
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrathin δ-MnO2 nanoflakes with Na+ intercalation as a high-capacity cathode for aqueous zinc-ion batteries†
Haijun Peng a,
Huiqing Fan*a,
Chenhui Yangb,
Yapeng Tianc,
Chao Wanga and
Jianan Suia
a,
Huiqing Fan*a,
Chenhui Yangb,
Yapeng Tianc,
Chao Wanga and
Jianan Suia
aState Key Laboratory of Solidification Processing, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, China. E-mail: hqfan@nwpu.edu.cn; hqfan3@163.com
bSchool of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an 710129, China
cElectronic Materials Research Laboratory, International Center for Dielectric Research, Key Laboratory of the Ministry of Education, School of Electronic & Information Engineering, Xi'an Jiaotong University, Xi'an 710049, China
First published on 6th May 2020
Abstract
Pristine δ-MnO2 as the typical cathode for rechargeable zinc-ion batteries (ZIBs) suffers from sluggish reaction kinetics, which is the key issue to prepare high-performance manganese-based materials. In this work, Na+ incorporated into layered δ-MnO2 (NMO) was prepared for ZIB cathodes with high capacity, high energy density, and excellent durable stability. By an effective fabricated strategy of hydrothermal synthesis, a three-dimensional interconnected δ-MnO2 nanoflake network with Na+ intercalation showed a uniform array arrangement and high conductivity. Also, the H+ insertion contribution in the NMO cathode to the discharge capacity confirmed the fast electrochemical charge transfer kinetics due to the enhanced ion conductivity from the insertion of Na+ into the interlayers of the host material. Consequently, a neutral aqueous NMO-based ZIB revealed a superior reversible capacity of 335 mA h g−1, and an impressive durability over 1000 cycles, and a peak gravimetric energy output of 459 W h kg−1. As a proof of concept, the as-fabricated quasi-solid-state ZIB exhibited a remarkable capacity of 284 mA h g−1 at a current density of 0.5 A g−1, and good practicability. This research demonstrated a significant enhancement of the electrochemical performance of MnO2-based ZIBs by the intercalation of Na+ to regulate the microstructure and boost the electrochemical kinetics of the δ-MnO2 cathode, thus providing a new insight for high-performance aqueous ZIBs.
1. Introduction
Energy storage technologies featuring high performance, cost effectiveness, and high safety are highly desirable to develop large-scale prepared and diverse electronic devices.1–6 Thanks to their high energy density, lithium-ion batteries (LIBs) have been commercialized successfully over the past decades. However, the consumption of scarce lithium resources restricts their large-scale use to meet future energy power requirements.7 Recently, rechargeable aqueous zinc-ion batteries (ZIBs) based on a two-electron transfer mechanism have exhibited huge potential as promising alternatives to LIBs due to their operational safety, facile fabrication, and environmental benignity, as well as a high ionic conductivity of the nonflammable aqueous electrolyte. Among the pre-existing aqueous electrolyte systems in ZIBs, the selected ZnSO4-based neutral electrolytes can alleviate the formation of Zn dendrites and avoid the irreversible formation of by-products (ZnO or Zn(OH)2) on the Zn anode, thus making metallic Zn an ideal anode.8–12 Furthermore, the Zn metal anode applied in aqueous ZIBs possesses the merits of abundant resource of Zn on earth, high theoretical capacity (820 mA h g−1), high output voltage (>1.8 V), relative low redox potential (−0.76 V vs. standard hydrogen electrode), and excellent stability in water.6,13,14As a typical cathode material for aqueous ZIBs, MnO2 is a compelling candidate and consequently, has been initially studied.15–18 For example, Lu et al. demonstrated an ultrahigh energy density and long durability Zn/MnO2 battery with a PEDOT buffer layer and a Mn2+-based neutral electrolyte.19 The flexible rechargeable battery delivered a discharge specific capacity of 366.6 mA h g−1 at a current density of 0.74 A g−1. Another aqueous Zn/MnO2 battery was fabricated by Xue et al. using a neutral electrolyte with oxygen-deficient manganese oxide as the cathode, achieving a high capacity of 345 mA h g−1 at 0.2 A g−1.20 As a matter of fact, there is a vital consideration for choosing suitable MnO2 hosts as the cathode in ZIBs. Regardless of the original crystal structure, MnO2 with polymorphs transforms into layered manganese oxide phases for structural transformation during the charge/discharge processes, which leads to the capacity fading of a MnO2 cathode in rechargeable aqueous ZIBs.16 Therefore, the layered δ-MnO2 can be a promising option as a cathode material for aqueous ZIBs to refrain from phase transformation. As an aqueous ZIB cathode, the layered δ-MnO2 polymorph possesses a large interlayer spacing, which could be appealing for high-efficiency Zn2+ insertion/extraction. The Zn2+ intake and release processes are associated with the 2D crystal network of δ-MnO2, which allows a fast transport pathway with an alleviated crystal phase change.21,22 Although there are some reports about high energy density coupled with excellent long-term stability being achieved for the δ-MnO2 cathode in a mild aqueous electrolyte, most δ-MnO2 electrode materials still present a very low utilization of the theoretical capacity.15,23 Hence, there is a great potential to develop a high-performance MnO2 cathode for ZIBs in mild aqueous electrolytes.
In general, Zn-ion storage in layered δ-MnO2 could principally fall into two categories: a diffusive component, which refers to the insertion/extraction of Zn2+ into the interlayer, and a capacitive part, involving the surface redox reaction: xZn2+ + 2xe− + MnO2 → MnOOZnx.20 Therefore, Zn2+-storage enhancement is dependent on structural modifications or surface chemistry modification, such as generating oxygen vacancies in the δ-MnO2 lattice.18,20 In the previous literature, embedding large metallic ions, such as Ca2+,12 Na+,24 and Zn2+ (ref. 25) into vanadium/manganese oxide was performed to bring about the formation of vanadates or manganates, which could enhance the electrochemical performance in aqueous ZIBs. Furthermore, at the device-level for ZIBs, how to develop an active material by its growth on a flexible conducting substrate is another problem to assemble an integrated cathode without a binder and conductive additive, which would be highly expected to show superior specific capacity and would be more convenient in practice application.
In this work, a facile and scalable approach was carried out through the incorporation of Na+ into layered δ-MnO2 immobilized on nickel foam. The prepared Na+-intercalated layered δ-MnO2 as a binder-free cathode, Zn plate as the anode, and neutral electrolyte (2.0 M ZnSO4 + 0.2 M MnSO4) were assembled together to fabricate a high-performance rechargeable aqueous zinc-ion battery. As a result, the Na+-intercalated δ-MnO2 (NMO) cathode in ZIB demonstrated a high capacity (335 mA h g−1), remarkable energy density (459 W h kg−1), and durable lifespan. Benefiting from the promoted H+-diffusion kinetics and the mild neutral electrolyte containing a moderate Mn2+-ions content, the rechargeable Zn/NMO battery presented an outstanding capacity retention of 93% after 1000 cycles with a high coulombic efficiency. For demonstration, a flexible quasi-solid-state ZIB was fabricated to power 23 light emitting diodes (LEDs) and showed the excellent practical application potential of the rechargeable aqueous ZIBs. Therefore, our work presents an inspired solution to develop high-capacity rechargeable aqueous zinc batteries by tuning the layer atomic structure of the cathode material.
