
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous niobia spheres with large surface area: alcothermal synthesis and controlling of their composition and phase transition behaviour†
Yoshitaka Kumabea,
Hitomi Tagaa,
Kai Kan abc,
Masataka Ohtani*abc and
Kazuya Kobiro*abc
abc,
Masataka Ohtani*abc and
Kazuya Kobiro*abc
aSchool of Environmental Science and Engineering, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: ohtani.masataka@kochi-tech.ac.jp; kobiro.kazuya@kochi-tech.ac.jp
bLaboratory for Structural Nanochemistry, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan
cResearch Center for Material Science and Engineering, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan
First published on 13th April 2020
Abstract
Submicron-sized niobia (Nb2O5) porous spheres with a high specific surface area (300 m2 g−1) and nano concave–convex surfaces were synthesized via a rapid one-pot single-step alcothermal reaction. Prolonged reaction time or high reaction temperatures resulted in a morphology change of Nb2O5 from amorphous sphere to rod crystals with hexagonal crystal phase. A similar alcothermal reaction yielded TiO2–Nb2O5 composite porous spheres, whose Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nb molar ratio was controlled by changing the precursor solution component ratios. A simple thermal treatment of amorphous TiO2–Nb2O5 porous spheres consisting of 1:2 (molar ratio) Ti:Nb at 600 °C for 2 h induced crystal phase transfer from amorphous to a monoclinic crystal phase of submicron-sized TiNb2O7 porous spheres with a specific surface area of 50 m2 g−1.
:Nb molar ratio was controlled by changing the precursor solution component ratios. A simple thermal treatment of amorphous TiO2–Nb2O5 porous spheres consisting of 1:2 (molar ratio) Ti:Nb at 600 °C for 2 h induced crystal phase transfer from amorphous to a monoclinic crystal phase of submicron-sized TiNb2O7 porous spheres with a specific surface area of 50 m2 g−1.
Introduction
Porous materials, including activated carbon, SiO2, metal oxides, zeolites, and metal–organic frameworks (MOFs), are promising functional materials because of their large surface area that can be used to adsorb several materials and achieve material storage-release for material transfer. These unique properties allow for applications such as gas storage and separation, catalysis, and catalyst supports.1 In particular, metal oxides are intriguing because of their intrinsic high-temperature stability and durability.2To obtain porous materials, several methods, including solution combustion,3 dealloying,4 electrochemical deposition,5 spray pyrolysis,6 sol–gel method,7 as well as hydrothermal and solvothermal methods,8 have been developed. In particular, the solvothermal method using high-temperature non-aqueous solvents as reaction media is a novel approach to prepare size-, morphology-, and stoichiometry-controlled metal oxides. In this context, a unique one-pot and single-step solvothermal method was developed to afford SiO2, TiO2, ZnO, ZrO2, and CeO2 with porous spherical morphologies, where numerous few nm-sized primary particles aggregate to yield a spherical secondary morphology with a few hundred nm in diameter.9 These porous spheres were named meso/macroporously architected roundly integrated metal oxides (MARIMOs). The large surface areas of MARIMOs with micro- and mesopores and nano concave–convex surface structure impart their unique abilities. Both solid and hollow MARIMOs can be obtained with TiO2. In addition, composite MARIMOs, including CeO2–ZrO2,10 Al2O3–TiO2,11 ZnO–TiO2,11 SiO2–TiO2,12 Co/Mn oxide,13 Ni/Mn oxide,13 and Co/Mn/Fe oxide13 have been prepared via the solvothermal method with tightly controllable stoichiometry. Thus, solvothermal method is a versatile approach for obtaining metal oxides with a porous spherical structure, large surface area, and nano concave–convex surface structure.
Nb2O5 has been used as a photocatalyst,14,15 solid acid,16 catalyst support,17,18 and anode material for lithium and sodium ion batteries.19 Several preparation methods are available for these materials.20 For example, Li et al. prepared monodisperse glycolated niobium oxide spheres via a simple antisolvent precipitation approach. Mesoporous niobium oxide spheres with a high specific surface area of 312 m2 g−1 were obtained by hydrothermal treatment.21 Li et al. synthesized Nb2O5 hollow nanospheres by heating a water solution of niobium oxalate and sodium dihydrogen phosphate dihydrate at 220 °C for 48 h.22 Dai et al. prepared monodispersed Nb2O5 microspheres via a facile one-pot solvothermal route using an isopropyl alcohol solution of niobium pentachloride and tetrabutyl ammonium hydroxide at 200 °C for 24 h.23 However, simpler preparation methods should be developed for further acceleration of their practical uses. Thus, niobium oxide and variants of MARIMOs with large surface area were prepared herein using a facile synthesis method for Nb2O5, TiO2–Nb2O5 composite, and TiNb2O7 MARIMOs.
