
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of azole drugs on energetics, kinetics, and ligand migration pathways of CO photo-dissociation in bacterial flavohemoglobins†
David Butcher‡
a,
Myriam Moussaouib,
Laura Bacioub and
Jaroslava Miksovska *ac
*ac
aDepartment of Chemistry and Biochemistry, Florida International University, Miami, FL, USA. E-mail: miksovsk@fiu.edu; Tel: +1-305-348-7406
bLaboratoire de Chimie Physique, UMR8000, Université Paris Sud, CNRS, Université Paris Saclay, 91405 Orsay, France
cBiomolecular Sciences Institute, Florida International University, Miami, FL, USA
First published on 10th May 2020
Abstract
Flavohemoglobins (fHbs) are heme proteins found in prokaryotic and eukaryotic microbes. They are involved in NO detoxification through an NO˙ dioxygenase mechanism. The N-terminal heme globin domain allows for binding of gaseous ligands whereas a C-terminal NADH/FADH binding domain facilitates association of redox cofactors necessary for ligand reduction. The NO˙ dioxygenase function is important in facilitating immune resistance by protecting the cell from nitrosative stress brought about by a host organism; as a result, bacterial flavoHbs have recently been considered as targets for the development of new antibiotics. Here, photoacoustic calorimetry and transient absorption spectroscopy have been used to characterize energetics, structural dynamics, and kinetics of CO migration within bacterial flavoHbs from Ralstonia eutropha (FHP) and Staphylococcus aureus (HMPSa) in the presence and absence of antibiotic azole compounds. In FHP, the ligand photo-release is associated with ΔH = 26.2 ± 7.0 kcal mol−1 and ΔV = 25.0 ± 1.5 mL mol−1 while in HMPSa, ΔH = 34.7 ± 8.0 kcal mol−1 and ΔV = 28.6 ± 17 mL mol−1 were observed, suggesting distinct structural changes associated with ligand escape from FHP and HMPSa. In the presence of ketoconazole, the CO escape leads to a more negative enthalpy change and volume change whereas association of miconazole to FHP or HMPSa does not impact the reaction volume. These data are in agreement with the computational results that propose distinct binding sites for ketoconazole and miconazole on CO bound FHP. Miconazole or ketoconazole binding to either protein has only a negligible impact on the CO association rates, indicating that azole drugs do not impact flavoHbs interactions with gaseous ligands but may inhibit the NOD activity through preventing the electron transfer between FAD and heme cofactors.
Introduction
Flavohemoglobins (fHbs) belong to a family of heme proteins found in a large number of modern microbes. fHb structure consists of the natural fusion of an α-helical heme-binding domain with a typical heme globin structure and a reductase domain which contains the FAD cofactor and the binding site of the co-substrate NADH. fHbs exhibit a nitric oxide dioxygenase (NOD) activity which render the microorganism resistant to NO˙ toxicity by converting it, at the heme active site, into nitrate (NO3−) in the presence of O2 as co-substrate. This is facilitated by the electron-donating cofactors bound to the reductase domain.1 The NOD function has primarily been implicated in NO˙ signaling modulation and in protection of pathogenic bacteria from NO˙ produced by phagocytes within the immune systems of host organisms.2 NO˙ is a free radical that plays an important role in mammalian innate immunity by damaging the bacterial cell by direct nitrosylation of enzymes and nucleic acids3 or by decomposition to peroxynitrite and other reactive oxygen species (ROS).4 Therefore, NOD activity of fHbs is responsible for enhanced bacterial survival under oxidative stress conditions.3 Having no counterpart in humans, their protective role against nitrosative stress makes microbial flavohemoglobins attractive as targets for antibiotic design.Two NOD mechanisms have been proposed for fHbs. According the first mechanism, O2 diffuses into the heme binding pocket and binds to the ferrous heme iron, after which NO binds into the active site where dioxygenation occurs, oxidizing the heme iron to the ferric form and forming an unstable peroxynitrite intermediate which decays to nitrate.2 According to the second mechanism, NO binds to the ferric heme iron first, forming Fe(III)–NO intermediate that is then reduced to Fe(II)–NO and reacts with oxygen to form nitrate.5 In both mechanisms, the diffusion of gaseous diatomic molecules through the protein matrix into the heme binding pocket and reduction of the heme iron by NADH and FAD are essential for the NOD activity of fHbs.
The enzymatic active site is located in the N-terminal globin domain that provides a binding site for heme b through coordination to a proximal histidine residue, His 85. The overall fold of the globin domain strongly resembles to well-studied mammalian globins. However, as reported by Ermler et al., the angle between the distal and proximal helices is increased and the helix E is displaced from the G and H helices leading to a larger distal cavity that is occupied by a cyclopropanated phospholipid.6 In addition, non polar residues Phe28, Met32, Phe43 and Val98 form a hydrophobic cavity in the vicinity of the active site that may serve as a binding site for gaseous ligands, namely NO7.
NOD activity of fHbs is strongly inhibited by azole-based antifungal drugs including ketoconazole, miconazole, and econazole and combination of such inhibitors with quinones constitute a potential antibacterial chemotherapy.8,10 Azole drugs bind to fHbs from Cupriavidus necator (FHP) – formerly known as Ralstonia eutropha – and Staphylococcus aureus (HMPSa) with a relatively high affinity.9 The crystal structures for ketoconazole, miconazole, and econazole bound to the Fe(III) form of the protein demonstrate that azole drugs (Fig. 1) are able to enter the distal heme pocket where the imidazole group of the drug forms a coordination bond with the heme iron.11 Interestingly, structural analyses also showed that FHP can adopt two distinct conformations depending on the azole drug. While the miconazole-bound FHP shares the same structure with the ligand free protein (so-called open state with respect to the NADH accessibility), in the ketoconazole or econazole-bound fom, FHP undergoes a rigid body motion of the NADH binding domain accompanied by conformational changes in the C-terminal arm of the protein and the helix E and CE loop from the heme binding domain leading to a so-called closed state.
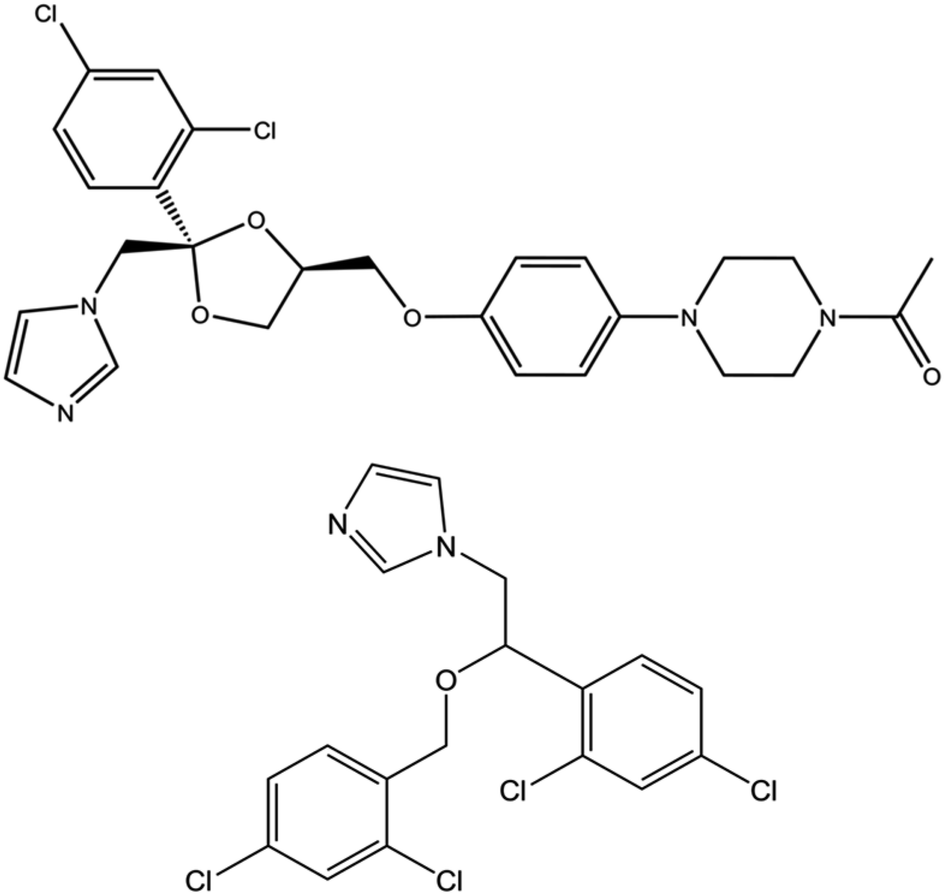 | ||
| Fig. 1 Chemical structures of ketoconazole (top) and miconazole (bottom). | ||
As the enzymatic activity of fHb involves migration of gaseous substrate into the heme bindin pocket, here we aim to provide insight into the impact of azole drugs on the conformational dynamics and structural energetics of fHbs. With this in aim we have combined experimental and molecular dynamic approach to characterize the mechanism of ligand migration between the heme binding pocket and surrounding solvent in the drug free and drug bound fHbs.
Methods and materials
Expression and purification of wild-type FHP (P39662) and HMPSa (A0A0E1ACT8) was carried out as described previously9,11 and the enzymatic activity of the purified proteins was assessed by probing quinone reduction.10 Ketoconazole and miconazole nitrate were obtained from Sigma-Aldrich and used without further purification. Protein samples were stored at −20 °C prior to sample preparation.Protein samples were prepared by diluting a freshly-melted aliquot of the protein stock with the sample buffer, 10 mM sodium phosphate and 10 mM KCl buffer, pH 7.5. For samples in the presence of ketoconazole and miconazole, the final drug concentrations in the sample buffer were 8.5 μM for ketoconazole and 408 μM for miconazole13 at 20 °C. Low molar solubility of ketoconazole in aqueous solution at neutral pH, 1.4 × 10−5,12 restricted the amount of the drug used in this study. The concentration of miconazole was selected to have 99% of fHbs in the miconazole bound form as described below. Final protein concentrations were 20 μM for drug-free and miconazole-containing samples and 9 μM for samples with ketoconazole. Protein samples were purged of dissolved gases by a flow of Ar run over the surface of the liquid (Airgas, Inc.) reduced using a few microliters of freshly-prepared sodium dithionite solution (1 mM) and thoroughly flushed with carbon monoxide (Airgas, Inc.). Formation of the reduced and CO-bound protein forms was confirmed by UV-vis spectroscopy (single beam spectrophotometer Cary50, Varian).