2. Experimental section
All the reagents were analytical grade and used without further purification.2.1. Synthesis of the sodium-intercalated δ-MnO2 nanoflakes
The δ-MnO2 nanoflakes on nickel foam (NF) with sodium intercalation were synthesized by a hydrothermal method. First, 5 mmol L−1 of KMnO4 was dissolved in 40 mL deionized water in a beaker, followed by stirring for 30 min, and then transferred to a 50 mL Teflon container at 180 °C for 3 h with NF immersed into the solution around the reactor wall.26 The resulting product was washed with ethanol and deionized water three times, respectively. Second, the K+ in the as-synthesized δ-MnO2 nanoflakes was extracted by an electrochemical method in 2 M ZnSO4 electrolyte with Zn as both the counter and reference electrodes. The galvanostatic charge upper cut-off voltage was set up at 1.95 V (versus metallic Zn) and maintained for half an hour at this voltage. Eventually, the Na+-intercalated δ-MnO2 (abbreviated as NMO) sample was obtained after hydrothermal treatment with 0.5 mol L−1 of Na2SO4 at 180 °C for 3 h followed by desiccation in an oven at 60 °C for 10 h.For comparison, the layered δ-MnO2 nanoflakes (abbreviated as MO) were obtained by using the same procedure as above without the additional hydrothermal treatment for the ions intercalation. Also, the K+-intercalated δ-MnO2 sample was also synthesized the same way as for Na+ intercalation to replace Na2SO4 with K2SO4, for which the product was correspondingly abbreviated as KMO. The three samples (MO, NMO, and KMO) on nickel foams could be used directly as an electrode with a loading mass of nearly 1.05 mg. The loading mass was evaluated by calculating the difference in the mass before and after ultrathin δ-MnO2 nanoflakes were grown on the nickel foam.
2.2. Materials characterization
The phases of the as-prepared samples were characterized by X-ray diffraction spectrometry (XRD; 7000S/L, Shimadzu, Japan) with Co Kα radiate at a scanning rate of 1° min−1. Thermogravimetric (TG) analysis was performed in a porcelain crucible using an STA Instruments STA 449F3 in an argon atmosphere. Before and after the charge/discharge tests, the morphologies of the samples were characterized by scanning electron microscopy (SEM; JSM-6700F, JEOL, Tokyo, Japan), transmission electron microscopy (TEM; Talos F200X, FEI, Hillsboro, OR, USA), and high-resolution transmission electron microscopy (HRTEM). The thickness of a few-layer NMO nanosheets coating was deposited on the clean silicon wafers and measured by atomic force microscopy (AFM) using a Bruker Dimension 3100 instrument under a MicroMasch NSC16 tip with 40 N m−1 of spring constant. The valence band spectra of Mn in δ-MnO2 were analyzed using X-ray photoelectron spectroscopy (VG ESCALAB 220i-XL, Thermo Scientific, Waltham, MA, USA) with an Al Kα source (E = 1486.6 eV). All the binding energies were referenced to the C 1s peak (284.6 eV) arising from adventitious carbon.2.3. Electrochemical measurements
An aqueous ZIB was assembled with the as-prepared δ-MnO2 cathode and Zn metal plate anode, using 2 M ZnSO4 and 0.2 M MnSO4 as the electrolyte. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed using an electrochemical testing system (CHI660B, Chenhua Instrument Co. Ltd., Shanghai, China) at ambient temperature. The EIS tests were conducted in the frequency range between 100 kHz and 10 mHz with an amplitude of 5 mV at an open-circuit potential. Galvanostatic charging/discharging was conducted on a LANHER battery tester (Wuhan) with the voltage range of 0.9 V to 1.8 V (vs. Zn/Zn2+).2.4. Preparation of flexible quasi-solid-state ZIBs
The quasi-solid-state Zn/MnO2 battery was constructed by sandwiching a piece of NKK separator (Nippon Kodoshi Corporation) and PVA/ZnSO4 gel electrolyte between the NMO cathode and a novel flexible Zn anode. The fabrication of the flexible Zn electrode was conducted with a three-electrode setup electrochemical deposition (electrochemical workstation, CHI 660B)-treated carbon cloth. Briefly, a piece of treated carbon cloth (CC) was chosen as the working electrode, with Pt wire and a Ag/AgCl/3 M KCl electrode as the counter and reference electrodes, respectively. Next, 12.5 g of zinc sulfate, 12.5 g of sodium sulfate, and 2.0 g of boric acid were dissolved in 100 mL distilled water and used as the electrolyte. The electrodeposition was set at a constant current density of −40 mA cm−2 with a time of 3 h. The mass loading of the electrodeposited zinc on carbon cloth was around 3.0–4.0 mg cm−2. For the gel electrolyte, 6 g of PVA and 50 mg of lignocellulose were dissolved in 60 mL DI water, followed by mixing with 20 mL of 2.0 M ZnSO4 and 0.2 M MnSO4 solution under vigorous stirring at 85 °C to obtain a transparent gel electrolyte.2.5. Computational method
The Vienna Ab Initio Package (VASP)27,28 was employed to perform density functional theory (DFT) calculations within the generalized gradient approximation (GGA) using the PBE29 formulation. The projected augmented wave (PAW) potentials were chosen30,31 to describe the ionic cores, taking the valence electrons into account using a plane wave basis set with a cutoff kinetic energy of 400 eV. Partial occupancies of the Kohn–Sham orbitals were allowed by using the Gaussian smearing method under a width of 0.05 eV. The electronic energy was considered as self-consistent when the energy change was smaller than 10–7 eV. A geometry optimization was considered convergent when the energy change was smaller than 10–6 eV. The spin polarization was considered in all the calculations.The equilibrium lattice constants of the δ-MnO2 unit cell were calculated by using a Gamma-point centered 7 × 13 × 5 k-point grid for Brillouin zone sampling with a = 4.8532 Å, b = 2.6919 Å, c = 4.6139 Å, α = 90°, β = 101.9°, and γ = 90°. The equilibrium lattice constants of the δ-MnO2 unit cell intercalated with a sodium (Na) layer between two layers of the host framework were calculated by using the same k-point grid for Brillouin zone sampling with a = 4.9236 Å, b = 2.8095 Å, c = 5.8103 Å, α = 90°, β = 106.5°, and γ = 90°.
