
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn approach to new chiral bicyclic imines and amines via Horner–Wadsworth–Emmons reaction†
Jakub Iwanejkoa,
Mateusz Sowińskia,
Elżbieta Wojaczyńska *a,
Tomasz K. Olszewski*a and
Marcin Góreckib
*a,
Tomasz K. Olszewski*a and
Marcin Góreckib
aFaculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: elzbieta.wojaczynska@pwr.edu.pl; tomasz.olszewski@pwr.edu.pl
bInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka St 44/52, 01-224 Warsaw, Poland
First published on 9th April 2020
Abstract
New chiral bicyclic imines, enamines and amines were prepared via Horner–Wadsworth–Emmons reaction of hexahydroquinoxalin-2(1H)-one-derived phosphonate, as the source of a phosphonate carbanion, and a wide range of structurally diverse carbonyl substrates. The simplicity of the synthetic protocol, high selectivity, and broad substrate scope are the main advantages of the presented methodology.
Introduction
Chiral cyclic imines, particularly derivatives with five- and six-membered rings, have arisen as an important class of nitrogen containing heterocycles, useful as synthetic intermediates of drugs, agrochemicals and pharmacologically relevant compounds.1 Among the latest examples, Zhang et al. described the preparation of an enantioenriched cyclic imine using Bosch's chiral lactam; the imine was applied for the construction of an important component in the total asymmetric syntheses of drugs, (−)-rhynchophylline and (+)-isorhynchophylline.2 Wu et al. reported a rhodium-catalyzed asymmetric 1,2-addition of arylboronic acids to six-membered 1,2,6-thiadiazinane 1,1-dioxide-type cyclic imines to afford highly optically active sulfamides (95–99% ee).3 High diastereoselectivity and enantioselectivity was observed in a [4 + 2] reaction between cyclic sulfonic imine and Boc-protected indole aldehyde yielding polycyclic pyridoindoles.4 Aza-Darzens reaction of cyclic N-sulfonylimines with α-halogenated ketones provided a variety of tri- and tetrasubstituted aziridines, including benzofused heterocycles as well as spiro-structures in high yields, diastereo- and enantioselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and >99.9% ee).5 Among the known bicyclic heterocycles, the hexahydroquinoxaline-2(1H)-one derivatives represent an interesting new class of compounds with promising biological activity (Fig. 1),6 and can be considered as direct analogues of quinoxalin-2(1H)-ones, well known for their broad pharmacological properties e.g. inhibitors against Hepatitis C Virus (HCV) and potent candidates for antitumor therapeutics.7
:1 dr and >99.9% ee).5 Among the known bicyclic heterocycles, the hexahydroquinoxaline-2(1H)-one derivatives represent an interesting new class of compounds with promising biological activity (Fig. 1),6 and can be considered as direct analogues of quinoxalin-2(1H)-ones, well known for their broad pharmacological properties e.g. inhibitors against Hepatitis C Virus (HCV) and potent candidates for antitumor therapeutics.7
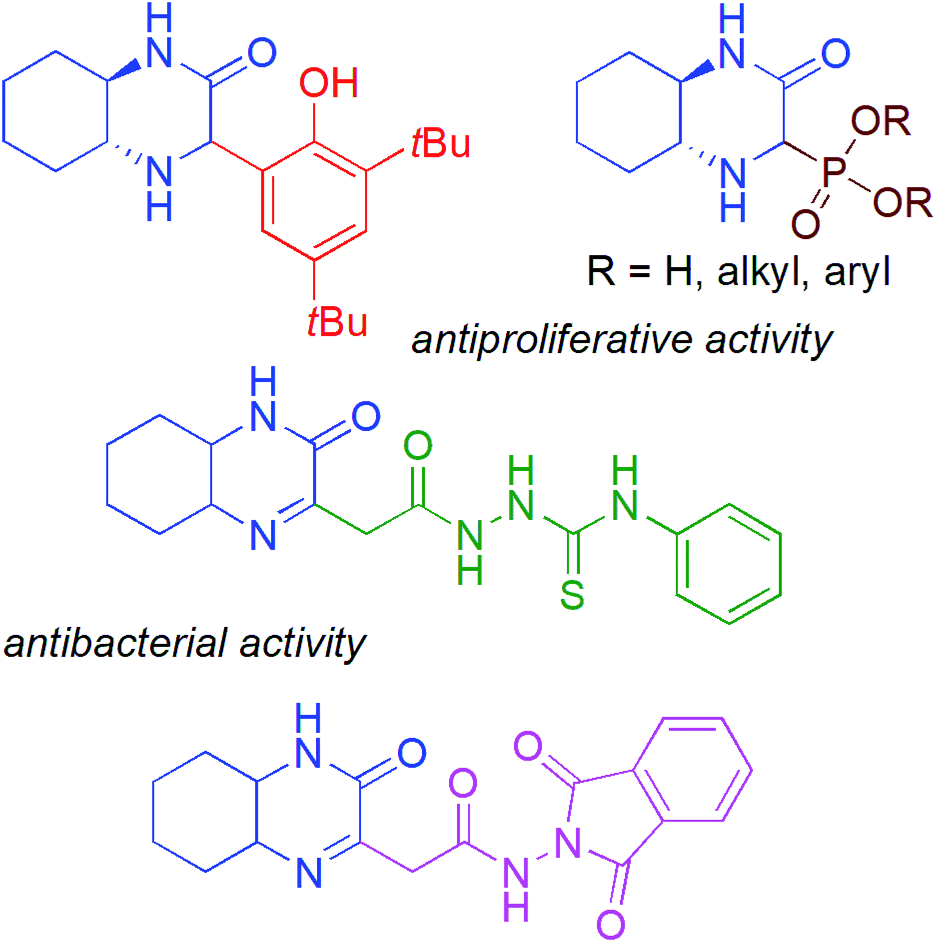 | ||
| Fig. 1 Examples of biologically active hexahydroquinoxaline-2(1H)-one derivatives. | ||
Surprisingly in spite of interesting biological properties, preparation of hexahydroquinoxaline-2(1H)-one derivatives still remain problematic and there are only few synthetic strategies that allow access to those molecules (Scheme 1). Unfortunately, each of them suffers from some drawbacks and limitations. The classical approach is based on condensation of cyclohexane-1,2-diamine with pyruvates (the Hinsberg reaction) (Scheme 1a).8a,b Unfortunately the chemical diversity using the latter methodology is limited by not always easy access to a wide range of pyruvates. Recently, a solid supported methodology was introduced by the group of Bräse using immobilized oxazolones in combination with difunctional nucleophiles and yielding heterocyclic products (Scheme 1b).8c This method, however, requires preparation in each case of new substituted oxazolone fragment and its subsequent introduction onto the resin which cannot be considered as straightforward process. Finally, palladium-catalyzed aminocarbonylation/cyclization synthetic strategy based on the use of carbon monoxide, cyclohexane-1,2-diamine and iodoindoles was introduced (Scheme 1c).8d The need of using metal catalyst as well as narrow substrates scope are among the major limitations of this method.
