
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning nanocavities of Au@Cu2O yolk–shell nanoparticles for highly selective electroreduction of CO2 to ethanol at low potential†
Bin-Bin Zhang,
Ya-Hui Wang,
Shan-Min Xu,
Kai Chen,
Yu-Guo Yang* and
Qing-Hua Kong *
*
Department of Chemistry, School of Science, Beijing Jiaotong University, Beijing 100044, China. E-mail: ygyang@bjtu.edu.cn; qhkong@bjtu.edu.cn
First published on 20th May 2020
Abstract
The electrosynthesis of high-value ethanol from carbon dioxide and carbon monoxide addresses the need for the large-scale storage of renewable electricity and reduction of carbon emissions. However, the electrosynthesis of ethanol by the CO2 reduction reaction (CO2RR) has suffered from low selectivity and energy efficiency. Here, we report a catalyst composed of Au nanoparticles in Cu2O nanocavities (Au@Cu2O) that is very active for CO2 reduction to ethanol through the confinement of the CO intermediate. The architecture shows tandem catalysis mechanisms in which CO2 reduction on Au yolks produces CO filling Cu nanocavities, where a sufficiently high CO concentration due to the confinement effect promotes ethanol formation and then results in an ethanol faradaic efficiency of 52.3% at −0.30 V versus the reversible hydrogen electrode (vs. RHE) via regulating the hollow size of the Cu2O nanocavities. Such a strategy provides a new way of fabricating various tandem catalysts with high selectivity and efficiency for the CO2RR.
Introduction
The electrocatalytic reduction of CO2 to valuable carbon-based fuels and chemicals offers a route to reduce CO2 emissions and facilitate the long-term storage of renewable electricity.1–6 In particular, C2 and C2+ products from the CO2 reduction reaction (CO2RR) have attracted considerable attention due to their relatively high energy density, added-value and widespread use as feedstocks in polymer synthesis, etc.7–9 Excellent electrocatalysts have been developed to boost the activity and selectivity of the CO2RR towards C2 and C2+ productions.10–14 Among currently available materials, Cu-based catalysts have been reported as the most promising electrodes for producing C2 and C2+ compounds under aqueous conditions in CO2RR, and have been extensively studied.8,12,15–21 However, there still present some scientific challenges, such as poor selectivity, low faradaic efficiency and durability, which need to be managed primarily in further work.To date, several avenues have been employed to improve the selectivity of Cu-based catalysts for CO2RR to specific products, including altering size, structure, composition, surface state, and so on.17–20 Hori et al. declared that the product selectivity of CO2RR shifted greatly with the crystal orientation, where Cu(100) yielded mainly C2H4 and Cu(111) benefited CH4 production.22 Sargent's group constructed a core–shell vacancy engineering catalyst (Cu2S–Cu–V) to steer products beyond alkenes and toward ethanol at certain potentials.23 More recently, theoretical and experimental investigations demonstrate that the activity and selectivity for C2 and C2+ of Cu-based catalysts can be greatly advanced by bimetallic strategy or constraining the local CO concentration at the catalyst–electrolyte interface.24,25 Au/Cu bimetallic electrocatalyst was obtained with improved activity and selectivity for electrochemical transformation of CO2 to alcohols over hydrocarbons at low overpotentials, and a tandem catalysis mechanism has been proposed where Au nanoparticles (Au NPs) reduce CO2 to CO near the copper surface, driving a high CO coverage.25 When supplied directly with CO instead of CO2 as a feedstock, oxide-derived nanocrystalline copper electrodes produce ethanol with up to 30% faradaic efficiency at modest overpotentials under alkaline conditions.26 In addition, the confinement effect is known to alter thermodynamic and transport properties of fluids.27–30 Prior studies of porous catalysts exploited confinement effect to implement a selectivity shift by extending the retention of C1 or C2 species, and then boost C2 or C3 production,16,24 which lack in-depth exploration. As such, developing a new strategy of combining bimetallic strategy and confinement effect is highly imperative for boosting the selectivity to ethanol in CO2 electroreduction and its practical application.
Herein, we further apply the confinement effect with a bimetallic yolk–shell structure of Au nanoparticle in Cu2O nanocavity (Au@Cu2O), whereas Au shows high catalytic activity for converting CO2 to CO at low potentials,31,32 and Cu is able to catalyze the second step of the tandem reaction, the reduction of CO to C2 products at low potentials.33–35 The Au@Cu2O catalyst has higher selectivity for ethanol compared with hydrocarbons at lower overpotentials. We propose that the selectivity shifting from C1 to C2 originates from a tandem catalysis mechanism, where Au NPs reduce CO2 to CO in the Cu nanocavities, producing a high CO concentration, thus boosts the ethanol production due to the confinement of CO intermediate. Meanwhile, we adjust the CO concentration in the nanocavities by regulating the hollow size of the Cu2O nanocavities, and then achieve an ethanol faradaic efficiency of 52.3% at −0.30 V vs. RHE.