Results & discussion
Synthesis and morphology changes of Nb2O5 MARIMO
Nb2O5 MARIMO was prepared according to a similar method reported previously (entry 1 in Table 1).9 Simple heating of a methanol solution of Nb(OEt)5 and formic acid at 300 °C for 10 min afforded Nb2O5 MARIMO in high yield (>90%). The scanning electron microscopy (SEM) images revealed that the obtained Nb2O5 MARIMO was perfectly monodispersed (Fig. 1a(i)), and their shape was approximately spherical with a diameter of 630 ± 50 nm (Fig. 1a(ii), 1b(i) and S1,† red line). Also, the obtained Nb2O5 MARIMO was composed of many amounts of a few nm sized primary particles (Fig. 1b(ii)). The formation mechanism of Nb2O5 MARIMO was expected to be similar to a proposed mechanism in our previous report.9 In the initial steps of the reaction, primitive niobium alkoxide oligomers are formed in high-temperature methanol and interconnected via surface interactions with carboxyl groups. As the further heating reaction of precursor mixture proceeds, niobium alkoxide oligomers react with the water, which generated by esterification of formic acid and methanol and/or dehydration condensation of methanol, to form Nb2O5. The pore structures are formed by gaseous products, such as CO2 and hydrocarbons generated by decomposition of alkoxide and formic acid, trapped inside the sphere. The Nb2O5 MARIMO obtained without formic acid was polydisperse and obtained in low yield (<40%; Fig. S2a†). In addition, when the solvent was changed to water, spherical shapes were not formed (Fig. S2b†). From the results of the control experiment, the combination of Nb(OEt)5, formic acid, and methanol is essential for the formation of a spherical morphology and high yield.| Entry | Sample name of MARIMOa | Molar ratio of Ti and Nb | Specific surface areac [m2 g−1] | Pore sized [nm] | TiO2 crystalline sizee [nm] | |
|---|---|---|---|---|---|---|
| Precursor solution | Productb | |||||
| a Denotes the atomic% of Nb to total number of Nb and Ti atoms.b Evaluated by STEM/EDX analysis.c BET method.d BJH plot.e Estimated using the Scherrer equation. | ||||||
| 1 | Nb2O5 | 0:100 |
— | 302 | 3.3 | — |
| 2 | TiO2–Nb2O5-75 | 25:75 |
21.7:78.3 |
274 | 5.9 | 34.7 |
| 3 | TiO2–Nb2O5-50 | 50:50 |
50.1:49.9 |
200 | 8.9 | 13.6 |
| 4 | TiO2–Nb2O5-25 | 75:25 |
74.3:25.7 |
250 | 3.4 | 10.0 |
| 5 | TiO2 | 100:0 |
— | 234 | 3.6 | 9.6 |
 | ||
| Fig. 1 (a) SEM and (b) TEM images of the prepared Nb2O5 MARIMO at (i) low magnification and (ii) high magnification. | ||
The direct evidence of the formation of a pore in Nb2O5 spheres was confirmed by nitrogen adsorption–desorption measurements (Fig. S3a(i)†). If the obtained Nb2O5 MARIMO has no pores, the specific surface area should be a few m2 g−1 estimated from the diameter (ca. 630 nm) and specific density (d = 4.6 g cm−3). Contrary to this expectation, the obtained Nb2O5 MARIMO showed a high specific surface area (302 m2 g−1) and porous structure less than 10 nm-sized (Fig. S3a(ii)†). This fact strongly supports the presence of the penetrated pore inside the spherical particles.