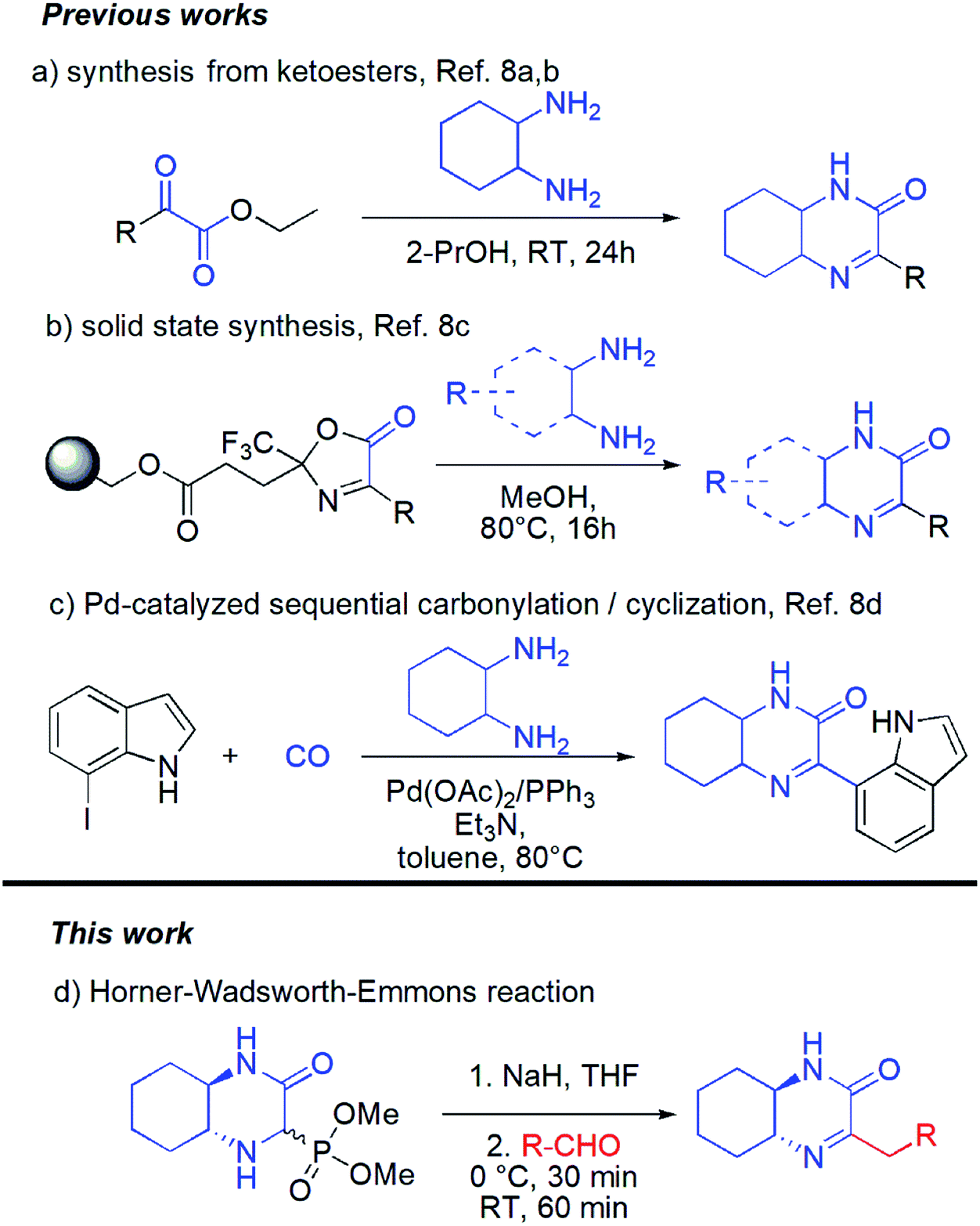 | ||
| Scheme 1 Possible routes to hexahydroquinoxaline-2(1H)-one derivatives. | ||
In continuation of our interest in preparation of new heterocyclic compounds,9 we have recently reported on the synthesis of chiral hexahydroquinoxalin-2(1H)-one-derived phosphonates and identified them as intermediates for the asymmetric synthesis of hexahydroquinoxalin-2(1H)-one derivatives, especially bicyclic nitrogen-containing phosphonic acids but also imines and enamines.10 Importantly, the latter application, in contrast to the known approaches toward the preparation of those heterocycles, allows for an easy modification of the substituents around the carbon–nitrogen bond thus providing a selective and simple way to access those valuable building blocks. Herein, we present a detailed study on the scope and limitations of the application of hexahydroquinoxalin-2(1H)-one-derived phosphonate, as the source of phosphonate carbanion, in the Horner–Wadsworth–Emmons reaction11 leading selectively to chiral heterocyclic imines, and amines as well as the use of the obtained compounds as building blocks in the preparation of other interesting bicyclic derivatives.
Results and discussion
The core intermediate in our approach, chiral hexahydroquinoxalin-2(1H)-one derived dimethyl phosphonate 2, can be easily accessed, even on a multigram scale, via a simple two-component phospha-Mannich reaction between chiral imine 1 and dimethyl H-phosphonate (Table 1).10a Subsequent Horner–Wadsworth–Emmons (HWE) reaction performed in the presence of NaH in THF at 0 °C with a variety of aldehydes gave after total 2 h reaction time an access to structurally diverse products as tautomeric mixture of imine and enamine in which, in most cases, the imine form (3) predominated (Table 1). Composition of reaction mixtures in each case could be unambiguously established by 1H NMR spectroscopy; measurements were performed after an extraction step on the crude products (see ESI† for details of experimental procedures).
|
|||||
|---|---|---|---|---|---|
| Entry | R | Imine, % | Enamine, % | Imine:enamine ratio |
Imine product |
| a Pure imine isolated. | |||||
| 1 | 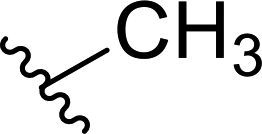 |
40 | 25 | 61:39 |
3a |
| 2 | 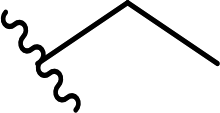 |
72a | 3 | 96:4 |
3b |
| 3 | 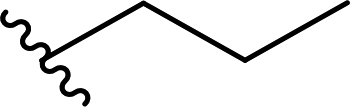 |
50 | 31 | 62:38 |
3c |
| 4 |  |
70a | 24 | 75:25 |
3d |
| 5 | 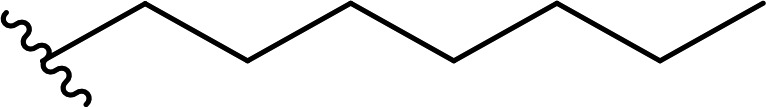 |
90a | 4 | 96:4 |
3e |
| 6 | 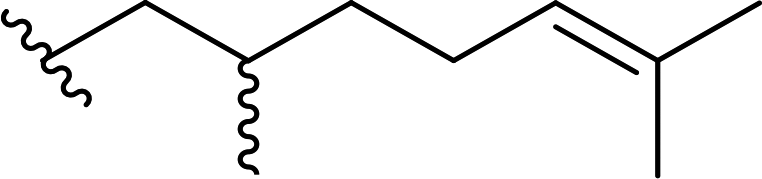 |
90 | 6 | 94:6 |
3f |
| 7 |  |
96a | 3 | 97:3 |
3g |
| 8 |  |
83a | 6 | 93:7 |
3h |
| 9 | 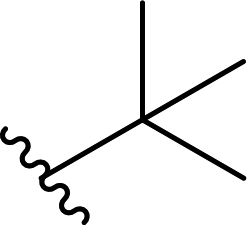 |
63a | <1 | 99:1 |
3i |
| 10 | 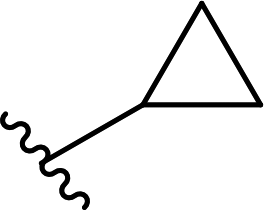 |
33 | 22 | 60:40 |
3j |
| 11 | 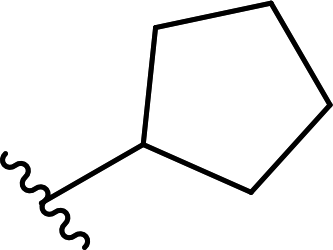 |
