DOI:
10.1039/D0RA02913K
(Paper)
RSC Adv., 2020,
10, 18553-18559
Synthesis, crystal structure and antiproliferative mechanisms of gallium(III) complexes with benzoylpyridine thiosemicarbazones†
Received
31st March 2020
, Accepted 1st May 2020
First published on 18th May 2020
Abstract
We have prepared six thiosemicarbazone ligands and synthesized the corresponding Ga(III) complexes. The antitumor activity of the ligand increases with its lipophilicity, and the antitumor activity of the Ga(III) complexes is affected by the ligands. Since C6 has the highest anticancer proliferative activity (0.14 ± 0.01 μM) against HepG-2 (Human hepatocarcinoma cell line), we characterized its structure by X-ray single crystal diffraction and explored its antiproliferation mechanism. Anti-tumor mechanism results show that Ga(III) complex (C6) promoted HepG-2 cell cycle arrest in the G1 phase by regulating the expression of cell cycle-associated proteins (Cdk 2, cyclin A and cyclin E). Ga(III) complex (C6) promotes apoptosis by consuming intracellular iron, enhancing intracellular reactive oxygen species (ROS), activating caspase-3/9, releasing cytochromes and apoptotic protease activating factor-1 (apaf-1).
Introduction
Gallium nitrate was introduced into clinical trials in the 1980s due to its good anticancer potential.1 Ga(III) complexes are important anti-tumor drugs that are superior to platinum due to certain aspects of their biological effects.2–6 Reports on the detection of poly-phosphates using Ga(III) complexes indicate that gallium has a strong binding affinity for ATP.7,8 Therefore, Ga(III) complexes may have the potential to inhibit ATP. Similar to iron, Ga(III) enters cells by binding to transferrin, so the solution to increase activity is to increase the bioavailability of Ga.9–11 The ligand-modified Ga(III) complexes exhibit advantageous pharmaceutical properties in various aspects, such as pharmacokinetics, affinity for malignant cells, and anti-tumor proliferative effect.12–14 Ga(III) complexes induce apoptosis in cancer cells by activating caspase activity and modulating cyclin to inhibit the cell cycle.15–18
The complexation of Ga(III) with thiosemicarbazone enhances the biological activity of Ga(III) and is considered to be an important strategy for the development of cytotoxic drugs.10,18–20 Lipophilicity plays a leading role in the rate at which molecules enter cells, and complexation can increase the lipophilicity of Ga(III).21 At the same time, the biological activity of thiosemicarbazone is closely related to the ability of forming complex with the metal and the lipophilicity of the molecule.21–24 Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone) is a potent ribonucleotide reductase inhibitor and is the most prominent representative of this family.25–27 It is currently used in phase I and phase II clinical trials for a variety of malignancies.25 Another star molecule (di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone, DPC) has entered clinical trials for drug-resistant cancer treatment in 2016.28 The synthesis and application of thiosemicarbazone and its gallium complex have been paid attention to and become one of the most promising research fields.
In order to further improve the anti-tumor mechanism of thiosemicarbazone-Ga(III) complexes, the following studies were carried out: (1) six thiosemicarbazone ligands and their corresponding Ga(III) complexes are synthesized (Scheme 1); (2) the structure and activity relationships of these compounds have been studied; (3) intracellular ROS, mitochondrial membrane potential, caspase-3/9 activation, and expression of cyt-c and apaf-1 were analyzed and used to investigate the mechanism behind gallium complexes promoted apoptosis; (4) expression of cell cycle-associated proteins (cdk2, cyclin A, cyclin E) which are also related with the apoptosis process, was further examined to better understand the underlying mechanism.
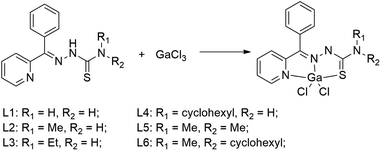 |
| | Scheme 1 Synthetic route of Ga(III) complexes. | |
Results and discussion
Synthesis and crystal structure description
The thiosemicarbazone ligands (L1–L6) were prepared as described in the literature, and the prepared ligands were subjected to NMR, mass spectrometry and elemental analysis. Coordination of an equimolar amount of ligand with Ga(III) chloride to synthesize a series of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand/Ga(III) complexes, and products were crystallized from methanol in high purity without purification (Scheme 1). The crystal data of C6 was displayed in Table S1,† and the selected angles and bond lengths are listed in Table S1.† C6 crystallized in the monoclinic system, and space group P21/n. The Ga(III) metal center of C6 is coordinated by one S atoms, two Cl atoms (Cl1 and Cl2) and two N atoms (N1 and N2), which formed a deformed trigonal bipyramid (Fig. 1). The lengths of Ga1–N1 [2.098(2) Å] and Ga1–N2 [2.0581(17) Å] are similar. There is no significant difference between the length of Ga1–Cl1 [2.2264(7) Å] and Ga1–Cl2 [2.2070(7) Å] bonds. The Ga–Cl and Ga–N bonds were shorter than the length of Ga1–S1 [2.3291(7) Å]. In Ga(III) complex, the anion Cl and tridentate thiosemicarbazone coordinate with the central copper ion(II) to form an approximately planar structure. The bond angles and bond lengths in C6 are identical to other known compounds in published articles.10,11,29
:1 ligand/Ga(III) complexes, and products were crystallized from methanol in high purity without purification (Scheme 1). The crystal data of C6 was displayed in Table S1,† and the selected angles and bond lengths are listed in Table S1.† C6 crystallized in the monoclinic system, and space group P21/n. The Ga(III) metal center of C6 is coordinated by one S atoms, two Cl atoms (Cl1 and Cl2) and two N atoms (N1 and N2), which formed a deformed trigonal bipyramid (Fig. 1). The lengths of Ga1–N1 [2.098(2) Å] and Ga1–N2 [2.0581(17) Å] are similar. There is no significant difference between the length of Ga1–Cl1 [2.2264(7) Å] and Ga1–Cl2 [2.2070(7) Å] bonds. The Ga–Cl and Ga–N bonds were shorter than the length of Ga1–S1 [2.3291(7) Å]. In Ga(III) complex, the anion Cl and tridentate thiosemicarbazone coordinate with the central copper ion(II) to form an approximately planar structure. The bond angles and bond lengths in C6 are identical to other known compounds in published articles.10,11,29
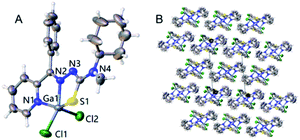 |
| | Fig. 1 (A) A molecular structure of C6 showing the environment about the Ga(III) atom. (B) The pack of Crystal. | |
Anticancer properties of Ga(III) complexes
A liver hepatocellular carcinoma cell line (HepG-2) was used to evaluate the anticancer activity of the thiosemicarbazide ligand and the Ga(III) complexes. All of the thiosemicarbazide ligands (L1–L6) and Ga(III) complexes (C1–C6) showed potent anti-proliferative activity and results were listed in Table 1. The anticancer activity of GaCl3 (50 μM) was not observed. More importantly, these chemical agents showed very low toxicity to normal cells (LO-2) compared to tumor cells.