Experimental
Chemicals
Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 99.99%), citric acid, trisodium salt (98%), copper(II) nitrate trihydrate (99%), hydrazine hydrate (hydrazine, 64%), polyvinylpyrrolidone (PVP, average M.W 58000), 2-propanol (99.7+%), Nafion (5%). All chemicals were obtained directly without further purification.Preparation of Au NPs
The Au NPs with a diameter of about 22 ± 2 nm were synthesized and used as the core materials for the Au@Cu2O yolk–shell particle fabrication.36,37 Au NPs were prepared by a standard citrate reduction procedure. Typically, 150 mL of 0.025 M HAuCl4 solution was added into a three-necked round bottom flask and heated to boiling under continuously magnetic stir with condensing and refluxing conditions. Then 7.5 mL of 0.02 M sodium citrate solution was added. After reaction for 30 min, heating resource was removed and the solution was allowed to cooled down naturally to room temperature. Finally, the Au NPs were collected from the solution by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), washed with deionized water several times and redispersed in 10 mL of deionized water.
000 rpm), washed with deionized water several times and redispersed in 10 mL of deionized water.
Preparation of Au@Cu2O yolk–shell NPs with different hollow sizes
Au@Cu2O yolk–shell NPs was prepared by hydrazine hydration reduction method.36,38 Typically, 1 g of PVP powders was added into 10 mL of 0.01 M Cu(NO3)2 solution under constant stirring until the PVP powders dissolved completely. Then a certain amount of as-obtained Au NPs solution was added, followed by immediate introduction of trace hydrazine hydrate. After reaction, the Au@Cu2O yolk–shell NPs were synthesized and centrifuged (10000 rpm), washed three times with water and isopropyl alcohol, and then dried in an oven at 60 °C for later use. Au@Cu2O NPs with different hollow sizes can be controlled through adjusting the amount of reducing agent and the reaction time (Fig. 1a).
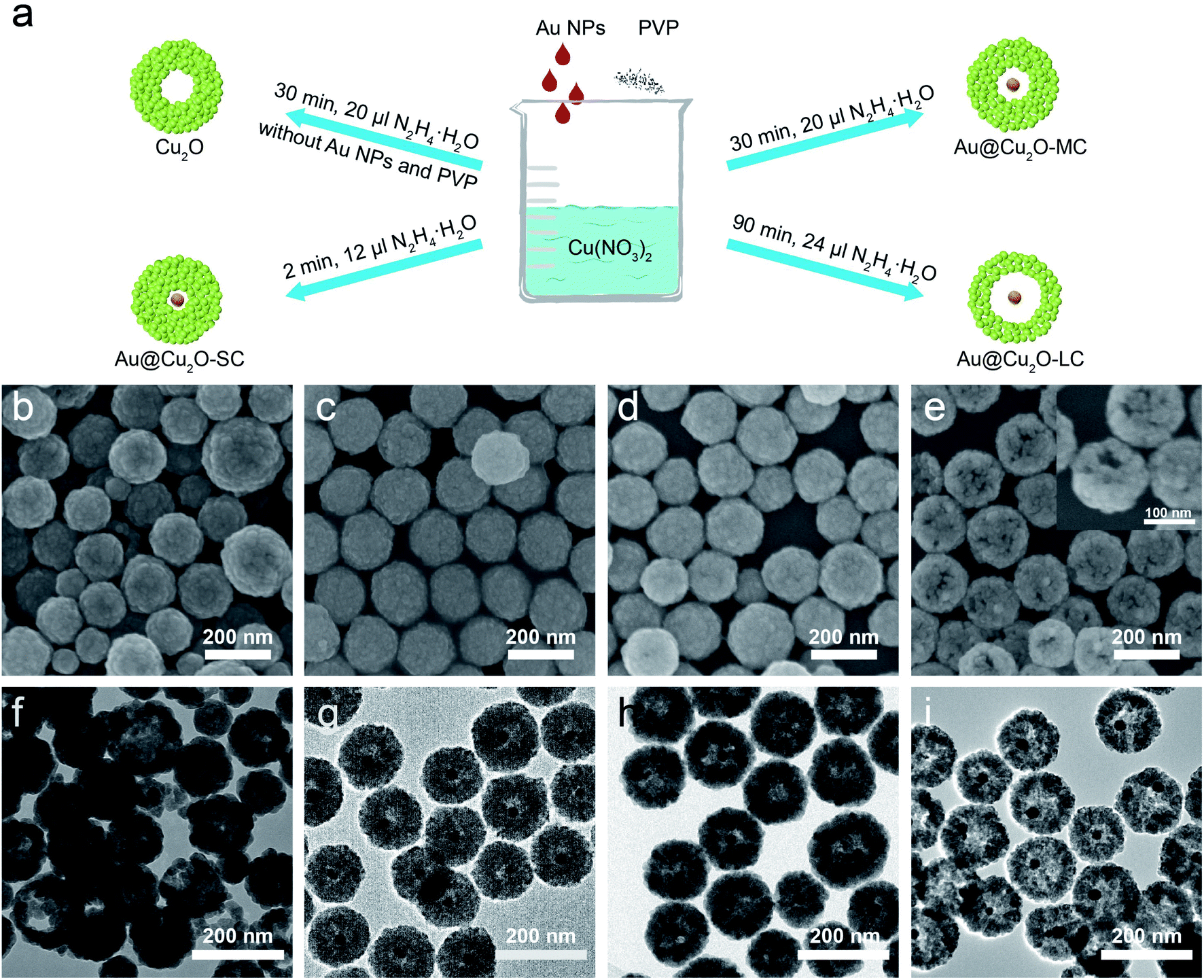 | ||
| Fig. 1 (a) Schematic illustration of the synthetic process of different samples. (b–e) SEM, (f–i) TEM images of Cu2O, Au@Cu2O-SC, Au@Cu2O-MC, Au@Cu2O-LC, respectively. | ||
Preparation of Cu2O NPs
Cu2O NPs was prepared in parallel by the same method as Au@Cu2O yolk–shell NPs except for no addition of Au NPs and PVP powders.Preparation of working electrode
To prepare the catalyst inks, 3 mg of catalyst powder, 10 μL of Nafion solution (5%) and 1200 μL of isopropyl alcohol were mixed and treated under sonication for 30 min. Then, 600 μL of the as-prepared ink was pipetted onto the two sides of a carbon cloth with area of 1 × 1.5 cm2 giving a catalyst loading of 1 mg cm−2.Characterizations