79a | 13 | 86:14 |
3k |
| 12 | 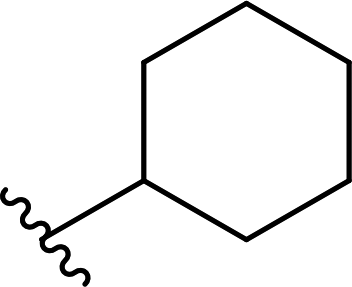 |
68a | 2 | 97:3 |
3l |
| 13 | 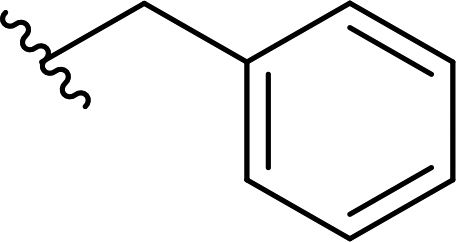 |
77a | 1 | 99:1 |
3m |
| 14 | 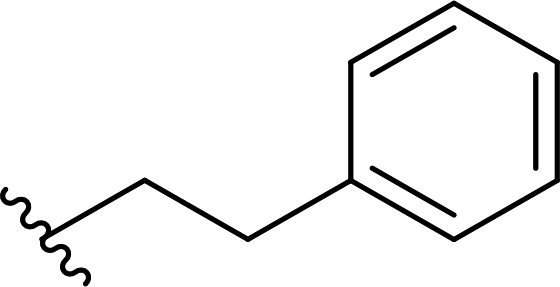 |
98a | 2 | 98:2 |
3n |
| 15 | 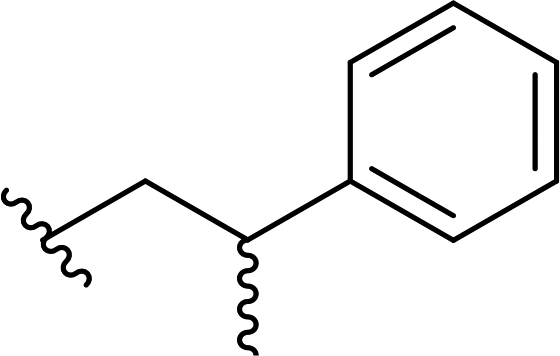 |
69a | 3 | 96:4 |
3o |
| 16 | 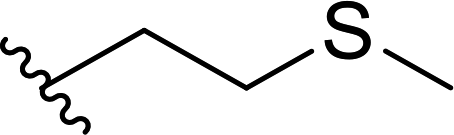 |
33 | 48 | 41:59 |
3p |
| 17 | 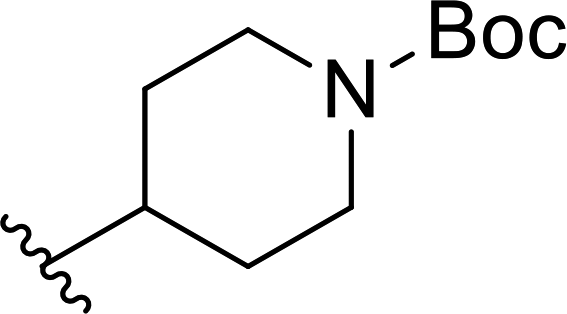 |
53 | 20 | 72:28 |
3r |
| 18 |  |
36 | 55 | 40:60 |
3s |
When simple aliphatic aldehydes, acetaldehyde and butyraldehyde (Table 1, entry 1 and 3) were used, imine:enamine ratio of ca. 3:2 was found. The use of reactants with longer aliphatic chains significantly improved the selectivity of the reaction, leading predominantly to stable imines with a chemical yield of up to 90% (Table 1, entries 5 and 6).
Application of branched aldehydes provided stable substituted imines with high selectivity and yield (entries 7–9). Importantly, the desired imines were also obtained in case of cyclic aldehydes (entries 10–12). It was observed that the selectivity of the HWE reaction increased with ring enlargement. The derivative with the cyclopropyl substituent proved to be unstable (Table 1 entry 10). While the use of benzaldehyde led mainly to enamine in 98% yield,10a benzyl aldehyde afforded practically only imine product in 77% yield (Table 1, entry 13). Similarly, high chemoselectivity was observed for other aldehydes bearing phenyl substituent at the alkyl chain (entries 14 and 15). Since citronellal and 3-phenylbutyraldehyde were used as racemic mixtures (entries 6 and 15), the resulting products bear an additional asymmetric center.
Lower selectivity was noted for aldehydes containing heteroatoms in their structure (Table 1, entries 16–18). The presence of sulfenyl group resulted in the reversed selectivity (imine:enamine ratio of ca. 2:3); also enantiomerically pure bicyclic aldehyde-2-azabicycloalkane derivative resulted in unstable unsaturated products, predominantly enamine (Table 1, entry 18).
The crude mixtures were separated using column chromatography on silica gel. In general, we observed that the mixtures of tautomers containing above 90% of imine form were much easier separable. Newly synthesized and isolated imines were found to be stable both in air atmosphere as well as in common organic solvents. On the other hand, mixtures containing at least 20% of enamine were inseparable neither by column chromatography nor crystallization, and were unstable. We observed slow decomposition of products (in solutions) into complex mixtures, and isolation of pure imine or enamine was not possible. However, immediate reduction of such reaction mixture obtained from HWE reaction could be performed yielding chiral amines (vide infra) (Table 2).