Table 1 IC50 values of ligands and Ga(III) complexes toward cell lines for 48 h
| |
IC50 (μM) |
| HepG-2 |
LO2 |
SI |
| L1 |
11.35 ± 0.21 |
43.64 ± 2.84 |
3.84 |
| L2 |
4.56 ± 0.11 |
35.75 ± 2.19 |
7.84 |
| L3 |
2.43 ± 0.10 |
32.97 ± 3.64 |
13.57 |
| L4 |
1.32 ± 0.09 |
30.32 ± 2.16 |
22.97 |
| L5 |
0.57 ± 0.04 |
23.85 ± 2.00 |
41.84 |
| L6 |
0.32 ± 0.02 |
11.54 ± 0.54 |
36.06 |
| C1 |
5.75 ± 0.54 |
33.76 ± 2.13 |
5.87 |
| C2 |
2.76 ± 0.39 |
25.83 ± 2.93 |
9.36 |
| C3 |
1.23 ± 0.09 |
26.64 ± 3.86 |
21.66 |
| C4 |
0.97 ± 0.02 |
20.43 ± 1.13 |
21.06 |
| C5 |
0.34 ± 0.02 |
18.86 ± 1.64 |
55.47 |
| C6 |
0.14 ± 0.01 |
10.65 ± 1.69 |
76.07 |
| Cisplatin |
22.43 ± 2.21 |
21.27 ± 2.35 |
— |
| GaCl3 |
>50 |
>50 |
— |
As shown in Table 1, the anticancer activity of L1–L6 increased sequentially after HepG-2 cells were treated for 48 hours. Among them, the antiproliferative activity of L1 (IC50, 11.35 ± 0.21 μM) is effectively lower than that of L6 (IC50, 0.32 ± 0.02 μM). Similarly, the cytotoxicity of the Ga(III) complex increased synchronously with ligand numbers after 48 hours of treatment on HepG-2. These data indicate that the antiproliferative activity of Ga(III) complexes is determined by the corresponding ligand, and the anticancer activity of the ligand is enhanced by coordination with gallium. L6 (IC50, 0.32 ± 0.02 μM) and C6 (IC50, 0.14 ± 0.01 μM) has the most excellent antiproliferative activity against HepG-2 in ligands and complexes, respectively. After 48 h of treatment, the cytotoxicity of L1–L6 increases in turn, and the toxicity of the complex is higher than that of the corresponding ligand. However, L6 has the best selectivity indices (SI) in ligands, and the presence of Ga(III) has a better impact on this parameter. The antitumor activity of all Ga(III) complexes was higher than that of cisplatin.
Cell apoptosis assay
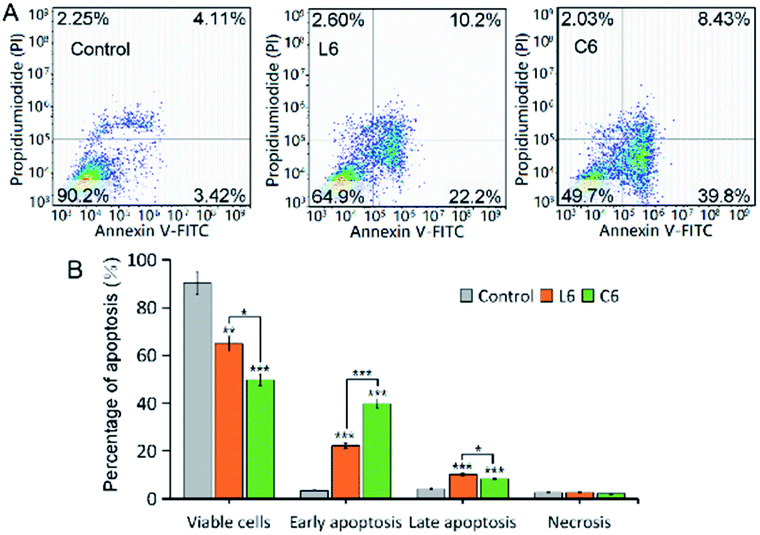
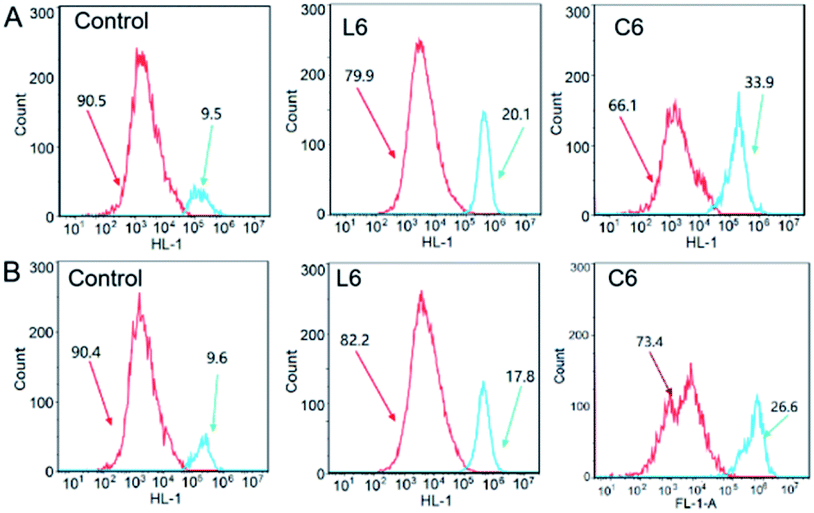
Apoptosis is physiological and programmed death process, a basic biological phenomenon of cells.30–32 Lack of apoptosis in cell proliferation is a crucial factor in tumor development.33–35 Therefore, an effective way to treat tumors is to stimulate and restore apoptosis of tumor cells. To assess the effect of concentration on the ability of gallium complexes (C6) to promote apoptosis in HepG-2 cells, Annexin v-fitc/PI was used to stain the cells and flow cytometry was used to detect apoptosis (Fig. 2). After incubation for 24 hours at 5 μM of L6 and C6, the percentage of early apoptosis and late apoptosis increased to 24.87% and 40.7% relative to the control.