To further characterization of the obtained Nb2O5 MARIMO, powder X-ray diffraction (XRD) and electron microscopic measurements were performed. No peaks were observed in the corresponding XRD pattern (Fig. 2c, red line), and no fringe structure in the Nb2O5 MARIMO primary particle was observed in the high-resolution transmission electron microscopy (HR-TEM) image (Fig. 1b(ii)). This indicated that the obtained Nb2O5 MARIMO was not crystalline but amorphous.
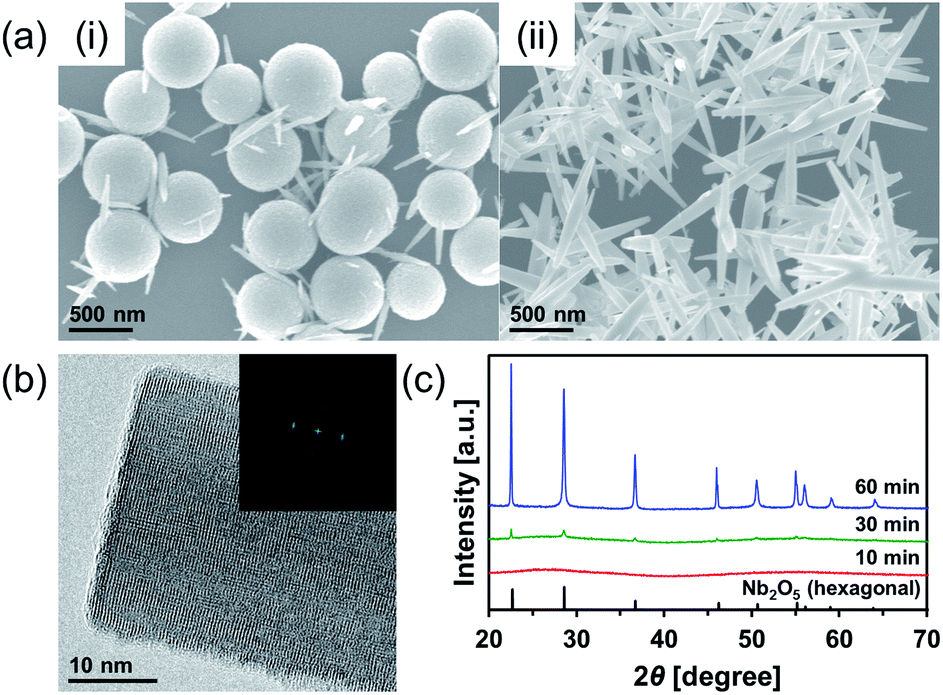 | ||
| Fig. 2 (a) SEM images of the Nb2O5 obtained at 300 °C with different solvothermal-reaction times (i) 30 min and (ii) 60 min. (b) HR-TEM image of the rod-like Nb2O5; corresponding FFT image is inset. (c) XRD patterns of the powdery products obtained using different solvothermal-reaction times (blue, 60 min; green, 30 min; red, 10 min). Reference peaks of hexagonal Nb2O5 (JCPDS 00-007-0061) are represented by the black line. | ||
To date, almost 15 polymorphic forms of Nb2O5 have been reported; however, hexagonal, orthorhombic, and monoclinic phases are relatively common.20 The crystal phases significantly affect the activity of the photocatalyst24 and solid acid.25 Thus, it is important to control of the crystal phase formation in the obtained Nb2O5 MARIMO. The easiest method to improve the crystallinity is further heating. When the heating time of the precursor solution was prolonged to 60 min, the crystal phase dramatically changed from amorphous to the hexagonal phase of Nb2O5 (Fig. 2c). This was concomitant with a drastic morphology change from spherical to rod-shaped (Fig. 2a(ii)). The corresponding HR-TEM image (Fig. 2b) showed that the crystal lattice distances were 0.39 nm. In addition, the fast Fourier transform (FFT) pattern in the HR-TEM image indicated that the direction of rod growth was along the [001] facet. When a 30 min heating procedure was used, spheres and rods coexisted. Compared to 10 min heating, a volume reduction of approximately 40% was observed for the 30 min heating process, where the diameter of the obtained Nb2O5 MARIMO decreased to 530 ± 50 nm (Fig. 2a(i) and S1,† blue line). Ostwald ripening was likely responsible for this shrinkage.26,27 Similarly, higher reaction temperatures (325 and 350 °C) with a fixed reaction time (10 min) resulted in rod-like hexagonal phase nanocrystals (Fig. S4†). Although the crystallinity was successfully improved by extending the reaction time or using a high reaction temperature, the porosity was lost due to the morphology change, and the specific surface area was greatly reduced.