3. Results and discussion
The Na+-intercalated layered δ-MnO2 (NMO) nanoflakes immobilized on nickel foam (NF) were prepared through a hydrothermal synthesis to obtain pristine layered δ-MnO2, and an electrochemical method to remove the K+ ions from the precursor solution, followed by the Na+-intercalation process under hydrothermal treatment. In order to effectively contrast the promoted overall performance caused by the Na+ intercalation, layered δ-MnO2 (MO) and K+-intercalated δ-MnO2 (KMO) samples were prepared, as described in the experimental protocol. The crystalline phase for the as-synthesized MO, NMO, and KMO samples confirmed by X-ray diffraction (XRD) are provided in Fig. S1,† which was in good agreement with the structure of the layered δ-MnO2 (JCPDS no. 86-0666). The reinforced intensity and symmetric peak of the (003) planes for the NMO and KMO samples manifested high crystallinity after the insertion of alkali cations (Na+ and K+). As presented in Fig. 1 and S2,† the scanning electron microscopy (SEM) images showed that all three samples consisted of two-dimension (2D) nanoflakes aligned vertically on the NF, which formed the three-dimensional (3D), conductive, and interconnected network structure. With the introduction of a moderate amount of alkali cations (Na+ and K+), the micromorphology of the intercalated δ-MnO2 nanoflakes displayed a more uniform arrangement, compared with the irregular arrays belonging to the MO sample. Interestingly, the NMO nanoflakes possessed more dense arrays and a favorable structure in the three samples, which means that the incorporation of Na+ gave rise to an effective modulation of the morphology of the pristine δ-MnO2 nanoflakes. The quantitative energy-dispersive X-ray spectroscopy (EDS) results in Fig. 1g and S3† illustrate the uniform spatial distribution of the involved elements for the three samples. The distinctly contrasted EDS mapping demonstrated the substantial homogeneous intercalation of Na+ and K+ into the interlayer of the δ-MnO2. To accurately detect the content of the inserted Na+, the NMO sample was surveyed by inductively coupled plasma optical emission spectroscopy (ICP-OES). Also, the stoichiometry of the inserted Na to Mn was confirmed to be 0.17. The corresponding high-resolution transmission electron microscopy (HRTEM) images in Fig. 1h and i confirmed the resolved lattice fringes with a lattice spacing of 0.245 nm for the (101) plane of the δ-MnO2 phase. The inset of Fig. 1i exhibits the selected area diffraction (SAED) patterns of the NMO flakes, which could be indexed to the (006) and (101) planes of the δ-MnO2 phase.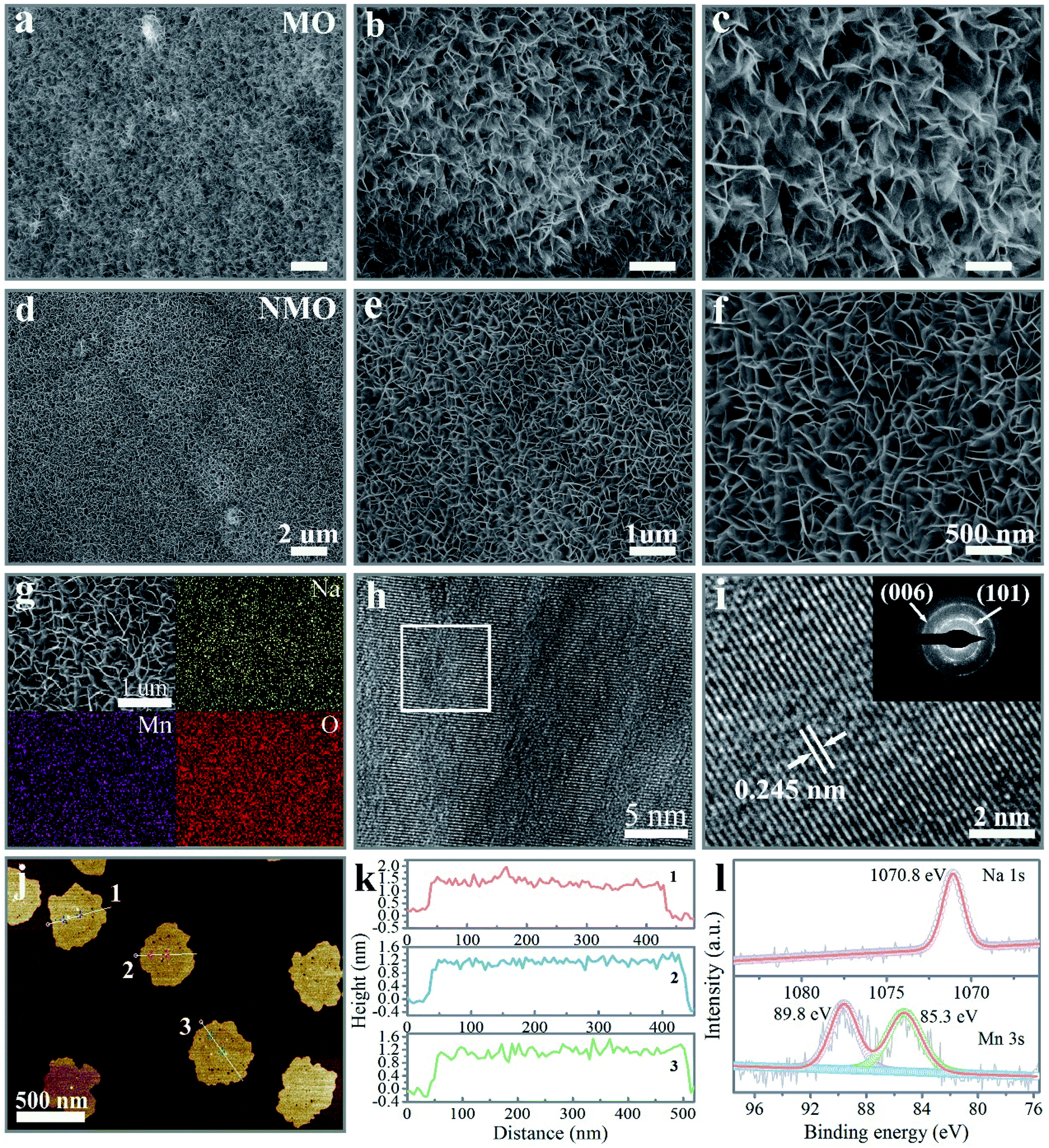 | ||
| Fig. 1 Morphological and structural characterization of the Na+-intercalated layered δ-MnO2 nanoflakes. SEM images of the MO (a)–(c) and NMO (d)–(f) nanoflakes. (g) SEM and EDX element mapping images of the NMO nanoflakes. (h) and (i) HRTEM images of the NMO nanoflakes (the inset is the SAED of (c)). (j) AFM image of the NMO nanoflakes. (k) The corresponding height profiles of the NMO nanoflakes. (l) High-resolution XPS spectra of Mn 3s and Na 1s of the NMO nanoflakes. | ||
Atomic force microscopy (AFM) was performed to further identify the surface morphology and thickness of the NMO nanoflakes. As illustrated in Fig. S4† and 1j, the NMO nanoflakes had a nano-size lateral scale of ∼500 nm with warped edges, which presented thicknesses of about 1.5 nm, corresponding to two monolayers along the (001) direction, distinguishing the few-layers characteristic of the prepared δ-MnO2 samples (Fig. 1k). Therefore, with the synergetic effect of high vapor pressure during hydrothermal treatment and the moderate concentration of Na+ cations, NMO nanoflakes were formed successfully by Na+ cations incorporation between the adjacent MnO6 polyhedron layers. The NMO nanoflakes confirmed by SEM, TEM, and AFM could be expected to supply a larger active surface area and multi-channel ion-diffusion path for cation diffusion during the reaction process of the ZIB system.26,32
The Raman spectrum of the NMO nanoflakes in Fig. S5† can be designate as showing the fundamental vibrational modes of δ-MnO2. The band peaks positioned at 627 and 582 cm−1 could be separately assigned to v2 (Mn–O) symmetric stretching vibrational modes of MnO6 groups and v3 (Mn–O) stretching within the basal plane of the MnO6 sheets. Another band recorded at 505 cm−1 corresponds to the v4 (Mn–O) stretching vibration.20,33,34 The detailed composition and valence state of the Mn element in the NMO sample were studied by X-ray photoelectron spectroscopy (XPS), as elucidated in Fig. S6a† and 1l. The deconvoluted high-resolution Mn 2p spectrum peaks positioned at 654.1 and 642.2 eV were considered to be the fingerprints of Mn 2p1/2 and Mn 2p3/2 for NMO, respectively.35–37 The spin-energy separation between the two Mn 3s doublet was 4.5 eV, from which the average oxidation state was deduced to be 3.87 for the as-obtained NMO sample from eqn (S1)† (Fig. 1l). Specifically, an additional Na 1s peak located at 1070.8 eV could be observed, further confirming the successful intercalation of Na+ into the pristine layered δ-MnO2 nanosheets.5
To explore the benefit of the incorporation of Na+ ions, full cells were assembled and assessed using the δ-MnO2-based network electrodes (MO, NMO, and KMO) as the cathodes and high pure Zn metal plate as the anode, with an aqueous electrolyte consisting of 2 M ZnSO4 and 0.2 M MnSO4. Fig. 2a presents the cyclic voltammetry (CV) curves of the Zn/MO and Zn/NMO batteries at a scan rate of 0.1 mV s−1, indicating the larger current response of the Zn/NMO battery than that of the Zn/MO battery. The extremely larger curve area and significantly enhanced peak current density of the NMO electrode indicate that the insertion of Na+ ions endowed the cathode with a higher capacity and boosted the reaction kinetics. This should be related to the accelerated ion-diffusion coefficient from the reversible and uninterrupted redox reactions that occur during the charge/discharge processes.38