|
||||
|---|---|---|---|---|
| Entry | R | Yield, % | drc | Amines 5b |
| a A tautomeric imine/enamine mixture was used.b In each case major diastereomer was isolated in pure form and absolute configuration was established (see ESI for details).c The value of dr established based on the 1H NMR of crude reaction product.d Equimolar mixture of epimers. | ||||
| 1 |  |
98 | 81 (S):19 (R) |
5a |
| 3 |  |
85 | 61 (S):39 (R) |
5b |
| 4 | 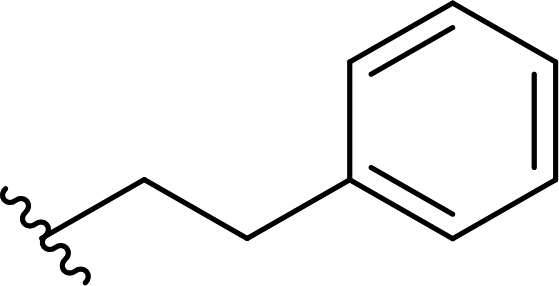 |
77 | 55 (R):45 (S) |
5c |
| 5a | 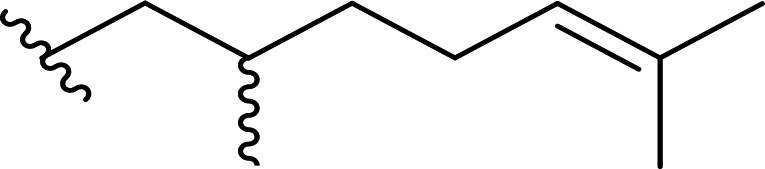 |
80 | 66 (R):34 (S) |
5dd |
| 6a | 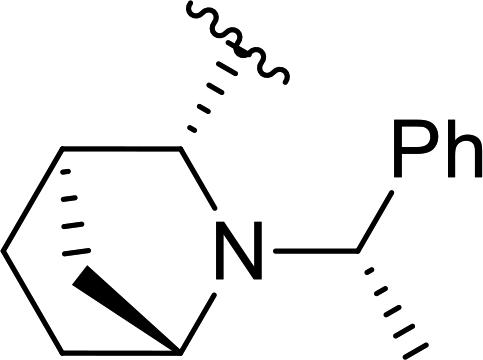 |
78 | 93 (S):7 (R) |
5e |
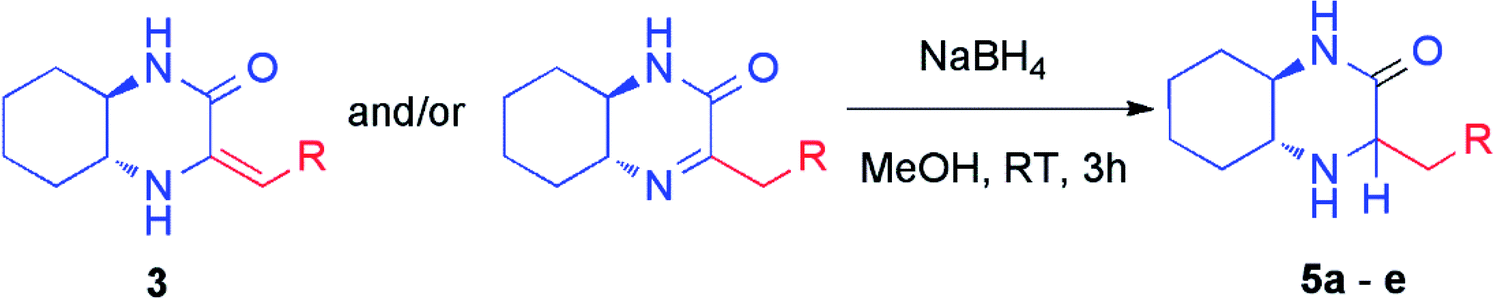
Additionally, HWE reaction was also performed with ketones. The use of acetophenone in the reaction with phosphonate 2 led to a complex mixture of olefination products that was reduced in situ and from the four possible diastereomeric products (two new stereogenic centers were formed) the pure main diastereomer was isolated in 32% overall yield (Scheme 2). The configuration of this product was determined on the basis of correlations found in 2D 1H NMR spectra (see ESI† for details). To our delight, a nearly quantitative reaction was observed with acetone. Unfortunately, imine and enamine were formed in practically equimolar amounts (53:47 ratio) and we found them to be inseparable. Again, subsequent reduction of the crude reaction mixture was performed leading to mixture of chromatographically separable epimeric amines (71:29 diastereomeric ratio) from which both diastereomers was isolated in pure form with 63% and 25% overall yields (Scheme 2).
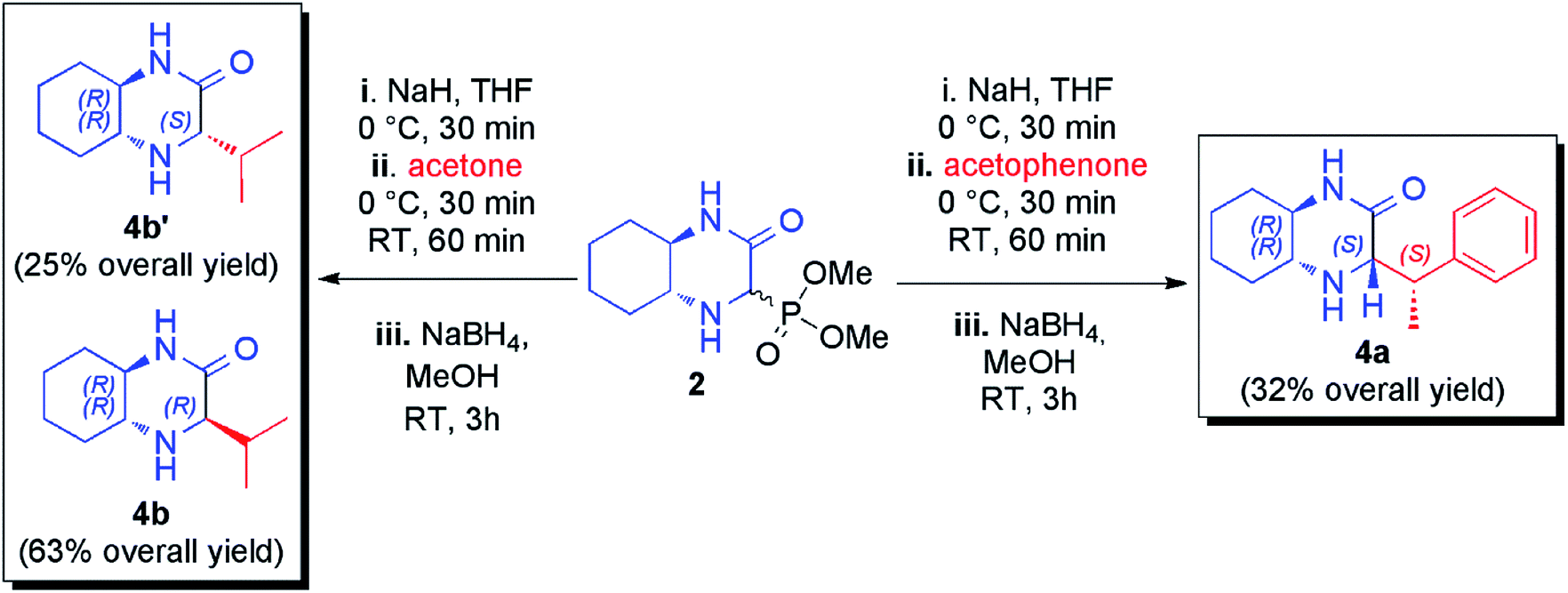 | ||
| Scheme 2 HWE reactions involving acetophenone and acetone. | ||
Subsequently, selected pure imines and also crude reaction mixtures containing imine and enamine were reduced with sodium borohydride (Table 2). An emphasis was put on the reduction of substrates bearing diverse substituents such as alkyl, cycloalkyl, alkylaryl groups as well as additional functional groups and heteroatoms. These reactions led to the formation of novel chiral amines with an additional stereogenic center. The resulting amines were purified by column chromatography and diastereomers were separated. Their configuration was established based on 2D NMR spectroscopy (Table 2). Reduction proceeded smoothly with a variable stereoselectivity which could be correlated with the steric hindrance exerted by the R substituent. This way, several enantiomerically pure amines 5a–e bearing three or more stereogenic centers were isolated with moderate to good diasteroselectivity. It is noteworthy that there was no external source of chirality used – we have observed an induction of diastereoselectivity based on (R,R)-diaminocyclohexane fragment.
Finally, we decided to demonstrate the utility of the obtained hexahydroquinoxaline-2(1H)-one derivate imines as chiral building blocks in asymmetric preparation of other molecules. The imine 3i was selected as model substrate (Scheme 3). Addition of phosphorus nucleophiles to imine 3i provided corresponding aminophosphonic acid 6e and aminophosphonate 6d in high yield and excellent diastereoselectivity. In turn, reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in 3i provided a mixture of epimeric amines 5a (ca. 4:1 ratio) and the major epimer of 5a could be separated by chromatography in 79% yield (Scheme 3). The latter was further subjected for reduction with lithium aluminum hydride to yield chiral cyclic diamine 6f in 67% yield (66% overall yield from 5a, Scheme 3). In turn, to introduce sulfur functionality, cyclic imine 3i and amine 5a were treated with Lawesson's reagent (Scheme 3). Thioamide 6b containing imine function was obtained with 63% yield from imine 3i and also from amine 5a, albeit in lower yield (34%). The reactivity of imine bond in this derivative was also checked. The aza-Pudovik reaction with dimethyl H-phosphonate appeared to proceed with a high diastereoselectivity comparable to 6d, but with a low chemical efficiency (6a, 6% yield, not isolated). Finally, reduction of 6b with sodium borohydride yielded amine 6c bearing thiolactam fragment that was isolated in the form of two diastereomers that were isolated in pure form with 51% and 24% yield respectively (Scheme 3).