Calcination of the as-synthesized amorphous Nb2O5 MARIMO was used as an alternative method to change the crystal phase. Generally, amorphous Nb2O5 crystallizes at 500 °C into hexagonal or orthorhombic phase, tetragonal phase at 800 °C, and monoclinic phase at >1000 °C.28 An exothermic phenomenon was observed at approximately 240 and 560 °C in the thermogravimetric-differential thermal analysis (TG-DTA, Fig. 3a). The exothermic peak at approximately 240 °C originated from the combustion of organic residues included in MARIMO. In addition, the exothermic peak with negligible weight loss at approximately 560 °C was assigned to a crystalline phase transfer.29 Calcination of the as-synthesized amorphous Nb2O5 MARIMO yielded a hexagonal phase at 500 °C and an orthorhombic phase at >600 °C, as determined from their XRD patterns (Fig. 3b). The spherical morphology was maintained at ≤600 °C, but collapsed at 700 °C (Fig. 3c(i) and S5†). Thus, Nb2O5 MARIMO with improved crystallinity was successfully obtained via calcination at 500 or 600 °C for 2 h (Fig. 3b, c and S5a†). The specific surface area of Nb2O5 MARIMO calcined at 500 °C was 88 m2 g−1 and average pore size expanded from 3.3 to 6.4 nm upon calcination (Fig. S3b†).
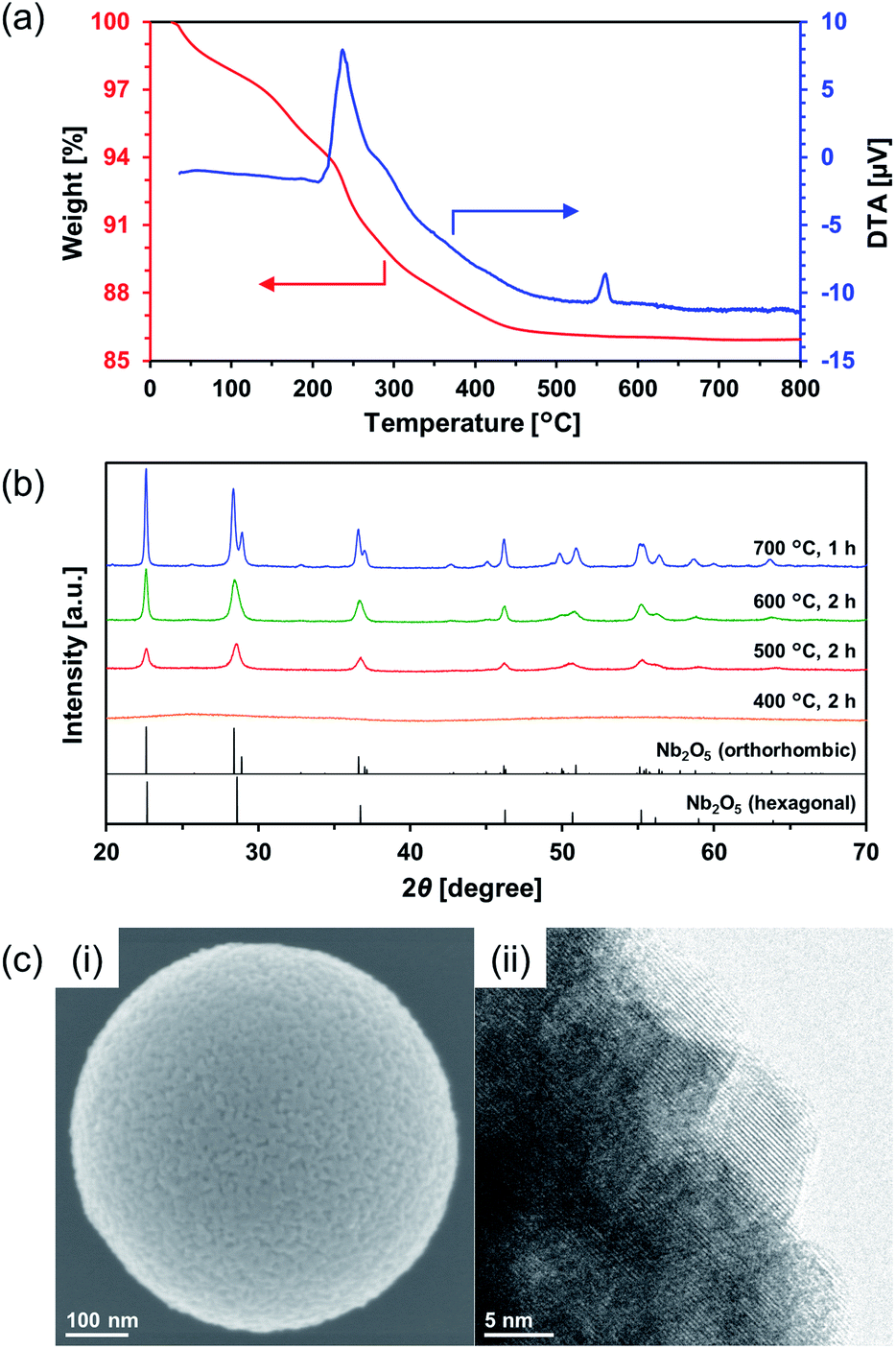 | ||
| Fig. 3 (a) TG-DTA profiles of the as-synthesized Nb2O5 MARIMO measured in air at a heating rate of 2 °C min−1. (b) XRD patterns of calcined Nb2O5 MARIMO at 400–700 °C (blue, 700 °C; green, 600 °C; red, 500 °C; orange, 400 °C). Reference peaks of orthorhombic Nb2O5 (JCPDS 01-071-0336) and hexagonal Nb2O5 (JCPDS 00-007-0061) are represented by black lines. (c) SEM (i) and HR-TEM (ii) images of the Nb2O5 MARIMO calcined at 500 °C for 2 h. The crystal lattice distances were 0.31 nm. | ||
Synthesis of TiO2–Nb2O5 composite MARIMO
Ti–Nb binary oxides have attracted significant attention since their discovery as they exhibit reduced electrical resistance,30 high photocatalytic activity under visible light irradiation,31 and controlled Brønsted/Lewis acidity.32 In particular, using Ti–Nb binary oxides in lithium and sodium ion batteries19,33,34 has attracted great attention because of its improved electronic conductivity,35 ion diffusion path expansion,35 and high theoretical capacity.36 As mentioned in the introduction, composite MARIMOs consisting of plural metal oxides have been successfully synthesized.10–13 Herein, a one-pot and single-step technique was used to obtain TiO2–Nb2O5 composite MARIMOs.A methanol solution of Ti(OiPr)4 and Nb(OEt)5 with formic acid was heated at 300 °C for 10 min. The molar ratio of Ti to Nb in the product MARIMO was easily controlled by changing the ratio in the corresponding precursor solution (entries 2–4 in Table 1). Nearly equivalent Ti/Nb ratios between the precursor solutions and products were obtained. As shown in Fig. 4a(i–iii), Ti and Nb atoms were equally distributed at the nano-level in the TiO2–Nb2O5-75, TiO2–Nb2O5-50, and TiO2–Nb2O5-25 samples, where the numbers denote the atomic percentage of Nb among the total number of Nb and Ti atoms. In addition, the TiO2–Nb2O5 composite MARIMOs were highly porous with specific surface areas of >200 m2 g−1 (Table 1 and Fig. S6†).