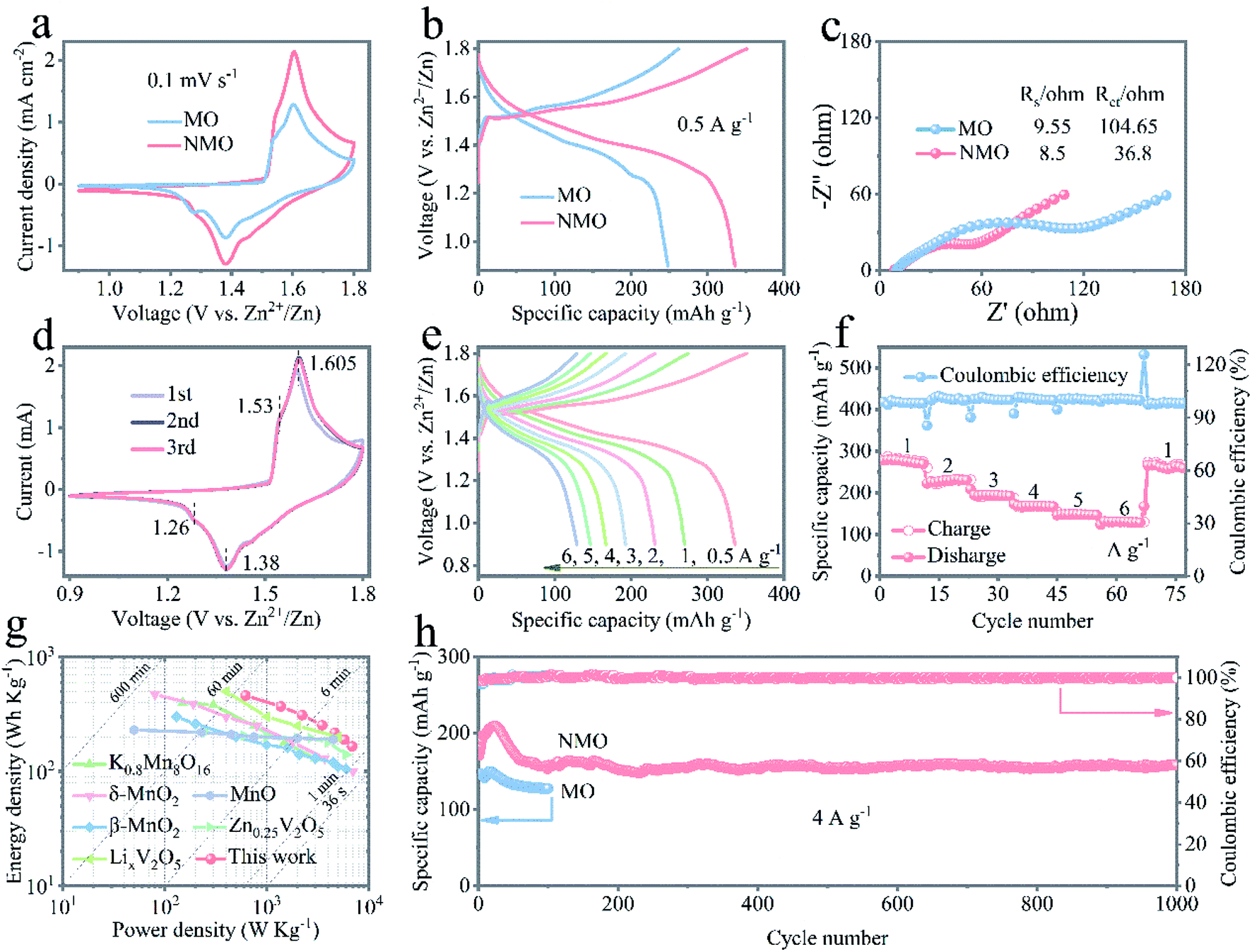 | ||
| Fig. 2 Electrochemical performance of the Zn/NMO and Zn/MO batteries in 2 M ZnSO4 and 0.2 M MnSO4 aqueous electrolyte. (a) Cyclic voltammetry curves of the Zn/NMO and Zn/MO batteries at a scan rate of 0.1 mV s−1. (b) Typical discharge/charge profiles at a current density of 0.5 A g−1 under a potential between 0.9 and 1.8 V. (c) Nyquist plots of the Zn/NMO and Zn/MO batteries. (d) Cyclic voltammetry curves of the Zn/NMO battery at a scan rate of 0.1 mV s−1. (e) Discharge/charge profiles at different current densities varying from 0.5 to 6 A g−1 of the Zn/NMO battery. (f) Rate capability from 1 to 6 A g−1 of the Zn/NMO battery. (g) Ragone plots of the Zn/NMO battery. (h) Long-life cycling performance of the Zn/NMO at a current density of 4 A g−1. | ||
The significant acceleration in kinetics could also be observed by the associated discharge curves at the current density of 0.5 A g−1 in Fig. 2b. The Zn/NMO battery achieved a high capacity of 335 mA h g−1 with two prolonged plateaus, whereas the Zn/MO battery delivered a low capacity of 249 mA h g−1. Also in contrast, the Zn/KMO battery exhibited a slightly increased specific capacity of 257 mA h g−1 at a current density of 0.5 A g−1, superior to that of the Zn/MO (Fig. S7†). The recorded galvanostatic charge/discharge profiles at the current density of 0.5 A g−1, which involve two sloping plateaus, agreed with the voltammogram observation for the Zn/MO and Zn/NMO batteries in Fig. 2b and are in accordance with the typical Zn/MnO2 battery systems.39,40 Both of the MO and NMO cathodes delivered a sloping plateau at around 1.35 V, followed by a flat platform at about 1.2 V. As investigated in previous literature about different ion-insertion processes for the aqueous Zn/MnO2 battery, the first discharge plateau could be mainly attributed to the insertion of H+ and the second one was dominated by the Zn2+ insertion.41–44 Reasonably, the prolonged first plateaus of the Zn/NMO battery in Fig. 2b and S7c† signify an enhanced H+-insertion contribution to the discharge capacity in the NMO cathode. These results confirm the striking advancement in the discharge capacity and the increased favorable H+-ions electrochemical kinetics of the NMO electrode due to the accelerated kinetics of the H+ in the layered NMO skeleton and the higher ion conductivity for the insertion of Na+ into the δ-MnO2 nanoflakes. Therefore, the fast transportation insertion/extraction of H+ ions were proved to be the main charge transport carriers in this work. Similarly, the relevant charge/discharge profiles of the KMO electrode showed a decent discharge capacity compared to the MO electrode for the inserted K+ as promoters for the ions in the δ-MnO2 nanoflakes (Fig. 2b and S7†). Specifically, the NMO electrode showed a better coulombic efficiency than that of the MO electrode at a current density of 0.5 A g−1 (Fig. S7c†), which suggests a significant improvement in the charge/discharge reversibility induced by the insertion of Na+ ions.
Due to the incorporation of Na+ into the pristine layered δ-MnO2 nanosheets, the NMO electrode could be expected to deliver faster ion-diffusion transmission and to further acquire an advanced capacity. Fig. 2c and S8† manifest the electrochemical impedance spectroscopy (EIS) results of the three batteries. The equivalent circuit based on Nyquist plots used for simulation of the EIS results is shown in Fig. S9.† The semicircle in the high–medium frequency region reflects the charge-transfer resistance (Rct) at the electrode/electrolyte interface and the straight line in the low-frequency region is related to the ion diffusion (Zw).45,46 The Zn/NMO battery demonstrated a decreased series resistance of 45.3 Ω (36.8 Ω for Rct) compared to that of 114.2 Ω for the Zn/MO battery (104.65 Ω for Rct). Correspondingly, the Rs (internal resistance of electrode) and Rct of the NMO and KMO electrodes, tabulated in Fig. S8 and Table S2,† show contracted semicircle values resulting from the intercalation of Na+ and K+. The smaller Rct values indicate that the incorporation of alkali cations (Na+ and K+) into the layered skeleton had a promotion effect in providing increased ion conduction and in lowering the interfacial and transfer resistance drastically, thus facilitating the fast kinetics of H+-ion diffusion intake and release from the NMO electrode.24
Fig. 2d displays the first to third CV charge/discharge profiles of the Zn/NMO battery at a scan rate of 0.1 mV s−1, which reveal two reversible redox peaks. Obviously, the cathodic peaks around 1.26 and 1.38 V were related to Zn2+ insertion into the NMO flakes and the reduction of Mn4+ to lower oxidation states, respectively. Two anodic peaks at 1.61 and 1.53 V were assigned to Zn2+ extraction and the reinstatement of Mn4+ during the anodic sweep.47 The current intensity of the anodic peaks at 1.61 V was clearly increased, demonstrating an activation process in the following cycles. The rate capabilities at various current densities for the three batteries are illustrated in Fig. 2e, f and S10.† The Zn/NMO battery possessed comparatively higher capacities than those of the Zn/KMO and Zn/MO at each rate. Also, specific capacities of 335, 281, 229, 194, 169, 148, and 131 mA h g−1 were recorded for the Zn/NMO battery for reversible discharge capacities at 0.5, 1, 2, 3, 4, 5, 6, and 263 mA h g−1 after reverting back to 1 A g−1, respectively. It is noteworthy that the average capacity at 6 A g−1 retained 39.1% of the capacity at 0.5 A g−1, owing to the favorable layered framework that benefits the fast insertion/extraction of electrolyte ions, showing a strong tolerance to the high-speed conversion reaction of the Zn/NMO battery.