Photoacoustic calorimetry technique (PAC)
The set-up of the PAC instrumentation has been described previously.14 For PAC measurements, samples were transferred to a 1.0 cm by 0.5 cm Spectrosil quartz cuvette (Starna Cells) and placed in a temperature-controlled sample holder (TC 125, Quantum Northwest). A piezoelectric transducer (Panametrics RV 103, 1 MHz) was attached to the side of the cuvette using a thin layer of honey. A frequency-doubled Nd:YAG laser operating at 532 nm with a 7 ns pulse width (Continuum Surelite I) was used to excite the sample through the 0.5 cm cuvette path with a power of <150 μJ. The resulting photoacoustic wave was amplified by an ultrasonic preamplifier (Panametrics 5662) and sent to a digital oscilloscope (WaveSurfer 42Xs). The same process was carried out for the reference compound Fe(III)meso-tetra(4-sulfonatophenyl)porphine chloride (Fe[III]4SP, Frontier Scientific).The details of PAC data analysis have been published previously.14 Briefly, the following equation is used to plot the PAC data:
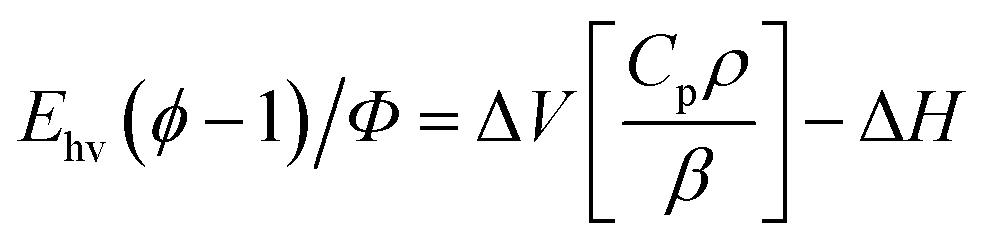 | (1) |
Transient absorption spectroscopy (TA)
Kinetics and quantum yields for CO rebinding to FHP and HMPSa were determined using a custom transient absorption instrument. Samples were transferred to a 1.0 cm by 0.2 cm Spectrosil quartz cuvette (Starna Cells) and placed in a temperature-controlled sample holder (TC 125, Quantum Northwest). Photo-dissociation of CO was initiated by the 532 nm output from Nd:YAG laser (7 ns pulse width, 20 mJ laser power). A 447 nm diode-pumped solid state laser (MDL-III-447, Changehun New Industries Optoelectronics Tech Co. Ltd.) was used as the probe beam, being directed through the sample cell and a monochromator (Jobin-Yvon H20). The probe beam intensity was detected using an amplified photodiode (PDA 10A, Thorlabs) and digitized. Kinetics for CO rebinding were determined by fitting the data with multiple exponentials decay model (OriginPro 8, OriginLab Corp.). Quantum yields for bimolecular rebinding were determined by comparison to CO-bound horse myoglobin as previously described.15Molecular dynamics simulations
We conducted cMD simulations of FHP in NAMD 2.11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 using CHARMM22 force field parameters.17 All simulations were run on a custom Fedora 23 molecular dynamics PC with an overclocked Core i7-6700K processor and Nvidia GeForce GTX 970 graphics card. FHP structures are only known in complex with bulky phospholipid or azole inhibitors which bias the size/shape of the substrate binding pocket.8 To generate the structure for simulation of ketoconazole bound to CO-FHP, the crystal structure containing ketoconazole (PDB ID 3OZW)11 was chosen in which the conformation of the C-terminal arm and helix E causes the more encapsulated heme distal pocket, known as the closed state. For simulation of miconazole bound to CO-FHP, the crystal structure of FHP containing miconazole (PDB ID 3OZU)11 in the open state was used. Carbon monoxide was added to the heme pocket in close proximity (∼2.4 Å) to the heme iron using YASARA.18 Structures were placed into a cubic water box with ≥5 Å margins around the protein surface and periodic boundary conditions. Final structures were generated using the PSFgen module of VMD 1.9.2.19 Protein structures were minimized, followed by slow heating to 310 K and short equilibrations in the canonical (NVT) and isothermal–isobaric (NPT) ensembles. Finally, a 20 ns production run was conducted in the NPT ensemble.
16 using CHARMM22 force field parameters.17 All simulations were run on a custom Fedora 23 molecular dynamics PC with an overclocked Core i7-6700K processor and Nvidia GeForce GTX 970 graphics card. FHP structures are only known in complex with bulky phospholipid or azole inhibitors which bias the size/shape of the substrate binding pocket.8 To generate the structure for simulation of ketoconazole bound to CO-FHP, the crystal structure containing ketoconazole (PDB ID 3OZW)11 was chosen in which the conformation of the C-terminal arm and helix E causes the more encapsulated heme distal pocket, known as the closed state. For simulation of miconazole bound to CO-FHP, the crystal structure of FHP containing miconazole (PDB ID 3OZU)11 in the open state was used. Carbon monoxide was added to the heme pocket in close proximity (∼2.4 Å) to the heme iron using YASARA.18 Structures were placed into a cubic water box with ≥5 Å margins around the protein surface and periodic boundary conditions. Final structures were generated using the PSFgen module of VMD 1.9.2.19 Protein structures were minimized, followed by slow heating to 310 K and short equilibrations in the canonical (NVT) and isothermal–isobaric (NPT) ensembles. Finally, a 20 ns production run was conducted in the NPT ensemble.
Autodock Vina version 1.1.2 was used to dock ketoconazole and miconazole to a structure derived from CO-FHP simulations. The search space for docking was a cube with 40 Å sides centered on the distal heme pocket and encompassing the majority of the globin domain and the portion of the reductase domain adjacent to the heme binding pocket. The lowest-energy binding conformations for both drugs were extracted and combined with the CO-FHP 10 ns structure using VMD. CHARMM force field parameters for the two drugs were generated using SwissParam.21 The simulations of azole drugs bound to CO-FHP were run for 20 ns and interactions between the protein and bound azole drug were analyzed using the Protein–Ligand Interaction Profiler web server22 and VMD.