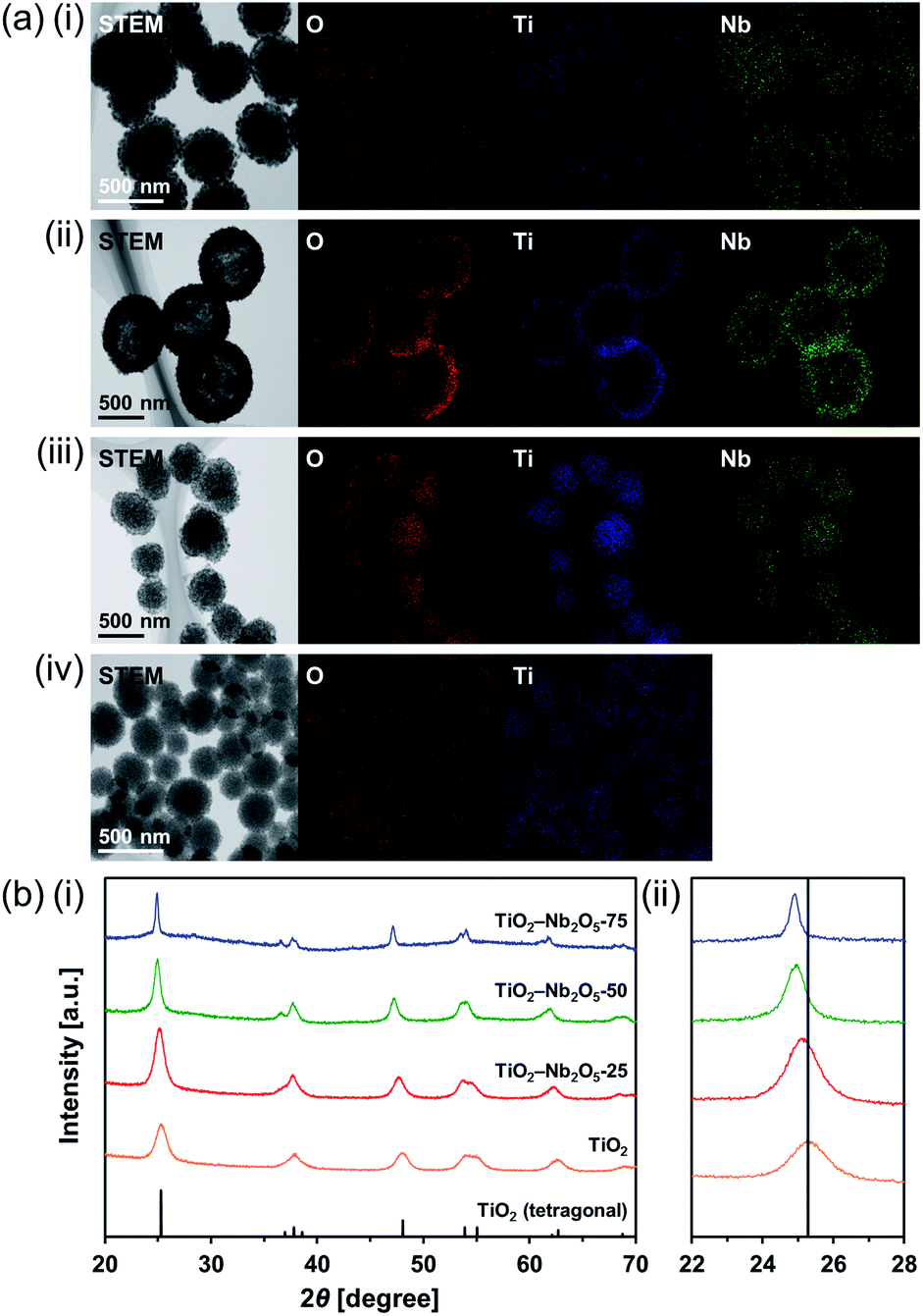 | ||
| Fig. 4 (a) STEM/EDX analysis of the prepared TiO2–Nb2O5 composite MARIMOs; (i) TiO2–Nb2O5-75, (ii) TiO2–Nb2O5-50, (iii) TiO2–Nb2O5-25, and (iv) TiO2. (b) XRD patterns of the obtained powdery products prepared with different molar ratios of Ti/Nb (blue, TiO2–Nb2O5-75; green, TiO2–Nb2O5-50; red, TiO2–Nb2O5-25; orange, TiO2). Reference peaks corresponding to tetragonal TiO2 (JCPDS 00-021-1272) are represented by the black line. The 2θ range: (i) 20–70°, (ii) 22–28°. | ||
Only the tetragonal (anatase-type) TiO2 peaks were observed in the XRD profiles when Nb atoms were included in the product MARIMOs (Fig. 4b(i)). Lower angle shifts of the diffraction peaks were also observed in the XRD profiles (Fig. 4b(ii)), indicating that the composite MARIMOs consisted of solid solutions and the crystal lattice was enlarged by the doped Nb atoms with larger ionic radii (Nb5+ = 0.64 Å, Ti4+ = 0.61 Å).37 Additionally, precursor solutions containing larger amounts of Nb(OEt)5 resulted in the larger primary particle sizes of TiO2 in the resulting MARIMOs (Table 1). However, this crystalline growth of TiO2 was not observed for the Al2O3–TiO2, ZnO–TiO2, and SiO2–TiO2 composites.11,12 Thus, the observed crystalline growth of TiO2 can be ascribed to the catalytic effect of solid Nb2O5 as a solid acid that promoted crystalline growth.