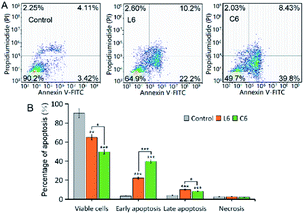 |
| | Fig. 2 (A) Representative dot plots of PI and Annexin V double staining on the HepG-2 cells; (B) comparison of apoptosis distributions. | |
Cell cycle analysis
We used flow cytometry to evaluate the effect of thiosemicarbazide ligands and Ga(III) complexes on cell cycle, and propidium iodide (PI) was used to stain DNA of HepG-2 cells as well as the cell cycle distribution was performed by flow cytometry (Fig. 3). After 24 h of co-cultivation of L6 and C6 with the cells, quantification of the results showed that the percentage of G2 phase cells were reduced compared to the vehicle-treated control. The increase in G1 phase cell proportion was consistent with other data.
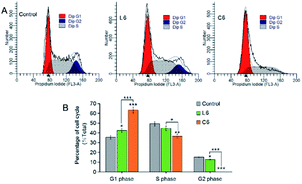 |
| | Fig. 3 (A) Effect on the cell cycle of the HepG-2 cells; (B) comparison of cell cycle distributions. | |
Mitochondrial membrane potential assay
Mitochondria are the main site for cellular aerobic respiration, known as “power house”, and the dissipation of mitochondrial membrane potential may be one of the causes of cell apoptosis.36 To examine the effect of the ligands and Ga(III) complexes on mitochondrial membrane potential (Δψm), changes in mitochondrial membrane potential were monitored by flow cytometry using JC-1 fluorescent probe (5,5,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanineiodide). Red fluorescence (JC-1 aggregates) occurs when the mitochondrial membrane potential is high, whereas green fluorescence (JC-1 monomer) is observed when the mitochondrial membrane potential is low (Fig. 4A). HepG-2 was cultured with 5 μM of L6 and C6, the Δψm was significantly (p < 0.001) decrease to 22.5% and 73.5% relative to the control, respectively.
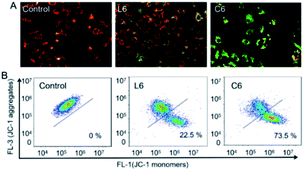 |
| | Fig. 4 Assay of HepG-2 cells mitochondrial membrane potential with JC-1 as fluorescence indicator. (A) Observed with a fluorescent inverted microscope; (B) examined by flow cytometry. | |
The expression of transferrin receptor-1 (TfR1) and ferritin proteins affected by Ga(III) complex
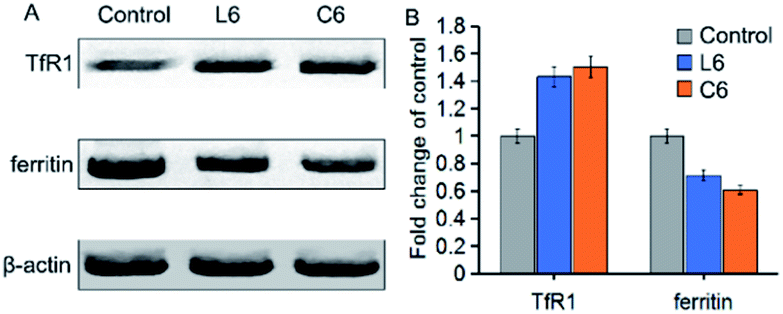
To further investigate the effects of thiosemicarbazone and gallium ions on iron metabolism in cells, we tested the effects of L6 and C6 on ferritin and transferrin receptor-1 (TfR1) expression (Fig. 5). After 24 h of co-cultivation of L6 and C6 with the cells, quantification of the results showed that the expression of TfR1 were increased by about 1.43 fold and 1.50 fold compared to the vehicle-treated control. While the expression of ferritin reduced 0.72 fold and 0.61 fold relative to the control group. Both ligand and Ga(III) complex down-regulated ferritin expression and up-regulated TfR1 expression, indicating cell response to iron consumption. The results on TfR1 and ferritin expression being very similar for the Ga(III) complex C6 and the corresponding ligand L6 could suggest that in both cases the ligand can chelate iron which would mean that an exchange between gallium and iron occurs (Fig. 5).
