
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the true MOF-5 luminescence†
Vincent Villemota,
Matthieu Hamel a,
Robert B. Pansub,
Isabelle Lerayb and
Guillaume H. V. Bertrand*a
a,
Robert B. Pansub,
Isabelle Lerayb and
Guillaume H. V. Bertrand*a
aCEA, LIST, Laboratoire Capteurs et Architectures Électroniques, F-91191, Gif-sur-Yvette Cedex, France. E-mail: guillaume.bertrand@cea.fr
bENS Paris Saclay, UMR CNRS 8531, F-94235, Cachan, France
First published on 14th May 2020
Abstract
Highly pure millimeter-sized MOF-5 single crystals were synthesized and characterized. Photoluminescence (PL) spectroscopy and time-correlated single photon counting (TCSPC) demonstrate a solvent-guest dependency of MOF-5 emission and its ligand-centred nature. These results allow measuring the true MOF-5 luminescence free of solvent at a wavelength of 355 nm, a significantly lower wavelength than previously published. MOF-5 emission was also evaluated with different solvents and various degrees of water intake, explaining previously published observations. Comparison between lifetimes shows the fluorophore stabilization within the frameworks and demonstrates the progressive influence of the Zn4O subunits on the fluorescence during hydration. Overall, this work highlights the necessity to obtain phase-pure material, especially when moisture sensitivity can play a role, before ascribing electronic transitions. This study is a rigorous new take on the iconic MOF-5 and on its photoluminescence properties.
Metal–organic frameworks (MOFs) are crystalline hybrid materials, composed of metal clusters that are linked by organic ligands. Due to their unique topology, these materials offer promising applications such as separation, catalysis, drug delivery or gas storage among others.1 While many contributions have demonstrated the use of MOFs as luminescent sensors their photophysical properties still draw attention after many years.2–6 Indeed, due to their hybrid nature, photoluminescence (PL) can arise from various sites: metallic nodes, organic linkers or interaction between both of them such as MLCT, LMCT, LLCT and MMCT (M: metal, L: ligand, and CT: charge transfer). Due to their near thermodynamic density of defect,7 the luminescence of these are negligible in crystalline phase. Furthermore, molecules trapped in the porosity can also emit, influence or induce luminescence, giving an additional complexity to photophysical comprehension.8 The present work demonstrates the influence of frameworks (especially the one based on Zn4O subunits) on linker PL and attests the necessity to obtain pure phase material to conclude on mechanisms leading to MOF luminescence. The well know MOF-5 (: IRMOF-1) was the subject of numerous studies during the past two decades.9–12 However, to the best of our knowledge there is no report on a ligand centered (LC) emission. Moreover, there is still no consensus on MOF-5 emission, with reported fluorescence between 390 nm11,12 and 525 nm9,10 and different assigned transitions. Thus, we chose MOF-5 as a case study to explain how coherently interpret photophysical properties of MOFs based on Zn4O subunits, and conclude on the luminescence of this iconic MOF. MOF-5 luminescence was first studied by Bordiga et al., with a reported maximum of emission centred at 525 nm. They assigned this emission as a charge transfer from benzene dicarboxylic acid to the Zn4O13 metal clusters.9 Later, Tachikawa et al. studied MOF-5 nanoparticles luminescence and found that the responsible transition for the green emission was similar to that of ZnO quantum dots (QDs). Therefore, they attributed the emission to MOF-trapped ZnO nanoparticles instead of MOF.10 Allendorf and co-workers were the first team to recognize the discrepancy between published results and underlined the presence of impurities within the frameworks. They synthesized MOF-5 with different processes, generating more or less pure materials and studied their PL properties. They were able to reproduce previous results from Bordiga and Tachikawa on frameworks including ZnO impurities. Above all, they highlighted the misunderstanding of previous reports which interpret emissions from impurities due to poor preparation. MOF-5 emission was then ascribed to a transition occurring at 397 nm upon excitation at 345 nm.11 A 395 nm emission would imply a strong electronic interaction between the benzene-1,4-dicarboxylic acid (H2BDC) ligand and the Zn cluster. Recent DFT studies compare geometry and IR spectra between electronically excited state and ground state of MOF-5.12 These investigations reveal the rigidity of Zn4O13 clusters compared to non-rigid BDC2− ligands which leads to similar calculated emission band for MOF-5 and free H2BDC. Results were quite similar with Allendorf's work and their interpretation suggests that the luminescence was carried out by LLCT mechanism with little contribution of the metallic cluster.