Interestingly, hollow MARIMO were formed at a 50:50 Ti/Nb molar ratio (Fig. 4a(ii) and S7†). However, solid TiO2–Nb2O5-50 MARIMO was obtained (Fig. S8†) when the reaction was immediately quenched at 0 min (the reaction temperature was 300 °C). From the XRD patterns, the longer reaction time yielded better crystallinity (Fig. S9†). Ostwald ripening was likely the cause of the hollow MARIMO formation.38 These unique hollow structures imparted large surface area, low density, and high loading capacity. Therefore, the prepared materials offer promising potential for use in advanced micro-/nanoreactors, catalysis, energy storage/conversion, biomedicine, sensors, and environmental remediation applications.39–41
Formation of TiNb2O7 MARIMO by crystallization of amorphous TiO2–Nb2O5 composite MARIMO
TiNb2O7 as an anode material for lithium ion batteries has a high theoretical capacity of 387.6 mA h g−1 derived from its 5-electron transfer mechanism (Ti4+/Ti3+, Nb5+/Nb4+, and Nb4+/Nb3+). This value is approximately two times higher than that of the commonly used Li4Ti5O12 (175 mA h g−1).36 Thus, TiNb2O7 has attracted significant attention is promising for application in electric vehicles.19,33,34 However, TiNb2O7 suffers from low ionic and electric conductivity. Tailoring special nanostructures of TiNb2O7 can be used to harness the advantages of nanomaterials, including high electrode/electrolyte contact area and short ion diffusion distances. In particular, microscale spherical materials with hierarchical nanoscale structure are promising for solving the drawbacks of pure nanoscale porous materials with low tap densities and poor coulombic efficiency. Although, the syntheses of TiNb2O7 porous spheres have been previously reported, those with larger surface areas remain elusive (Table S1†). Thus, to obtain TiNb2O7 porous spheres with large surface area, the synthetic conditions of the precursor composite TiO2–Nb2O5 as well as the calcination conditions were closely examined.First, a similar alcothermal treatment of a 1:2 (mol ratio) Ti(OiPr)4 and Nb(OEt)5 mixed methanol solution including formic acid at 300 °C afforded a precursor composite MARIMO with tetragonal (anatase-type) TiO2 and amorphous Nb2O5 as determined from its XRD pattern (Fig. S10,† blue line). The corresponding SEM images revealed characteristically large primary particles on the MARIMO surface (Fig. S11a(i)†). The second step involved calcination of the precursor MARIMO at 700 °C for 2 h and yielded monoclinic TiNb2O7 MARIMO (Fig. S11a(ii) and S12a†, blue line). However, the XRD patterns showed that the calcined MARIMO contained mainly monoclinic TiNb2O7 with a small amount of tetragonal (anatase-type) TiO2 and orthorhombic Nb2O5 (Fig. S12b†, blue line). This indicates that a pure TiNb2O7 MARIMO was not afforded by the calcination of the as-synthesized TiO2–Nb2O5 MARIMO obtained at 300 °C.
Therefore, amorphous TiO2–Nb2O5 composite MARIMO was selected as an alternative precursor (Fig. S10, red line and S11b(i)†). The material was prepared using a lower temperature (200 °C) alcothermal reaction. As shown in Fig. 5a, calcination of the amorphous TiO2–Nb2O5 composite MARIMO at >600 °C for 2 h produced the desired TiNb2O7 MARIMO with a pure monoclinic phase. In addition, tetragonal (anatase-type) TiO2 and orthorhombic Nb2O5 peaks were not observed even in case of the obtained sample at 700 °C (Fig. S12b†, red line). Fringe structures were observed in the HR-TEM image (Fig. 5b(i)) of the primary particles, clearly showing that the MARIMO was composed of single crystals of TiNb2O7. The TiNb2O7 porous morphology was clearly demonstrated by high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM; Fig. 5b(ii)). The specific surface area of the obtained monoclinic TiNb2O7 MARIMO was smaller (50 m2 g−1) than that before calcination (Fig. S13†, 497 m2 