X-ray diffraction (XRD) pattern was carried out on a Rigaku D/Max-2500 diffractometer equipped with a Cu Kα1 radiation (λ = 1.54 Å). Scanning electron microscopic images (SEM) were collected on a JEOL scanning electron microscope (S-4800, Japan). Transmission electron microscopic images (TEM) were obtained by a JEM-2100F microscope (JEOL, Japan) equipped with an EDS detector (Oxford Instrument, UK). X-ray photoelectron spectroscopy (XPS) was performed on an ESCALab220i-XL electron spectrometer (VG Scientific, UK) with a monochromatic Al Kα source. The gas products for CO2 reduction were measured on a gas chromatography (GC, Agilent Technologies 7890B). The liquid products were analyzed with a Bruker AVANCE 600 using dimethyl sulphoxide (DMSO) as an internal standard.Electrochemical performance test
Electrochemical measurements were performed on a CHI 660E electrochemical workstation in a typical H-type electrolysis cell under ambient pressure and room temperature using platinum mesh (1 × 1 cm2) and Ag/AgCl electrode (saturated KCl) as counter and reference electrodes, respectively. The cathode and anode compartments were separated by a proton-exchange membrane (Nafion 117). The 0.1 M KHCO3 aqueous solution was used as electrolyte directly without any purification. The electrode potentials were converted to the reversible hydrogen electrode reference (RHE) scale using the following Nernst equation:| E(RHE) = E(Ag/AgCl) + 0.197 V + 0.0591 × pH. |
Before test, the catalyst was measured at −0.2 V vs. RHE for 30 min in Ar-saturated 0.1 M KHCO3 solution to ensure all Cu2O-shells had been reduced to Cu-shells. Then, the catalyst was immediately transfer into CO2-saturated 0.1 M KHCO3 solution to perform linear scanning voltammetry (LSV) test from 0.2 to −1.1 V vs. RHE until the performance of the electrode became stable. Finally, CO2RR experiment at a constant applied voltage spanned over 3600 seconds. The electrochemically surface area (ECSA) was determined by measuring the double layer capacitance (Cdl), which was derived from the CV curves at various scan rates.
Analysis of CO2 reduction products
We measure the reduction performance of the catalyst by calculating the faradaic efficiency (FE) of the CO2 reduction product. The FE of products can be calculated using the following formula:39i: the specific reduction product, CO, HCOO−, C2H5OH or H2; Qtotal: the total charge of CO2RR, C; Qi: charge used for the reduction of certain product, C; Ni: number of moles for certain product, mol; n: number of electrons transferred for the CO2-to-CO, HCOO− and C2H5OH conversion or water-to-H2 reduction, which is 2, 2, 12, 2 for CO, HCOO−, C2H5OH and H2, respectively; F: faradaic constant, which is 96
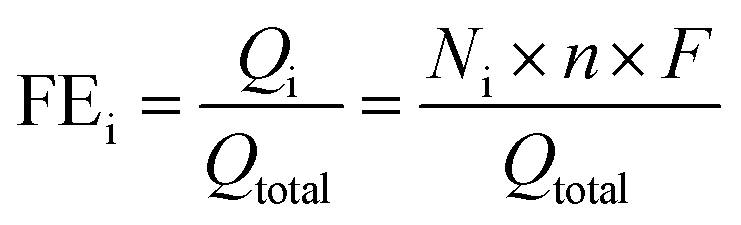 485 C mol−1.