N bond in 3i provided a mixture of epimeric amines 5a (ca. 4:1 ratio) and the major epimer of 5a could be separated by chromatography in 79% yield (Scheme 3). The latter was further subjected for reduction with lithium aluminum hydride to yield chiral cyclic diamine 6f in 67% yield (66% overall yield from 5a, Scheme 3). In turn, to introduce sulfur functionality, cyclic imine 3i and amine 5a were treated with Lawesson's reagent (Scheme 3). Thioamide 6b containing imine function was obtained with 63% yield from imine 3i and also from amine 5a, albeit in lower yield (34%). The reactivity of imine bond in this derivative was also checked. The aza-Pudovik reaction with dimethyl H-phosphonate appeared to proceed with a high diastereoselectivity comparable to 6d, but with a low chemical efficiency (6a, 6% yield, not isolated). Finally, reduction of 6b with sodium borohydride yielded amine 6c bearing thiolactam fragment that was isolated in the form of two diastereomers that were isolated in pure form with 51% and 24% yield respectively (Scheme 3).
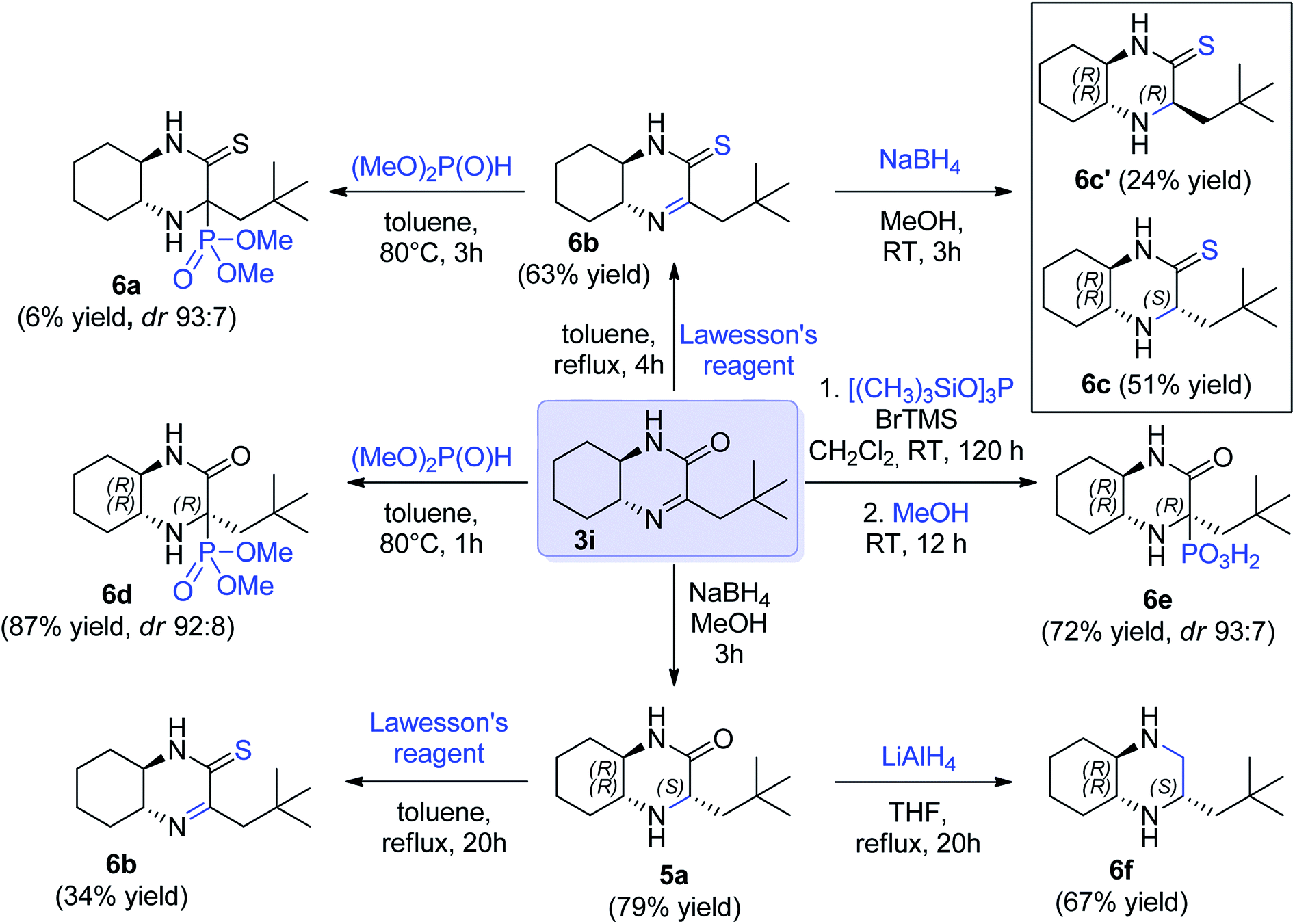 | ||
| Scheme 3 The utility of imine 3i as model substrate in further transformations. | ||
Determination of the absolute configuration of 6d and 6e
The assignment of absolute configuration (AC) of the main epimers 6d and 6e obtained in high diastereomeric excess was based on the comparison of their experimental and calculated electronic circular dichroism (ECD) spectra simulated using the time-dependent density functional theory (TDDFT). This combined methodology has illustrated its great efficiency in structural studies for many organic compounds having very different nature and origin.12 The ECD spectra of 6d and 6e were measured in acetonitrile. They exhibit very similar profile which is recognized by positive bands at ca. 240 and 200 nm, and negative one at ca. 220 nm (Fig. 2). This observation clearly indicates the same AC of newly formed chirality centre at C-4.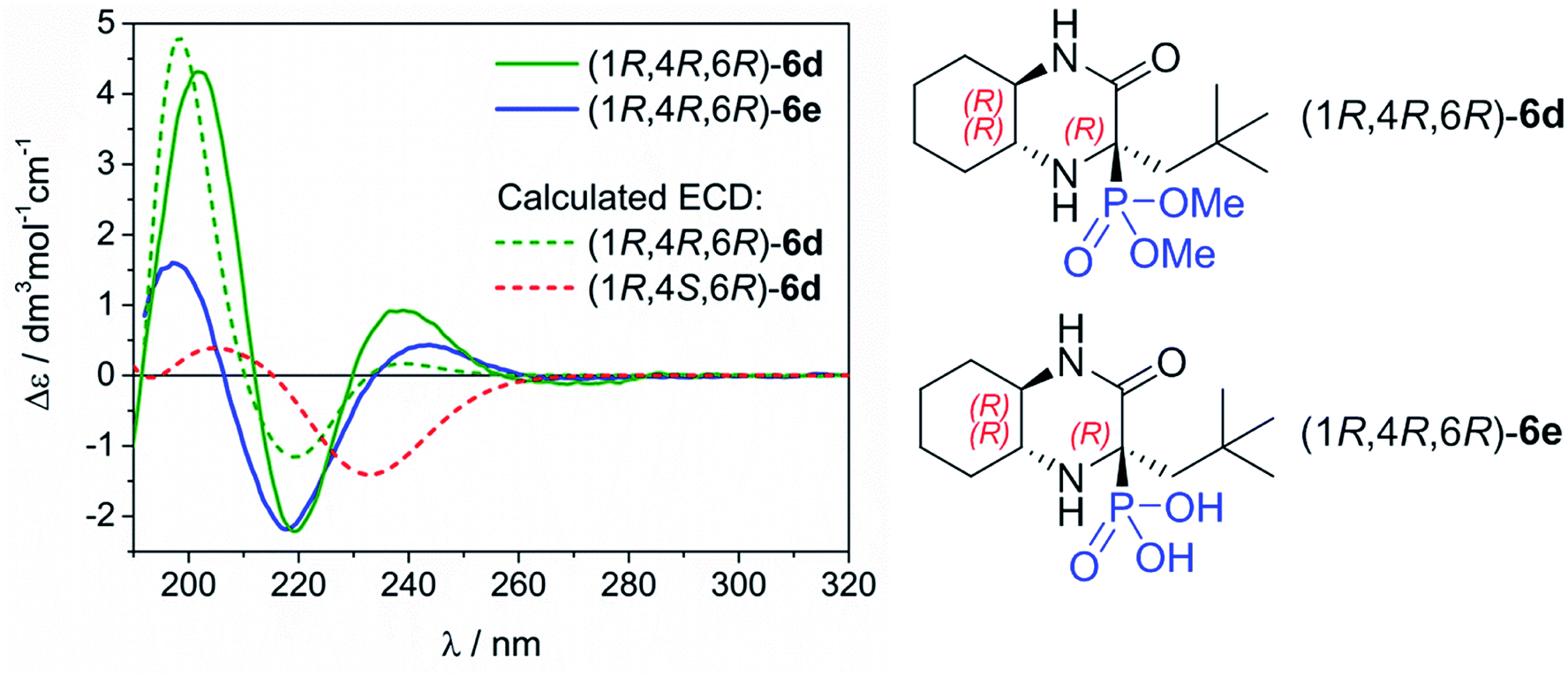 | ||
| Fig. 2 Experimental ECD spectra of the main epimers of 6d and 6e recorded in CH3CN compared to simulated spectrum for (1R,4R,6R)-6d and (1R,4S,6R)-6d using CAM-B3LYP/def2-TZVP/PCM(CH3CN) level of theory (UV shift = +17 nm, σ = 0.3 eV). | ||
To rationalize the experimental observations and assign the AC at C-4, the TDDFT calculations were performed for (1R,4R,6R)-6d using CAM-B3LYP/def2-TZVP level of theory including a polarizable continuum solvent model (PCM) for CH3CN. These simulations were preceded by a detailed conformational search at the molecular mechanic level, and further re-optimization of the resultant structures at the DFT level by using the following combination: ωB97X-D/6-311+G(d,p)/PCM/CH3CN. For computational details, see Experimental part and Table S1/Fig. S1 and S2 in ESI† with structures of the most abundant conformers and their ECD spectra. As can be seen in Fig. 2, the calculated TDDFT spectrum perfectly stays in line with the experimental spectra. On the strength of this, we can assign the AC of newly formed chirality centre for epimers obtained in high diastereomeric excess as 4R. At the same time, we confirmed that the AC at C-4 for the minor epimer is (4S). To improve this statement the same computational procedure was applied for simulating ECD spectrum of (1R,4S,6R)-6d. As a result, the obtained spectrum is contradictory to (1R,4R,6R)-6d, which further supports our assignment.
Conclusions