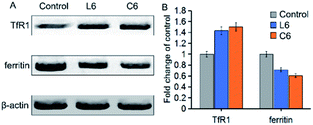 |
| | Fig. 5 (A) Western blot analysis of TfR1 and ferritin proteins in HepG-2 cells treated with 5 μM of L6 and C6 for 24 h; β-actin was used as the internal control. (B) Expression levels of TfR1 and ferritin proteins shown as percentages; the values are relative to the control. | |
Intracellular ROS measurements
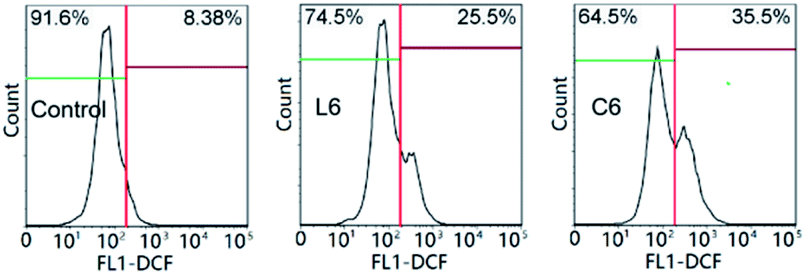
The loss of mitochondrial function may cause disorder of anti-free radical metabolism and free radical oxidative metabolism in cells.37 Drug invasion leads to excessive intracellular reactive oxygen species (ROS) production or decreased clearance capacity, resulting in oxidative stress and apoptosis.38 After incubation for 24 hours at 5 μM of L6 and C6, the percentage of intracellular ROS level increased to 17.12% and 27.12% relative to the control (Fig. 6). At the same concentration, C6 promoted the ability of HepG-2 cells to produce ROS significantly stronger than (Fig. 6) L6.
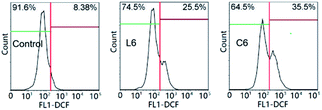 |
| | Fig. 6 The intracellular reactive oxygen species (ROS) of HepG-2 cells. | |
Detection of changes in protein levels of apaf-1 and cyt-c
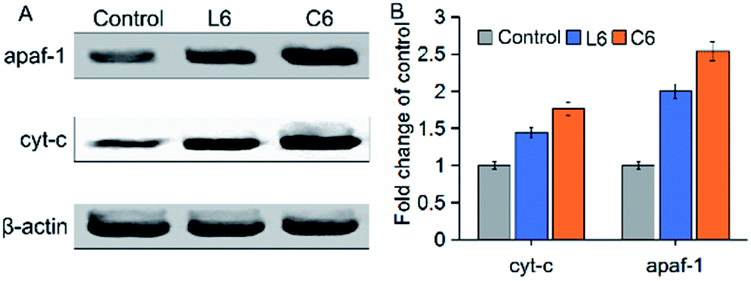
The apoptotic active substances (cyt-c) will release from the mitochondria through the permeability transition (PT) pore under the action of the mitochondrial membrane potential (Δψm).39,40 To confirm this phenomenon, the influence of L6 and C6 on the expression of apaf-1 and cyt-c in HepG-2 cells were performed by western blot (Fig. 7). The results showed that 5 μM of L6 had significant effect on the expression of apaf-1 and cyt-c. After treated with 5 μM C6, induced a significant (p < 0.05) increase in apaf-1 and cyt-c protein levels than L6 incubated cells. The increase in pro-apoptotic protein levels implied the occurrence of apoptosis.
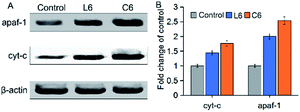 |
| | Fig. 7 (A) Western blot was used to determine the protein levels of apaf-1 and cyt-c; (B) densitometric analysis of apaf-1 and cyt-c: the density of each protein bands was normalized with that of the β-actin band shown in part A. | |
Caspase-3/9 activation assay
Upon initiation of apoptosis, cyt-c is released into the cytoplasm and subsequently activates caspase-3/9 protein to form apoptotic bodies.41–43 Thus, the percentage of activated caspase-3/9 was analyzed when HepG-2 cells were treated with L6 and C6, respectively (Fig. 8). After incubation with 5 μM of L6 and C6 for 24 hours, the percentage of activated caspase-3 protein level increased to 10.6% and 24.4% compared to the control, and the activated caspase-9 protein level increased to 8.2% and 17%. The results showed that L6 and C6 are potent activators of caspase-3/9, and up-regulation of these pro-apoptotic protein is an important pathway for apoptosis of HepG-2 cells induced by thiosemicarbazide ligands and their gallium complexes.
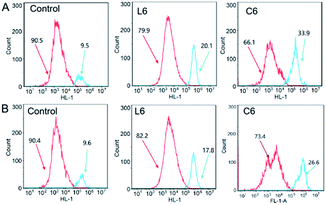 |
| | Fig. 8 (A) The activation of caspase-3 in HepG-2 cells; (B) the activation of caspase-9 in HepG-2 cells. | |
Detection of changes in protein levels of cell cycle related proteins
Regulation of the cell cycle is influenced by cyclin, cyclin-dependent kinase (Cdk), Cdk inhibitor, DNA, and the core mechanism is the activation and regulation of Cdk inhibitors.44 Therefore, L6 and C6 treated cells were analyzed to reveal the cyclin-dependent kinases (Cdk 2) and cyclins (cyclin A and cyclin E) expression (Fig. 9). HepG-2 cells treated with 5 μM L6 and C6 induced significant (p < 0.05) decreased in these proteins' expression level.
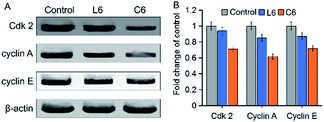 |
| | Fig. 9 (A) Western blot was used to determine the protein levels of Cdk 2, cyclin A and cyclin E in HepG-2 cells; (B) densitometric analysis of Cdk 2, cyclin A and cyclin E: the density of each of these protein bands was normalized with that of the β-actin band shown in part A. | |
Experimental
Material
Chemicals and solvents were purchased from Innochem Co., Ltd. (Shanghai, China), all of which are of analytical grade and used without further purification. Deionized water was used throughout the experiment. The HepG-2 and LO2 cell lines were obtained from Chinese Academy of Sciences. At 298 K, NMR spectra were recorded on a Bruker AVANCE III HD 600 MHz spectrometer. Proton chemical shifts were referenced to the central peak of DMSO-d6. Electrospray ionization mass (ESI-MS) spectrometry were performed on an Agilent 1260-6224 ion trap spectrometer, working in aqueous solution.