The excellent performance could be further seen in the Ragone plots in Fig. 2g. Encouragingly, our battery exhibited a high energy density, while it could also fully discharge within 59 min, which is much faster than conventional Zn/MnO2 batteries, which usually require hours.36 More importantly, the Zn/NMO battery was able to deliver a maximum energy density of 459 W h kg−1 based on the active mass of the NMO nanoflakes at 0.5 A g−1, which considerably exceeded those of recently reported cathode materials in aqueous ZIBs, such as K0.8Mn8O16 nanorods,18 σ-MnO2,20 α-MnO2,15 β-MnO2,17 LixV2O5·nH2O,5 Zn0.25V2O5·nH2O,48 ZnMn2O4,25 and so on.49,50 Also, the Zn/NMO battery still accommodated a relatively high value of 165 W h kg−1 with a large power density of 6890 W kg−1 at a current density of 6 A g−1. The maximum power density of the Zn/NMO battery was ultimately much higher those of the previous report (2760 W kg−1)6 and comparable to the Zn/CMOP battery (6600 W kg−1).51
Additionally, the long-term cycling stability of the Zn/MO, Zn/NMO, and Zn/KMO batteries is evaluated by GCD measurements at a current density of 4 A g−1. As described in Fig. 2h and S10b,† an increment of capacity in the beginning sections of the cycling was observed based on the initial discharge capacity, which was attributed to the gradual activation process of the electrodes, which is very common for Zn/MnO2 batteries in ZnSO4 electrolyte with the addition of MnSO4.18,52,53 Through the EIS measurement results, the reduced charge-transfer impedance (Rct) could be responsible for the electrochemical activation of the Zn/NMO battery during the charge/discharge process, as shown in Fig. S11†.24,53 Remarkably, the reversible discharge capacity retained 157 mA h g−1 with 93% capacity retention of the initial capacity (171 mA h g−1) over 1000 cycles, while a coulombic efficiency around 100% in all cycles substantiated the impressive durability of the NMO cathode in the mild electrolyte. However, the Zn/MO battery showed a capacity fading, which maintained only 135 mA h g−1 after 100 cycles. Therefore, the combination of the superior capacity, energy density, high-rate performance, long cycle life, environmentally friendly nature, and low cost of the NMO electrode materials in the Zn/MnO2 batteries suggests them as an excellent alternative solution for energy storage.
To provide further insights into the ion transport and charge-storage mechanism behind the expansive performance, the electrochemical kinetics process of the Zn/NMO battery was analyzed according to CV experiments. CV curves under different scan rates were well reproducible with cathodic peaks located at 1.26 and 1.38 V and anodic peaks at 1.605 and 1.53 V, respectively (Fig. 3a and S12†). Based on eqn (S2) to (S5),† the electrochemical performance of the Zn/NMO, Zn/KMO, and Zn/MO batteries could be deconvoluted into an ionic diffusion-controlled contribution and surface capacitive contribution to the total charge storage. Fig. 3b and S13† show the linear regression fitted between log(peak i) and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ν from 0.1 to 1 mV s−1. According to the calculation results, the derived b values for peaks 1, 2, and 3 were determined to be 0.51, 0.63, and 0.59, which were near 0.5, respectively. These result identified that the electrochemical reaction of the NMO electrode was controlled by a diffusion process. Consequently, the diffusion-controlled portion contributed 84.1% to the total capacity at a scan rate of 0.1 mV s−1 (Fig. S14a†), providing a fast reaction kinetics. In addition, the diffusion-controlled contribution percents of the NMO and KMO electrodes were almost higher than that of the MO electrode at all scan rates (Fig. S14†). The larger diffusion contribution ratios of the NMO electrode could be attributed to the enhanced ion conduction caused by the intercalated Na+ ions, which was responsible for the high performance of the Zn/NMO battery.
ν from 0.1 to 1 mV s−1. According to the calculation results, the derived b values for peaks 1, 2, and 3 were determined to be 0.51, 0.63, and 0.59, which were near 0.5, respectively. These result identified that the electrochemical reaction of the NMO electrode was controlled by a diffusion process. Consequently, the diffusion-controlled portion contributed 84.1% to the total capacity at a scan rate of 0.1 mV s−1 (Fig. S14a†), providing a fast reaction kinetics. In addition, the diffusion-controlled contribution percents of the NMO and KMO electrodes were almost higher than that of the MO electrode at all scan rates (Fig. S14†). The larger diffusion contribution ratios of the NMO electrode could be attributed to the enhanced ion conduction caused by the intercalated Na+ ions, which was responsible for the high performance of the Zn/NMO battery.
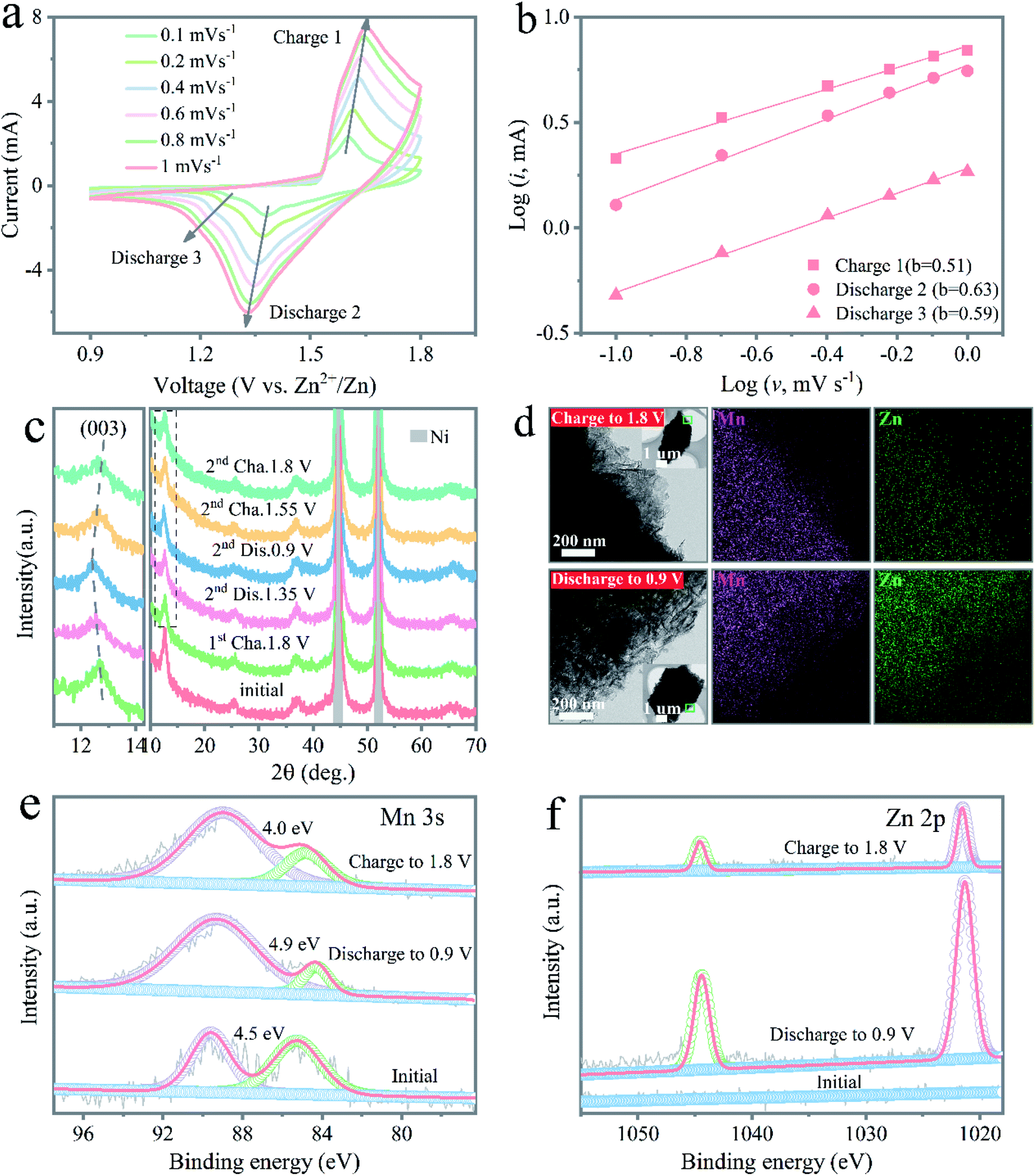 | ||
| Fig. 3 (a) CV curves of the Zn/NMO battery with scan rates from 0.1 to 1 mV s−1. (b) The log(peak current) vs. log(scan rate) plots of three peaks in the CV curves. (c) Ex situ XRD patterns at the selected potential states under a current density of 0.5 A g−1. (d) TEM and EDX element mapping images in the fully discharged and charged states of the NMO electrode. High-resolution XPS spectra of Mn 3s (e) and Zn 2p (f) under initial, fully discharged, and charged states, respectively. | ||
To further explore the reaction mechanism during the charge/discharge process, the structural evolution of the NMO electrode was carefully investigated. Obviously, the ex situ SEM images in Fig. S15† evidence that the NMO nanoflakes retained their original morphology at the discharged state of 0.9 V in the first cycle. Fig. 3c displays the typical charge and discharge curve of the NMO electrode and the selected charge/discharge state for the ex situ XRD measurements in the second cycle at a current density of 0.5 A g−1. Examination of the XRD results revealed that the layered structure could be well maintained with no prominent change for the peaks of the layered NMO electrode during the charge and discharge process, suggesting the stable crystal structure in the course of the charging/discharging, which coincided with previous reports for layered δ-MnO2 cathodes. Fig. S15c† shows that an additional small peak appeared, which refers to the ZnMn2O4 phase in the discharge process. In the charge process, the peak intensity of the ZnMn2O4 phase reversibly disappears at the charge voltage of 1.8 V. The formation of ZnMn2O4 clearly confirmed the insertion of Zn2+ ions into the δ-MnO2 layers. Also, the disappearance of the ZnMn2O4 phase indicated the reversibility and the stable structure of the NMO electrode during charge/discharge cycling.