The coordinates of the initial drug-free CO-FHP simulation after 10 ns were also used to generate the initial structure for a simulation in which CO was no longer bound to the heme iron and allowed to move freely within the protein matrix. A locally enhanced sampling (LES)23 technique was used to duplicate the CO ligand 20 times to allow for the observation of multiple ligand diffusion pathways. A 30 ns production run was carried out in the NPT ensemble, during which the dissociation of all 20 enhanced ligands from the protein matrix was observed. Further analyses were conducted using VMD.
Results
UV-vis spectroscopy analyses of CO-bound fHbs
The absorption spectra for met-, deoxy-, and CO-FHP in the absence of azole drugs are shown in Fig. 2, top panel. The spectra resemble the absorption spectra of other penta-coordinate heme proteins with the Soret band of met-FHP located at 395 nm and additional Q bands situated at 458 nm, 486 nm and 646 nm. Upon heme iron reduction, the Soret band shifts to 437 nm and a new band appears at 563 nm. The Soret band for CO-FHP is narrow with an absorption maximum at 424 nm and two Q bands at 540 nm and 572 nm consistent with heme iron coordination to the proximal histidine with CO at the distal position. Upon ketoconazole addition, the Soret band for met-FHP partially undergoes a slight red shift to 411 nm with a shoulder located at 396 nm (Fig. 2, central panel) compared to the spectrum without any drug. Such shift indicates heme iron coordination with the imidazole group of ketoconazole. The coordination of ketoconazole with the heme iron is more evident from the absorption spectrum of deoxy-FHP that exhibits the Soret band maximum at 430 nm and Q band at 560 nm with a shoulder at 530 nm, as observed previously for vertebrate hexa-coordinate globins with histidine as the proximal and distal ligands. This absorption spectrum is consistent with a fraction of deoxy-FHP containing heme iron in a low spin, hexa-coordinated state. Binding of CO to deoxy-FHP in the presence of ketoconazole leads to an absorption spectrum that is identical to that of CO-FHP in the absence of the drug, indicating that the presence of the azole drug has a negligible impact on the electronic structure of the CO-bound FHP heme group.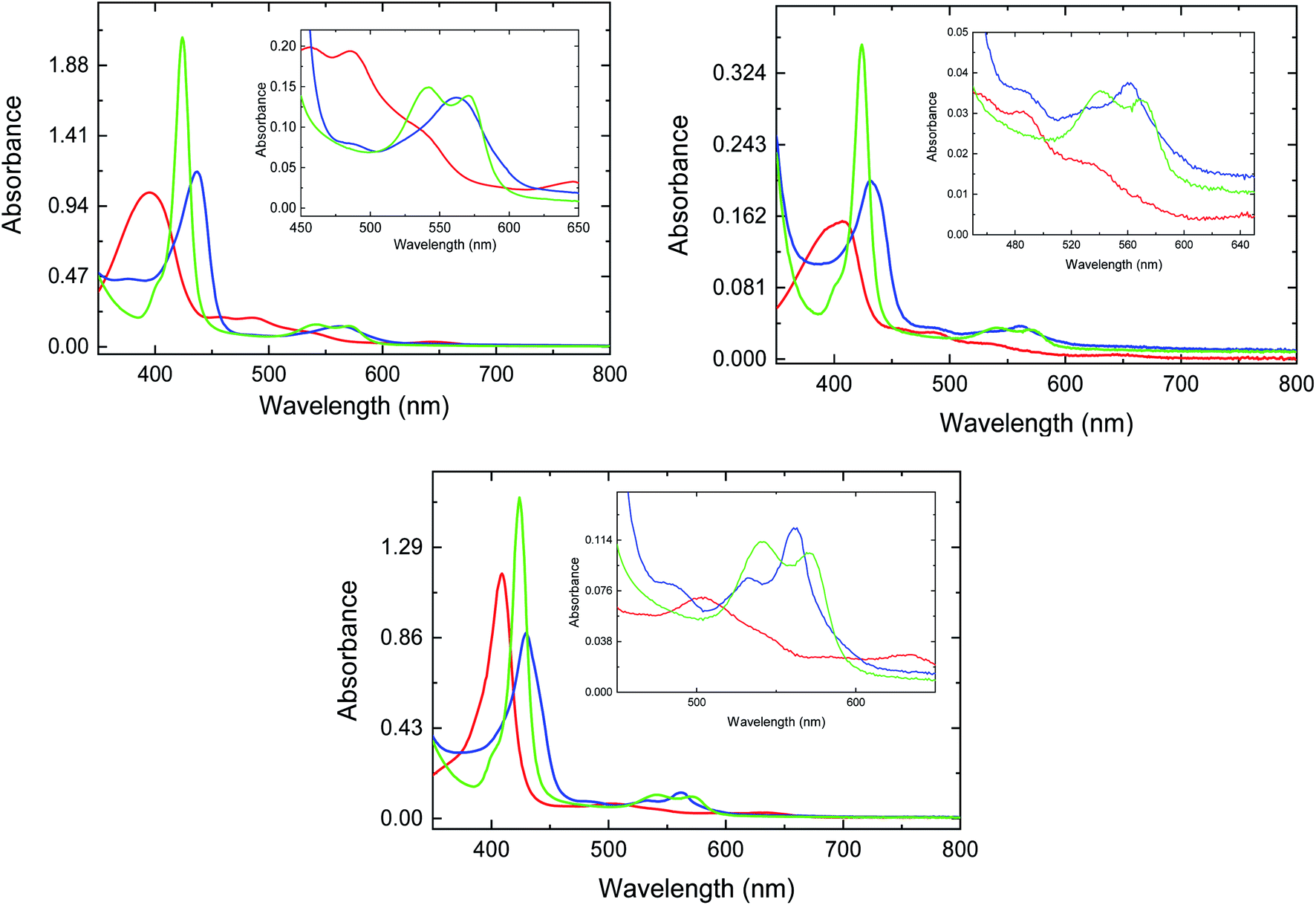 | ||
| Fig. 2 UV-vis traces for met- (red line), deoxy- (blue line), and CO-FHP (green line) in the absence (top panel) and presence of ketoconazole (center panel) and miconazole (bottom panel). | ||
The absorption spectra of FHP measured in the presence of 408 μM miconazole (Fig. 2, bottom panel) are analogous to those determined for FHP in the presence of ketoconazole. Notably, the absorption spectrum of deoxy-FHP shows a Soret band maximum at 430 nm and two Q bands at 532 nm and 562 nm, indicating that the major fraction of protein contains hexa-coordinated heme iron with the imidazole group of miconazole as the sixth axial ligand. Based on the dissociation constants (KD) that have been determined previously for ketoconazole and miconazole,10 it can be estimated that approximately 34% and 99% of FHP is in the drug-bound form, respectively. The absorption spectra of HMPSa in the absence and presence of the azole drugs are analogous to those determined for FHP indicating similar interactions between the azole drugs and the heme iron (data not shown).
Thermodynamic characterization of CO photodissociation
To characterize the impact of azole drug association to the heme distal pocket on the interactions of FHP and HMPSa with diatomic ligands, the dynamic and energetics of structural changes associated with ligand dissociation form heme proteins on nanosecond to microsecond timescale were investigated using PAC. Photoacoustic traces for CO photo-release from FHP in the absence of azole drugs are shown in Fig. S1.† The absence of a phase shift between the acoustic wave for the sample and the acoustic wave for the reference indicates that the photolysis of the CO–Fe bond and the subsequent ligand escape from the protein matrix into surrounding solvent is fast (τ < 50 ns) for drug free proteins as well as for FHP and HMPSa in the presence of ketoconazole and miconazole (data not shown).As described in the Methods and materials section, the volume and enthalpy change associated with ligand escape from FHP and HMPSa was determined by plotting the ratio of the amplitude of the sample acoustic wave as a function of the temperature dependent factor (Cpρ/β) as shown for CO photo-release from FHP and HMPSa in the presence and absence of azole drugs in Fig. 3.
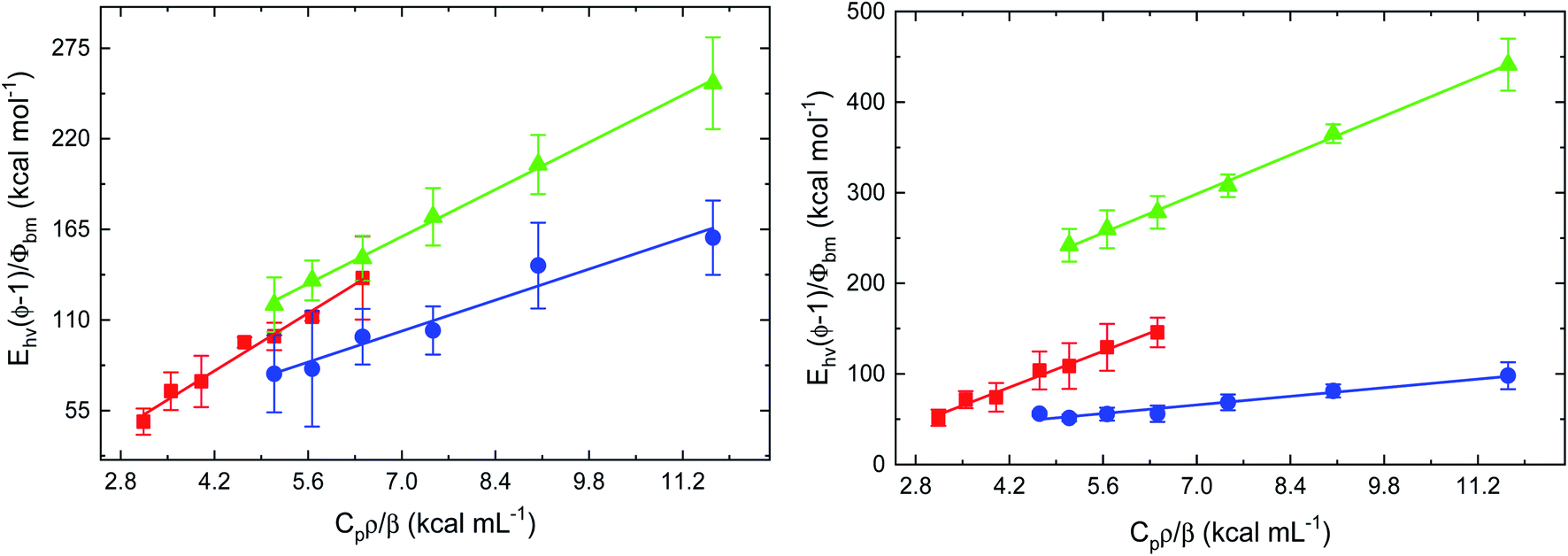 | ||
| Fig. 3 Plots of Ehν(ϕ − 1)/Φbm vs. Cpρ/β for photo-dissociation of CO from CO-FHP (left) and CO-HMPSa (right) in the absence of drug compounds (red), with 8.5 μM ketoconazole (blue), and with 408 μM miconazole (light green). | ||
For both proteins, the plots were analyzed according to eqn (1) and the extrapolated volume and enthalpy changes are listed in Table 1. For FHP and HMPSa, the volume and enthalpy changes accompanying CO photo-release from the heme iron and subsequent ligand escape from the protein matrix are large and positive, pointing towards similar structural changes due to the transition from CO bound, hexa-coordinate protein to ligand free, penta-coordinate protein (see below). CO photo-dissociation in the presence of ketoconazole leads to an exothermic enthalpy change of −9.0 ± 11.7 kcal mol−1 and a smaller volume change of 13.5 ± 1.5 mL mol−1.