g−1), but 50 m2 g−1 is the highest value reported for this type of material to the best of our knowledge (Fig. 5c(i) and Table S1†). As expected, the average pore size was enlarged from 2.5 to 8.8 nm (Fig. 5c(ii)), which is favourable for guest materials to access deep into the porous spheres. In addition, the pore space between the TiNb2O7 MARIMO can buffer volume changes derived from the phase transitions occurring during lithiation/delithiation, which may result in good cycling performance via enhanced structural stability.42 Thus, TiNb2O7 porous spheres were successfully prepared with a monoclinic crystal phase and wide pores by calcination of the amorphous TiO2–Nb2O5 composite MARIMO precursor.
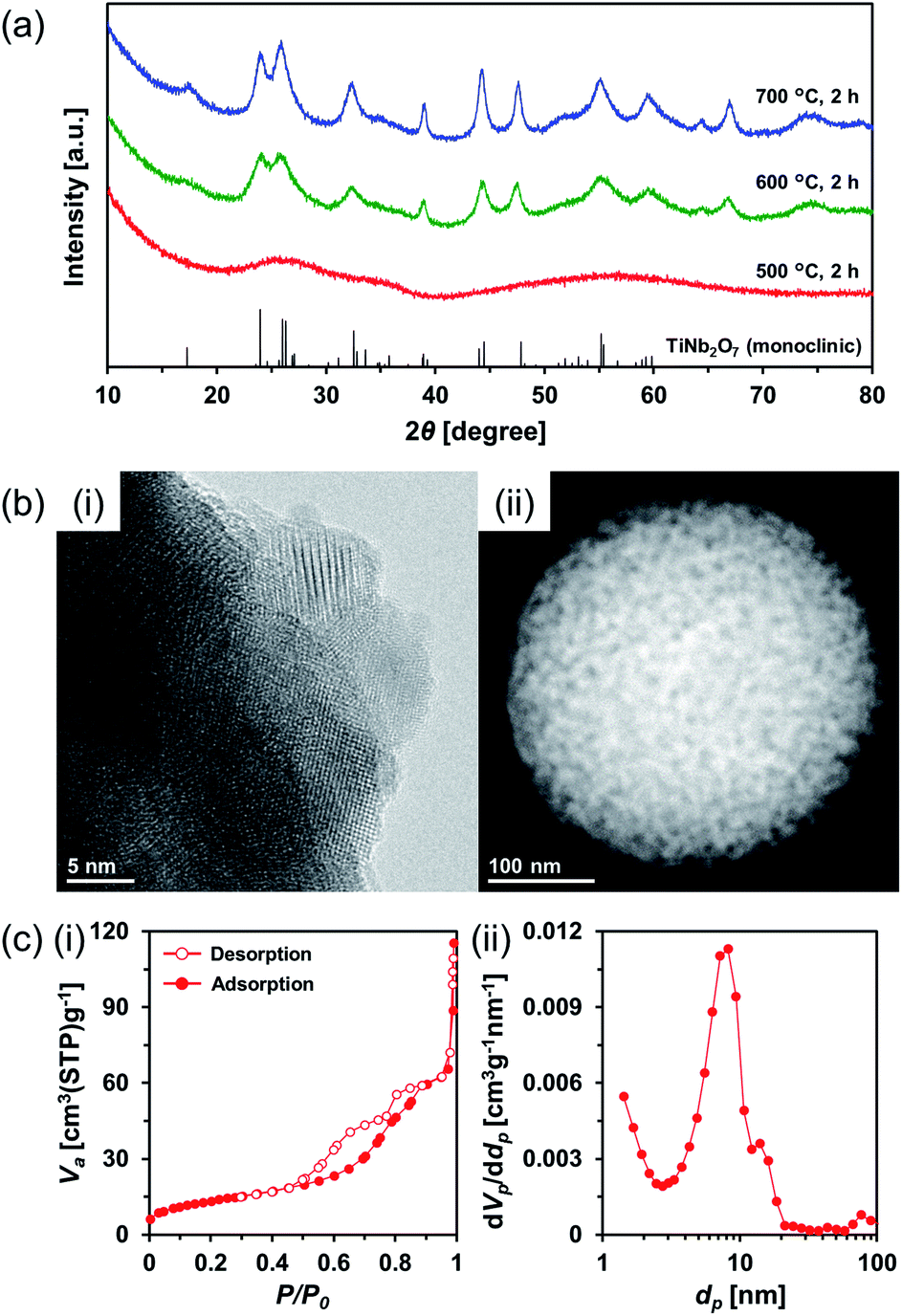 | ||
| Fig. 5 (a) XRD patterns of the calcined amorphous TiO2–Nb2O5 composite MARIMO at 500–700 °C (blue, 700 °C; green, 600 °C; red, 500 °C). Reference peaks of monoclinic TiNb2O7 (JCPDS 00-039-1407) are indicated by the black line. (b) HR-TEM (i) and HAADF-STEM (ii) images of the amorphous TiO2–Nb2O5 composite MARIMO calcined at 600 °C. (c) Nitrogen adsorption–desorption isotherms (i) and pore size distributions (ii) of the amorphous TiO2–Nb2O5 composite MARIMO calcined at 600 °C. | ||
Conclusions
Monodispersed Nb2O5 porous spheres (Nb2O5 MARIMO) with 630 nm in diameter were prepared via the simple alcothermal reaction of a methanol solution of Nb(OEt)5 with formic acid. The crystallinity and pore size of the prepared Nb2O5 MARIMO were improved by prolonging the reaction time or simple calcination of the product. TiO2–Nb2O5 composite MARIMO was obtained using a similar alcothermal reaction of Ti(OiPr)4, Nb(OEt)5, and formic acid in methanol, with a product Ti:Nb atomic ratio that was controlled by changing molar ratio of Ti(OiPr)4 and Nb(OEt)5 in the precursor solution. A two-step synthetic approach for the generation of TiNb2O7 porous spheres was developed via an alcothermal treatment of a 1:2 (molar ratio) mixed solution of Ti(OiPr)4 and Nb(OEt)5 in methanol followed by simple calcination. Considering the fact that the obtained submicron-sized porous spheres of niobia and their composites have a large surface area, high performance can be expected in the field of catalysts and batteries. To evaluate catalyst and battery performance, the research for a practical application is undergoing.
Experimental section
Materials
Methanol, formic acid, niobium(V) ethoxide [Nb(OEt)5], and titanium tetraisopropoxide [Ti(OiPr)4] were obtained from FUJIFILM Wako Pure Chemical Corporation. All reagents were used as received unless otherwise noted.Synthetic procedure
Characterization methods
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Creation of New Business and Industry Program through the Kochi Prefectural Industry-Academia-Government Collaboration Research Promotion Operation and funding from JSPS KAKENHI Grant Number 15K06560, 17K14858, and 19K05143. The authors also wish to thank the Research Center for Nanotechnology in Kochi University of Technology for performing TEM and SEM analyses.References
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed.
- Y. Boyjoo, M. Wang, V. K. Pareek, J. Liu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 6013–6047 RSC.
- A. Varma, A. Mukasyan, A. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- H.-J. Qiu, L. Peng, X. Li, H. T. Xu and Y. Wang, Corros. Sci., 2015, 92, 16–31 CrossRef CAS.
- A. Lahiri and F. Endres, J. Electrochem. Soc., 2017, 164, 597–612 CrossRef.
- J. Leng, Z. Wang, J. Wang, H.-H. Wu, G. Yan, X. Li, H. Guo, Y. Liu, Q. Zhang and Z. Guo, Chem. Soc. Rev., 2019, 48, 3015–3072 RSC.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- M. K. M. Lane and J. B. Zimmerman, Green Chem., 2019, 21, 3769–3781 RSC.
- P. Wang, K. Ueno, H. Takigawa and K. Kobiro, J. Supercrit. Fluids, 2013, 78, 124–131 CrossRef CAS.