During the second cycle, the (003) peak was shifted positively at the state of 1.35 V, which was attributed to the insertion of H+ ions, leading to the shrinkage of the crystal lattice, as revealed in Fig. 3c. It was also found that the (003) peak was shifted slightly to a smaller angle after discharge at 0.9 V, due to the insertion of Zn2+ ions, leading to the lattice expansion as the ionic radius of Zn2+ (0.74 Å) is larger than those of Mn ions (0.66 Å of Mn2+ and 0.39 Å of Mn4+).6 Interestingly, the diffraction peaks gradually turned back to the original position after charging, testifying to the highly reversible insertion/extraction of the NMO electrode. These results further confirmed the first H+ and second Zn2+ insertion during discharge. Next, ex situ TEM was also conducted to exhibit the (de)intercalation behaviors of Zn2+ in the NMO, as shown in Fig. 3d. There was no significant change in the nanoflakes for the electrode in the fully discharged and charged states. Therefore, the ex situ XRD and TEM characterizations confirmed that the NMO electrode had a high reversibility in the charge/discharge process.
The high-resolution XPS spectra for the NMO electrode depicted the valence status change of Mn during the charge and discharge process, as shown in Fig. 3e. The Mn 3s core level spectra showed the energy separations of the peaks at 4.5, 4.0, and 4.9 eV for the original, charge, and discharge states, respectively. The distinct change in peak splitting during the charge and discharge process indicated the oxidation state of Mn varied to the reduced states. The corresponding high-resolution spectra of Zn 2p were further used to verify the insertion and extraction process (Fig. 3f). No Zn 2p peak could be found in the pristine NMO electrode, while the two peaks located at 1044.7 and 1021.7 eV were assigned to Zn 2p1/2 and Zn 2p3/2 in the NMO electrode after discharge, which were ascribed to the intercalation of Zn ions. In the following charge process, the reduced intensity of Zn 2p confirmed the successful extraction of Zn2+ from the host framework containing the residue of partial Zn2+ after full charging in the electrode. Obviously, the augmented intensity of the Zn 2p core level spectra in the discharge state demonstrated the successful insertion of Zn2+ after the discharge process, as illustrated in Fig. S16.†
Density functional theory (DFT) calculations were carried out to investigate the influence of the incorporated Na+ on the electrochemical performance of the layered δ-MnO2. As depicted in Fig. 4a and b, a modified interlayer charge of δ-MnO2 by intercalating Na+ was exhibited by the different charge distribution, in which the yellow and cyan electrons describe the charge accumulation and charge depletion, respectively. Through the calculated density of state (DOS) and band structures of the δ-MnO2 and Na+-inserted δ-MnO2 structures, indirect band gaps of 1.23 eV for δ-MnO2 and 0.77 eV for Na+-inserted δ-MnO2 could be observed from the shallow energy level. Fig. 4c and d show the electronic status near the Fermi energy, which mainly consists of Mn 3d orbital states. Also, the electronic density of states close to the Fermi level revealed a new energy state to present a decreased band gap of 37% after intercalating the Na+ (Fig. 4d). The conducted result identified that the bandgap of Na+-inserted δ-MnO2 was narrowed compared with the δ-MnO2 because of the doped alkali cation, which is conducive to the electron transportation from the valence band to the conduction band. That is, there was a significant enhancement of the conductivity for the Na+-inserted δ-MnO2.
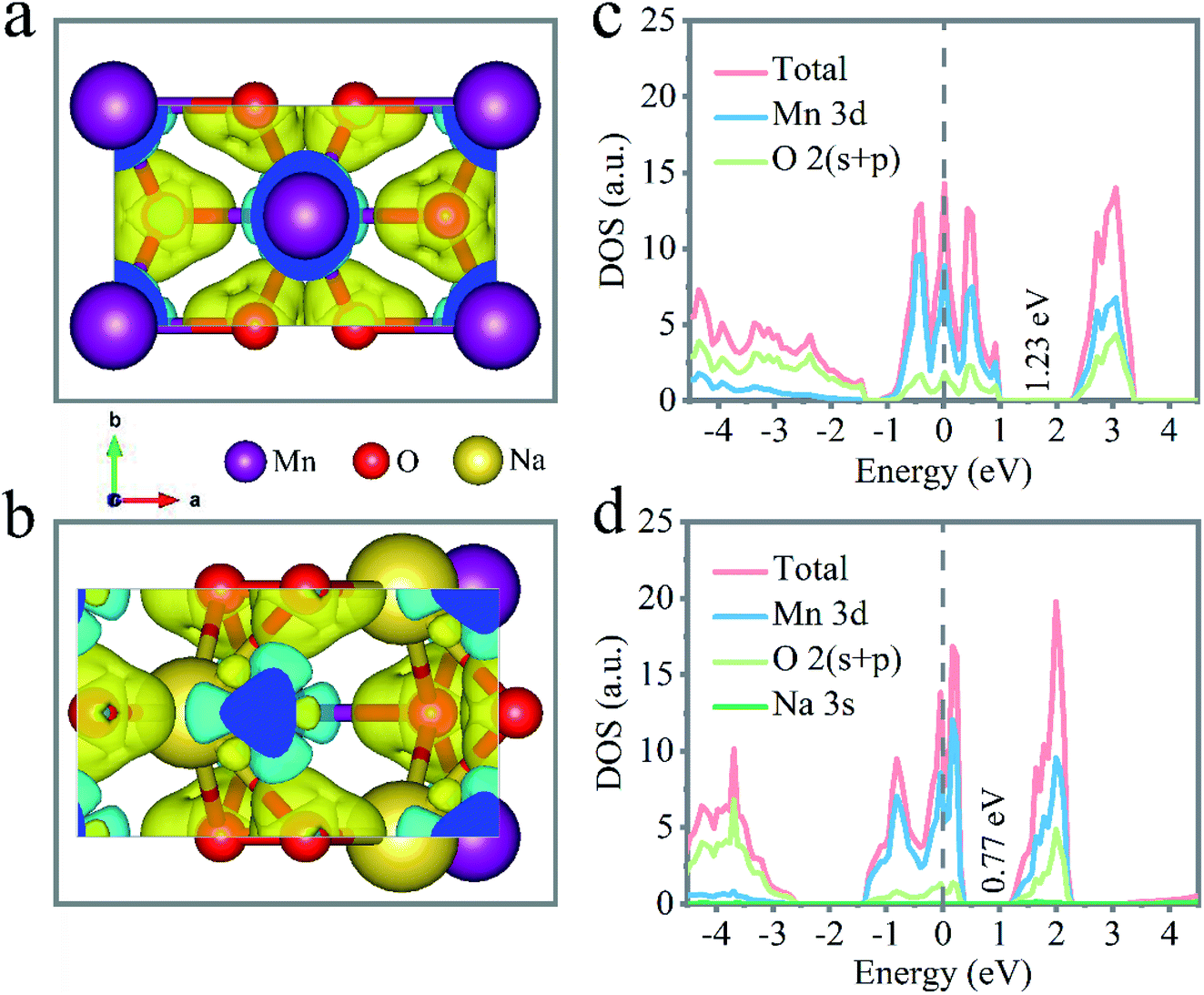 | ||
| Fig. 4 Density functional theory calculations of the layered δ-MnO2 and Na+-intercalated δ-MnO2. Electron density difference of the layered δ-MnO2 (a) and Na+-intercalated δ-MnO2 (b), charge accumulation and depletion described as yellow and cyan, respectively. Total and local density of states of the layered δ-MnO2 (c) and Na+-intercalated δ-MnO2 (d). | ||