Motivated by both experimental and theoretical reports lacking of consistency,6 the following work goal is to reproduce, explain and unify previous results. Hence this study focuses on obtaining highly pure phase material before studying PL properties and to ascribe a mechanism to MOF-5 emission. We were thus able to observe a UV emission from MOF-5 single crystals, and the PL dependence with solvents and water molecules which slowly diffuse within the crystal and collapse its structure.
As discussed above, poor preparation may be responsible for misunderstanding of PL properties requiring the synthesis of pure phase material.11 A focus was made in our laboratory to obtain large millimeter-sized MOF-5 single crystals in a reproducible manner with high purity. They were synthesized (see ESI†) from zinc nitrate hexahydrate (Zn(NO3)2·6H2O) and benzene-1,4-dicarboxylic acid (H2BDC) in N,N-diethylformamide (DEF) using a solvothermal procedure which was slightly different from the one reported by Yaghi et al.13,14 Colorless, large single crystals were immediately preserved from moisture inside a N2 glovebox and washed with anhydrous solvents to avoid any framework decomposition as we will see below. The purity was assessed by several structural characterizations: X-ray diffraction, thermal and infrared analyses (ESI†). Fig. 1 represents powder X-ray diffraction (PXRD) data. Refinement on single crystal X-ray data permits to calculate the associated PXRD pattern (black) that is in perfect agreement with previous reports on MOF-5.15,16 Following activation, thermogravimetric analysis (TGA) demonstrated characteristic thermal decomposition of the frameworks with three distinct regions assigned to H2O/solvent desorption and structural decomposition of the framework.16 More importantly, the weight loss of our sample upon decomposition into ZnO is 58% which perfectly fits the theoretical calculated weight loss of original MOF-5, 57.7% (ESI, Fig. S1†) indicating that there is no remaining ZnO or other guest impurities in our crystals. Raman and BET analysis were also performed and show results in complete agreement with previous reports (ESI, Fig. S2 and S3†).
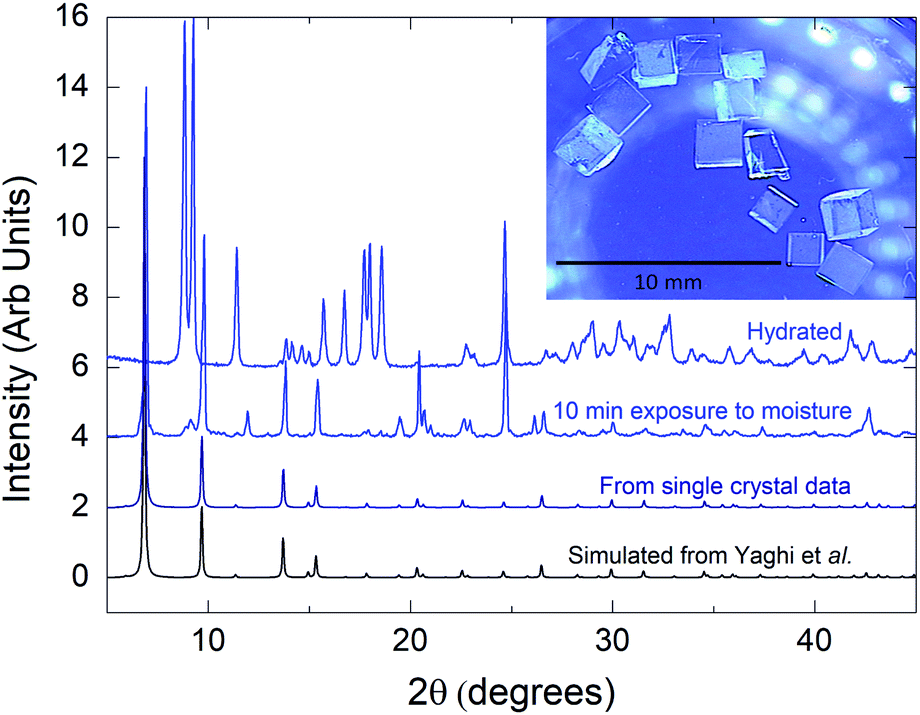 | ||
| Fig. 1 Powder X-ray diffraction data. Intensities are normalized to the maximum. The pattern for MOF-5 was calculated from single crystal structural data (see ESI†). Inset is a picture of single crystals. | ||
Photoluminescence investigations were performed (Fig. 2) on a pure single crystal soaked in different solvents and compared to the H2BDC building unit dissolved in these same solvents. Reasons to collect PL data on MOF full of solvent was motivated by the observations of strong solvent effect on the H2BDC fluorescence. Results show indeed different maximum wavelengths for MOF-5 from one solvent to another. When impregnated with DMF or DMSO, the framework exhibits a maximum of emission at 342 or 363 nm, respectively. The smaller solvatochromic shift observed in DMF comes with a reduced broadening of the band that is observed in DMF for the free ligand and the MOF.2–17 MOF emission shows a 5 nm bathochromic shift compared to the ligand for DMF. These observations are substantially the same for MOF-5 in DMSO, which has the same spectrum profile than H2BDC, with the difference that framework emission presents a hypsochromic shift of 4 nm. It is well known that the dipole moment of the solvent, size of the solute molecules and their interaction with the solvent play a major role on the system's global fluorescence. The solvatochromism is due