| Protein | Drug identity | Drug conc. 20 °C (μM) | Percent drug-bound | ΔH (kcal mol−1) | ΔV (mL mol−1) | Φbm (20 °C) |
|---|---|---|---|---|---|---|
| CO-FHP | None | 0 | 0 | 26.2 ± 7.0 | 25.0 ± 1.5 | 0.50 |
| Ketoconazole | 8.5 | 34 | −9.0 ± 11.7 | 13.5 ± 1.5 | 0.41 | |
| Ketoconazole | 500* | 98* | −27.1* | 7.8* | 0.36* | |
| Miconazole | 409 | 99 | −17.4 ± 3.4 | 20.4 ± 0.4 | 0.64 | |
| CO-HMPSa | None | 0 | 0 | 34.7 ± 8.0 | 28.6 ± 1.7 | 0.31 |
| Ketoconazole | 8.5 | 32 | −12.9 ± 3.4 | 7.4 ± 0.4 | 0.52 | |
| Ketoconazole | 500* | 98* | −35.5* | −2.7* | 0.62* | |
| Miconazole | 409 | 99 | −83.9 ± 5.4 | 30.7 ± 0.7 | 0.34 |
The fraction of the protein in the drug bound form, f, depends on the equilibrium dissociation constant, KD, and the amount of drug used, [Lt]. Assuming a single binding site for azole drug binding, the fraction (f) was calculated employing an eqn (2):
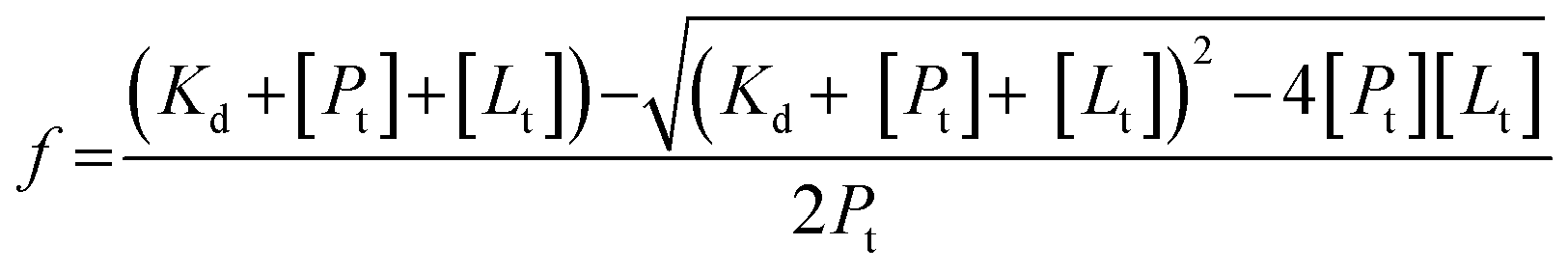 | (2) |
Using the values for equilibrium dissociation constant for ketoconazole binding to FHP and HMPSa 10 μM and 12 μM, respectively,9 and values for miconazole binding to FHP and HMPSa 2.6 μM and 3.7 μM, respectively,9 33% of FHP and HMPSa is in the ketoconazole bound form and 99% of both protein is in the azole drug bound form. In such case, the observed volume or enthalpy change can be expressed using eqn (3) and (4), assuming a single binding site for ketoconazole as observed in the crystal structure:10
| ΔV = ffreeΔVfree + fboundΔVbound | (3) |
| ΔH = ffreeΔHfree + fboundΔHbound | (4) |
Using the equations above, the reaction volume and enthalpy change for CO photo-dissociation from FHP or HMPSa saturated (ΔVbound & ΔHbound) with ketoconazole were calculated and are listed in Table 1. Interestingly, the volume and enthalpy changes associated with CO photo-dissociation from FHP or HMPSa in the presence of miconazole differ from those determined in the presence of ketoconazole suggesting a difference between the structures of drug-bound FHP and HMPSa in the deoxy- and/or CO-bound form. Association of ketoconazole leads to a negative enthalpy change and smaller volume change for CO dissociation from the protein, whereas in the presence of miconazole, the enthalpy change is more negative whereas the volume change for CO photo-dissociation is analogous to that observed in the absence of drug.
Kinetics of geminate and bimolecular rebinding of CO
In addition to characterizing the reaction thermodynamic parameters, the rate constants for bimolecular rebinding of CO to FHP or HMPSa in the absence/presence of azole drugs were determined using TA (Table 2). The transient absorption traces at 447 nm for CO rebinding to fHbs (CO rebinding to deoxy-FHP in the absence of azole drugs is presented in Fig. S2†) were analyzed using a multi-exponential decay model. For FHP, CO rebinding to the heme iron is tri-exponential with rate constants for bimolecular ligand rebinding of 2.00 μM−1 s−1, 0.18 μM−1 s−1, and 0.06 μM−1 s−1. Association of miconazole does not impact these rates while, the presence of ketoconazole increases the rates for CO rebinding two times. These results suggest that association of ketoconazole decreases the activation barrier for ligand rebinding likely through increasing accessibility of the heme-binding pocket. The heterogeneous ligand rebinding in the absence and presence of the drugs indicates that the protein can adopt multiple conformations with each conformation having a distinct rate constant for CO rebinding and/or there are several ligand migration pathways with distinct rate constants for CO rebinding (vide infra).| Protein | Drug identity | Drug conc. (μM) | Percent drug-bound | agem | kgem (μs−1) | a1 | k1 (μM−1 s−1) | a2 | k2 (μM−1 s−1) | a3 | k3 (μM−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CO-FHP | None | 0 | 0 | N/A | 0.26 ± 0.07 | 2.00 ± 0.07 | 0.50 ± 0.01 | 0.18 ± 0.02 | 0.24 ± 0.05 | 0.06 ± 0.01 | |
| Ketoconazole | 8.5 | 34 | 0.15 ± 0.02 | 3.43 ± 0.45 | 0.27 ± 0.07 | 0.49 ± 0.13 | 0.57 ± 0.09 | 0.12 ± 0.02 | |||
| Miconazole | 409 | 99 | 0.24 ± 0.02 | 2.00 ± 0.10 | 0.36 ± 0.04 | 0.26 ± 0.01 | 0.39 ± 0.02 | 0.05 ± 0.001 | |||
| CO-HMPSa | None | 0 | 0 | 0.39 ± 0.01 | 0.11 ± 0.002 | 0.32 ± 0.01 | 2.72 ± 0.05 | 0.29 ± 0.01 | 0.93 ± 0.01 | N/A | |
| Ketoconazole | 8.5 | 32 | 0.41 ± 0.01 | 0.14 ± 0.02 | 0.12 ± 0.01 | 6.54 ± 0.96 | 0.47 ± 0.02 | 1.5 ± 0.05 | |||
| Miconazole | 409 | 99 | 0.38 ± 0.01 | 0.11 ± 0.004 | 0.32 ± 0.01 | 1.24 ± 0.03 | 0.30 ± 0.01 | 0.31 ± 0.01 | |||
In the case of CO rebinding to HMPSa, an additional phase with a time constant of ∼9 μs was detected. This phase represents a geminate CO rebinding to the heme iron as the time constant for this phase is independent of CO concentration (data not shown). In a similar way as observed for FHP, the presence of azole drugs has only a minor impact on the rate constants for CO rebinding to HMPSa, with CO rebinding being faster for ketoconazole-bound HMPSa and approximately two times slower in the presence of miconazole.
CO migration pathways investigated by classical molecular dynamics
Using locally enhanced sampling in NAMD 2.11, migration of CO ligands from the heme binding pocket into the protein exterior was observed. All enhanced 20 CO molecules were tracked throughout the 30 ns MD simulation to determine their exit pathway from the protein matrix. 21 separate CO escape events and 2 entries of CO into the protein matrix were observed via six different ligand migration pathways, designated A through F. Details about these pathways are found in Table 3 and the two pathways with the most escape events, A and E, are shown in Fig. 4. Pathway A directly leads from the distal heme cavity to the bulk solvent and passes between residues Leu103 and Tyr126 on the G and H helices, respectively. These residues are located in the vicinity of the ring A of the prosthetic group. Pathway A is by far the most prominent with 13 of 21 escape events occurring this way. Pathway B passes between the A-helix and GH-loop in the vicinity of Thr13, Val16, Leu100, and Ile118 with one escape through this path. Pathway C is between the AB-loop and E-helix with two escape events. Pathway D is close to the EF-loop and passes between Ala63 and Val77 close to Arg59 with one escape event. Pathway E passes between the heme propionate groups and CD loop and resembles to the canonical “distal His gate” escape pathway described for proteins such as hemoglobin and myoglobin.24 The critical residues for this pathway include Asn44 and Arg48 and the nearby heme-6-propionate group. Three escapes are seen to occur through pathway E, with escaped ligands temporarily residing in a hydrophobic area between the globin and reductase domains adjacent to Ile371, Val395, and Pro398. From here, the pathway becomes bifurcated with one CO molecule escaping directly to the bulk solvent while two move into a hydrophobic pocket in the reductase domain bordered by Phe396, Pro280, Phe259, Ser209, Gly188, Met284, Tyr190, Glu394, and Cys368 and escape to the solvent later. The final pathway F is close to the N-terminus, adjacent to the H-helix and EF-loop with only one escape event.| Pathway designation | Critical residues | Number of CO escapes from globin domain | Escape percentage |