485 C mol−1.
Results and discussion
The Au NPs with uniform size and morphology were obtained according to previous literature (Fig. S1†).36,37 Subsequently, the Au nanoparticle was encapsulated within a porous Cu2O shell with an average size of 140 nm and the hollow size was precisely controlled by regulating the reaction time and the amount of reducing agents, as depicted in Fig. 1a. Meanwhile, Fig. S2† shows the geometry of the multilayer particle we simulated, in which the radius of the Cu2O NPs is donated as R and the radius of the hollow size is donated as R1. In the structure, C2 chemical selectivity can be tuned by systematically altering the R1 of 14, 35, 48 nm, which are named small cavity Au@Cu2O (Au@Cu2O-SC), middle cavity Au@Cu2O (Au@Cu2O-MC) and large cavity Au@Cu2O (Au@Cu2O-LC), respectively. Scanning electron microscopy (SEM) images in Fig. 1b–e reveal that the surfaces of all obtained samples are uneven, and as reaction time went on, a clear porous structure was seen on the surface of Au@Cu2O-LC. Corresponding to SEM images, transmission electron microscopy (TEM) images in Fig. 1f–i show all the samples have a porous Cu2O shell. And the inner hollow size became larger along with the increased reaction time and the shell structure gradually grew looser. In addition, the samples except porous Cu2O NPs belong to yolk–shell structure and exhibit an average diameter of 140 ± 10 nm with an average Au-cores diameter of 22 ± 2 nm. Moreover, the statistical analysis based on over 100 nanoparticles indicates that they are in narrow size distribution (Fig. S3†), agreeing well with SEM and TEM results. Take Au@Cu2O-MC catalyst for example, more detailed structural information was characterized in Fig. 2. High-resolution TEM image (HRTEM) clearly shows the lattice fringes in spacings of 0.30 and 0.25 nm corresponding to (110) and (111) planes of cubic Cu2O with a characteristic interplanar angel of 90° on the shell (Fig. 2a).40 Dark-field scanning transmission electron microscopy (DF-STEM) image (Fig. 2b) exhibits the loose and porous structure of the shell material, which will promise efficient mass transport for potential electrochemical applications. Meanwhile, the corresponding energy dispersive X-ray spectroscopic (EDS) elemental mapping images (Fig. 2c–f) clearly display that Au distributes homogeneous on the core part while Cu and O are distributed mainly across the nanoparticle shell section, confirming the yolk–shell structure of Au@Cu2O.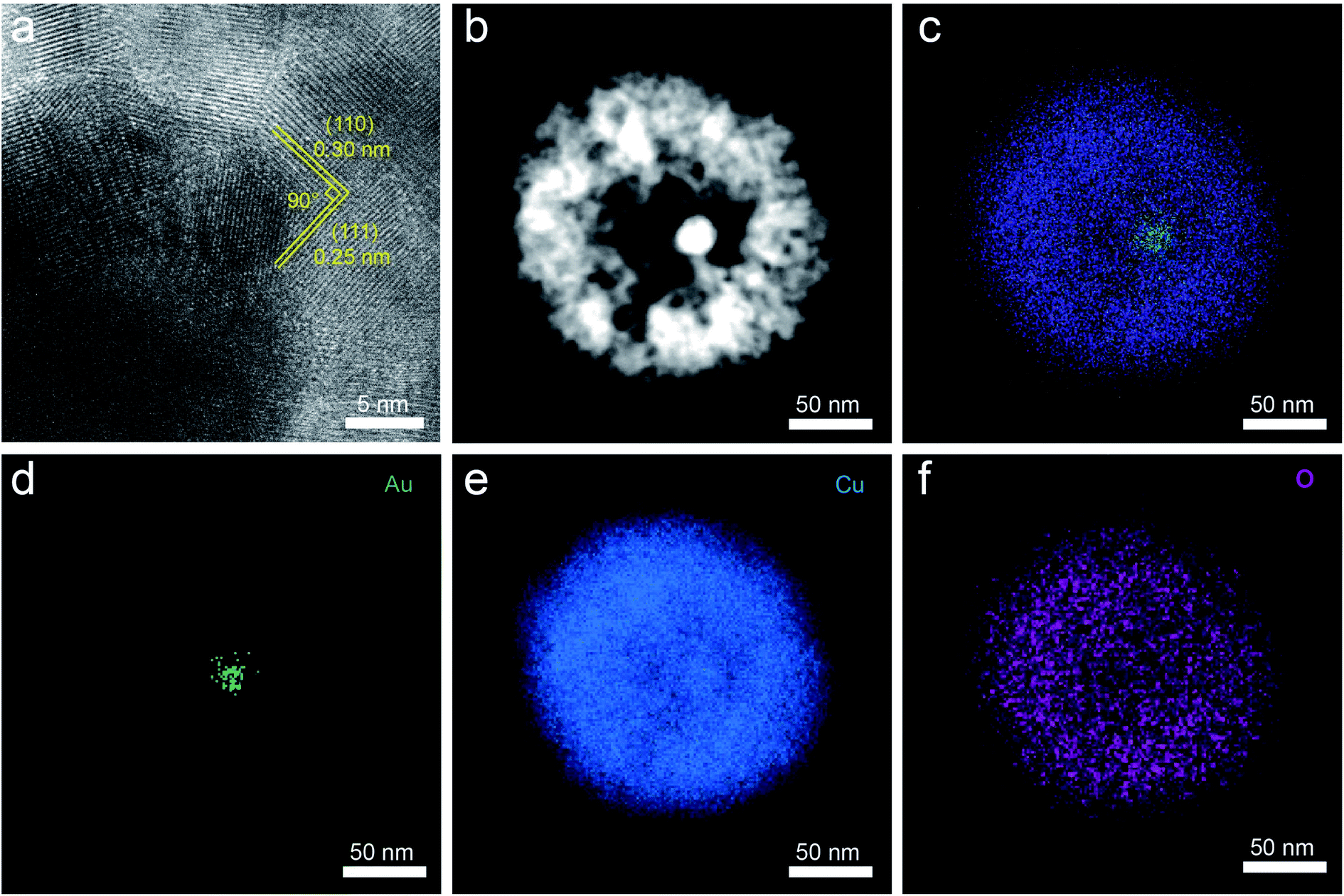 | ||