In summary, we have developed an efficient protocol based on the Horner–Wadsworth–Emmons reaction of easily available hexahydroquionoxalin-2(1H)-one-derived phosphonate and a wide range of carbonyl substrates leading to new diastereomerically pure bicyclic imines and amines. In most cases, reaction led selectively to the desired imines that were isolated in good to excellent yields. Subsequent reduction of the pure imines or tautomeric imine/enamine mixtures provided corresponding chiral amines in very good yields. We have also demonstrated that the obtained chiral heterocyclic compounds could be applied as useful synthetic intermediates in the synthesis of other useful derivatives in highly asymmetric manner and using simple transformations.Experimental section
General information
Melting points were determined on the Schmelzpunkt Bestimmer Apotec melting-point apparatus using the standard open capillary method and are uncorrected. 1H, 13C and 31P NMR spectra were collected on Jeol 400yh, Bruker Avance III 500 and Bruker Avance II 600 instruments. NMR spectra recorded in CDCl3, D2O and were referenced to the respective residual 1H or 13C signals of the solvents. The reported J values are those observed from the splitting patterns in the spectrum and may not reflect the true coupling constant values. NOESY experiments were carried out at 293 K. Infrared spectra (4000–400 cm−1) were collected on a PerkinElmer 2000 FTIR spectrophotometer. High resolution mass spectra were collected using electrospray ionization on Waters LCT Premier XE TOF instrument. Optical rotations were measured using an Optical Activity Ltd. Model AA-5 automatic polarimeter; [α]D values are given in 10−1 deg cm2 g−1. Chromatographic separations were performed on silica gel 60 (70–230 mesh). Thin layer chromatography was carried out using silica gel 60 precoated plates.General procedure A for synthesis of cyclic imines 3a–s
Sodium hydride (60% dispersion in mineral oil, 1.30 mmol, 52.0 mg, 1.30 equiv.) was dispersed in anhydrous THF (10 mL) under argon atmosphere. The mixture was cooled to 273 K in an ice bath and then dimethyl-[(1R,6R)-3-oxo-2,5-diazabicyclo[4.4.0]dec-4-yl]phosphonate 2 (1.10 mmol, 288 mg, 1.10 equiv.) was added. The mixture was stirred for 30 minutes. The aldehyde was added (1.00 mmol, 1.00 equiv.) and the reaction continued for 30 minutes in 273 K and then for 60 minutes in a room temperature. Reaction mixture was washed with Et2O (20 mL) and saturated NaHCO3 solution (20 mL). The organic layer was dried (Na2SO4), filtered and evaporated under reduced pressure. Depending on yield and purity of crude product, the imines were described without additional purification or purified by silica gel column chromatography (eluent:CH2Cl2/MeOH 97:3 v/v).
General procedure B for synthesis of amines 4a–b
Sodium hydride (60% dispersion in mineral oil, 1.30 mmol, 52.0 mg, 1.30 equiv.) was dispersed in anhydrous THF (10 mL) under argon atmosphere. The mixture was cooled to 273 K in an ice bath and then dimethyl-[(1R,6R)-3-oxo-2,5-diazabicyclo[4.4.0]dec-4-yl]phosphonate 2 (1.10 mmol, 288 mg, 1.10 equiv.) was added. The mixture was stirred for 30 minutes. The ketone (acetophenone or acetone) was added (1.00 mmol, 1.00 equiv.) and the reaction continued for 30 minutes in 273 K and then for 60 minutes in a room temperature. Reaction mixture was washed with Et2O (20 mL) and saturated NaHCO3 solution (20 mL). The organic layer was dried (Na2SO4), filtered and evaporated under reduced pressure. The crude product was then dissolved in methanol (10 mL) at room temperature and sodium borohydride (1.30 mmol, 49.2 mg, 1.30 equiv. was added). The mixture was stirred for 3 hours. Solvent was evaporated under reduced pressure and the residue was subjected to column chromatography (eluent:CH2Cl2/MeOH 95:5 v/v) to obtain pure amines.