|---|---|---|---|
| A | L103, Y126 | 13 | 62 |
| B | T13, V16, L110, I118 | 1 | 5 |
| C | G21, Q54 | 2 | 10 |
| D | A63, V77, R59 | 1 | 5 |
| E | N44, Q48, I371, V395, P398 | 3 | 14 |
| F | M1, M75, L78, L129 | 1 | 4 |
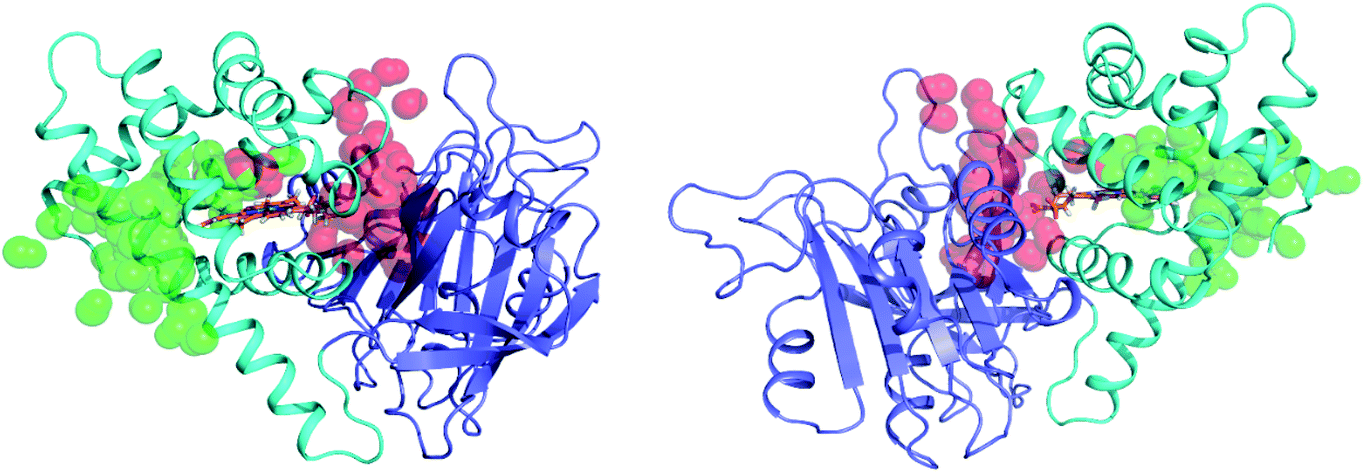 | ||
| Fig. 4 Coordinates of LES-enabled CO molecules immediately before and after escape from FHP. CO molecules escaping through pathways A and E are shown in green and red respectively. The globin domain is shown in cyan, the reductase domain is shown in blue, and heme is shown in orange. | ||
Binding sites of ketoconazole and miconazole in FHP
Autodock Vina20 was used to predict the binding sites for ketoconazole and miconazole in the CO-bound FHP and the residues forming binding sites are shown in Fig. S3 and S4,† respectively. The highest-affinity binding sites for ketoconazole are found in the hydrophobic cleft between the globin and reductase domains, directly alongside the heme propionate groups and directly adjacent to the flavin group of FAD (Fig. 5). Ketoconazole is in close proximity with the hydrophobic residue Ile371 and the oxygen of the terminal acetyl group is well-positioned to form a hydrogen bond with Gln55 while the dichlorophenyl group appears to form cation-π interactions with Arg206. This position is adjacent to the binding site of ketoconazole resolved in the crystal structure of ligand-free FHP in the met form, in which the drug was located partially inside the distal heme pocket with its imidazole group directly coordinating the heme iron.11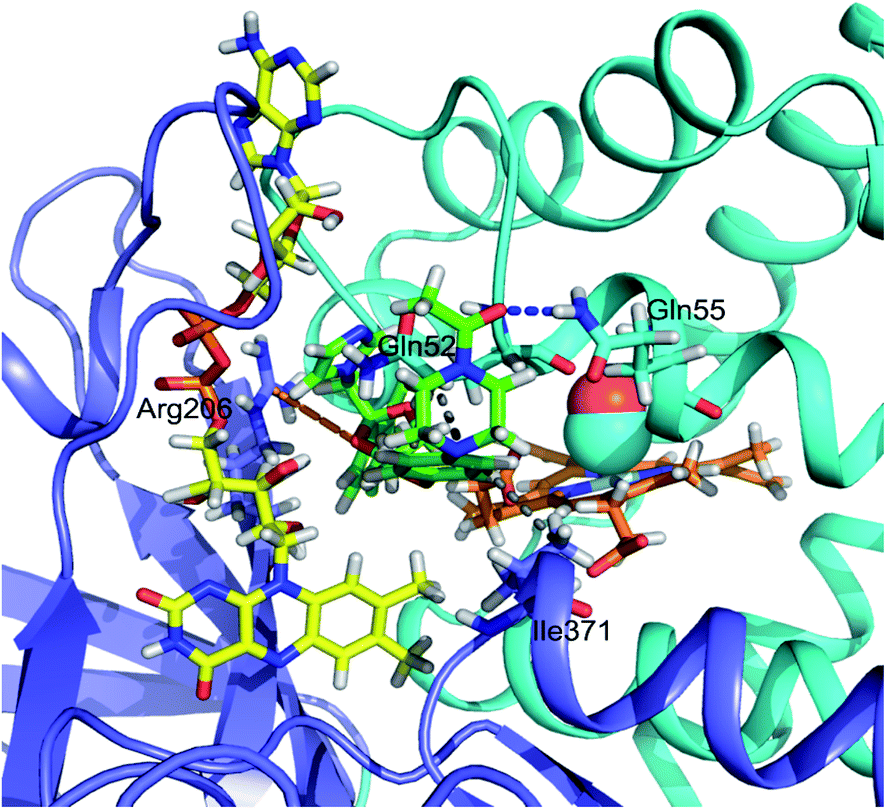 | ||
| Fig. 5 A view of the heme binding pocket of CO-FHP with ketoconazole after 20 ns of MD simulation. Hydrophobic interactions are shown as gray lines, cation-π interactions are shown as orange lines, and hydrogen bonds are shown as dark blue lines. FAD is colored in yellow and ketoconazole is in green. The globin domain is shown in cyan, the reductase domain is shown in blue, and heme is shown in orange. | ||
In the case of miconazole the highest-affinity binding site predicted by Autodock Vina places miconazole in the distal heme pocket, in contact with the hydrophobic cluster which includes Ala56, Leu102, Trp122, and Tyr126. After 20 ns of MD simulation, miconazole repositions slightly to form hydrophobic contacts with Ile25, Phe28, Ala56, Leu102, and Tyr126 as shown in Fig. 6 (an alternate view in Fig. S5†). Over the course of the simulation, the imidazole group of the drug moves from its initial position to being just above the plane of the heme group on the distal side, in close proximity to the heme iron (∼7 Å) with the surrounding of the drug molecule being almost entirely hydrophobic. The position of the drug is similar to that of miconazole found in the crystal structure.11
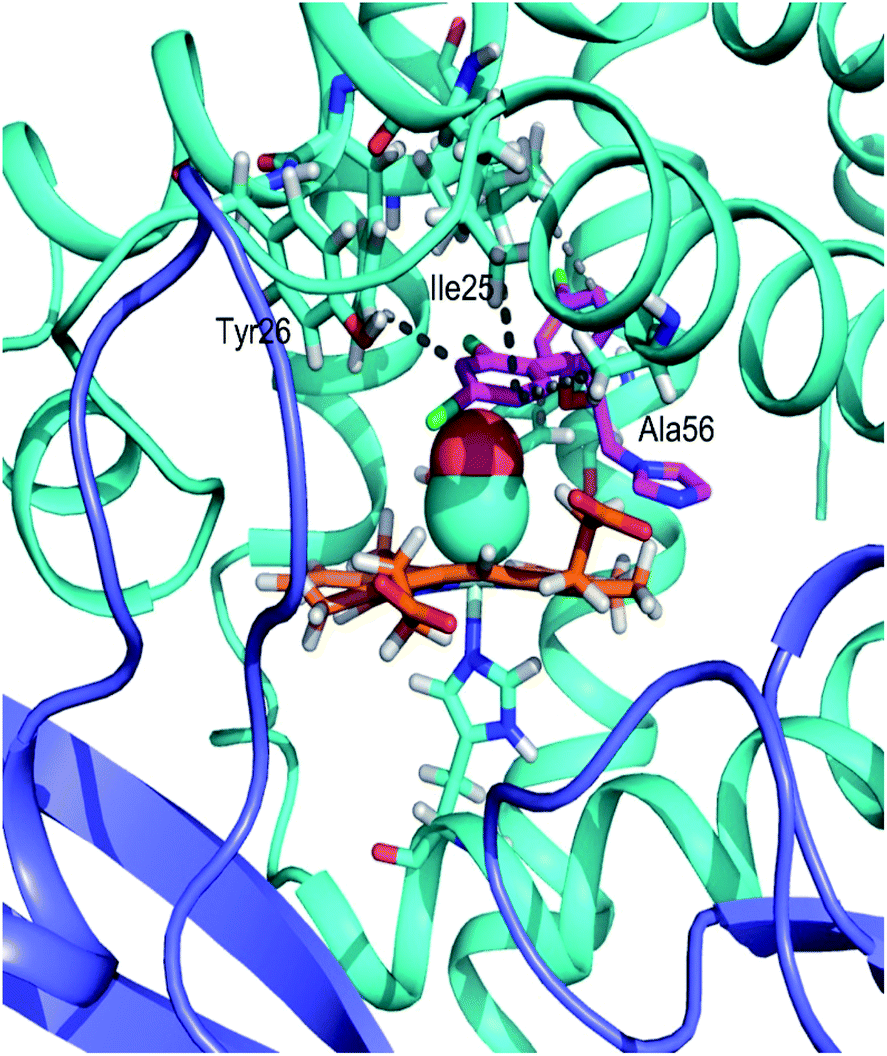 | ||
| Fig. 6 A view of the heme binding pocket of CO-FHP with miconazole (in magenta) after 20 ns of MD simulation. Hydrophobic interactions are shown as gray lines. The globin domain is shown in cyan, the reductase domain is shown in blue, and heme is shown in orange. | ||
Discussion
Ligand migration pathways in FHP
The LES-enabled simulation of unbound CO inside the protein matrix of FHP reveals a number of potential pathways for ligand migration within the protein. These pathways are essential to the NOD activity of the protein in vivo, which requires migration of gaseous ligands into the heme active site. The most prevalent pathway A passes between residues Leu103 and Tyr126 allowing direct movement of ligands between the distal heme pocket and bulk solvent. A majority, 13 out of 21 CO escape events occur via this pathway. No large-scale changes in tertiary structure or major fluctuations of critical residues are associated with the migration of CO molecules into or out of the protein matrix through this pathway. The positions of the critical residues Leu103 and Tyr126 do not change significantly during the LES-enabled simulation. The position of Leu103 is stable due to it being part of a hydrophobic cluster formed from neighboring residues which include Leu102, Ile106, Trp122, and Ala123. This hydrophobic cluster has been previously identified in the crystal structure of FHP.6,23 The position of the side chain of Tyr126 is stabilized by a hydrogen bond to the backbone oxygen of Tyr95. In the LES-enabled simulations, CO molecules can easily pass through the openings between these residues, suggesting a minimal energetic barrier for ligand migration. This is consistent with PAC data which shows fast ligand migration from the protein (τ < 50 ns) after photo-cleavage of iron CO bond.Pathway E passes between the CD loop (residues 40–55) and heme propionate groups. The segment between helices C and E termed CD loop is the second most common escape pathway and is involved in 14% of CO escapes. The geometry of the CD loop is altered in FHP compared to more well-known eukaryote globins, such as myoglobin, hemoglobin or the yeast fHb where it is structured in the so called D-helix loop. As a result in FHP, the position of the E-helix is shifted away from the G- and H-helices resulting in a larger volume on the distal side of the heme group.6,23 The CD loop constitutes a variable segment that balances different distances between helices C and E allowing ligand migration. Nevertheless this pathway bears some resemblance to that found in proteins which have a distal His gating mechanism; i.e. ligand migration between the bulk solvent and heme binding pocket occurs between the E-helix/CD-loop and heme propionate groups as shown in computational24 and experimental25 studies. Though other pathways for ligand escape are observed, they are collectively responsible for 19% of CO dissociation events. Given that LES ligands experience significantly lower energetic barriers for migration in the protein matrix, the role of these pathways in the ligand migration may be negligible.