Synthesis and characterization of ligands (L1–L6)
The synthesis of ligands (L1–L6) was performed by previous methods.11 One mole equivalent of 2-benzoyl pyridine and 5 drops of CH3COOH were added to 10 mL ethanol (1 mM) solution of thiosemicarbazide and refluxed for 4 h. The formed precipitate was collected by filtration and washed with ethanol, and dried in vacuo. 2-(Phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide (L1) and N,N-dimethyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide (L5) have been reported in our previous literature.15,20
N-Methyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide (L2). Yield 69%. Anal. Calcd for C14H14N4S: C, 62.20; H, 5.22; N, 20.72. Found: C, 62.34; H, 5.20; N, 20.76. 1H NMR (600 MHz, DMSO-d6) δ 12.83 (s, 1H), 8.87 (ddd, J = 5.0, 1.7, 0.9 Hz, 1H), 8.73 (d, J = 4.7 Hz, 1H), 8.02 (td, J = 7.8, 1.8 Hz, 1H), 7.66–7.63 (m, 2H), 7.61 (ddd, J = 7.7, 4.9, 1.1 Hz, 1H), 7.49–7.44 (m, 3H), 7.36 (dd, J = 8.0, 1.0 Hz, 1H), 3.05 (d, J = 4.6 Hz, 3H). MS m/z (%) 269.09 (M − H, 100).
N-Ethyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide (L3). Yield 76%. Anal. Calcd for C15H16N4S: C, 63.35; H, 5.67; N, 19.70. Found: C, 63.39; H, 5.61; N, 19.66. 1H NMR (600 MHz, DMSO-d6) δ 12.76 (s, 1H), 8.87 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 8.78 (s, 1H), 8.02 (td, J = 7.8, 1.8 Hz, 1H), 7.65–7.63 (m, 2H), 7.61 (ddd, J = 7.7, 4.9, 1.1 Hz, 1H), 7.48 (qd, J = 3.1, 1.2 Hz, 3H), 7.35 (dt, J = 8.1, 1.1 Hz, 1H), 3.62 (dd, J = 7.2, 6.1 Hz, 2H), 1.15 (t, J = 7.1 Hz, 3H). MS m/z (%) 283.11 (M − H, 100).
N-Cyclohexyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide (L4). Yield 71%. Anal. Calcd for C19H22N4S: C, 67.42; H, 6.55; N, 16.55. Found: C, 67.49; H, 6.51; N, 16.50. 1H NMR (600 MHz, DMSO-d6) δ 12.82 (s, 1H), 8.87 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 8.25 (d, J = 8.6 Hz, 1H), 8.02 (td, J = 7.8, 1.9 Hz, 1H), 7.62 (t, J = 1.8 Hz, 1H), 7.62–7.59 (m, 2H), 7.48 (dd, J = 5.0, 2.3 Hz, 2H), 7.34 (dd, J = 8.1, 1.1 Hz, 1H), 4.20–4.16 (m, 1H), 1.88 (dd, J = 12.4, 4.0 Hz, 2H), 1.72 (dt, J = 13.3, 3.5 Hz, 2H), 1.62–1.58 (m, 1H), 1.46 (dd, J = 11.7, 3.5 Hz, 2H), 1.28 (dt, J = 12.9, 3.5 Hz, 2H), 1.20–1.04 (m, 2H). MS m/z (%) 337.16 (M − H, 100).
N-Cyclohexyl-N-methyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide (L6). Yield 79%. Anal. Calcd for C20H24N4S: C, 68.15; H, 6.86; N, 15.89. Found: C, 68.21; H, 6.81; N, 15.85. 1H NMR (600 MHz, DMSO-d6) δ 13.01 (s, 1H), 8.93–8.86 (m, 1H), 8.06–8.02 (m, 1H), 7.67 (ddt, J = 7.1, 3.7, 1.6 Hz, 2H), 7.65–7.62 (m, 1H), 7.56–7.53 (m, 1H), 7.49 (dd, J = 3.5, 1.4 Hz, 2H), 7.37–7.30 (m, 1H), 3.03 (s, 3H), 1.77 (d, J = 13.1 Hz, 2H), 1.65 (d, J = 12.2 Hz, 2H), 1.59 (s, 1H), 1.47 (dd, J = 12.3, 3.6 Hz, 2H), 1.30 (dt, J = 13.2, 3.4 Hz, 2H), 1.17–1.06 (m, 2H). MS m/z (%) 351.16 (M − H, 100).
Synthesis and characterization of Ga(III) complexes
The synthesis of Ga(III) complexes (C1–C6) was performed by previous methods.11 A solution of thiosemicarbazone in methanol (10 mL, 1 mM) was added GaCl3 (one mole equivalent) with stirring and set still, waiting for the solvent to slowly emanate. After a few days, the Ga(III) complex crystals were obtained and washed with ethanol, followed by vacuum drying.
Dichlorido[2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide-N,N′,S]gallium(III) (C1). Yield 81%. Anal. Calcd for C13H11Cl2GaN4S: C, 39.43; H, 2.80; N, 14.15. Found: C, 39.50; H, 2.84; N, 14.13. ESI-MS, m/z (r. i.): 392.93 (M − H, 100).
Dichlorido[N-methyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide-N,N,S]-gallium(III) (C2). Yield 85%. Anal. Calcd for C14H13Cl2GaN4S: C, 41.01; H, 3.20; N, 13.67. Found: C, 41.07; H, 3.16; N, 13.69. ESI-MS, m/z (r. i.): 406.95 (M − H, 100).
Dichlorido[N-ethyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide-N,N,S]-gallium(III) (C3). Yield 83%. Anal. Calcd for C15H15Cl2GaN4S: C, 42.49; H, 3.57; N, 13.21. Found: C, 42.43; H, 3.51; N, 13.27. ESI-MS, m/z (r. i.): 420.97 (M − H, 100).
Dichlorido[N-cyclohexyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide-N,N,S]-gallium(III) (C4). Yield 82%. Anal. Calcd for C19H21Cl2GaN4S: C, 47.73; H, 4.43; N, 11.72. Found: C, 47.76; H, 4.40; N, 11.77. ESI-MS, m/z (r. i.): 475.01 (M − H, 100).
