can be related to the Ti3+ centers with different environments. They appear as a result of the partial reduction of the TiO2 surface layer present on the surface of the initial TiC sample. Exposure to H2 probably generates defects like (Ti3+–VO–Ti3+); however, they were not registered by EPR method. The appearance of these defects is indicated by the change in the TiC sample color from dark grey to rich black during heat treatment under H2. The black color of titanium dioxides has been explained by the formation of similar structures comprised of two Ti3+ ions.33
TiC powder after thermal treatment under vacuum
Synthesis of the Ti3AlC2 MAX phase from TiC, Ti and Al was performed under vacuum at 1573 K. To trace the possible structural-phase transformations in TiC under these conditions, the sample was heated in an evacuated ampoule without adding Ti and Al powders under conditions similar to those used for the synthesis of Ti3AlC2. According to the XRD data, the phase composition of the sample during such treatment remains unaltered (Fig. 5b). Only cubic TiC phase with a slightly reduced parameter a = 4.324(3) Å was observed in the XRD spectrum. A change in the relative intensities of the lines was also observed. Thus, the reflection from the (200) plane increased in relation to the other reflections of the TiC face-centered cubic lattice. The observed changes in the lattice parameter and peak intensities of the sample thermally treated in vacuum might be explained by an increase in the defect concentration in the carbon sublattice. In spite of the thermal stability of its structural phase state, TiC undergoes decarbonization at high temperatures.2The recording of EPR spectra was only successful using a small sample portion (2.4 mg), since the electrical conductivity of the material increased considerably after annealing in vacuum. In the EPR spectrum, broadened signals with the following g-factors were registered: 2.054, 2.002, 1.933 and 1.879, as well as a wide rise at giso = 2.15. The actual observed signals of oxygen centers, carbon vacancies and Ti3+ are identical to those detected in other TiC samples. However, the g parameters vary slightly, which could be a consequence of increased defect concentration in the TiC crystal lattice after high-temperature treatment in vacuum.
4 Conclusions
The application of a series of complementary methods allowed us to characterize the structural features of TiC and to study the defect- and phase-forming processes taking place at the surface and in the bulk of titanium carbide under thermal treatment in air, hydrogen and vacuum. The presence of oxidized states of both titanium and carbon in the forms of TiO2−x and carbonate–carboxylate structures (CO32−, COO−) at the surface of the untreated TiC sample was confirmed experimentally. Thermal stability of the TiC crystal lattice during the treatments in hydrogen (473 K) and in vacuum (1573 K) was ascertained, in spite of a considerable increase in the concentration of structural defects. The oxidation of TiC with the formation of isolated anatase and rutile TiO2 phases takes place when annealing TiC in air (723 K). The paramagnetic defects typical of both TiC and TiO2−x were revealed. Carbon vacancies play the most important role in determining the electro-physical properties of TiC. The interactions and electron transfer in carbon vacancies are responsible for the metallic characteristics, high electrical conductivity and superconductivity of the substance. The other centers (Ti3+, O−, O−⋯O−) registered by EPR method are typical of a titanium carbide surface layer, where titanium oxides (TiO, TiO2−x, TiO2) are formed in considerable quantities. The nature of defects and the revealed changes in the structure at the TiC/TiOx interface are similar to the processes observed at the Ti3C2Tx(MXene)/TiOx interface. The obtained results contribute to a better understanding of the formation of promising MXene materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 777810; Belarusian Republican Foundation for Fundamental Research (grant no. X20CJIKT-004); grant no. APVVSK-BY-RD-19-0011 (Slovakia); V. G. thanks Project HPC-EUROPA3 (INFRAIA-2016-1-730897) for the support.References
- L. E. Toth, Transition Metal Carbides and Nitrides, Academic Press Inc., New York, 1971 Search PubMed.
- E. K. Storms, The Refractory Carbides, Academic Press, New York and London, 1967 Search PubMed.
- H. D. Goldschmidt, Interstitial Alloys, Butterworth-Heinemann, Oxford, UK, 1st edn, 1967 Search PubMed.