- E. K. C. Pradeep, T. Habu, H. Tooriyama, M. Ohtani and K. Kobiro, J. Supercrit. Fluids, 2015, 97, 217–223 CrossRef CAS.
- E. K. C. Pradeep, M. Ohtani and K. Kobiro, Eur. J. Inorg. Chem., 2015, 5621–5627 CrossRef CAS.
- H. T. T. Nguyen, T. Habu, M. Ohtani and K. Kobiro, Eur. J. Inorg. Chem., 2017, 3017–3023 CrossRef CAS.
- M. Ohtani, T. Muraoka, Y. Okimoto and K. Kobiro, Inorg. Chem., 2017, 56, 11546–11551 CrossRef CAS PubMed.
- S. Furukawa, Y. Ohno, T. Shishido, K. Teramura and T. Tanaka, ACS Catal., 2011, 1, 1150–1153 CrossRef CAS.
- H. Liu, N. Gao, M. Y. Liao and X. S. Fang, Sci. Rep., 2015, 5, 7716 CrossRef CAS PubMed.
- S. M. A. H. Siddiki, M. N. Rashed, M. A. Ali, T. Toyao, P. Hirunsit, M. Ehara and K. Shimizu, ChemCatChem, 2018, 10, 1–15 CrossRef.
- T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef CAS PubMed.
- E. S. Gnanakumar, N. Chandran, I. V. Kozhevnikov, A. Grau-Atienza, E. V. R. Fernandez, A. Sepulveda-Escribano and N. R. Shiju, Chem. Eng. Sci., 2019, 194, 2–9 CrossRef CAS.
- Q. Deng, Y. Fu, C. Zhu and Y. Yu, Small, 2019, 15, 1804884 CrossRef PubMed.
- R. A. Rani, A. S. Zoolfakar, A. P. O'Mullane, M. W. Austin and K. K. Zadeh, J. Mater. Chem. A, 2014, 2, 15683–15703 RSC.
- C. C. Li, J. Dou, L. W. Chen, J. Y. Lin and H. C. Zeng, ChemCatChem, 2012, 4, 1675–1682 CrossRef CAS.
- L. Li, J. Deng, R. Yu, J. Chen, Z. Wang and X. Xing, J. Mater. Chem. A, 2013, 1, 11894–11900 RSC.
- Z. F. Dai, H. Dai, Y. Zhou, D. L. Liu, G. T. Duan, W. P. Cai and Y. Li, Adv. Mater. Interfaces, 2015, 2, 1500167 CrossRef.
- H. Zhang, Q. Wu, C. Guo, Y. Wu and T. Wu, ACS Sustainable Chem. Eng., 2017, 5, 3517–3523 CrossRef CAS.
- K. Nakajima, J. Hirata, M. Kim, N. K. Gupta, T. Murayama, A. Yoshida, N. Hiyoshi, A. Fukuoka and W. Ueda, ACS Catal., 2018, 8, 283–290 CrossRef CAS.
- J. Xue, R. Wang, Z. Zhang and S. Qiu, Dalton Trans., 2016, 45, 16519–16525 RSC.
- J. Chen, H. Wang, G. Huang, Z. Zhang, L. Han, W. Song, M. Li and Y. Zhang, J. Alloys Compd., 2017, 728, 19–28 CrossRef CAS.
- E. I. Ko and J. G. Weissman, Catal. Today, 1990, 8, 27–36 CrossRef CAS.
- H. Kominami, K. Oki, M. Kohno, S. Onoue, Y. Kera and B. Ohtani, J. Mater. Chem., 2001, 11, 604–609 RSC.
- G. Li, X. Zhang, H. Lu, C. Tan, K. Chen, H. Lu, J. Gao, Z. Yang, G. Zhu, G. Wang and Z. He, Sens. Actuators, B, 2019, 283, 602–612 CrossRef CAS.
- L. Kong, C. Wang, H. Zheng, X. Zhang and Y. Liu, J. Phys. Chem. C, 2015, 119, 16623–16632 CrossRef CAS.
- M. Akizuki and Y. Oshima, J. Supercrit. Fluids, 2018, 141, 173–181 CrossRef CAS.
- S. Lou, Y. Zhao, J. Wang, G. Yin, C. Du and X. Sun, Small, 2019, 15, 1904740 CrossRef CAS PubMed.
- J. Ma, X. Guo, H. Xue, K. Pan, C. Liu and H. T. Pang, Chem. Eng. J., 2020, 380, 122428 CrossRef CAS.
- H. Usui, S. Yoshioka, K. Wasada, M. Shimizu and H. Sakaguchi, ACS Appl. Mater. Interfaces, 2015, 7, 6567–6573 CrossRef CAS PubMed.
- J.-T. Han, Y.-H. Huang and J. B. Goodenough, Chem. Mater., 2011, 23, 2027–2029 CrossRef CAS.
- Y. Lv, B. Cai, Q. Ma, Z. Wang, J. Liu and W.-H. Zhang, RSC Adv., 2018, 8, 20982–20989 RSC.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492–3495 CrossRef CAS.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS PubMed.
- L. Yu, H. Hu, H. B. Wu and X. W. Lou, Adv. Mater., 2017, 29, 1604563 CrossRef PubMed.
- G. Liu, L. Zhao, R. Sun, W. Chen, M. Hu, M. Liu, X. Duan and T. Zhang, Electrochim. Acta, 2018, 259, 20–27 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01704c |
| This journal is © The Royal Society of Chemistry 2020 |