Dichlorido[N,N-dimethyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide-N,N,S]-gallium(III) (C5). Yield 86%. Anal. Calcd for C15H15Cl2GaN4S: C, 42.49; H, 3.57; N, 13.21. Found: C, 42.44; H, 3.52; N, 13.29. EI-MS, m/z (r. i.): 4.6.94 (100), [M − CH3]+; 387.00 (19.5), [M − Cl]+.
Dichlorido[N-cyclohexyl-N-methyl-2-(phenyl(pyridin-2-yl)methylene)hydrazinecarbothioamide-N,N,S]-gallium(III) (C6). Yield 89%. Anal. Calcd for C20H23Cl2GaN4S: C, 48.81; H, 4.71; N, 11.38. Found: C, 48.85; H, 4.75; N, 11.42. EI-MS, m/z (r. i.): 475.01 (100), [M − CH3]+; 455.06 (31.2), [M − Cl]+; 371.98 (19.5), [M − phenyl]+.
Determination of structure of Ga(III) complexes
The single crystal of C6 was placed on a glass fiber. The cell constants and crystal data are accurately measured by RAPID IP diffractometer or Rigaku AFC5S (Mo Ka radiation, k = 0.71069 A). The crystal structures were solved directly and differential Fourier calculations were performed, followed by full matrix least squares refinement using the SHELXL97 and TEXSAN package. All non-hydrogen atoms were refined anisotropically. Crystal data, structural refinement and data collection are shown in Table 1.
Cytotoxicity assay (MTT)
The cell lines were cultured in an incubator (ThermoFisher) with 5% CO2, 95% air at 37 °C. We used a MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) assay to investigate the cell cytotoxicity. Dissolving the thiosemicarbazone ligand and the Ga(III) complex in dimethyl sulfoxide (DMSO) to form a 10 mM stock solution. The DMSO solution in which the complexes were dissolved was diluted with different volumes of cell culture medium to ensure a DMSO concentration of <0.1% so as not to affect cell proliferation. 100 μL of medium containing cells was seeded in 96-well plates and adhered for 12 h at 37 °C, in 5% CO2/95% air. The seeding densities used were 5 × 104 cells per well. Different concentrations of ligand or Ga(III) complex (20, 10, 5 1, 0.5 μM) were added to the wells and incubated for 48 h, then 20 μL 5 mg mL−1 of MTT was added to the wells for another 4 hours. Further added with 100 μL of DMSO, and the optical density (OD) of living cells was measured at a wavelength of 570 nm. The IC50 (half maximum inhibitory concentration) values were calculated using the nonlinear multi-purpose curve fitting program (GraphPad Prism).
Cell cycle distribution analysis
HepG-2 cells (5 mL, 1 × 105 cells per mL) were incubated in a 70 mm culture dish, and then the cells were cultured at 37 °C, 5% CO2/95% air for 12 hours. After treating the cells with L6 (5 μM) or C6 (5 μM) for 24 hours, respectively, the collected cells were washed three times with cold phosphate buffer (PBS, pH = 7.4). Cells were immobilized with 75% EtOH for 12 h at −20 °C and washed with iced PBS. The cells were incubated with Ribonuclease (RNase, 2.5 mg mL−1) and stained with 50 mg mL−1 propidium iodide for 0.5 hours and analyzed by flow cytometry (BD, Accuri™ C6). Data was analyzed for 10000 events per sample using MFL 32 software.
Cell apoptosis assay
Apoptosis events in HepG-2 cells induced by L6 (5 μM) or C6 (5 μM) were assessed by Annexin V staining and PI according to the procedure of the Detection Kit (Bection Dickinson). We used 2 mL 1 × 106 cells per mL, which were incubated with L6 (5 μM) or C6 (5 μM) for 24 h at 5% CO2 and 37 °C. HepG-2 cells were dispersed in 300 μL of mixed buffer (140 mM NaCl, pH 7.4, 2.5 mM CaCl2, 10 mM Hepes/NaOH), followed by the addition of 5 μL of PI and Annexin V. Finally, the samples were incubated for 20 minutes at room temperature and assayed by flow cytometry (BD, Accuri™ C6) and analyzed by FlowJo software.
The mitochondrial membrane potential assay
JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide) staining of mitochondrial membrane potential of HepG-2 cells induced by L6 (5 μM) or C6 (5 μM) was performed according to the JC-1 test kit (Biyuntian, Suzhou) method. HepG-2 cells were incubated with L6 (5 μM) or C6 (5 μM) for 0.5 hours at 37 °C for 24 h. The cells washed with iced PBS were incubated with 1 μg mL−1 of JC-1 for 0.5 h at 37 °C in the dark. After centrifugation, the supernatant was rinsed three times with cold PBS (phosphate buffer, pH = 7.4). Data was obtained by flow cytometry (BD, Accuri™ C6) and FlowJo software was performed to analyze the results.
Intracellular ROS measurements
Evaluation of intracellular ROS was performed by the procedure of active oxygen assay kit (Beyotime, Suzhou) and flow cytometry. Incubate HepG-2 cells with L6 (5 μM) or C6 (5 μM) in serum-free cell culture medium for 24 h at 37 °C. 2 μM 2′,7′-dichlorodihydro-fluorescein diacetate (H2DCF-DA) was added to the cells, and the samples were washed with serum-free medium. Data was obtained by flow cytometry (BD, Accuri™ C6) and FlowJo software was used to analyze the results.
Caspase-3 and caspase-9 activity analysis
The activities of caspase-3 and caspase-9 were detected by CaspGLOW™ Fluorescein Active Caspase-3 and Caspase-9 Staining Kits. L6 (5 μM) or C6 (5 μM) was added to HepG-2 cells for 24 hours. The controlled cells were harvested from RPMI 1640 medium containing 10% fetal calf serum (FBS), then centrifuged and washed three times with PBS. The cells were uniformly mixed with 300 mL of the culture broth, followed by the addition of 1 μL of FITC-DEVD-FMK, and the sample was allowed to stand at 37 °C for an hour. Samples were tested on a BD Accuri™ C6 flow cytometer. The experimental results are expressed as a percentage change in activity between the sample and the untreated control.
Western blot analysis
5 mL of HepG-2 cells (1 × 105 cells per mL) were seeded on a 100 mm diameter dish and incubated for 24 h at 37 °C, in 5% CO2/95% air atmosphere. Cells were harvested and washed 3 times with ice-cold PBS after treatment of L6 (5 μM) or C6 (5 μM) for 24 hours. The cells were disrupted with buffer and centrifuged at 10000g for 15 minutes under 4 °C to obtain a supernatant. The protein concentration was determined using a BSA Assay Kit (Beyotime, China), and the samples were detected by SDS-polyacrylamide gel electrophoresis. Under ice bath conditions, the protein was loaded on the polyvinylidene fluoride membrane for 3 hours, followed by preparation of skim milk (5%) for the presence of protein-blocked 1 h in TBST buffer at 25 °C. Membranes and primary antibodies were incubated overnight at 4 °C and washed three times with TBST buffer (20 mM Tris–HCl, 0.05% Tween 20, 150 mM NaCl, pH 8.0). After the membranes were incubated with the secondary antibody for 1 h at 25 °C, they were washed three times in the same manner. Immunoreactivity was imaged by Amersham ECL Plus (Amersham) and the amount of protein in each lane was determined by β-actin.
Statistical analysis
The significance of the differences was assessed by the Student's t test. The experimental results are shown as mean ± SD, and when p < 0.05, it is indicated that the data is significant.
Conclusions
Herein, we report a series of thiosemicarbazide ligands and gallium complexes with potential antiproliferative activity. The structure–activity relationship results indicate that the modification of the lipophilic group on the ligand significantly improve its antiproliferative activity, and the anti-cancer activity after coordination with gallium is further improved. Ga(III) complex disrupts cellular iron metabolism by up-regulating the expression of TfR1 and down-regulating ferritin. Studies on the apoptosis mechanism of cells show that Ga(III) complex promotes the apoptosis of HepG-2 cells by regulating pro-apoptotic factors (apaf-1, cyt-c and caspase) through the mitochondrial apoptosis pathway. Ligands and gallium complexes inhibit cell cycle distribution by regulating the expression of cyclin-dependent kinases (Cdk 2) and cyclins (cyclin A and cyclin E). The results of the anticancer mechanism indicate that the increased cytotoxicity of the Ga(III) complexes compared with ligands is a result of a combination of multiple mechanisms related to the ability to promote apoptosis and inhibition of cell cycle transition in HepG-2 cells. Overall, these findings are highly beneficial for the development of Ga(III)-thiosemicarbazone complexes as potential anti-cancer therapeutics.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81571812), Pingdingshan College PhD Startup Fund under Grant PXY-BSQD-202003; Key Specialized Research and Development breakthrough in Henan Province (202102310476 and 182102310181); and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions under Grant 1107047002.
Notes and references
- P. Collery, B. Keppler, C. Madoulet and B. Desoize, Critical Reviews in Oncology/Hematology, 2002, 42, 283–296 CrossRef PubMed.
- C. R. Chitambar, Met. Ions Life Sci., 2018, 18 DOI:10.1515/9783110470734-016.
- D. J. Straus, Semin. Oncol., 2003, 30, 25–33 CrossRef CAS PubMed.
- V. Nikolova, S. Angelova, N. Markova and T. Dudev, J. Phys. Chem. B, 2016, 120, 2241–2248 CrossRef CAS PubMed.
- K. Kumar, S. Schniper, A. González-Sarrías, A. A. Holder, N. Sanders, D. Sullivan, W. L. Jarrett, K. Davis, F. Bai, N. P. Seeram and V. Kumar, Eur. J. Med. Chem., 2014, 86, 81–86 CrossRef CAS PubMed.
- J. Qi, Q. Yao, K. Qian, L. Tian, Z. Cheng and Y. Wang, J. Inorg. Biochem., 2018, 186, 42–50 CrossRef CAS PubMed.
- X. Zhang, Y. Jiang and N. Xiao, Chem. Commun., 2018, 54, 12812–12815 RSC.
- L. Xiao, S. Sun, Z. Pei, Y. Pei, Y. Pang and Y. Xu, Biosens. Bioelectron., 2015, 65, 166–170 CrossRef CAS PubMed.
- J. A. Lessa, G. L. Parrilha and H. Beraldo, Inorg. Chim. Acta, 2012, 393, 53–63 CrossRef CAS.
- I. C. Mendes, M. A. Soares, R. G. Dos Santos, C. Pinheiro and H. Beraldo, Eur. J. Med. Chem., 2009, 44, 1870–1877 CrossRef CAS PubMed.
- J. Qi, Q. Yao, K. Qian, L. Tian, Z. Cheng, D. Yang and Y. Wang, Eur. J. Med. Chem., 2018, 154, 91–100 CrossRef CAS PubMed.
- A. Mohsen, C. Saby, P. Collery, G. M. Sabry, R. E. Hassan, A. Badawi, P. Jeannesson, D. Desmaële and H. Morjani, J. Biol. Inorg. Chem., 2016, 21, 837–849 CrossRef CAS PubMed.
- A. Salem, E. Noaman, E. Kandil, A. Badawi and N. Mostafa, Tumor Biol., 2016, 37, 11025–11038 CrossRef CAS PubMed.
- M. Pribisko, J. Palmer, R. H. Grubbs, H. B. Gray, J. Termini and P. Lim, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, E2258–E2266 CrossRef CAS PubMed.
- J. Qi, K. Qian, L. Tian, Z. Cheng and Y. Wang, New J. Chem., 2018, 42, 10226–10233 RSC.
- J. Qi, J. Deng, K. Qian, L. Tian, J. Li, K. He, X. Huang, Z. Cheng, Y. Zheng and Y. Wang, Eur. J. Med. Chem., 2017, 134, 34–42 CrossRef PubMed.
- J. Qi, Y. Zheng, K. Qian, L. Tian, G. Zhang, Z. Cheng and Y. Wang, J. Inorg. Biochem., 2017, 177, 110–117 CrossRef PubMed.
- C. R. Chitambar and W. E. Antholine, Antioxid. Redox sign., 2013, 18, 956–972 CrossRef.
- J. A. Lessa, M. A. Soares, R. G. Dos Santos, I. C. Mendes, L. B. Salum, H. N. Daghestani, A. D. Andricopulo, B. W. Day, A. Vogt and H. Beraldo, BioMetals, 2013, 26, 151–165 CrossRef.
- C. R. Kowol, R. Berger, R. Eichinger, A. Roller, M. A. Jakupec, P. P. Schmidt, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 1254–1265 CrossRef PubMed.
- M. Pribisko, J. Palmer, R. H. Grubbs, H. B. Gray, J. Termini and P. Lim, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2258–E2266 CrossRef CAS PubMed.
- C. B. Scarim, D. H. Jornada, M. G. M. Machado, C. M. R. Ferreira, J. L. Dos Santos and M. C. Chung, Eur. J. Med. Chem., 2019, 162, 378–395 CrossRef CAS PubMed.
- D. B. Lovejoy, D. M. Sharp, N. Seebacher, P. Obeidy, T. Prichard, C. Stefani, M. T. Basha, P. C. Sharpe, P. J. Jansson, D. S. Kalinowski, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2012, 55, 7230–7244 CrossRef CAS PubMed.
- C. Stefani, P. J. Jansson, E. Gutierrez, P. V. Bernhardt, D. R. Richardson and D. S. Kalinowski, J. Med. Chem., 2012, 56, 357–370 CrossRef PubMed.
- C. R. Kowol, W. Miklos, S. Pfaff, S. Hager, S. Kallus, K. Pelivan, M. Kubanik, É. A. Enyedy, W. Berger, P. Heffeter and B. K. Keppler, J. Med. Chem., 2016, 59, 6739–6752 CrossRef PubMed.
- J. Kolesar, R. C. Brundage, M. Pomplun, D. Alberti, K. Holen, A. Traynor, P. Ivy and G. Wilding, Cancer Chemoth. Pharm., 2011, 67, 393–400 CrossRef.
- É. A. Enyedy, M. F. Primik, C. R. Kowol, V. B. Arion, T. Kiss and B. K. Keppler, Dalton Trans., 2011, 40, 5895 RSC.
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 4965–4984 CrossRef.
- J. G. Da Silva, L. S. Azzolini, S. M. S. V. Wardell, J. L. Wardell and H. Beraldo, Polyhedron, 2009, 28, 2301–2305 CrossRef.
- J. Loscalzo, Cell Metab., 2008, 8, 182–183 CrossRef CAS PubMed.
- H. Y. Ryu, J. K. Emberley, J. J. Schlezinger, L. L. Allan, S. Na and D. H. Sherr, Mol. Pharmacol., 2005, 68, 1087–1096 CrossRef CAS PubMed.
- A. Amsterdam, R. Sasson, I. Keren-Tal, D. Aharoni, A. Dantes, E. Rimon, A. Land, T. Cohen, Y. Dor and L. Hirsh, Biochem. Pharmacol., 2003, 66, 1355–1362 CrossRef CAS PubMed.
- H. Tuerxun and J. Cui, Clin. Transl. Oncol., 2019, 21, 695–701 CrossRef CAS PubMed.
- H. S. Chin, M. X. Li, I. Tan, R. L. Ninnis, B. Reljic, K. Scicluna, L. F. Dagley, J. J. Sandow, G. L. Kelly, A. L. Samson, S. Chappaz, S. L. Khaw, C. Chang, A. Morokoff, K. Brinkmann, A. Webb, C. Hockings, C. M. Hall, A. J. Kueh, M. T. Ryan, R. M. Kluck, P. Bouillet, M. J. Herold, D. Gray, D. Huang, M. F. van Delft and G. Dewson, Nat. Commun., 2018, 9, 4976 CrossRef PubMed.
- Y. Liang, B. Mafuvadze, C. Besch-Williford and S. M. Hyder, Breast Cancer, Dove Med Press, 2018, vol. 10, pp. 53–67 Search PubMed.
- H. Han, J. Hemp, L. A. Pace, H. Ouyang, K. Ganesan, J. H. Roh, F. Daldal, S. R. Blanke and R. B. Gennis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14109–14114 CrossRef CAS PubMed.
- C. Chen, Y. Liu, Y. Liu and P. Zheng, Cell Cycle, 2009, 8, 1158–1160 CrossRef CAS PubMed.
- T. Paul, A. Banerjee, S. Reddy, S. K. Mahato and N. Biswas, Anticancer Drugs, 2019, 30, 167–178 CrossRef CAS PubMed.
- M. D. Ramamoorthy, A. Kumar, M. Ayyavu and K. N. Dhiraviam, Anticancer Agents Med. Chem., 2018, 18, 1313–1322 CrossRef CAS.
- D. G. Lee, B. K. Choi, Y. H. Kim, H. S. Oh, S. H. Park, Y. S. Bae and B. S. Kwon, Cancer Lett., 2016, 382, 186–194 CrossRef CAS.
- R. Tamura, M. Takada, M. Sakaue, A. Yoshida, S. Ohi, K. Hirano, T. Hayakawa, N. Hirohashi, K. Yura and K. Chiba, Sci. Rep., 2018, 8, 1611 CrossRef.
- H. Bardak, A. C. Uguz and Y. Bardak, Physiol. Int., 2017, 104, 301–315 CAS.
- T. Kanno and T. Nishizaki, J. Cell. Physiol., 2011, 226, 2329–2337 CrossRef CAS PubMed.
- P. Xia, Y. Liu, J. Chen, S. Coates, D. X. Liu and Z. Cheng, J. Biol. Chem., 2018, 293, 19672–19685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1899688. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02913k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Taichen Liub,
Wei Zhaob,
Xinhua Zheng*b and
Yihong Wang
ab,
Taichen Liub,
Wei Zhaob,
Xinhua Zheng*b and
Yihong Wang