- N. Guskos, T. Bodziony, M. Maryniak, J. Typek and A. Biedunkiewicz, J. Alloys Compd., 2008, 455, 52–54 CrossRef CAS.
- T. Bodziony, N. Guskos, A. Biedunkiewicz, J. Typek, R. Wróbel and M. Maryniak, Mater. Sci.-Pol., 2005, 23, 899–907 CAS.
- M. V. Klein, J. A. Holy and W. S. Williams, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 17, 1546 CrossRef CAS.
- L. F. Matteis, Phys. Rev. B: Solid State, 1972, 5, 315 CrossRef.
- L. G. Radosevich and W. S. Williams, J. Am. Ceram. Soc., 1970, 53, 30–33 CrossRef CAS.
- R. G. Lye and E. M. Logothetis, Phys. Rev., 1966, 147, 622 CrossRef CAS.
- J. F. Houlihan and L. N. Mulay, Inorg. Chem., 1974, 13, 745–747 CrossRef CAS.
- N. Guskos, J. Typek, T. Bodziony, G. Zolnierkiewicz, M. Maryniak and A. Biedunkiewiczc, J. Alloys Compd., 2009, 470, 51–54 CrossRef CAS.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- B. Scheibe, K. Tadyszak, M. Jarek, N. Michalak, M. Kempiński, M. Lewandowski, B. Peplińska and K. Chybczyńska, Appl. Surf. Sci., 2019, 479, 216–224 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- G. Li, L. Tan, Y. Zhang, B. Wu and L. Li, Langmuir, 2017, 33, 9000–9006 CrossRef CAS PubMed.
- X. Sang, Y. Xie and M.-W. Lin, ACS Nano, 2016, 10, 9198–9200 CrossRef PubMed.
- Y. Yoon, T. A. Le, A. P. Tiwari, I. Kim, M. W. Barsoum and H. Lee, Nanoscale, 2018, 10, 22429–22438 RSC.
- C. Poole, Electron spin resonance, Intersci. Publisheers, New York, 1967 Search PubMed.
- M. A. Augustyniak-Jabłokow, Y. V. Yablokov, B. Andrzejewski, W. Kempiński, S. Łoś, K. Tadyszak, M. Y. Yablokov and V. A. Zhikharev, Phys. Chem. Miner., 2010, 37, 237–247 CrossRef.
- J. Halim, K. M. Cook, M. Naguib, P. Eklund, Y. Gogotsi, J. Rosen, W. Michel and M. W. Barsoum, Appl. Surf. Sci., 2016, 362, 406–417 CrossRef CAS.
- C. Peng, X. Yang, Y. Li, H. Yu, H. Wang and F. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 6051–6060 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, John Wiley & Sons, New York, 3rd edn, 2001 Search PubMed.
- G. Busca, G. Ramis, J. M. Gallardoá Amores, V. Sanchezá Escribano and P. Piaggio, J. Chem. Soc., Faraday Trans., 1994, 90, 3181–3190 RSC.
- J. E. Wertz and J. R. Bolton, Electron spin resonance: elementary theory and practical applications, McGrawHill, New York, 1972 Search PubMed.
- Y. Li, D.-S. Wang, N. H. Lee and S.-J. Kim, Chem. Phys. Lett., 2005, 404, 25–29 CrossRef CAS.
- F. Silveri, M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. Catlow, Phys. Chem. Chem. Phys., 2019, 21, 5335–5343 RSC.
- B. M. Khabibullin and E. G. Kharakhash'yan, Soviet Physics - Uspekhi, 1974, 16, 806–818 CrossRef.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2018, 7, 6791–6795 RSC.
- V. Pfeifer, T. E. Jones, J. V. Vélez, C. Massué, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, J. Allan and M. Hashagen, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- E. G. Avvakumov, V. F. Anufrienko and S. V. Vosel, Izv. Sibir. Otd. Akad. Nauk SSSR, Ser. Khim., 1987, 1, 41–48 Search PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |