
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu catalyst supported on nitrogen-doped nanotubes as high efficiency electrocatalysts for hydrogen evolution in alkaline media†
Qinglei Liu,
Lehao Yang,
Peng Sun,
Haigang Liu,
Jiahua Zhao,
Xiankun Ma,
Yongfei Wang * and
Zhiqiang Zhang*
* and
Zhiqiang Zhang*
School of Chemical Engineering, University of Science and Technology Liaoning, Anshan, 114041, China. E-mail: wyf8307@ustl.edu.cn
First published on 11th June 2020
Abstract
Due to the potential application in the future energy conversion system, there is an increasing demand for efficient, stable and cheap platinum-free catalysts for hydrogen evolution. However, it is still a great challenge to develop electrocatalysts with high activity similar to platinum or even higher, especially those that can work under alkaline conditions. Ruthenium (Ru), as a cheap substitute for platinum, has been studied as a feasible substitute for (HER) catalyst for hydrogen evolution reaction. In this paper, we designed and developed a novel Ru catalyst (Ru@CNT) supported on nitrogen-doped carbon nanotubes. Electrochemical tests show that even under alkaline conditions (1 M KOH), Ru@CNT still shows excellent catalytic performance and good durability. It only needs 36.69 mV overpotential to reach a current density of 10 mA cm−2, and its Tafel slope is 28.82 mV dec−1. The catalytic performance of the catalyst is comparable to that of 20% Pt/C. The significant activity is mainly attributed to the chelation of highly dispersed ruthenium atoms on nitrogen-doped carbon nanotubes. Secondly, the one-dimensional pore structures supported by nitrogen heterocarbon nanotubes can provide more opportunities for active centers. Excellent HER performance makes Ru@CNT electrocatalyst have a broad application prospect in practical hydrogen production.
Introduction
Hydrogen evolution reaction (HER) plays an important role in the electrochemical decomposition of water to achieve clean and sustainable development of hydrogen energy.1,2 However, it is difficult to produce hydrogen from water because the cracking reaction of water requires a high overpotential. Energy-efficient HER must use catalysts to trigger proton reduction with minimal overpotential and enhance kinetics. In practice, room temperature water electrolysis can be carried out not only in acidic electrolyte but also in alkaline electrolyte, but a lot of research has been carried out in acidic medium. The high cost of proton exchange membrane and the slow electron transfer kinetics of oxygen evolution reaction in acidic medium greatly hinder the wide application of acid hydrolyser. If the reaction is carried out in alkaline medium, these problems can be alleviated.3–7 However, due to the large kinetic barrier of the previous hydrolysis separation step (Volmer step), the kinetics of hydrogen evolution in alkaline environment is two to three times slower than that of all electrocatalysts including Pt8–10 in acidic electrolyte. Therefore, it is of great significance and challenge to develop HER catalysts with high activity and stability in alkaline medium. Platinum as an electrocatalyst for hydrogen evolution reaction has almost zero overpotential and excellent long-term durability, so it is still the best HER catalyst.11 Unfortunately, the high price of platinum based electrocatalysts hinders their widespread commercialization.12 Therefore, it is of great significance to find a cheap substitute for platinum electrocatalyst. So far, a certain number of durable and efficient alternative catalysts have been reported to replace platinum-based electrocatalysts in alkaline media.Ruthenium (Ru) has a bonding strength similar to that of hydrogen (≈65 kcal mol−1),13 which can be used as a cheap substitute for Pt and is widely used in many important chemical reactions. In particular, ruthenium is much cheaper (290$ per oz) than platinum group metals, such as platinum (835$ per oz), palladium (2107$ per oz) or iridium (1733$ per oz). Although there are many reports of ruthenium-based electrocatalysts, there are few reports of ruthenium-based electrocatalysts with Pt-like activity. Wang et al. A binderless catalyst anchored by a single Ru atom on the surface of MoS2 nanosheet array supported by carbon cloth was synthesized. The η10 of the catalyst in 0.1 M KOH is −41 mV.14 In another study, Zheng et al. prepared ruthenium/carbon nitride (C3N4) nanocomposites by high temperature annealing, which η10 in 0.1 M KOH is −79 mV.15 Li et al. Graphene nanocrystals (CGnP), functionalized by edge carboxylic acid were formed by the mechanochemical reaction of graphite and dry ice, and then the precursors of Ru and CGnP were mixed and coordinated in aqueous medium. After subsequent annealing, uniform Ru nanoparticles (≈2 nm) were anchored on GnP matrix (Ru@GnP). The η10 of the catalyst in 0.1 M KOH is −22 mV.16 Although Ru, with electrocatalytic activity has been obtained, the over potential and stability of these materials still need to be improved. In addition, the preparation process of these catalysts is also complex, which limits the practical application of this metal/carbon-based catalyst.
Here, we show a new strategy to prepare the catalyst sample as shown in Fig. 1, chelating Ru atoms on nitrogen-doped carbon nanotubes. The mass loading of Ru in the prepared Ru@CNT-500 catalyst is as low as 0.068 wt%. Under alkaline condition (1 M KOH), Ru@CNT still showed excellent catalytic performance with an initial overpotential of 0 mV, Tafel, a slope of 28.82 mV dec−1, and good durability. Most importantly, it takes only 36.69 mV of overpotential to achieve a current density of 10 mA cm−2. The catalytic performance of the catalyst is similar to that of 20% Pt/C. The significant activity is mainly attributed to the highly dispersed ruthenium atoms embedded in nitrogen heterocarbon nanotubes. Secondly, the three-dimensional pore structures supported by nitrogen heterocarbon nanotubes can provide more opportunities for active centers. These one-dimensional pore structures can provide higher specific surface area and larger pore volume, which not only maximize the effectiveness of electron transfer in the surface area of nano-electrocatalysts, but also provide better mass transfer from reactants to electrocatalysts. Third, proper calcination has little effect on the morphology of the catalyst, and improves the conductivity of the catalyst.
 | ||
| Fig. 1 Schematic diagram of the steps for preparing Ru@CNT samples. | ||
Results and discussion
Structure and morphology of catalysts
XRD and Raman tests of CNT, carbonized CNT, Ru@CNT-800, Ru@CNT-700, Ru@CNT-600, Ru@CNT-500 and Ru@CNT-400 were carried out to understand the bulk structure of the prepared samples. Fig. 2a shows the XRD curve of polypyrrole CNT before and after calcination. It can be seen that there is a (002) characteristic diffraction peak of graphite lattice at 2θ = 26° before and after carbonization, and the peak shape is more obvious after carbonization. After carbonization of hollow carbon nanotubes, a weak peak is carbon (100) at about 42.8°, so the CNT obtained by carbonization at high temperature produces graphitized carbon.17 Fig. 2b shows the XRD curve of Ru@CNT samples at different calcination temperatures. It can be found that the (002) characteristic diffraction peak of carbon almost does not change with temperature, indicating that the carbon skeleton of CNT is highly stable at high temperature. When the diffraction angle is about 43.8°, the peak not only reflects the (100) crystal plane of carbon material, but also related to the (101) diffraction peak of Ru.18 With the increase of temperature, the diffraction peak of (101) becomes steeper, and the metal nanoparticles inevitably agglomerate with the increase of calcination temperature, forming larger particles, which makes the diffraction peak of Ru (101) become obvious. Fig. 2c shows the Raman spectra of hollow carbon nanotubes before and after carbonization. There are two distinct peaks at 1350 cm−1 and 1577 cm−1. The corresponding D and G characteristic peaks of carbon materials refer to disordered and defective carbon atoms and graphitized carbon atoms, respectively.19,20 The peak strength ratio (ID/IG) before and after carbonization is 1.0748 and 1.1602 respectively. High temperature carbonization increases the disorder, defect and graphitization of carbon nanotubes, improves the electrochemical active center of carbon nanotubes and the adhesion point and conductivity of ruthenium, which is beneficial to the improvement of electrocatalytic hydrogen evolution performance. Fig. 2d shows the Raman spectra of Ru@CNT samples calcined at different temperatures. From the peak intensity ratio (ID/IG) of Ru@CNT (Fig. 2e), it can be found that the degree of disorder and graphitization of the samples increase significantly when the temperature is higher than 500 °C. This can be attributed to the shedding and agglomeration of Ru atoms on carbon nanotubes and the graphitization of carbon nanotubes. This is consistent with the test results of XRD.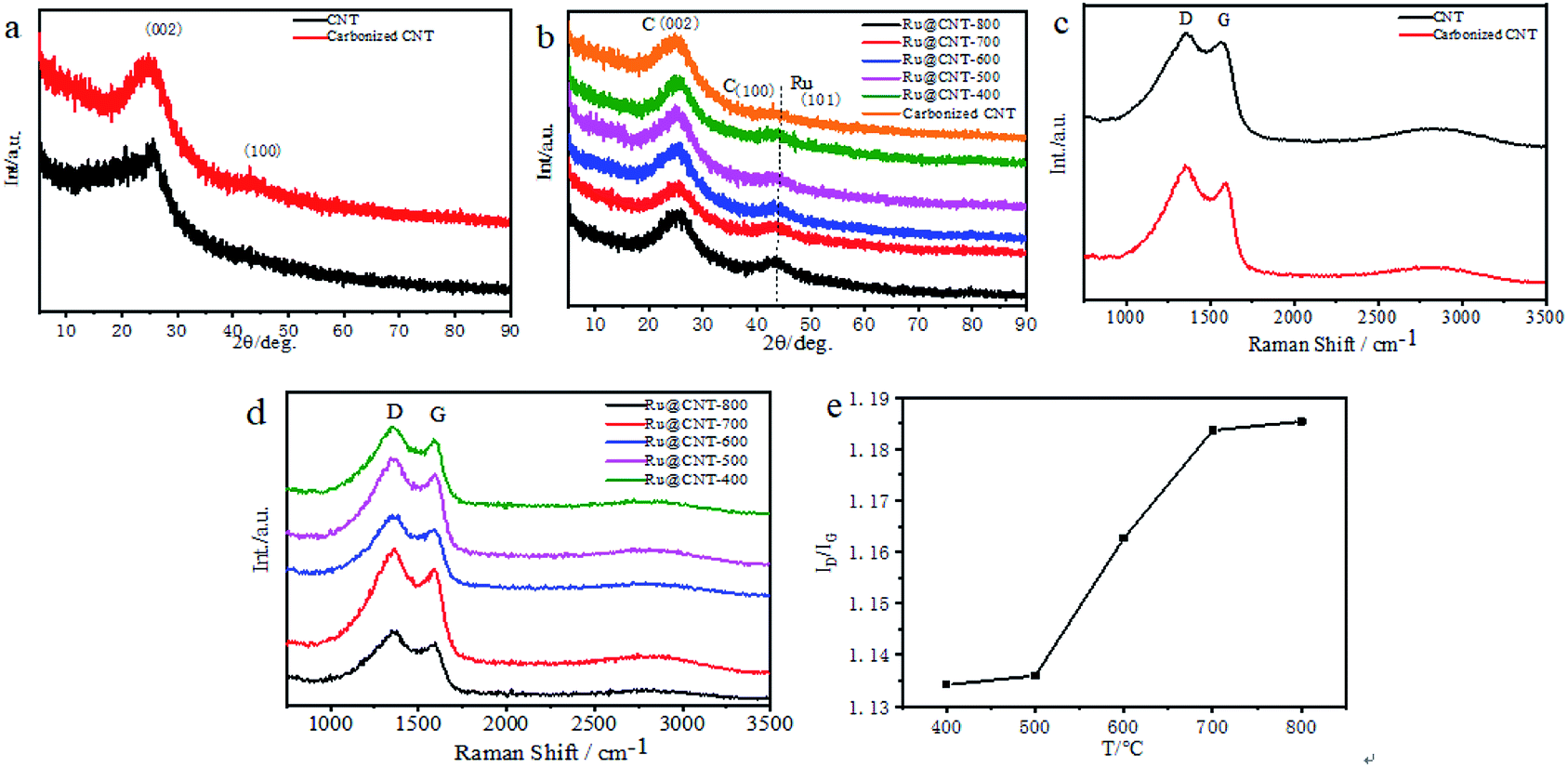 | ||
| Fig. 2 (a) XRD spectra before and after carbonization of CNT; (b) XRD spectra of Ru@CNT samples with different annealing temperatures; (c) Raman spectrum before and after carbonization of CNT; (d) Raman spectrum of Ru@CNT sample at different annealing temperatures; (e) peak-to-earth ratio (ID/IG) of Ru@CNT samples at different annealing temperatures. | ||
The morphology of the samples was observed by SEM. Fig. 3a and b show images of hollow carbon nanotubes before and after carbonization. The results show that after the first carbonization, the shape of CNT becomes regular, forming amorphous carbon, and has a certain degree of graphitization.21,22 The SEM analysis of Ru@CNT samples calcined at different temperatures shows that the CNTs still retain the characteristics of one-dimensional hollow nanotubes after adsorbing ruthenium, such as Fig. 3c. The calcination temperature has little effect on the morphology of nitrogen-doped carbon nanotubes, and its hollow structure makes the catalyst have a large contact area, which is beneficial to the electrocatalytic hydrogen evolution.23 In order to further understand the morphology and structure of the catalyst we analysed the sample Ru@CNT-500 by TEM. Fig. 3d and e show that the sample has a hollow tubular structure with an outer diameter of 110 nm and an inner diameter of 45 nm. The prepared CNT shows the same fibre characteristics, and the hollow structure can provide a large electrode–electrolyte contact area to ensure the high availability of catalytic active sites.24,25 High resolution TEM (HRTEM) images further reveal the distribution of Ru nanoparticles in CNTs. It can be seen from Fig. 3f and S1† that the Ru nanoparticles of Ru@CNTs increase and agglomerate with the increase of temperature. Although the ruthenium nanoparticles of Ru@CNT-400 and Ru@CNT-600 are uniformly distributed, the loading is low, which is the highly active part of HER, and the hydrogen evolution effect is poor. The loading amount of ruthenium nanoparticles of Ru@CNT-700 and Ru@CNT-800 is higher, but the particle size distribution is uneven and the particle size is larger, so the hydrogen evolution performance is reduced. The Ru nanoparticles of Ru@CNT-500 have high loading rate and are uniformly dispersed on the carbon nanotube skeleton with an average particle size of 2.1 nm. Therefore, Ru@CNT-500 has good catalytic performance.26 HRTEM images also show that the lattice spacing of 0.21 nm corresponds to the (002) plane of hexagonal Ru (Fig. 3g). EDS mapping confirms the uniform distribution of N and Ru elements on CNT (Fig. 3h).27,28 It was found that N and Ru elements were uniformly dispersed on CNT, which proved that ruthenium nanoparticles were dispersed on hollow carbon nanotubes. Therefore, no other characteristic peaks of ruthenium were found in the XRD spectrum. In addition, inductively coupled plasma atom emission spectrometry (ICP-AES) analysis shows that the total Ru content is 0.068 wt%.
 | ||
| Fig. 3 (a and b) SEM image of CNT and carbonized CNT; (c–h) SEM, TEM, HRTEM and HAADF-STEM of Ru@CNT-500 and corresponding EDS element map. ((c) is SEM; (d and e) is TEM; (f and g) is HRTEM, and (h) is HAADF-STEM and STEM-EDS diagram). | ||
The element composition, chemical state and molecular structure of Ru@CNT-500 surface were measured by XPS. Fig. 4a shows the full spectrum information of the sample Ru@CNT. The spectrum shows that there are four elements of C, N, O and Ru in the sample. Fig. 4b shows the high resolution XPS fitting map of C 1s. It can be seen that the C 1s orbitals include the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond at 284.38 eV, the C–C bond at 284.97 eV, the C–N bond at 286.15 eV and the CO bond at 288.57 eV. The C–C, C–C and C–N bonds are derived from the carbon skeleton of polypyrrole carbon nanotubes, and the existence of C–N bond proves that there is N atom doping in the carbon skeleton.29,30 The weak peak of CO bond is caused by the absorption of oxygen or carbon dioxide and the oxidation of a small amount of surface metal ruthenium particles.31 Three typical types of nitrogen elements can be observed in Fig. 4c, such as pyridine type at 398.17 eV, pyrrole type at 400.65 eV and graphite type at 404.37 eV, in which pyrrole nitrogen plays a dominant role in the sample. Fig. 4d shows the fitting spectrum of Ru 3d. The coexistence of Ru0 and Rux+ can be observed. The characteristic peaks of Ru0 3d5/2 and Ru0 3d3/2 are located in 280.37 eV and 282.68 eV corresponding to ruthenium, respectively. The characteristic peaks at 281.19 eV and 286.51 eV can be classified as other fitting peaks near Rux+ 3d5/2 and Rux+ 3d3/2287.0 eV. Because the Ru 3d orbital coincides with the carbon standard peak, it interferes with the detection of ruthenium.32 We further analyzed the Ru 3p orbital to determine the existence form and valence state of ruthenium nanoparticles. As shown in Fig. 4e, the peaks at 461.75 eV and 484.19 eV belong to the characteristic peaks of Ru0 3d5/2 and Ru0 3d3/2, and 464.54 eV and 487.21 eV are the characteristic peaks of Rux+ 3d5/2 and Rux+ 3d3/2. Rux+ is attributed to the oxidation of ruthenium particles on the sample surface. By comparing the peak area, it is found that although Ru0 and Rux+ coexist in the sample, Ru0 dominates.33
C bond at 284.38 eV, the C–C bond at 284.97 eV, the C–N bond at 286.15 eV and the CO bond at 288.57 eV. The C–C, C–C and C–N bonds are derived from the carbon skeleton of polypyrrole carbon nanotubes, and the existence of C–N bond proves that there is N atom doping in the carbon skeleton.29,30 The weak peak of CO bond is caused by the absorption of oxygen or carbon dioxide and the oxidation of a small amount of surface metal ruthenium particles.31 Three typical types of nitrogen elements can be observed in Fig. 4c, such as pyridine type at 398.17 eV, pyrrole type at 400.65 eV and graphite type at 404.37 eV, in which pyrrole nitrogen plays a dominant role in the sample. Fig. 4d shows the fitting spectrum of Ru 3d. The coexistence of Ru0 and Rux+ can be observed. The characteristic peaks of Ru0 3d5/2 and Ru0 3d3/2 are located in 280.37 eV and 282.68 eV corresponding to ruthenium, respectively. The characteristic peaks at 281.19 eV and 286.51 eV can be classified as other fitting peaks near Rux+ 3d5/2 and Rux+ 3d3/2287.0 eV. Because the Ru 3d orbital coincides with the carbon standard peak, it interferes with the detection of ruthenium.32 We further analyzed the Ru 3p orbital to determine the existence form and valence state of ruthenium nanoparticles. As shown in Fig. 4e, the peaks at 461.75 eV and 484.19 eV belong to the characteristic peaks of Ru0 3d5/2 and Ru0 3d3/2, and 464.54 eV and 487.21 eV are the characteristic peaks of Rux+ 3d5/2 and Rux+ 3d3/2. Rux+ is attributed to the oxidation of ruthenium particles on the sample surface. By comparing the peak area, it is found that although Ru0 and Rux+ coexist in the sample, Ru0 dominates.33
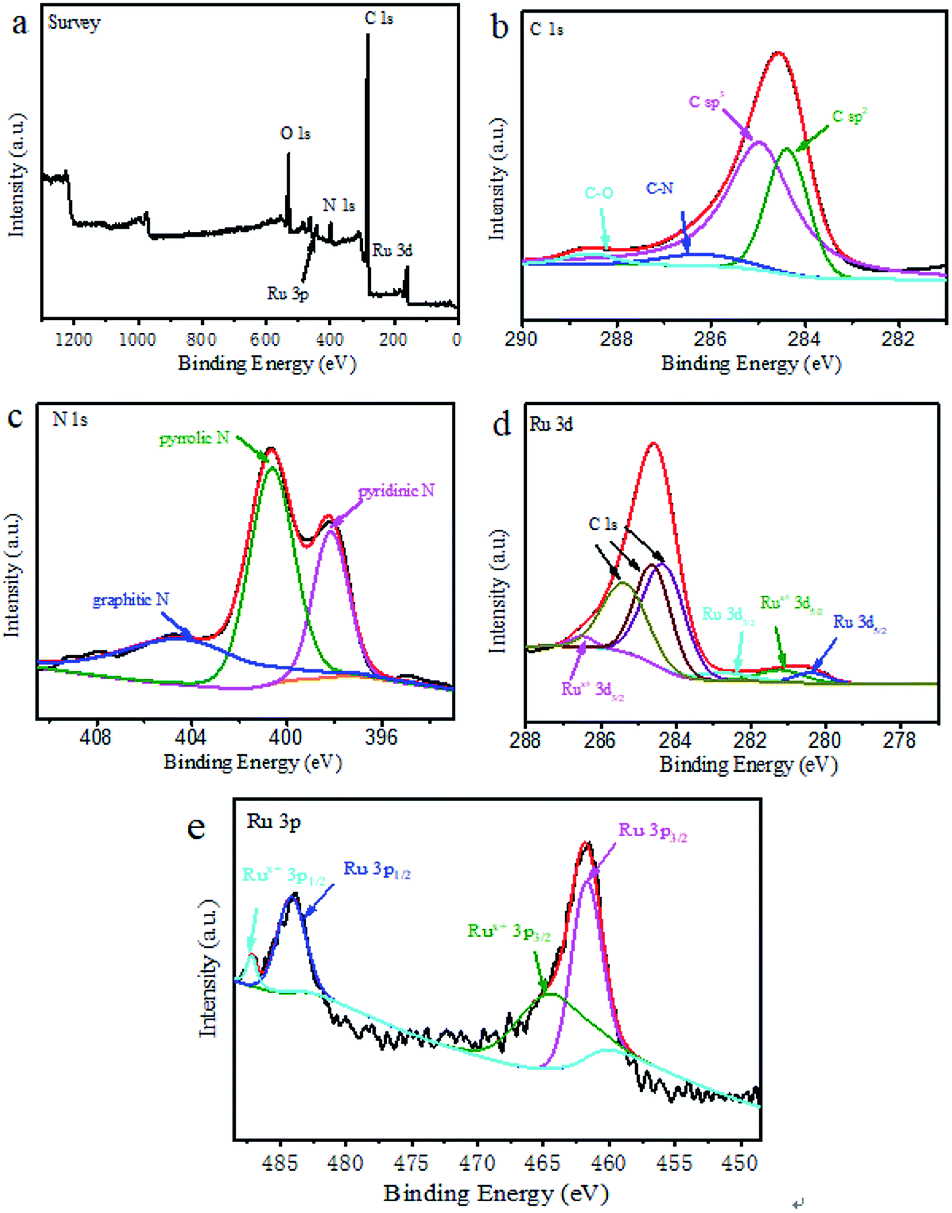 | ||
| Fig. 4 (a) XPS full spectrum of Ru@CNT-500; (b, c, d and e) are the high-resolution spectra of C 1s, N 1s, Ru 3d, Ru 3p of sample Ru@CNT-500. | ||
The XPS analysis of all Ru@CNT samples showed that the Ru content of Ru@CNT-400 was 1.76%, the Ru content of Ru@CNT-500 was 2.1%, the Ru content of Ru@CNT-600 was 1.6%, the Ru content of Ru@CNT-700 was 1.45%, and the Ru content of Ru@CNT-800 was 1.79%. Through the comparative analysis of Ru 3d peaks of all Ru@CNT samples (Fig. S2†), the Ru 3d peak of Ru@CNT samples moves to the direction of bond energy decreasing with the increase of calcination temperature, indicating that the proportion of Ru0 in total Ru in Ru@CNT samples increases with the increase of calcination temperature. The Ru contents of Ru@CNT-400 (1.76%) and Ru@CNT-800 (1.79%) are similar, but the catalytic performance of Ru@CNT-800 is better, which further indicates that Ru0 is the main contributor to the catalytic activity. The fitting analysis of Ru 3p peaks of all Ru@CNT samples can be seen (Fig. S3†). The higher the content of Ru0, the better the catalytic performance of Ru@CNT, which is consistent with the results of electrocatalytic performance of Ru@CNT samples.
The specific surface area and pore channel information of Ru@CNT-500 were analyzed by N2 adsorption and desorption method.34 Fig. 5a shows that the isotherm shows a typical type IV curve. According to the BET method, the specific surface area of the sample is 15.875 m3 g−1, and Fig. 5b shows the pore size distribution of the sample calculated by BJH method. It can be seen that the sample mainly includes two types: mesoporous and macroporous. The average pore volume and pore size calculated by BJH method are 0.131 cm3 g−1 and 57.417 nm, respectively, which are consistent with the results of SEM and TEM analysis. The detailed analysis of the adsorption isotherm shows that when the relative pressure P/P0 < 0.1, the isotherm increases slightly, indicating that the sample Ru@CNT-500 has a small amount of micropores, which can be attributed to the defects caused by nitrogen doping on CNTs. With the increase of P/P0, the isotherm suddenly increased in the high-pressure region (P/P0 ≥ 0.8), and the isotherm appeared obvious hysteresis ring,35 indicating that the sample has porous structure and the main pore size distribution is about 57 nm. The macropores can be used as the storage space of electrolytes. The existence of mesoporous pores shortens the diffusion path between electrolytes and ions, and the high specific surface area increases the contact between electrolytes and electrode materials and improves the ability of electron transport.
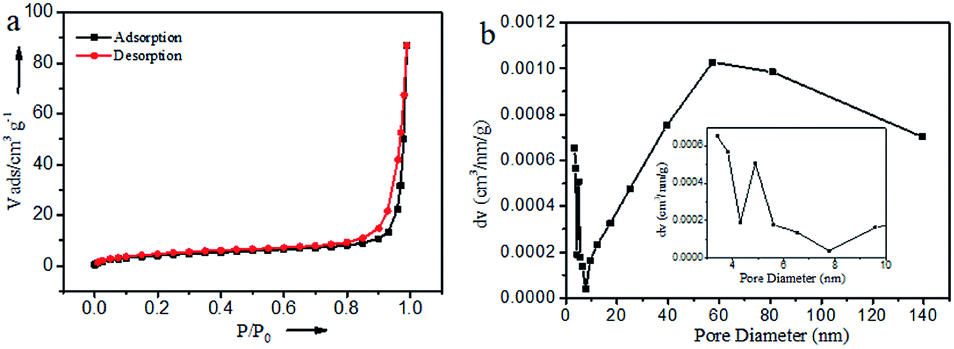 | ||
| Fig. 5 N2 adsorption/desorption isotherms and the BJH pore-size distribution curves of Ru@CNT-500. | ||
Electrocatalytic hydrogen evolution performance
In order to test the effect of different annealing temperature on the electrochemical properties of Ru@CNT samples, the HER properties of Ru@CNT-800, Ru@CNT-700, Ru@CNT-600, Ru@CNT-500 and Ru@CNT-400 were tested under alkaline conditions. From the polarization curve in Fig. 6a and Table 1, it can be seen that when the annealing temperature is 500 °C (sample Ru@CNT-500), the sample shows better HER properties. When the current density reaches 10 mA cm−2, the overpotential of Ru@CNT-500 is 36.69 mV, which is similar to that of commercial Pt/C. In order to explore the reaction mechanism of samples at different calcination temperatures, Tafel analysis was carried out. From the Tafel slope diagram (Fig. 6b), it can be seen that under alkaline conditions, the sample has excellent Tafel activity, especially the sample Ru@CNT-500 reaches 28.82 mV dec−1, is better than Pt/C (33.28 mV). It can be concluded that under alkaline conditions, the HER reaction of the sample Ru@CNT-500 follows the Volmer-Tafel reaction mechanism, in which the Tafel reaction (the binding of adsorbed hydrogen ions) is the rate control step.36 | ||
| Fig. 6 HER polarization curves (a) and Tafel plots (b) for the Ru@CNT samples and Pt/C catalyst. (c) Current density as a function of the scan rate for Ru@CNT samples. (d) Nyquist curves for the Ru@CNT samples and Pt/C catalyst at η = 10 mV. (e) Time dependent current density plot at potential of 37 mV. | ||
| Sample | Alkaline media (pH = 14) η10 (mV vs. RHE) |
|---|---|
| Ru@CNT-800 | 39.67 |
| Ru@CNT-700 | 78.11 |
| Ru@CNT-600 | 50.22 |
| Ru@CNT-500 | 36.69 |
| Ru@CNT-400 | 53.68 |
| 20% Pt/C | 30.68 |
In order to study the electrochemical active surface area of the sample, the cyclic voltammetry of the sample at different calcination temperature was measured at different scanning speed to obtain the double layer capacitance of the sample. The cyclic voltammetry curve was measured between 0.29 V and 0.39 V (vs. RHE). The scanning rate began with 20 mV s−1, then increased at the rate of 20 mV s−1, and finally to 100 mV s−1. Under alkaline conditions, the double layer capacitances of Ru@CNT-800, Ru@CNT-700, Ru@CNT-600, Ru@CNT-500 and Ru@CNT-400 are 3.890 mF cm−2, 5.195 mF cm−2, 4.900 mF cm−2, 6.600 mF cm−2 and 3.640 mF cm−2, respectively (Fig. 6c). It is known that the double layer capacitance of the electrocatalyst is proportional to the effective catalytic activity area of the catalyst, so the sample Ru@CNT-500 calcined at 500 °C has a larger effective catalytic activity area. That is to say, it has higher surface active site and higher HER current density, so its electrocatalytic hydrogen evolution performance is better than that of other samples.
In order to study the effect of calcination temperature on electrochemical properties, the electrochemical impedance spectroscopy of Ru@CNT samples was measured under alkaline conditions as shown in Fig. 6d. The equivalent circuit model shown in Fig. 6d is used to fit,37 where Rs is the solution resistance, Rct is the charge transfer resistance, and CPE represents the equivalent capacitance element. The charge resistance of Ru@CNT-500 calcined at 500 °C is the smallest, the charge transfer rate is the fastest, and the impedance spectrum shows the semicircle with the smallest radius. The results show that in the HER process, the sample Ru@CNT-500 has faster interface electron kinetics and higher charge transfer ability than commercial Pt/C. This can be attributed to the one-dimensional structure of hollow carbon nanotubes and the small and uniformly dispersed Ru nanoparticles increase the contact area with the electrolyte and increase the charge transfer rate, which is consistent with the electrochemical catalytic hydrogen evolution performance. It is also found that RCT increases with the increase of temperature, which is caused by the agglomeration of metal nanoparticles. The stability of electrocatalyst is another important criterion for evaluating electrocatalyst. Fig. 6e shows the constant voltage curve of Ru@CNT-500 under overpotential when the current density is 10 mA cm−2. It can be seen from the curve that after 10 hours of testing, the current density is still about 80% of the initial value (the current density increases from −10 mA cm−2 to −7.9 mA cm−2), indicating that the sample has good stability.38 Meanwhile, its durability was investigated by scanning 2000 cycles at a scanning rate of 5 mV s−1. As shown in Fig. S4,† there is a slight decline in the performance of the Ru@CNT-500 before and after 2000 cycles. This weak attenuation mainly occurred in the first 20 minutes of the test, which was caused by the shedding of a small amount of unstable Ru nanoparticles on the surface of carbon nanotubes, and showed good stability in the subsequent tests.
Experiment
Catalysts preparation
All chemicals are reagent grade and used without further purification.Preparation of CNT
Methyl orange (0.784 g, 2.4 mmol) was dissolved in 480 mL deionized water, and then pyrrole (0.838 g, 12.5 mmol) and FeCl3·6H2O (6.48 g, 25.3 mmol) were slowly added to the above solution under stirring. Their mixture will produce black precipitates. After aging for 24 hours, the precipitates were washed with water and ethanol for several times and dried in air at 80 °C for 24 hours. Finally, under the protection of nitrogen, the precipitates were carbonized at 800 °C for 3 h and the heating rate was 4 °C min−1.Preparation of Ru@CNT
The carbonized 50 mg CNT was dispersed in 10 mL (15.30 mmol L−1) RuCl3 aqueous solution, ultrasonic for 1 h, static for 24 h, filtered, washed with deionized water and dried in vacuum at room temperature. The dried samples were calcined twice at different temperatures, and the ruthenium elements were reduced and anchored at 800 °C (Ru@CNT-800), 700 °C (Ru@CNT-700), 600 °C (Ru@CNT-600), 500 °C (Ru@CNT-500) and 400 °C (Ru@CNT-400).Preparation of working electrodes
First, the 3.0 mg sample powder was dispersed with 30 μL 5 wt% Nafion solution in 1 mL deionized water/isopropanol (v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixed solvent, and the mixed solution was treated by ultrasonic for 30 minutes. Then, the 5 μL catalyst ink was loaded on the surface of the glass carbon electrode (GCE: diameter = 3 mm) with a liquid transfer gun, and the working electrode of the supported catalyst was obtained by natural drying.
:1) mixed solvent, and the mixed solution was treated by ultrasonic for 30 minutes. Then, the 5 μL catalyst ink was loaded on the surface of the glass carbon electrode (GCE: diameter = 3 mm) with a liquid transfer gun, and the working electrode of the supported catalyst was obtained by natural drying.
Characterization of catalysts
The crystal structure and phase composition of the catalyst were qualitatively and quantitatively analyzed by XRD (X'Pert Pro Cuk alpha X-ray diffractometer, test conditions: the tube current and voltage are set to 40 mA and 40 kV, respectively, and the scanning rate is 5° min−1 in the 5–90° 2θ range). The morphology of the samples was observed by scanning electron microscopy (SEM, SIGMA HD, acceleration voltage 5000 V) and transmission electron microscopy (TEM, JEM2100F, acceleration voltage 20000 V). The surface chemical composition of the samples was determined by X-ray photoelectron spectroscopy (XPS, AXIS Supra). The BET specific surface area of the sample was determined by the nitrogen adsorption–desorption isotherm of the 3H-2000PS1 analyzer at 77 K. Scattering Spectral Analysis of carbon Materials by Laser Raman Spectrometer (Raman HORIBA Scientific (Jobin), XploRA PLUS). The samples were measured by inductively coupled plasma atomic emission spectrometry (ICP-AES) and analyzed according to the characteristic lines obtained in order to obtain the types and contents of elements in the samples (Thermo Scientific, IRIS Intrepid II XSP). The electrocatalytic tests were carried out at room temperature using Dutch Ivium Vertex. One. EIS electrochemical workstation in a three-electrode system. Glassy carbon electrode (GCE, diameter: 3 mm), graphite rod and Ag/AgCl (3.5 M KCl) electrode were used as working electrode, opposite electrode and reference electrode, respectively. The electrolyte solution is 1 M KOH (alkaline, pH = 14). All electrochemical measurements were carried out at room temperature in N2 saturated atmosphere.
Conclusions
In summary, we have demonstrated a route to the facile fabrication of Ru@CNTs by a straightforward pyrolysis method. Ru@CNT-500 is an efficient electrocatalyst, which shows excellent catalytic behavior for HER in alkaline medium, and its catalytic activity is close to that of Pt/C. Equally important, the performance of the catalyst is better than that of Pt/C in terms of Tafel slope and electrochemical impedance. Ru is a Pt-group metal but its cost is only 4% that of Pt. Therefore, these insightful studies of Ru@CNT electrocatalysts provide clues for the exploration of electrocatalysts as alternatives to Pt catalysts in the field of energy conversion.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of Liaoning Province (No. 2019-ZD-0266).References
- K. Xu, et al., Adv. Mater., 2018, 30, 1703322 CrossRef PubMed.
- Y. Shi, et al., J. Am. Chem. Soc., 2017, 139(43), 15479–15485 CrossRef CAS PubMed.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 11741 CrossRef CAS PubMed.
- L. C. Seitz, et al., Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- J. Ryu, A. Wuttig and Y. Surendranath, Angew. Chem., Int. Ed., 2018, 57, 9300–9304 CrossRef CAS PubMed.
- V. Grozovski, et al., J. Electrochem. Soc., 2017, 164, E3171 CrossRef CAS.
- E. D. Carneiro-Neto, M. C. Lopes and E. C. Pereira, J. Electrochem. Soc., 2016, 765, 92–99 CAS.
- R. Subbaraman, et al., Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- N. M. Markovića, et al., J. Chem. Soc., Faraday Trans., 1996, 92, 3719–3725 RSC.
- W. C. Sheng, H. A. Gasteiger and S. H. Yang, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- J. W. Su, et al., Nat. Commun., 2017, 8, 14969 CrossRef PubMed.
- H. Zhang, M. S. Jin and Y. N. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- W. J. Mitchell, et al., J. Am. Chem. Soc., 1995, 117(9), 2606–2617 CrossRef CAS.
- D. W. Wang, et al., Appl. Catal., B, 2019, 249, 91–97 CrossRef CAS.
- Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- F. Li, et al., Adv. Mater., 2018, 30, 1803676 CrossRef PubMed.
- X. Liu, et al., Nanoscale, 2015, 7, 6136–6142 RSC.
- S. Y. Tee, et al., Nanotechnology, 2015, 26, 415401 CrossRef PubMed.
- S. W. Sun, et al., ACS Appl. Mater. Interfaces, 2019, 11, 19176–19182 CrossRef CAS PubMed.
- W. Y. Chen, et al., Sci. Bull., 2019, 64, 1095–1102 CrossRef.
- Z. L. Jin, Q. Y. Jian and Q. J. Guo, Int. J. Hydrogen Energy, 2019, 44, 26848–26862 CrossRef CAS.
- F. Li, et al., Adv. Mater., 2018, 30, e1803676 CrossRef PubMed.
- J. L. Dong, et al., Nano Lett., 2014, 14, 1228–1233 CrossRef PubMed.
- Z. L. Chen, et al., Adv. Mater., 2018, 30, e1802011 CrossRef PubMed.
- Q. Q. Yan, et al., Nat. Commun., 2019, 10, 4977 CrossRef PubMed.
- H. K. Sung, et al., Part. Part. Syst. Charact., 2019, 36, 1900090 CrossRef.
- J. Li, et al., J. Catal., 2019, 375, 351–360 CrossRef CAS.
- P. S. Li, et al., Small, 2019, 15, e1904043 CrossRef PubMed.
- M. Cheng, et al., Chem.–Eur. J., 2019, 25, 8579–8584 CrossRef CAS PubMed.
- X. Cheng, et al., ACS Sustainable Chem. Eng., 2018, 11487–11492 CrossRef CAS.
- M. H. Wei, et al., Energy Storage Materials, 2019, 17, 226–233 CrossRef.
- W. Wang, et al., Sci. Rep., 2014, 4, 4452 CrossRef PubMed.
- Z. L. Wang, et al., Angew. Chem., Int. Ed., 2018, 57, 5848–5852 CrossRef CAS PubMed.
- S. Ghosh and S. Ramaprabhu, J. Colloid Interface Sci., 2019, 559, 169–177 CrossRef PubMed.
- X. Tong, et al., RSC Adv., 2016, 6, 34261–34270 RSC.
- Y. G. Li, et al., J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- R. D. Armstrong and M. Henderson, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 81–90 CrossRef CAS.
- H.-Y. Chen, A.-J. Wang, L. Zhang and J.-J. Feng, ACS Appl. Energy Mater., 2019, 11, 7886–7892 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02894k |
| This journal is © The Royal Society of Chemistry 2020 |