The exceptional performance of the aqueous rechargeable Zn/NMO battery has motivated engineers to fabricate flexible quasi-solid-state pouch-type Zn/NMO batteries. With flexible NMO nanoflakes on NF and electrodeposited Zn nanoflakes on CC (Fig. S17†) as the cathode and anode, the electrochemical properties of the assembled Zn/NMO battery sandwiched with PVA/ZnSO4 gel were evaluated. Fig. 5a displays the rate performance of the flexible quasi-solid-state Zn/NMO battery, where a platform at 1.35 V in the charge/discharge profiles is clearly illustrated. It is noteworthy that the quasi-solid-state Zn/NMO battery showed a superior specific capacity of 284 mA h g−1 at 0.5 A g−1, and 120 mA h g−1 at 6 A g−1. The decent rate performance exhibited good reversibility, suggesting a promising application feasibility for energy storage for the solid-state Zn/NMO battery. The reason why the quasi-solid-state Zn/NMO battery possesses a relatively poor specific capacity is mainly due to the slower ion response of the Zn/NMO battery in polymer electrolytes, as certified by the lager charge-transfer resistance evaluated by the electrochemical impedance spectroscopy results (Fig. S18†). Furthermore, the quasi-solid-state Zn/NMO battery possessed promising long cyclic durability with 84.3% capacity retention of the initial capacity and a high coulombic efficiency after 100 cycles at a high current density of 4 A g−1 (Fig. 5b). The notable long-term cycle stability was mainly ascribed to the interfacial phase accessible between the PVA/ZnSO4 gel electrolyte and the electrode as well as the abundant substantial aqueous environment in the sealed pouch from the superior water possessing capability of the gel electrolyte.
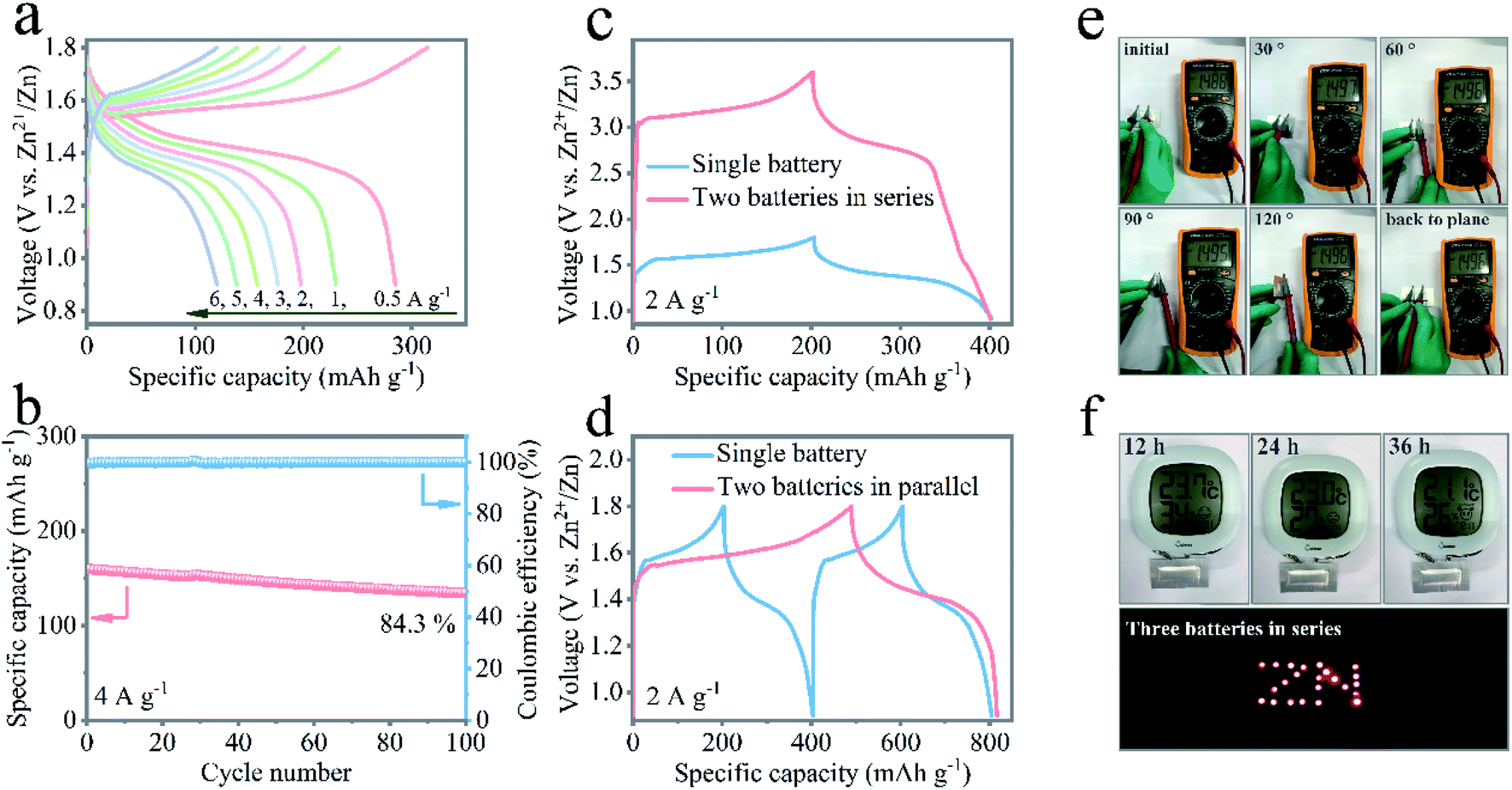 | ||
| Fig. 5 (a) Electrochemical performance of a quasi-solid-state Zn/NMO battery. (b) Cycling stability at a current density of 4 A g−1. Galvanostatic charge/discharge profiles of a single quasi-solid-state Zn/NMO battery and two batteries in series (c) and parallel (d) under a current density of 2 A g−1. (e) Digital photos of the evaluated open circuit potential of a single quasi-solid-state Zn/NMO battery under different bending states. (f) Electronic hygrometer powered by single pouch-type battery with a 4 cm length and 1 cm width for different dwelling hours. The photographs at the bottom right corner depict 23 LEDs powered by three integrated batteries connected in series. | ||
The as-fabricated batteries can be integrated circuits by assembling them in series or parallel to demonstrate a required voltage or current and for the integration of battery devices. While a single battery delivered an output voltage of 1.8 V, the charge/discharge curves of two batteries connected in series exhibited an enlarged 3.6 V potential window, which was double that of a single battery with a similar discharge time at the same current density, as shown in Fig. 5c. When two batteries were jointed in parallel, the discharge capacity was double that of a single device under an identical discharge current density (Fig. 5d). Moreover, when bent at different states, the charged devices displayed decent and unaffected open circuit voltages (Fig. 5e). In addition, a single pouch battery with the size of 4 cm length and 1 cm width was capable of driving an electronic hygrometer over 36 h. Also, three tandem devices could be charged to power 23 light-emitting diode (LED) indicators (Fig. 5f and Movie 1†). Eventually, the quasi-solid-state ZIBs afforded a high capacity output and stable open circuit voltage to deliver the indicators. These findings cast light on the potential of Na-ions modified δ-MnO2 cathode materials for rechargeable Zn/MnO2 batteries in practical applications.