Thermodynamic parameters for CO dissociation and rebinding in FHP
The reaction enthalpy (ΔH) of CO dissociation and rebinding in FHP can be expressed as a sum of several contributions according to the following equation:
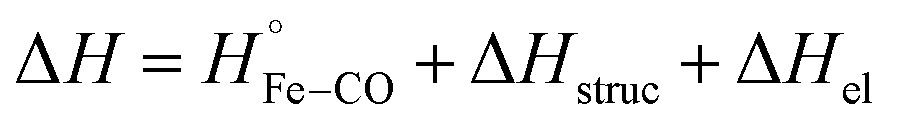 | (5) |
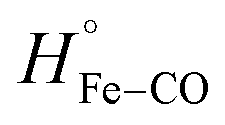 is equal to the bond dissociation enthalpy of the Fe–CO bond, ΔHstruc represents energetic changes in the protein structure including breakage/formation of hydrogen bonds or other dipole interactions and ΔHel includes enthalpy changes resulting from electrostriction which arises from the reorganization of solvent molecules due to exposure or removal or charged species from the bulk solvent.
is equal to the bond dissociation enthalpy of the Fe–CO bond, ΔHstruc represents energetic changes in the protein structure including breakage/formation of hydrogen bonds or other dipole interactions and ΔHel includes enthalpy changes resulting from electrostriction which arises from the reorganization of solvent molecules due to exposure or removal or charged species from the bulk solvent.
For photo-dissociation of CO from drug-free CO-FHP, the reaction enthalpy (ΔH) was found 26.2 ± 7.0 kcal mol−1 which is close to the predicted value of 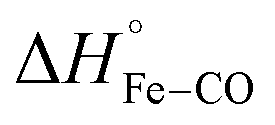 (26 kcal mol−1) according to DFT calculations of the myoglobin heme active center.26 This indicates that the observed enthalpy change is almost entirely due to photolysis of the Fe–CO bond. The observed reaction enthalpy change is consistent with the absence of a hydrogen bond between bound CO and nearby amino acid residues. Previously, a hydrogen bond network between the iron bound oxygen, a bridging water molecule, and TyrB10, –Tyr29 in FHP, – was predicted for E. coli fHb, based on QM/MM computations27 Similar hydrogen bonds interactions between Tyr29 and Gln53 sidechains and oxygen were observed in the crystal structure of yeast flavohemoglobin7 (PBD entry 4G1V). It is unclear if the presence of the hydrogen bond between the gaseous ligand and Tyr29 is ligand-dependent or protein-dependent. However, observed reaction enthalpy is consistent with the MD simulation of CO-FHP, where no bridged hydrogen bond is observed between Tyr29 and the bound CO, indicating the absence of these interactions in FHP.
(26 kcal mol−1) according to DFT calculations of the myoglobin heme active center.26 This indicates that the observed enthalpy change is almost entirely due to photolysis of the Fe–CO bond. The observed reaction enthalpy change is consistent with the absence of a hydrogen bond between bound CO and nearby amino acid residues. Previously, a hydrogen bond network between the iron bound oxygen, a bridging water molecule, and TyrB10, –Tyr29 in FHP, – was predicted for E. coli fHb, based on QM/MM computations27 Similar hydrogen bonds interactions between Tyr29 and Gln53 sidechains and oxygen were observed in the crystal structure of yeast flavohemoglobin7 (PBD entry 4G1V). It is unclear if the presence of the hydrogen bond between the gaseous ligand and Tyr29 is ligand-dependent or protein-dependent. However, observed reaction enthalpy is consistent with the MD simulation of CO-FHP, where no bridged hydrogen bond is observed between Tyr29 and the bound CO, indicating the absence of these interactions in FHP.
The reactions volume change (ΔV) can be described by the following equation:
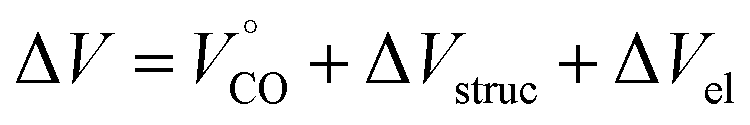 | (6) |
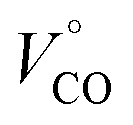 is the partial molar volume of the CO molecule (37.3 ± 0.5 mL mol−1)28 and ΔVel represents the volume change due electrostriction.
is the partial molar volume of the CO molecule (37.3 ± 0.5 mL mol−1)28 and ΔVel represents the volume change due electrostriction.
The observed ΔV of 25.0 ± 1.5 mL mol−1 for CO dissociation from FHP is ∼12 mL mol−1 smaller than expected for the escape of the CO molecule into the bulk solvent, considering the partial molar volume of CO ∼37 mL mol−1. A migration of water molecule(s) (V° = 18 mL mol−1) into the heme binding pocket may contribute to a small reaction volume change. Indeed, several water molecules were found in the heme binding pocket during MD simulation which is plausible given the large volume of the heme pocket in FHP (∼2000 Å3).11
Impact of azole drugs on thermodynamic parameters for CO dissociation and rebinding in FHP
The binding of ketoconazole to FHP results in a more exothermic enthalpy for CO photo-dissociation with ΔHbound ∼ −27.1 kcal mol−1. Assuming that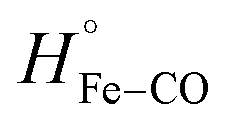 does not change, ΔHstruc + ΔHel is then −53.3 kcal mol−1 according to eqn (5). Our computational results predict that when CO is bound to the heme iron, ketoconazole binds outside the heme binding pocket with the imidazole group approximately 10 Å from the heme iron as shown in Fig. 5. The crystal structure of the met form of FHP in complex with ketoconazole shows that the drug binds within the heme binding pocket. This suggests that upon CO photolysis the drug may enters the heme binding pocket. Repositioning of the ketoconazole from into the heme binding pocket may leads to the displacement of water molecules from the protein interior into the surrounding solvent. Release of water molecules from the protein matrix is predicted to be exothermic due to the formation of hydrogen bonds between the displaced water molecules and those in the bulk solvent.29 Also, the ketoconazole transfer into the heme binding pocket may also result into exothermic solvation of sidechains of polar residues Gln55 and Arg 206 which closely interact with the ketoconazole in the CO bound FHP.
does not change, ΔHstruc + ΔHel is then −53.3 kcal mol−1 according to eqn (5). Our computational results predict that when CO is bound to the heme iron, ketoconazole binds outside the heme binding pocket with the imidazole group approximately 10 Å from the heme iron as shown in Fig. 5. The crystal structure of the met form of FHP in complex with ketoconazole shows that the drug binds within the heme binding pocket. This suggests that upon CO photolysis the drug may enters the heme binding pocket. Repositioning of the ketoconazole from into the heme binding pocket may leads to the displacement of water molecules from the protein interior into the surrounding solvent. Release of water molecules from the protein matrix is predicted to be exothermic due to the formation of hydrogen bonds between the displaced water molecules and those in the bulk solvent.29 Also, the ketoconazole transfer into the heme binding pocket may also result into exothermic solvation of sidechains of polar residues Gln55 and Arg 206 which closely interact with the ketoconazole in the CO bound FHP.
Additional exothermic contribution may be due to the coordination of the heme iron by the imidazole group of ketoconazole. Coordination of the heme iron by the imidazole group of histidine has been associated with a negative enthalpy change of approximately −16 kcal mol−1 as observed in horse methemoglobin.30 However, it is unclear whether ketoconazole is able to enter the heme binding pocket and form a coordination bond with heme iron within the ∼50 ns resolution of the PAC instrument, as this requires a larger displacement of the drug.
In this context, the decrease in ΔV observed for CO photo-dissociation in the presence of ketoconazole compared to the drug-free protein (ΔΔV = −17.2 mL mol−1) can be attributed to the same processes, i.e. movement of ketoconazole from being partially solvent-exposed in the cleft between the globin and reductase domains into the heme binding pocket and entrance of water molecules into the volume vacated by the drug.
One can consider also the different structures reported for FHP to explain the observed differences in ΔH and ΔV for the drug-free and ketoconazole-bound protein. Indeed, the crystallographic data show that FHP adopts an open conformation in the drug-free form and a closed conformation with bound ketoconazole.11 Since the crystal structure was determined for the met form of the protein, it is unclear if the open to close form transition takes place in the deoxy and CO bond form of the protein. We do not expect to observe a large scale closed to open state structural transition within the timescale of our PAC measurements (<10 μs). Nonetheless repositioning of ketoconazole after CO photolysis could destabilize the closed state promoting structural changes which contribute to the different ΔH and ΔV values observed in the presence and absence of the drug.