| Fig. 2 (a) HRTEM, (b) DF-STEM and (c–f) EDS elemental mapping images of Au@Cu2O-MC. | ||
The composition and crystalline structure of these samples were investigated by X-ray diffraction (XRD) technique. All of the recorded diffraction peaks in the typical XRD patterns (Fig. 3a) can be well indexed to cubic Au (JCPDS no. 89-3697) and cubic Cu2O (JCPDS no. 78-2076), corresponding to the HRTEM results. Further clues can be seen in X-ray photoelectron spectroscopic spectra (XPS) (Fig. 3b and c). By comparing the Cu 2p and Cu-LMM XPS peaks of Cu2O and Au@Cu2O-MC, it could be clearly seen that the Cu atom in Au@Cu2O-MC is Cu+, which is beneficial for CO2RR according to previous reports.15,16,26,41 In addition, since XPS is a surface analysis technique with investigation depth of 2–5 nm and the Au-core is coated by Cu2O, the signal of Au element is not detected.42 Based on these results, it can be concluded that Au-core encapsulates in porous Cu2O-shell particle, which constitute the tandem catalyst. Consequently, it is suggested that the active Cu sites of the catalyst during CO2RR are derived from Cu2O.
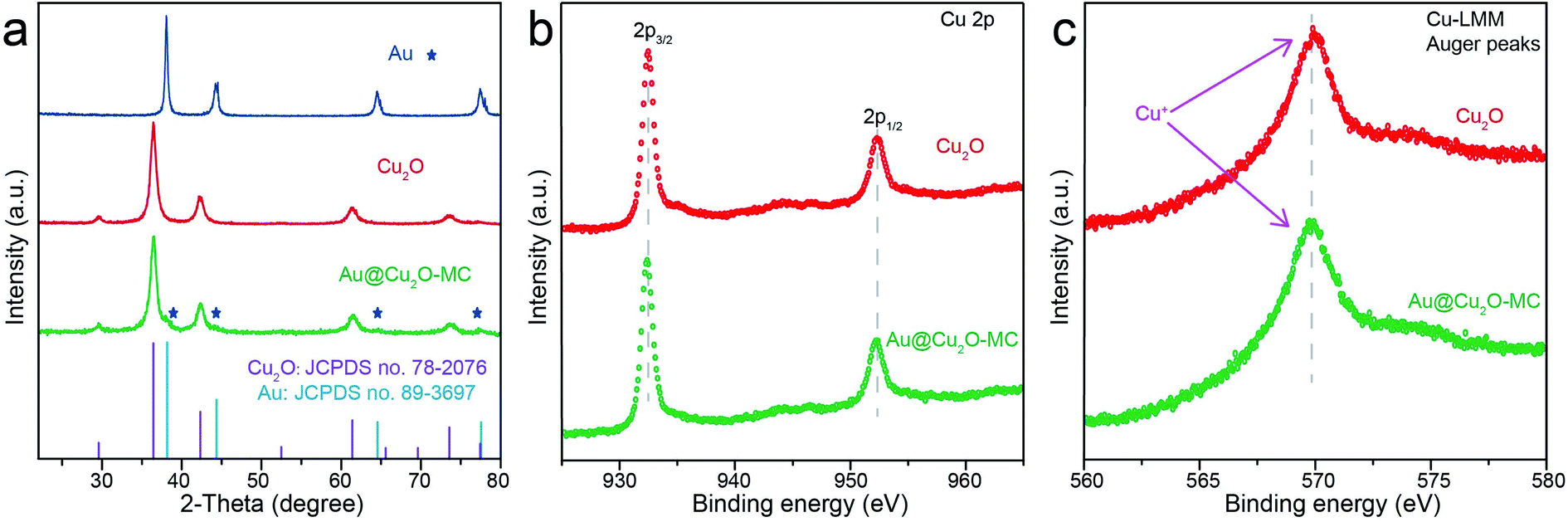 | ||
| Fig. 3 (a) XRD patterns of different samples, (b) high-resolution Cu 2p and (c) Cu-LMM XPS spectra for Cu2O and Au@Cu2O-MC. | ||
The CO2RR performances were evaluated over the as-obtained samples supported on the carbon cloth in 0.1 M KHCO3 solution saturated with CO2 using H-cell set-up. The products of CO2RR were analyzed and quantified by online gas chromatography (GC) for the gas products and 1H nuclear magnetic resonance spectroscopy (1H NMR) for the liquid-phase products. We can determine the electrochemical activity by linear sweep voltammetry (LSV) towards CO2RR, initially. Compared with the current density under Ar-saturated 0.1 M KHCO3, it has a significant increase under CO2-saturated electrolyte, indicating that Au@Cu2O-MC has superior CO2RR performance (Fig. S4a†). From Fig. S4b,† it can be seen the difference of current density between these samples, and Au@Cu2O-MC has the maximum current density at the same potential, showing the excellent CO2RR performance. For porous Cu2O NPs, at low applied cathodic potential of −0.3 and −0.35 V vs. RHE, the only detectable product is ethanol and the faradaic efficiency (FEC2H5OH) reaches 16% at −0.3 V vs. RHE (Fig. 4a), demonstrating the Cu2O NPs has intrinsic C–C coupling potential. At more cathodic potentials, a substantial difference in the product distribution is found with the products of C2 and significant increases in the CO, HCOO− production rates. After introducing Au NPs in the cavities (Fig. 4b), the selectivity shifts clearly and CO, HCOO−, C2H5OH products are generated at −0.3 and −0.35 V vs. RHE. The production of CO and HCOOH on the Au@Cu2O-SC as well as its absence on the pure porous Cu2O NPs, together with the promoted FEC2H5OH (36%) at low potentials, prove the effect of the suggested tandem reaction mechanism within the nano-confined space with both catalytic sites located in cavities. The Au-core is active for electroreduction of CO2 to CO, yet the Cu-shell is able to reduce the retention of CO in the cavities to improve ethanol production (Fig. 4e). When the hollow size of the catalyst was increased to 35 nm, the largest FEC2H5OH was improved to 52.3% at −0.3 V vs. RHE (Fig. 4c). The reduction of the FEC2H5OH at higher cathodic potentials from −0.45 to −0.6 V vs. RHE indicates that the previously described tandem mechanism is almost inactive in this potential window for Au@Cu2O-MC due to the overwhelmingly competitive HER from the exposed metal species (Fig. S5†). Further increasing the hollow size to 48 nm, the largest FEC2H5OH is 38% at −0.35 V vs. RHE (Fig. 4d), which is slightly less than that of Au@Cu2O-MC. However, the active potential window of tandem reaction mechanism for Au@Cu2O-LC is broader than that of Au@Cu2O-MC, and all of the FEC2H5OH at whole potentials from −0.27 to −0.45 V vs. RHE is higher than that of Au@Cu2O-SC, as well as a reduction of the detected CO at −0.27 and −0.3 V vs. RHE for Au@Cu2O-MC and Au@Cu2O-LC, suggesting the reaction mechanism of CO2RR can be monitored via regulating the concentration of CO intermediate by steering the hollow size of the Cu2O cavity.43 Therefore, advisable hollow size for tandem reaction drove by spatial confinement effect is very important. Moreover, the ECSA of all the samples have been determined from the CV curves at different scan rates from 10 to 50 mV s−1 (Fig. S6†) to estimate the effect from morphology. As shown in Fig. S7,† the Au@Cu2O-SC shows a Cdl value of 2.71 μF cm−2, similar to 2.08 and 2.23 μF cm−2 of Au@Cu2O-MC and Au@Cu2O-LC, further verify the reaction mechanism we have proposed previously.