General procedure C for synthesis of amines 5a–e and 6c
To a solution of an appropriate imine (1.00 mmol, 1.00 equiv.) in methanol (10 mL) at room temperature was added sodium borohydride (1.30 mmol, 49.2 mg, 1.30 equiv.). The mixture was stirred for 3 hours. Solvent was evaporated under reduced pressure and the residue was subjected to column chromatography (eluent:CH2Cl2/MeOH 95:5 v/v) to obtain pure diastereomers of amine.
Procedure D for the synthesis of 6b
To a solution of imine 3i (2.00 mmol, 444 mg, 2.00 equiv.) in toluene (15 mL), Lawesson's reagent (1.20 mmol, 485 mg, 0.600 equiv.) was added under an argon atmosphere. The reaction mixture was refluxed for 4 hours. Upon completion of the reaction (monitored by TLC), it was quenched with water (20 mL) and extracted with ethyl acetate (3 × 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (eluent:CH2Cl2/MeOH 98:2 v/v).
Procedure E for the synthesis of dialkyl aminophosphonic acid ester 6d and aminophosphonic acid 6e
Procedure E for the synthesis of dialkyl aminophosphonic acid ester 6d and aminophosphonic acid 6e can be found in the literature.10bProcedure F for reduction of 5a to bisamine 6f
To a solution of 5a (0.500 mmol, 112 mg, 1.0 equiv.) in anhydrous THF (5 mL) under argon atmosphere was added lithium aluminium hydride (0.750 mmol, 28.5 mg, 1.5 equiv.). The mixture was heated under reflux for 20 hours and then cooled down to room temperature. The reaction was quenched with addition of water (30 μL), NaOH (15% solution, 30 μL) and water (90 μL). Resulting suspension was filtered through Celite and washed with THF. The organic layer was dried (K2CO3), filtered and evaporated under reduced pressure and the resulting crude product was purified by column chromatography (eluent:CH2Cl2/MeOH 80:20 v/v).
:1; mp 93–95 °C; IR (KBr): 696, 760, 1369, 1452, 1493, 1628, 1684, 2858, 2927, 3027, 3062, 3195 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.13–7.28 (m, 5H), 5.96 (br. s, 1H), 2.91–3.00 (m, 2H), 2.74 (sext, J = 7.6 Hz, 1H), 2.50 (q, J = 6.1 Hz, 1H), 2.27–2.30 (m, 1H), 1.62–1.94 (m, 5H), 1.21–1.41 (m, 5H), 1.27 (dd, J = 7.0, 2.3 Hz, 3H), 13C NMR (100 MHz, CDCl3): δ 165.7, 158.5, 147.1, 128.4 (2C overlapped), 127.2 (2C overlapped), 126.0, 62.5, 54.3, 40.0, 34.9, 32.1, 31.9, 31.1, 25.3, 23.8, 22.3; HRMS (ESI-TOF) calcd for C18H24N2O [M + H]+ m/z: 285.1967, found: 285.1963.Full characteristic of compounds 3b, 3d and 3g can be found in our previous paper.10a
:1; IR (KBr): 812, 844, 1126, 1352, 1363, 1451, 1659, 2858, 2936, 3177, 3299, 3426 cm−1; 1H NMR (400 MHz, CDCl3, both isomers present): δ 6.03 (br. s, 2H), 5.05–5.10 (m, 2H), 3.42 (ddd, J = 10.0, 3.7, 2.8 Hz, 2H), 2.96–3.02 (m, 2H), 2.49–2.56 (m, 2H), 1.41–2.03 (m, 18H), 1.66 (s, 6H), 1.58 (s, 6H), 1.09–1.54 (m, 18H), 0.88 (d, J = 6.4 Hz, 3H), 0.878 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3, both isomers present): δ 173.3, 173.27, 131.2 (2C overlapped), 125.0, 124.9, 58.8, 58.77, 58.6 (2C overlapped), 53.3, 53.27, 37.2, 36.9, 34.2, 33.9, 32.6, 32.3, 31.6 (2C overlapped), 30.9 (2C overlapped), 30.5, 30.1, 25.8 (2C overlapped), 25.7, 25.5, 24.9 (2C overlapped), 23.8 (2C overlapped), 19.7, 19.5, 17.7 (2C overlapped); HRMS (ESI-TOF) calcd for C18H33N2O [M + H]+ m/z: 293.2593, found: 293.2603.:1; IR (KBr): 831, 1121, 1350, 1452, 1664, 2858, 2928, 3258, 3428 cm−1; 1H NMR (400 MHz, CDCl3, both isomers present): δ 6.08 (br. s, 2H), 5.04–5.08 (m, 2H), 3.49 (dd, J = 8.3, 3.4 Hz, 2H), 2.99–3.03 (m, 2H), 2.46–2.52 (m, 2H), 1.52–2.02 (m, 18H), 1.65 (s, 6H), 1.57 (s, 6H), 1.09–1.48 (m, 18H), 0.88 (d, J = 6.4 Hz, 3H), 0.87 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3, both isomers present): δ 171.8, 171.7, 131.21, 131.2, 124.93, 124.9, 59.8 (2C overlapped), 58.5 (2C overlapped), 57.8 (2C overlapped), 37.2, 36.9, 33.1, 33.0, 32.72, 32.7, 31.6 (2C overlapped), 30.4 (2C overlapped), 29.9 (2C overlapped), 25.8 (2C overlapped), 25.6 (2C overlapped), 24.7 (2C overlapped), 23.8 (2C overlapped), 19.6, 19.5, 17.8 (2C overlapped); HRMS (ESI-TOF) calcd for C19H33N2O [M + H]+ m/z: 293.2593, found: 293.2603.:7; IR (KBr): 557, 1081, 1207, 1261, 1345, 1470, 1601, 1651, 2949, 3051, 3180 cm−1; 1H NMR (400 MHz, CDCl3): δ 3.77–3.82 (m, 1H), 3.54–3.57 (m, 1H), 3.20 (s, 2H), 2.56 (dd, J = 15.9, 4.6 Hz, 1H), 1.51–2.05 (m, 5H), 1.16–1.30 (m, 2H), 0.92 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 167.9, 67.8 (d, J = 120.0 Hz), 57.0, 53.1, 48.9, 43.5, 30.3 (3C overlapped), 30.2, 26.8, 23.6, 22.4; 31P{1H} NMR (162 MHz, CDCl3): δ 9.9; HRMS (ESI-TOF) calcd for C13H26N2O4P [M + H]+ m/z: 303.1474, found: 303.1476.ECD measurement
ECD spectra were carried out using a Jasco J-815 spectrometer (Tokyo, Japan) at room temperature in spectroscopic grade CH3CN (6.0 × 10−4 M) in quartz cells with a path length 0.1, 0.2 or 1 cm. All spectra were measured using a scanning speed of 100 nm min−1, a step size of 0.2 nm, a bandwidth of 1 nm, a response time of 0.5 seconds, and an accumulation of 5 scans. The spectra were background-corrected using solvent recorded under the same conditions.Conformational search and simulations of ECD spectra
The conformational search was carried out using CONFLEX 7 software13 with the MMFF94s force fields within 10 kcal mol−1 energy window. Next, all the obtained structures were submitted to the Gaussian16 (version C.01) program for DFT re-optimisation at the B97XD/6-311+G(d,p) using PCM model for CH3CN. All conformers were confirmed to contain no imaginary frequencies. The final structures within 1.5 kcal mol−1 energy window were selected for subsequent simulations of ECD spectra. Boltzmann populations were calculated at 298 K. Theoretical ECD/UV spectra were simulated at the CAM-B3LYP/def2-TZVP level using the PCM model for CH3CN. Furthermore, the ECD spectra were calculated using aug-cc-pVDZ basis set with B3LYP functional; the obtained results are similar to CAM-B3LYP/def2-TZVP. Calculations were performed for the first 50 excited electronic states. Rotatory strengths were calculated using both the length and the velocity formalisms. The differences between these two were less than 5%, so only the velocity representations (Rvel) were taken into account. The UV correction was applied according the experimental data recorder under the same conditions as mentioned beforehand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. G. thanks the Wroclaw Centre for Networking and Supercomputing (WCSS) for the computational support.Notes and references
- (a) J. Iwanejko and E. Wojaczyńska, Org. Biomol. Chem., 2018, 16, 7296–7314 RSC; (b) E. Speich, L. Banfi, L. Moni, R. Riva, V. Rocca and A. Basso, Chem. Heterocycl. Compd., 2018, 54, 329–333 CrossRef CAS; (c) P. Capurro, L. Moni, A. Galatini, C. Mang and A. Basso, Molecules, 2018, 23, 2758–2763 CrossRef PubMed.