to the difference in the solvation of the excited state with respect to the ground state. It has been described as a stabilization of the electric dipole of both states with the dielectric constant and refractive index of the solvent.18 This phenomenon is also visible for highly pure single crystals. The impact of the solvent on MOF-5 structure is negligible19 compared to other breathing MOF, we discarded this hypothesis as a reason of the solvatochromism. Our interpretation is that the solvent behaviour within the framework is similar to its bulk. Solvent molecules can enter into the pores and form a solvation environment similar to a solution one. One can note here that we are treating the absorbed molecules as a solvation shell from the point of view of the BDC linker. Chemi- and physi-sorption interaction are similar in both cases, balancing hydrogen bonding and van der Waals forces. In case of DMF, DMSO and later water, we think H-bond between the MOF surface and the molecules, as well as between molecules themselves, are the predominant factor of molecules immobilisation inside a pore. As shown in Fig. 2, the similarity in wavelengths and vibronic structure points towards luminescence in MOF-5 arising from a single excited state S1 which is similar to the H2BDC in solution and thus, suggests a ligand-centred emission only. Decay time measurements allow us to confirm our hypothesis, revealing a monoexponential decay, characteristic of a single photophysical process, and the same lifetime for frameworks and free linker in both solvents (ESI, Fig. S4 and S5†).
 | ||
| Fig. 2 Steady-state emission spectra for H2BDC (dashed) and MOF-5 (solid), respectively dissolved and soaked, in DMF (top), DCM/CH2Cl2 (middle) and DMSO (bottom) under 300 nm excitation. | ||
To remove the effect of the solvent and investigate only MOF-5 fluorescence, solvent molecules were removed from porosity with extreme care as the framework can collapse due to capillary forces while activating.20 The resulting PLE and PL spectra (black) are shown in Fig. 3, revealing excitation and emission maxima at 313 nm and 358 nm, respectively. The symmetry of the excitation and emission coupled with vibronic structure suggest that the transition occurs from a single excited state S1 to the fundamental state S0 as seen above. However, and compared to Fig. 2, we can notice a broadening of the emission which we attribute to a partial collapse of the framework. We assume that the shift to shorter wavelengths of both emission and excitation maxima compared to H2BDC are the consequence of an increase of the band gap due to stabilization of linker within the framework and the absence of adsorbed molecules. Indeed, the decrease of the Stokes shift confirms this idea as the thermal states are getting closer to the fundamental and excited states (ESI, Fig. S7†). Moreover, a recent report21 on MOF-5 study by DFT shows the electronic band structure of cubic frameworks based on Zn4O units and finds that its band gap should be located in the 3.56–3.46 eV range, corresponding to an emission between 348 nm and 358 nm, which is consistent with our observation. Time-correlated Single Photon Counting (TCSPC) measurements were performed on solid sample and demonstrate that evacuated MOF-5 lifetime at 355 nm is comparable with benzenecarboxylic disodic salt (Na2BDC) with a heavier fast components. This fast component is closer to the solubilized H2BDC than the solid state confirming the idea that the framework stabilizes the fluorophore in the same way a solvation shell would. (ESI, Fig. S8†).