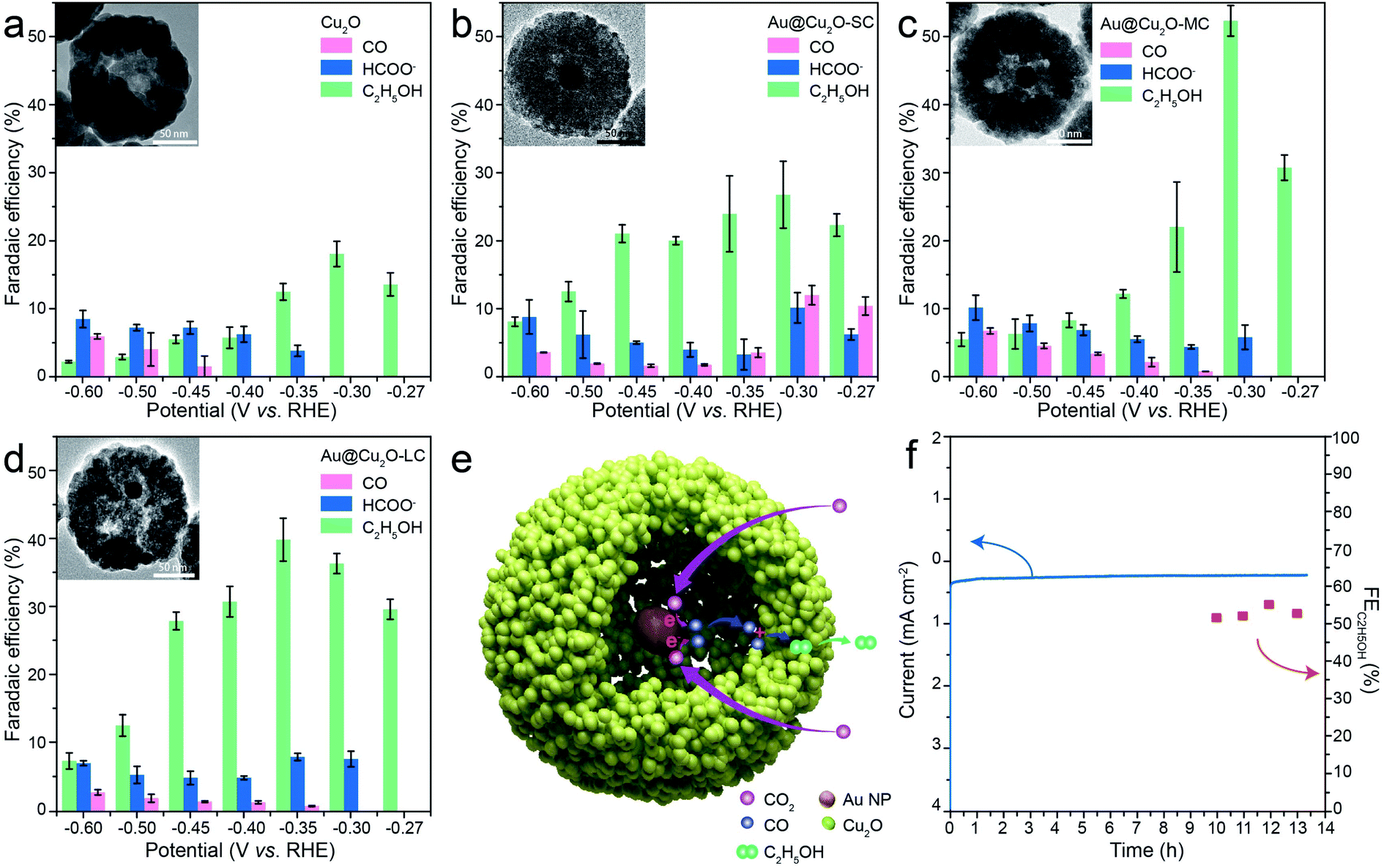 | ||
| Fig. 4 (a–d) The faradaic efficiencies of carbon monoxide, formic acid, and ethanol from the CO2RR products under a range of potentials of different samples. (e) Schematic illustration of tandem catalysis mechanism in the Au@Cu2O cavity. (f) Time-dependent current density curve and FEC2H5OH of Au@Cu2O-MC in CO2-saturated 0.1 M KHCO3 solution at −0.3 V vs. RHE. | ||
The durability of each catalyst was further assessed by chronoamperometry (CA) measurement since it is another important performance parameter for estimating an electrocatalyst. Fig. 4f exhibits the consecutive over 13 h CA curves at a constant potential of −0.3 V vs. RHE for Au@Cu2O-MC, and it outputs a steady current density for ethanol production with a nearly unchanged FEC2H5OH. The FEC2H5OH was retained >50% during the entire period, suggesting the excellent long-term durability of the catalyst, comparable with or outperforming most of other state-of-the-art Cu-based CO2RR catalysts (Table S1†). In addition, the catalysts after CO2RR tests were further characterized. TEM image (Fig. S8a†) and XRD pattern (Fig. S8b†) reveal that Au@Cu2O was in situ electroreduction to Au@Cu, hinting that the actual active sites are from Au-core and Cu-shell, which is in agreement with previously reported results.16,25,26
Conclusions
In summary, we have developed a bimetallic catalyst of Au@Cu2O yolk–shell with improved activity and selectivity for the electrochemical reduction of CO2 to ethanol at lower potential. It was demonstrated that the spatial confinement of different active sites with a tandem catalysis mechanism leads to the selectivity shift from C1 to C2. The Au-core can reduce CO2 to CO in the copper nanocavity, producing a high CO concentration, thus the Cu-shell transforms CO to ethanol production. In addition, we confine the CO concentration in the nanocavity by optimizing the hollow size of the Cu2O nanocavity, and then perform an ethanol faradaic efficiency of 52.3% at −0.30 V vs. RHE. These results suggest that the present strategy may shed light on the design and preparation of highly active tandem catalysts for other electrochemical reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (No. 2018JBM067) and the National Natural Science Foundation of China (No. 21603011).Notes and references