ΔH for CO photo-dissociation in the presence of miconazole is also more exothermic than in the absence of the drug (ΔΔH = −43.6 kcal mol−1), with ΔHstruc + ΔHel = −43.4 ± 7.0 kcal mol−1. The binding site for miconazole is directly within the heme binding pocket, therefore unlike ketoconazole, we do not expect to see an exothermic contribution from migration of the drug into the heme binding pocket and displacement of water molecules into the bulk solvent. The value of ΔHstruc + ΔHel for CO photo-dissociation in the presence of miconazole (−43.4 ± 7.0 kcal mol−1) can be attributed to the formation of a coordination bond between the heme iron and the imidazole group of miconazole. This occurs in the case of the miconazole-bound protein due to the close proximity of the drug to the heme iron, allowing it to bind to the heme iron within ∼50 ns of CO photodissociation. Only a small difference between ΔV values was observed for CO escape from FHP in the presence and absence of miconazole (ΔΔV = −4.6 mL mol−1) suggesting that neither position of the drug nor protein structure change greatly upon CO photo-dissociation. This is also consistent with the similar structure of the drug-free and miconazole-bound proteins that both adopt the open conformation.11
The impact of azole drugs on kinetic parameters for CO dissociation and rebinding on FHP
As compared to the thermodynamic parameters resolved using PAC, the rate constants for CO rebinding to FHP determined using TA reveal a minimal impact of drug presence. Presence of ketoconazole, which binds adjacent to the heme binding pocket in CO-FHP and within the heme binding pocket in the absence of gaseous ligands, does increase the rate of CO rebinding by approximately two times suggesting increased accessibility of the heme binding pocket. The increase of the rate for CO rebinding indicates that repositioning of ketoconazole within the protein does not represent a rate-limiting step for CO rebinding to heme iron. The presence of miconazole has only minor impact on the rates of CO rebinding. This is surprising given the miconazole binding site, which has been observed to be directly within the heme binding pocket in the crystal structure of met-FHP11 and is predicted to be unchanged in CO-FHP according to our Autodock Vina results (Fig. 6). In addition, this binding location places the drug directly within the most prevalent ligand migration pathway identified in our LES simulation, pathway A. These results suggest that distinct pathway(s) can be used for the CO migration in the presence of drugs without impeding ligand migration. Similar rate constants for CO rebinding to miconazole and ketoconazole-bound FHP also indicate that the transition between the open and closed state of the protein have minor impact on ligand migration rates. Overall, these results suggest that neither ketoconazole nor miconazole inhibit the accessibility of gaseous ligands to the heme active site in FHP.The impact of azole drugs on thermodynamic and kinetic parameters for CO dissociation and rebinding in HMPSa
In the case of HMPSa, CO photo-dissociation in the absence of azole drugs leads to an enthalpy increase of ΔH = 34.7 ± 8.0 kcal mol−1. This value is slightly higher than that observed for the same process in FHP, in which ΔH was attributed primarily to the breakage of the Fe–CO bond. If we assume the value of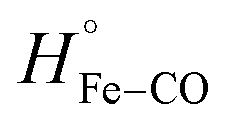 to be unchanged between the two proteins, it follows that there is an additional endothermic contribution of 8.7 kcal mol−1. This value is similar to that determined for the energy of the strong hydrogen bond between the distal His and bound O2 in myoglobin based on density functional theory studies.31 This suggests that a bridged hydrogen bond between TyrB10, a water molecule, and the bound ligand predicted based on QM/MM studies of E. coli fHb27 can also be found in CO-HMPSa. This is consistent with previous studies showing close catalytic activities between HMPSa and E. coli fHbs9 ΔV for CO photo-dissociation from HMPSa (28.6 ± 1.7 mL mol−1) is only slightly larger than that for FHP, indicating similar overall structural changes between the CO-bound and ligand free protein in FHP and HMPSa. Ezzine et al. have used a homology modeling to characterize structural properties of HMPSa and reported that resides Tyr29, Gln53 and Phe33 are displaced from the heme binding pocket compare to the FHP structure. However, such displacement may caused a local reorganization and does not affect overall changes in protein structure triggered by CO photodissociation.
to be unchanged between the two proteins, it follows that there is an additional endothermic contribution of 8.7 kcal mol−1. This value is similar to that determined for the energy of the strong hydrogen bond between the distal His and bound O2 in myoglobin based on density functional theory studies.31 This suggests that a bridged hydrogen bond between TyrB10, a water molecule, and the bound ligand predicted based on QM/MM studies of E. coli fHb27 can also be found in CO-HMPSa. This is consistent with previous studies showing close catalytic activities between HMPSa and E. coli fHbs9 ΔV for CO photo-dissociation from HMPSa (28.6 ± 1.7 mL mol−1) is only slightly larger than that for FHP, indicating similar overall structural changes between the CO-bound and ligand free protein in FHP and HMPSa. Ezzine et al. have used a homology modeling to characterize structural properties of HMPSa and reported that resides Tyr29, Gln53 and Phe33 are displaced from the heme binding pocket compare to the FHP structure. However, such displacement may caused a local reorganization and does not affect overall changes in protein structure triggered by CO photodissociation.
As for FHP, in the presence of drugs, in particular miconazole, CO photo-dissociation from HMPSa becomes more exothermic. Also, ΔV for CO photo-dissociation from HMPSa in the presence of ketoconazole and miconazole are distinct as compared to the drug-free protein. The lack of a structure for HMPSa complicates the analysis of the thermodynamic data somewhat; while the overall fold of FHP and HMPSa are likely very similar (as described in ref. 9), the low sequence identity (35%) suggests that there may be differences in the structure of the distal pocket and or composition of the drug binding sites which contribute the observed differences in thermodynamic parameters for CO photo-release.
Association of ketoconazole results in bimolecular rebinding rate constants approximately two times faster while the presence of miconazole has little impact. The presence of a slow geminate rebinding step in HMPSa indicates that a fraction of the photo-dissociated ligand is trapped within the protein matrix. These data also point out that the distribution of hydrophobic cavities is distinct between FHP and HMPSa, likely due to a more rigid structure in HMPSa and reinforce the idea that binding of the azole drug does not inhibit gaseous ligand access to the heme active site.
The mechanism of inhibition of NOD activity by azole drugs
We have determined that despite the binding of ketoconazole and miconazole in the vicinity of ligand migration pathways observed in our LES simulations of FHP, ligand migration in FHP and HMPSa is not significantly altered in the presence of azole drugs. Therefore, restriction of gaseous ligand access to the heme active site does not appear to be the mechanism by which NOD activity of fHbs is inhibited by azole drugs. On the other hand, the binding of ketoconazole to FHP, may impact an electron transfer between FAD and the heme iron. Several electron transfer pathways have been predicted between the heme group and bound electron-donating cofactors via structural water molecules based on computational studies in FHP32 and E. coli fHb27 In FHP, the most prevalent predicted pathway (∼75% of electron transfers) involves transfer of electrons from the C8M methyl group of FAD through one or two bridging water molecules and potentially the adjacent residue Lys84, ultimately reaching the heme via the heme-7-propionate group.32 Comparison of MD simulations of CO-FHP bound to FAD in the presence and absence of ketoconazole (Fig. 7) shows a significant dehydration of the interface between FAD and the heme propionate group, i.e. the bridging water molecules which facilitate electron transfer are no longer present. In addition, the presence of ketoconazole causes the side chain of Gln53 to rotate to form a hydrogen bond with the heme-6-propionate group, thereby altering its orientation. The heme-6-propionate group has been predicted to participate in the second most prevalent electron transfer pathway in FHP.32 Therefore, alteration of the electron transfer pathway may be the mechanism of inhibition of NOD activity by ketoconazole in FHP. For miconazole, inhibition of NOD activity is possibly due to an allosteric effect which is communicated from the miconazole binding site in the heme binding pocket to the interface between FAD and the residues/water molecules which participate in electron transfer. Overall, azole drugs do not restrict NO/O2 molecules from accessing the heme active site, but likely hinder electron transfer from bound NADH and FAD, preventing the reduction of the heme iron which is necessary to the NOD catalytic cycle.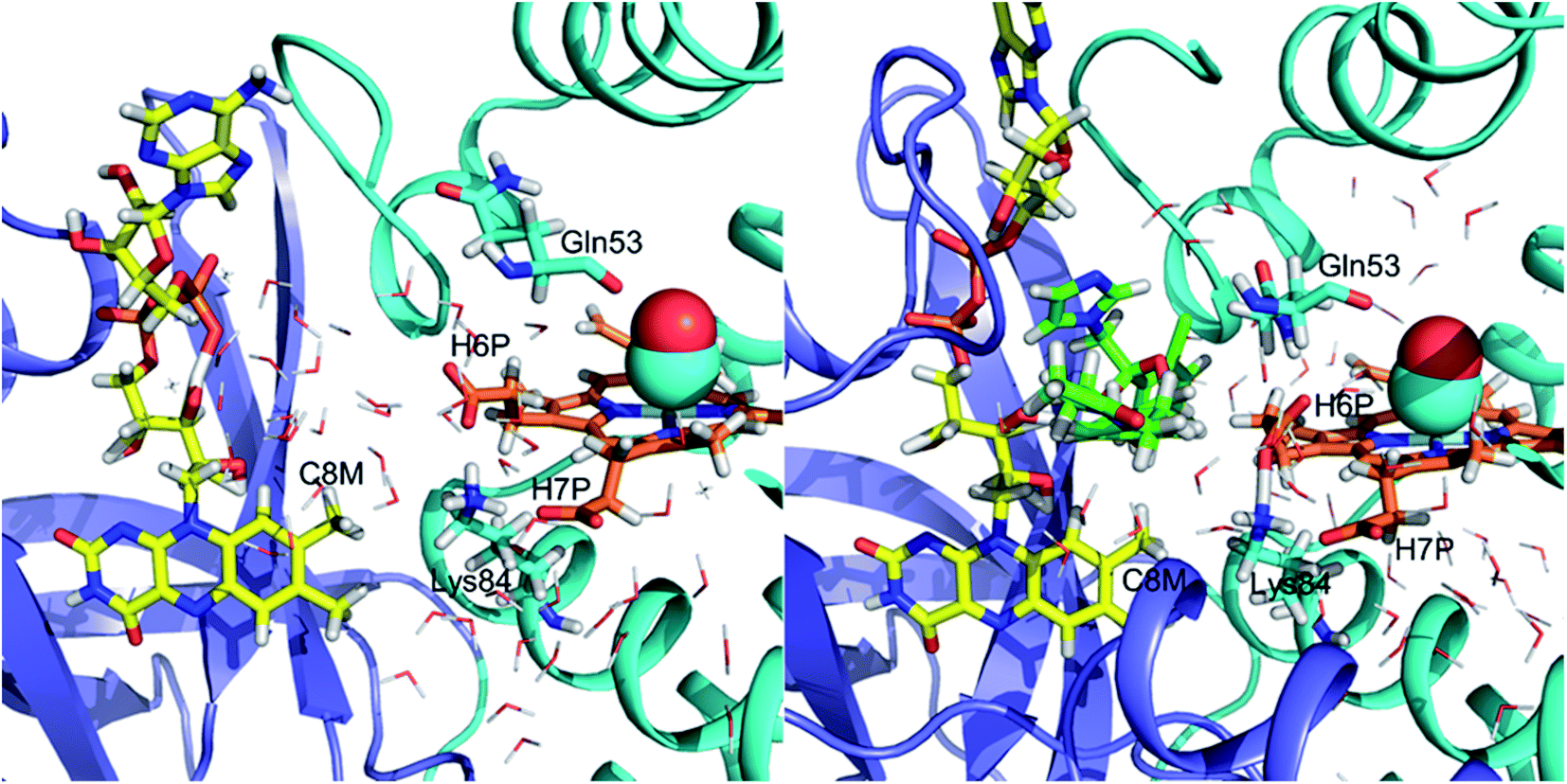 | ||
| Fig. 7 Interface between FAD and the heme group in the absence (left) and presence (right) of ketoconazole. Water molecules are represented as thin lines. C8M is the C8 methyl group of the flavin group of FAD. H6P and H7P stand for the heme-6-propionate and heme-7-propionate groups, respectively. Residues 50–58 of the E helix and 341–403 of the reductase domain are not shown to prevent obstruction of the view of the interface. The globin domain is shown in cyan, the reductase domain is shown in blue, heme is shown in orange, FAD is shown in yellow and ketoconazole is shown in green. | ||
Conclusion
The results presented here demonstrate the distinct nature of the interactions of azole drugs with FHP and HMPSa in the presence of the gaseous diatomic ligand bound to the heme iron. Likely due to its more compact structure, miconazole binds in the vicinity of the distal pocket whereas elongated structure of ketoconazole restricts its access to the heme binding pocket. The different binding pattern is evident from the thermodynamic parameters associated with the CO photo-release, likely due to the distinct changes overall protein structure and/or hydration. However, azole drug association has a minimal impact on ligand migration as the rate constant for CO binding to heme iron are not significantly modified in the presence of drugs. In addition, our simulations of CO-FHP with and without ketoconazole shown that a significant dehydration of the interface between FAD and the heme group occurs in the presence of ketoconazole. Further development of antibiotic compounds designed to inhibit the NOD activity of fHbs would likely benefit from a focus on inhibition of electron transfer in flavohemoglobins, specifically disruption of the electron transfer pathway ferries electrons from the flavin group of FAD to the heme propionate groups through adjacent structural water molecules.Accession codes
Flavohemoglobin from Cupriavidus necator (Uniprot ID: P39662).Flavohemoglobin from Staphylococcus aureus (Uniprot ID: A0A0E1ACT8).