 | ||
| Fig. 3 Comparison of excitation (dashed) and emission (solid) spectra for an activated MOF-5 (black) and solid H2BDC (blue). | ||
The above observation contrasts with previous reports on MOF-5 photoluminescence. However, this is not surprising regarding the architecture of this type of framework. The secondary building unit (SBU), composed of ZnII is, as well as CdII, favorable to have a luminescence carried out by linkers only. The reason resides in the d10 configuration of these ions. The absence of potential d–d transitions allows only luminescence from ligand.22 The SBU is not spectator so far as it varies the length of the ligand and so on the distribution of π-electron responsible of fluorescence.
As luminescence is carried out by the ligand only and dependent on the surroundings, we assume that any degradation affecting the structure will impact the associated PL spectra. As already observed in many contributions, MOF-5 is moisture sensitive.13,23,24 In Fig. 1, only 10 min moisture exposure results in the appearance of a new crystalline phase and longer exposure results in the appearance of a new emitting species which was never ascribed but nonetheless already observed.16 Allendorf and co-workers were one of the first to highlight the presence of a 445 nm emitting species.11 Despite no explanation, this phenomenon can be explained by water adsorption and insertion into the frameworks, involving the Zn4O SBU in the fluorescence process. As studied by Siegel and co-worker water adsorption and insertion in MOF-5 behaves in two steps. First stage consists of adsorption of H2O molecules close to Zn–O clusters. Then the second stage consists in water insertion via breaking of Zn–O bonds23 as shown in Fig. 4a. Fig. 4b shows different PL spectra of a MOF-5 crystal soaked in DMF and exposed to moisture. We can observe a rapid shift of the framework trident shaped emission from 342 nm to 365 nm and the apparition of a broad emission band centred at 445 nm. Then the crystal was evacuated from DMF and left several days in open air, leading to the observation of a pure 445 nm centred emission. Our interpretation, based on Siegel's work, is that the first shift is due to the adsorption of water and insertion in the carboxylate–zinc bond, bringing electron density toward the carboxylic group, resulting on shortening of bandgap. This could explain the displacement of fluorescence maximum towards longer wavelengths and emission broadening by molecular motion. The structure then collapses (totally or partially) due to the cleavage of Zn–O bonds (ESI, Fig. S3†). This collapse is also observed by gas adsorption analysis, with a decrease of the specific area from 2400 m2 g−1 to 1800 m2 g−1. At this point a complex of terephthalate–H2O–zinc most probably creates the luminescence emitting at 445 nm. Our hypothesis is also based on TCSPC measurement (Fig. 4c). Decay times of partially hydrated MOF-5 (red) taken at 350 and 450 nm show a large discrepancy indicating at least two different emissive species. One has a very close resemblance to the fully hydrated MOF-5 (blue) emitting at 445 nm, whereas the faster component could be attributed to the crystalline MOF emitting at 345 nm. The loss of vibronic structure at 445 nm, due to hydration, attests from a luminescence mechanism that becomes different than a ligand centred emission and thus results in an elongation of lifetime and interpretation as a probable MLCT or LMCT transition.