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- W. Bi, C. Wu and Y. Xie, ACS Energy Lett., 2018, 3, 624–633 CrossRef CAS.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- L.-q. He, H. Yang, J.-j. Huang, X.-h. Lu, G.-r. Li, X.-q. Liu, P.-p. Fang and Y.-x. Tong, RSC Adv., 2019, 9, 10168–10173 RSC.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna and O. S. Bushuyev, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- F. J. Gomez, G. Chumanov, M. F. Silva and C. D. Garcia, RSC Adv., 2019, 9, 33657–33663 RSC.
- Y. X. Duan, F. L. Meng, K. H. Liu, S. S. Yi, S. J. Li, J. M. Yan and Q. Jiang, Adv. Mater., 2018, 30, e1706194 CrossRef PubMed.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS PubMed.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. Garcia de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, e1804867 CrossRef PubMed.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- S. Zhong, X. Yang, Z. Cao, X. Dong, S. M. Kozlov, L. Falivene, J. K. Huang, X. Zhou, M. N. Hedhili, Z. Lai, K. W. Huang, Y. Han, L. Cavallo and L. J. Li, Chem. Commun., 2018, 54, 11324–11327 RSC.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- P. B. O'Mara, P. Wilde, T. M. Benedetti, C. Andronescu, S. Cheong, J. J. Gooding, R. D. Tilley and W. Schuhmann, J. Am. Chem. Soc., 2019, 141, 14093–14097 CrossRef PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- T. Le, A. Striolo, C. H. Turner and D. R. Cole, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- Y. Liu, X.-Y. Yu, Y. Fang, X. Zhu, J. Bao, X. Zhou and X. W. D. Lou, Joule, 2018, 2, 725–735 CrossRef CAS.
- P. Han, Z. Wang, M. Kuang, Y. Wang, J. Liu, L. Hu, L. Qian and G. Zheng, Adv. Energy Mater., 2018, 8, 1801230 CrossRef.
- L. Tang, X. Meng, D. Deng and X. Bao, Adv. Mater., 2019, 31, e1901996 CrossRef PubMed.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- S. Mezzavilla, S. Horch, I. E. L. Stephens, B. Seger and I. Chorkendorff, Angew. Chem., Int. Ed., 2019, 58, 3774–3778 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- M. Fan, Z. Bai, Q. Zhang, C. Ma, X.-D. Zhou and J. Qiao, RSC Adv., 2014, 4, 44583–44591 RSC.
- L. Zhang, D. A. Blom and H. Wang, Chem. Mater., 2011, 23, 4587–4598 CrossRef CAS.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
- J. Zhang, G. Liu, F. He, L. Chen and Y. Huang, RSC Adv., 2015, 5, 87903–87907 RSC.
- C. He, Y. Zhang, Y. Zhang, L. Zhao, L.-P. Yuan, J. Zhang, J. Ma and J.-S. Hu, Angew. Chem., Int. Ed., 2020, 59, 4914–4919 CrossRef CAS PubMed.
- Y. Shang, D. Sun, Y. Shao, D. Zhang, L. Guo and S. Yang, Chem.–Eur. J., 2012, 18, 14261–14266 CrossRef CAS PubMed.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- P. J. Cumpson, Appl. Surf. Sci., 1999, 144, 16–20 CrossRef.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02482a |
| This journal is © The Royal Society of Chemistry 2020 |