Abbreviations
| fHbs | Flavohemoglobins |
| NOD | Nitric oxide dioxygenase |
| ROS | Reactive oxygen species |
| NO | Nitric oxide |
| FHP | Flavohemoglobin from Cupriavidus necator |
| HMPSa | Flavohemoglobin from Staphylococcus aureus |
| PAC | Photoacoustic calorimetry |
| TA | Transient absorption spectroscopy |
| cMD | Classical molecular dynamics |
| LES | Locally enhanced sampling |
| CO | Carbon monoxide |
| Fe[III]4SP | Fe(III)meso-tetra(4-sulfonatophenyl)porphine chloride |
| ΔV | Volume change |
| ΔH | Enthalpy change |
Conflicts of interest
There are no conflicts to declare.References
- R. K. Poole and M. N. Hughes, New functions for the ancient globin family: bacterial responses to nitric oxide and nitrosative stress, Mol. Microbiol., 2000, 36, 775–783 CrossRef CAS PubMed.
- M. T. Forrester and M. W. Foster, Protection from nitrosative stress: a central role for microbial flavohemoglobin, Free Radical Biol. Med., 2012, 52, 1620–1633 CrossRef CAS PubMed.
- L. Liu, M. Zeng, A. Hausladen, J. Heltman and J. S. Stamler, Protection from nitrosative stress by yeast flavohemoglobin, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4672–4676 CrossRef CAS PubMed.
- S. Dupré-Crochet, M. Erard and O. NüBe, ROS production in phagocytes: why, when, and where?, J. Leukocyte Biol., 2013, 94, 657–670 CrossRef PubMed.
- A. Hausladen, A. Gow and J. S. Stamler, Flavohemoglobin denitrosylase catalyzes the reaction of a nitroxyl equivalent with molecular oxygen, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10108–10112 CrossRef CAS PubMed.
- U. Ermler, R. A. Siddiqui, R. Cramm and B. Friedrich, Crystal structure of the flavohemoglobin from Alcaligenes eutrophus at 1.75 Å, EMBO J., 1995, 14, 6067–6077 CrossRef CAS PubMed.
- E. El Hammi, E. Warkentin, U. Demmer, N. M. Marzouki, U. Ermler and L. Baciou, Active site analysis of yeast flavohemoglobin based on its structure with a small ligand or econazole, FEBS J., 2012, 279, 4565–4575 CrossRef CAS PubMed.
- R. A. Helmick, A. E. Fletcher, A. M. Gardner, C. R. Gessner, A. N. Hvitved, M. C. Gustin and P. R. Gardner, Imidazole Antibiotics Inhibit the Nitric Oxide Dioxygenase Function of Microbial Flavohemoglobin, Antimicrob. Agents Chemother., 2005, 49, 1837–1843 CrossRef CAS PubMed.
- A. Ezzine, M. Moussaoui, E. El Hammi, M. N. Marzouki and L. Baciou, Antimicrobial Agents Act Differently on Staphylococcus aureus and Ralstonia eutropha Flavohemoglobins, Appl. Biochem. Biotechnol., 2014, 173, 1023–1037 CrossRef CAS PubMed.
- M. Moussaoui, L. Misevičienė, Ž. Anusevičius, A. Marozienė, F. Lederer, L. Baciou and N. Čėnas, Quinones and nitroaromatic compounds as subversive substrates of Staphylococcus aureus flavohemoglobin, Free Radical Biol. Med., 2018, 123, 107–115 CrossRef CAS PubMed.
- E. El Hammi, E. Warkentin, U. Demmer, F. Limam, N. M. Marzouki, U. Ermler and L. Baciou, Structure of Ralstonia eutropha Flavohemoglobin in Complex with Three Antibiotic Azole Compounds, Biochemistry, 2011, 50, 1255–1264 CrossRef CAS PubMed.
- N. Hashemzadeh and A. Jouyban, Solubility of Ketoconazole in Ethanol + Water Mixtures at Various Temperatures, Chem. Eng. Commun., 2015, 202, 1211–1215 CrossRef CAS.
- S. Tenjarla, P. Puranajoti, R. Kasina and T. Mandal, Preparation, characterization, and evaluation of miconazole-cyclodextrin complexes for Improved oral and topical delivery, J. Pharm. Sci., 1998, 87, 425–429 CrossRef CAS PubMed.
- R. W. Larsen and J. Miksovska, Time resolved thermodynamics of ligand binding to heme proteins, Coord. Chem. Rev., 2007, 251, 1101–1127 CrossRef CAS.
- N. Belogortseva, M. Rubio, W. Terrell and J. Miksovska, The contribution of heme propionate groups to the conformational dynamics associated with CO photodissociation from horse heart myoglobin, J. Inorg. Biochem., 2007, 101, 977–986 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- R. B. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, CHARMM: a program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4, 187–217 CrossRef.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8q, Proteins: Struct., Funct., Bioinf., 2009, 77, 114–122 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- O. Trott and A. J.Olson, Autodock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, SwissParam: a fast force field generation tool for small organic molecules, J. Comput. Chem., 2011, 32, 2359–2368 CrossRef CAS PubMed.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, PLIP: fully automated protein-ligand interaction profiler, Nucleic Acids Res., 2015, 43, 443–447 CrossRef PubMed.
- A. Roitberg and R. Elber, Modeling side chains in peptides and proteins: application of the locally enhanced sampling and the simulated annealing methods to find minimum energy conformations, J. Chem. Phys., 1991, 95, 9277–9287 CrossRef CAS.
- M. S. Shadrina, G. H. Peslherbe and A. M. English, O2 and water migration Pathways between the solvent and heme pockets of hemoglobin with open and closed conformations of the distal HisE7, Biochemistry, 2015, 54, 5279–5289 CrossRef CAS PubMed.
- I. Birukou, R. L. Schweers and J. S. Olson, Distal histidine stabilized bound O2 and acts as a gate for ligand entry in both subunits of adult human hemoglobin, J. Biol. Chem., 2010, 2851, 8840–8854 CrossRef PubMed.
- C. Rovira and M. Parrinello, First-principles molecular dynamics simulations of models for the myoglobin active center, Int. J. Quantum Chem., 2000, 80, 1172–1180 CrossRef CAS.
- D. N. Ferreiro, L. Boechi, D. A. Estrin and M. A. Martí, The key role of water in the dioxygenase function of Escherichia coli flavohemoglobin, J. Inorg. Biochem., 2013, 119, 75–84 CrossRef CAS PubMed.
- J. C. Moore, R. Battino, T. R. Rettich, Y. P. Handa and E. Wilhelm, Partial molar volumes of “gases” at infinite dilution in water at 298.15 K, J. Chem. Eng. Data, 1982, 27, 22–24 CrossRef CAS.
- R. Baron, P. Setny and J. A. McCammon, Water in Cavity-Ligand Recognition, J. Am. Chem. Soc., 2010, 132, 12091–12097 CrossRef CAS PubMed.
- H.-J. Steinhoff, J. Schlitter, A. Redhardt, D. Husmeier and N. Zander, Structural fluctuations and conformational entropy in proteins: entropy balance in an intramolecular reaction in methemoglobin, Biochim. Biophys. Acta, 1992, 1121, 189–198 CrossRef CAS.
- E. Sigfridsson and U. Ryde, On the significance of hydrogen bonds for the discrimination between CO and O2 by myoglobin, J. Biol. Inorg Chem., 1999, 4, 99–110 CrossRef CAS PubMed.
- E. El Hammi, C. Houée-Lévin, J. Řezáč, B. Lévy, I. Demachy, L. Baciou and A. de la Lande, New insights into the mechanism of electron transfer within flavohemoglobins: tunnelling pathways, packing density, thermodynamic and kinetic analyses, Phys. Chem. Chem. Phys., 2012, 14, 13872–13880 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02529a |
| ‡ Present address: National High Magnetic Field Laboratory, Florida State University, Tallahassee, FL. |
| This journal is © The Royal Society of Chemistry 2020 |