 | ||
| Fig. 4 (a) Adsorption and insertion of water in MOF. Print with permission from Siegel et al. from ref. 23 (b) PL emission data and (c) decay time of pure single crystal impregnated with DMF (black), partially hydrated single crystal impregnated with DMF (red) and fully hydrated (blue) fitting see ESI, Fig. S6.† | ||
In order to confirm this idea, Fig. 5 represents the degradation of MOF-5 by moisture exposure monitored by PL. The spectra show two distinct new emissions, the first one is assigned to the hydrated frameworks luminescence and the second one is assigned to a framework decomposition product, at 365 nm and 445 nm respectively. The general trend is that both emission intensities increase as well as water concentrates within the framework. Comparing normalized intensities highlights that the maximum of emission shifts from 355 nm to 365 nm before the framework decomposes, giving thus the awaited final 445 nm emission. The surface normalized intensity (ESI, Fig. S9†) shows an isobestic point at 410 nm confirming the transformation from one luminescent species to another one.
 | ||
| Fig. 5 Emission spectra of MOF-5 during time (top) and associate normalized intensity (bottom). Dashed lines represents different contributions. | ||
To conclude, millimetric MOF-5 single crystals have been successfully synthesized by solvothermal path. Structural characterizations were carried out showing highly pure phase materials in good agreement with literature precedents. Hence photophysical investigations using PL and decay time allow demonstrating that fluorescence in MOF-5 is ligand centred and therefore has a solvent guest dependent behavior. If free from solvent, the maximum emission occurs at 355 nm. This experimental value are in excellent agreement with excited states energy calculated by previous DFT studies. We also show that the integrity of the framework plays a major role in the resulting fluorescence. Indeed, hydration and structure collapse causes the broadening of the fluorescence and Zn4O/BDC bond cleavage by water promotes two new luminescent species with different emission wavelengths and longer lifetime than the original crystalline MOF. These observations tend to be similar for the isoreticular series with same topology as MOF-5 and will be explored. We expect that these results, which are an extension to the works already done on MOF-5, will have utility in comprehension on luminescent mechanisms involved in MOFs composed of Zn4O building blocks.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to the grant given by the CEA for V. V. The authors are also very grateful to Dr M. D. Allendorf for the fruitful discussions and review on this manuscript, to Dr T.-H. Tran-Thi and C. Rivron for assistance with surface area measurements and to Dr P. Thuery and Dr S. Coste for X-ray diffraction measurements.Notes and references
- P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- R. B. Lin, S. Y. Liu, J. W. Ye, X. Y. Li and J. P. Zhang, Adv. Sci., 2016, 3, 1–20 Search PubMed.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- L. Wilbraham, F. X. Coudert and I. Ciofini, Phys. Chem. Chem. Phys., 2016, 18, 25176–25182 RSC.
- T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F. X. Coudert, Nat. Chem., 2016, 9, 11–16 CrossRef PubMed.
- N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincă, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS PubMed.
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2004, 10, 2300–2301 RSC.
- T. Tachikawa, J. R. Choi, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2008, 112, 14090–14101 CrossRef CAS.
- P. L. Feng, J. J. Perry Iv, S. Nikodemski, B. W. Jacobs, S. T. Meek and M. D. Allendorf, J. Am. Chem. Soc., 2010, 132, 15487–15489 CrossRef CAS PubMed.
- M. Ji, X. Lan, Z. Han, C. Hao and J. Qiu, Inorg. Chem., 2012, 51, 12389–12394 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- O. Shekhah, H. Wang, D. Zacher, R. A. Fischer and C. Wöll, Angew. Chem. Int. Ed., 2009, 48, 5038–5041 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- J. Hafizovic, M. Bjørgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- H. Laguitton-Pasquier, R. Pansu, J. P. Chauvet, A. Collet, J. Faure and R. Lapouyade, Chem. Phys., 1996, 212, 437–455 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- W. G. Shim, K. J. Hwang, J. T. Chung, Y. S. Baek, S. J. Yoo, S. C. Kim, H. Moon and J. W. Lee, Adv. Powder Technol., 2012, 23, 615–619 CrossRef CAS.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- H. Q. Pham and T. Mai, J. Phys. Chem. C, 2014, 118, 4567–4577 CrossRef CAS.
- J. Cepeda and A. Rodríguez-diéguez, CrystEngComm, 2016, 18, 8556–8573 RSC.
- Y. Ming, N. Kumar and D. J. Siegel, Omega, 2017, 2, 4921–4928 CrossRef CAS PubMed.
- J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 10678–10679 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02509g |
| This journal is © The Royal Society of Chemistry 2020 |