- Z. Zhang, W. Zhang, F. Kang, F. C. F. Ip, N. Y. Ip and R. Tong, J. Org. Chem., 2019, 84, 11359–11365 CrossRef CAS PubMed.
- C. Y. Wu and M. H. Xu, Org. Lett., 2019, 21, 5035–5039 CrossRef CAS PubMed.
- P. Zheng, S. Wu, C. Mou, W. Xue, Z. Jin and Y. R. Chi, Org. Lett., 2019, 21, 5026–5029 CrossRef CAS PubMed.
- J. Pan, J. H. Wu, H. Zhang, X. Ren, J. P. Tan, L. Zhu, H. S. Zhang, C. Jiang and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 7425–7430 CrossRef CAS PubMed.
- (a) O. I. El-Sabbagh, M. E. El-Sadek, S. M. Lashine, S. H. Yassin and S. M. El-Nabtity, Med. Chem. Res., 2009, 18, 782–797 CrossRef CAS; (b) J. Iwanejko, E. Wojaczyńska, J. Trynda, M. Maciejewska, J. Wietrzyk, A. Kochel and J. Wojaczyński, Tetrahedron, 2017, 73, 2276–2282 CrossRef CAS.
- (a) R. Liu, Z. Huang, M. G. Murray, X. Guo and G. Liu, J. Med. Chem., 2011, 54, 5747–5768 CrossRef CAS PubMed; (b) X. Xie, Y. Yan, N. Zhu and G. Liu, Eur. J. Med. Chem., 2014, 76, 67–78 CrossRef CAS PubMed; (c) L. Shi, H. Zhou, J. Wu and X. Li, Mini-Rev. Org. Chem., 2015, 12, 96–112 CrossRef CAS; (d) S. Tariq, K. Somakala and M. Amir, Eur. J. Med. Chem., 2018, 143, 542–557 CrossRef CAS PubMed; (e) Y. Ramli, A. Moussaif, K. Karrouchi and E. M. Essassi, J. Chem., 2014, 563406 Search PubMed.
- (a) S. N. Murthy, B. Madhav and Y. V. D. Nageswar, Helv. Chim. Acta, 2010, 93, 1216–1220 CrossRef CAS; (b) M. Periasamy, B. Venkanna, M. Nagaraju and L. Mohan, Synthesis, 2020, 52, 127–134 CrossRef CAS; (c) S. Gräßle, S. Vanderheiden, P. Hodapp, B. Bulat, M. Nieger, N. Jung and S. Bräse, Org. Lett., 2016, 18, 3598–3601 CrossRef PubMed; (d) L. Damas, R. M. B. Carrilho, S. C. C. Nunes, A. A. C. C. Pais, L. Kollár, M. Pineiro and M. M. Pereira, R. Soc. Open Sci., 2018, 5, 181140 CrossRef PubMed.
- (a) E. Wojaczyńska, J. Wojaczyński, K. Kleniewska, M. Dorsz and T. K. Olszewski, Org. Biomol. Chem., 2015, 13, 6116–6148 RSC; (b) T. K. Olszewski, E. Wojaczyńska, R. Wieczorek and J. Bąkowicz, Tetrahedron: Asymmetry, 2015, 26, 601–607 CrossRef CAS; (c) T. K. Olszewski, Tetrahedron: Asymmetry, 2015, 26, 393–399 CrossRef CAS; (d) E. Pięta, E. Podstawka-Proniewicz, B. Boduszek, T. K. Olszewski, M. Nattich-Rak and Y. Kim, Appl. Surf. Sci., 2015, 335, 167–183 CrossRef; (e) E. Podstawka-Proniewicz, E. Pięta, K. Zborowski, A. Kudelski, B. Boduszek, T. K. Olszewski, Y. Kim and L. M. Proniewicz, J. Phys. Chem. A, 2014, 118, 5614–5625 CrossRef PubMed; (f) P. Knapkiewicz, K. Skowerski, D. E. Jaskolska, M. Barbasiewicz and T. K. Olszewski, Org. Process Res. Dev., 2012, 16, 1430–1435 CrossRef CAS; (g) T. K. Olszewski and D. E. Jaskólska, Heteroat. Chem., 2012, 23, 605–609 CrossRef CAS; (h) A. Bulow, T. Meyer, T. K. Olszewski and M. Bols, Eur. J. Org. Chem., 2004, 323–329 CrossRef.
- (a) J. Iwanejko, A. Brol, B. Szyja, M. Daszkiewicz, E. Wojaczyńska and T. K. Olszewski, Tetrahedron, 2019, 75, 1431–1439 CrossRef CAS; (b) J. Iwanejko, A. Brol, B. Szyja, M. Daszkiewicz, E. Wojaczyńska and T. K. Olszewski, Org. Biomol. Chem., 2019, 17, 7352–7359 RSC.
- The Horner–Wadsworth–Emmons (HWE) reaction is widely used in organic synthesis among existing methodologies as one of the most reliable methods for the highly stereoselective olefination of carbonyl compounds with a broad substrate scope. Kobayashi et al. in a review described recent applications of the HWE reaction in natural product synthesis, highlighting its use for carbon chain elongation, coupling reactions of synthetic segments, ring-closing reactions, tandem reactions including HWE olefination, and asymmetric reactions: K. Kobayashi, K. Tanaka III and H. Kogen, Tetrahedron Lett., 2018, 59, 568–582 CrossRef CAS.
- (a) M. Woźnica, P. Kowalska, M. Łysek, M. Masnyk, M. Górecki, M. Kwit, F. Furche and J. Frelek, Curr. Org. Chem., 2010, 14, 1022–1036 CrossRef; (b) R. Kołodziejska, M. Górecki, J. Frelek and M. Dramiński, Tetrahedron: Asymmetry, 2012, 23, 683–689 CrossRef; (c) S. Qiu, E. De Gussem, K. Abbaspour Tehrani, S. Sergeyev, P. Bultinck and W. Herrebout, J. Med. Chem., 2013, 56, 8903–8914 CrossRef CAS PubMed; (d) G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS PubMed; (e) N. U. Rehman, H. Hussain, S. Al-Shidhani, S. K. Avula, G. Abbas, M. U. Anwar, M. Górecki, G. Pescitelli and A. Al-Harrasi, RSC Adv., 2017, 7, 42357–42362 RSC.
- (a) H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950–8951 CrossRef CAS; (b) H. Goto and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1993, 187–198 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02646h |
| This journal is © The Royal Society of Chemistry 2020 |