4. Conclusions
Through a facile hydrothermal preparation, three-dimensional interconnected δ-MnO2 nanoflakes with Na+ intercalation were synthesized successfully as a zinc-ion battery cathode. The modified Na+-intercalated δ-MnO2 nanoflakes cathode was endowed with a faster charge-transfer kinetics and greater H+-storage capacity than the pristine δ-MnO2 cathode. Consequently, the rechargeable aqueous Zn/NMO battery delivered a superior reversible capacity of 335 mA h g−1 at 0.5 A g−1, and a high gravimetric energy density, and impressive durability. The assembled quasi-solid-state Zn/NMO battery exhibited a high capacity of 284 mA h g−1 at a current density of 0.5 A g−1, and an excellent integration and practical ability. This research reveals the elevated electrochemical performance of zinc-ion batteries possible through the intercalation of Na+ cations to the δ-MnO2 cathode, providing insights for high-performance aqueous zinc-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51672220), the National Defense Science Foundation (32102060303), the Fundamental Research Funds for the Central Universities of NPU (3102019GHXM002), and the SKLSP Project (2019-TZ-04) of China. We would also like to thank the Analytical & Testing Center of Northwestern Polytechnical University for SEM and XRD test.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed
.
- Z. Wen, M.-H. Yeh, H. Guo, J. Wang, Y. Zi, W. Xu, J. Deng, L. Zhu, X. Wang, C. Hu, L. Zhu, X. Sun and Z. L. Wang, Sci. Adv., 2016, 2, 1600097 CrossRef PubMed
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed
- J. Liu, C. Xu, Z. Chen, S. Ni and Z. X. Shen, Green Energy Environ., 2018, 3, 20 CrossRef
- Y. Yang, Y. Tang, G. Fang, L. Shan, J. Guo, W. Zhang, C. Wang, L. Wang, J. Zhou and S. Liang, Energy Environ. Sci., 2018, 11, 3157 RSC
- C. Zhu, G. Fang, S. Liang, Z. Chen, Z. Wang, J. Ma, H. Wang, B. Tang, X. Zheng and J. Zhou, Energy Storage Mater., 2019, 24, 394 CrossRef
- J. M. Tarascon and M. Armand, Materials for Sustainable Energy, Macmillan Publishers Ltd, 2010, p. 171 Search PubMed
- Z. Le, F. Liu, P. Nie, X. Li, X. Liu, Z. Bian, G. Chen, H. B. Wu and Y. Lu, ACS Nano, 2017, 11, 2952 CrossRef CAS PubMed
- J. Yin, A. B. Brady, E. S. Takeuchi, A. C. Marschilok and K. J. Takeuchi, Chem. Commun., 2017, 53, 3665 RSC
- B. Lin, X. Zhu, L. Fang, X. Liu, S. Li, T. Zhai, L. Xue, Q. Guo, J. Xu and H. Xia, Adv. Mater., 2019, 31, 1900060 CrossRef PubMed
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543 CrossRef CAS PubMed
- C. Xia, J. Guo, P. Li, X. Zhang and H. N. Alshareef, Angew. Chem., Int. Ed., 2018, 57, 3943 CrossRef CAS PubMed
- Y. Cai, F. Liu, Z. Luo, G. Fang, J. Zhou, A. Pan and S. Liang, Energy Storage Mater., 2018, 13, 168 CrossRef
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef PubMed
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef
- Y. Zeng, X. Zhang, Y. Meng, M. Yu, J. Yi, Y. Wu, X. Lu and Y. Tong, Adv. Mater., 2017, 29, 1700274 CrossRef PubMed
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y.-W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518 CrossRef CAS PubMed
- Y. Jin, L. Zou, L. Liu, M. H. Engelhard, R. L. Patel, Z. Nie, K. S. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31, 1900567 CrossRef PubMed
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775 CrossRef CAS PubMed
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643 CrossRef CAS PubMed
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894 CrossRef CAS PubMed
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed
- S. Cai, D. Zhang, L. Shi, J. Xu, L. Zhang, L. Huang, H. Li and J. Zhang, Nanoscale, 2014, 6, 7346 RSC
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Solid State Ionics, 2003, 159, 345 CrossRef CAS
- A. Adomkevicius, L. Cabo-Fernandez, T.-H. Wu, T.-M. Ou, M.-G. Chen, Y. Andreev, C.-C. Hu and L. J. Hardwick, J. Mater. Chem. A, 2017, 5, 10021 RSC
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915 RSC
- W. Qiu, Y. Li, A. You, Z. Zhang, G. Li, X. Lu and Y. Tong, J. Mater. Chem. A, 2017, 5, 14838 RSC
- S. Chen, G. Liu, H. Yadegari, H. Wang and S. Z. Qiao, J. Mater. Chem. A, 2015, 3, 2559 RSC
- C. Yang, Y. Tang, Y. Tian, Y. Luo, M. Faraz Ud Din, X. Yin and W. Que, Adv. Energy Mater., 2018, 8, 1802087 CrossRef
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733 RSC
- M. Chamoun, W. R. Brant, C.-W. Tai, G. Karlsson and D. Noréus, Energy Storage Mater., 2018, 15, 351 CrossRef
- W. Chen, G. Li, A. Pei, Y. Li, L. Liao, H. Wang, J. Wan, Z. Liang, G. Chen, H. Zhang, J. Wang and Y. Cui, Nat. Energy, 2018, 3, 428 CrossRef CAS
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823 CrossRef CAS PubMed
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933 CrossRef CAS PubMed
- S.-D. Han, S. Kim, D. Li, V. Petkov, H. D. Yoo, P. J. Phillips, H. Wang, J. J. Kim, K. L. More, B. Key, R. F. Klie, J. Cabana, V. R. Stamenkovic, T. T. Fister, N. M. Markovic, A. K. Burrell, S. Tepavcevic and J. T. Vaughey, Chem. Mater., 2017, 29, 4874 CrossRef CAS
- D. D. Edwards, J. H. Hwang, S. J. Ford and T. O. Mason, Solid State Ionics, 1997, 99, 85 CrossRef CAS
- K. W. Nam, H. Kim, J. H. Choi and J. W. Choi, Energy Environ. Sci., 2019, 12, 1999 RSC
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121 CrossRef CAS
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS
- J. Zhao, H. Ren, Q. Liang, D. Yuan, S. Xi, C. Wu, W. Manalastas, J. Ma, W. Fang, Y. Zheng, C.-F. Du, M. Srinivasan and Q. Yan, Nano Energy, 2019, 62, 94 CrossRef CAS
- V. Soundharrajan, B. Sambandam, S. Kim, V. Mathew, J. Jo, S. Kim, J. Lee, S. Islam, K. Kim, Y.-K. Sun and J. Kim, ACS Energy Lett., 2018, 3, 1998 CrossRef CAS
- X. Zhang, S. Wu, S. Deng, W. Wu, Y. Zeng, X. Xia, G. Pan, Y. Tong and X. Lu, Small Methods, 2019, 1900525 CrossRef CAS
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef PubMed
- J. Hao, J. Mou, J. Zhang, L. Dong, W. Liu, C. Xu and F. Kang, Electrochim. Acta, 2018, 259, 170 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02556a |
| This journal is © The Royal Society of Chemistry 2020 |