
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of a direct Z-scheme BiOCl–phosphotungstic acid heterojunction for the improved photodegradation of tetracycline†
Haijuan Tonga,
Bingfang Shi*a and
Shulin Zhao *b
*b
aKey Laboratory of Regional Ecological Environment Analysis and Pollution Control of West Guangxi, College of Chemistry and Environmental Engineering, Baise University, Baise 533000, China. E-mail: shibingfang@126.com
bState Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources, Guangxi Normal University, Guilin, 541004, China. E-mail: zhaoshulin001@163.com
First published on 7th May 2020
Abstract
The fabrication of a Z-scheme heterojunction photocatalyst can effectively modulate the electron transfer and separation of photoinduced charge carriers to enhance photocatalytic performance. Here, we demonstrate a direct Z-scheme BiOCl–phosphotungstic acid (BiOCl–HPW) heterojunction, fabricated via a one-step hydrothermal synthesis, as a highly active and stable photocatalyst for the photodegradation of tetracycline. BiOCl–HPWs result in a dramatic improvement in visible-light utilization and photogenerated e− and h+ separation, as well as a decrease in the electrical resistance by the addition of HPW. Notably, the BiOCl–HPW heterojunction with optimized phosphotungstic acid (HPW) content achieved an outstanding photodegradation rate of tetracycline (0.0195 min−1). A reasonable Z-scheme photocatalytic mechanism based on the analysis of band structure and monitoring of active radicals is proposed. This study highlights the potential implications of the BiOCl–HPW heterojunctions in the photodegradation of other toxic chemicals and photocatalytic studies.
1. Introduction
Antibiotics left in the environment have become a worrying modern society problem because of their wide use as growth promoters and antibacterial drugs.1 Tetracycline (TC), as a typical antibiotic, causes serious problems for human health and the ecosystem due to its potential negative impact on human beings and the ecological environment.2,3 Excluding tetracycline from polluted waters has become imperative.4 Photodegradation has recently emerged as a promising technique for its green, efficient, and effective elimination of toxic chemicals.5 Traditional semiconductor photocatalysts such as TiO2 and ZnO have attracted increasing attention for the photodegradation of organic pollutants in water because of their nontoxicity, chemical stability, low cost, and wide availability.6–8 However, their practical application for photocatalytic water purification is hindered by their intrinsic wide bandgaps and low degradation efficiencies.9 Therefore, the development of photocatalysts with excellent photocatalytic activity that can utilize the maximum amount of solar light is the focus of environmental science.In this regard, bismuth oxyhalides (BiOX, X = F, Cl, Br and I) have been applied as promising photocatalysts because of their low toxicity, high thermal strength, and excellent electronic properties.10 BiOX exhibits larger photodegradation rate under visible-light irradiation, mainly attributed to the narrow bandgap resulting from O 2p and Bi 6s2 hybridized valence bands.11–13 Consequently, a series of BiOX with high photocatalytic activities have been synthesized and successfully applied in the degradation of organic dyes.14–16 Although the BiOX mentioned above demonstrates stronger photocatalytic performance under visible light, their practical photocatalytic proficiency is limited by poor optical absorption capacity and photogenerated charge carrier recombination.11 To improve the photocatalytic performance of BiOX, various synthesis strategies based on introducing defects,17 element doping,18 and constructing heterojunctions19 have been explored. Of these proposed approaches, fabricating BiOX-based Z-scheme heterojunction has proven to be one of the easiest techniques to prepare advanced photocatalysts because of the feasible and effective separation of the isolated photogenerated e− and h+, thus effectively reducing the recombination efficiency of the charged carriers. In recent years, researchers have prepared different BiOX – based Z-scheme heterojunctions such as N-GQDs/BiOCl,20 CdS/BiOCl,21 BiOCl/g-C3N4,22 and Bi2O3/BiOCl.23 Although these approaches for the synthesis of BiOCl-based heterojunctions have been successful and have somewhat effectively improved the photocatalytic performance compared with one-component catalysts, undesirable features still exist. Specifically, the electron transfer is still restricted because of the electrical resistance between the contact interfaces of the two semiconductors.24 Noble metals (Ag, Au, or Pt) and carbon quantum dots have been employed as decorated accessories in heterojunctions to effectively reduce the electric resistance because of their excellent electrical conductivity.25–28 However, the high temperature and lengthy fabrication procedures required as well as the high costs are drawbacks. Theoretically, electron mediators are prone to accelerate electron transfer. Hence, the introduction of cocatalysts on the BiOX surface that act as mediators and enable easier electronic migration may suppress the recombination of photogenerated carriers, which is a new challenge.
As an electron mediator, phosphotungstic acid (HPW) can promote electron transfer between cocatalysts due to its well-defined Keggin structure.29 Additionally, HPW can catalyze many organic synthesis reactions such as oxidation, esterification, and C–N bond formation because of its strong acidity and oxidative capacity.30–32 Inspired by the excellent properties of HPW, researchers have invested massive effort on the preparation of HPW based heterojunctions, such as copolymer–HPW,33 choline–HPW,34 and polyimide–HPW.9 These new successful approaches have shown that the synthesized HPW based heterojunctions have slightly enhanced electron–hole interactions and photocatalytic activity compared with pure HPW. However, they inevitably require a rigorous, complex synthesis process, including many steps under N2 gas flow,33 complex process and time consuming (slowly dropped and approximately 24 h),34 and high synthesis temperature (325 °C for 4 h),9 respectively.
Herein, a one-step hydrothermal approach for synthesizing BiOCl–phosphotungstic acid (BiOCl–HPW) heterojunctions using Bi(NO3)3·5H2O and HPW as precursors is proposed. The prepared BiOCl–HPW heterojunctions exhibited good stability. Moreover, the photocatalytic activity of BiOCl–HPWs was superior to those of HPW and as-prepared BiOCl. As expected, 92.2% tetracycline was removed by the BiOCl–HPW-3 after 120 min simulated solar light irradiation. We also propose a reasonable Z-scheme photocatalytic mechanism based on the analysis of band structure and monitoring of active radicals. Our results can offer new opportunities for the development and in-depth understanding of noble direct Z-scheme heterojunction photocatalysts for efficient photodegradation of organic pollutants.
2. Experimental section
2.1 Chemicals and reagents
Tetracycline (TC), phosphotungstic acid (HPW), polyvinylpyrrolidone, isopropanol (IPA), ammonium oxalate (AO), p-benzoquinone (BQ), ethylene glycol, Bi(NO3)3·5H2O, terephthalic acid (TA), and KCl were purchased from Aladdin Chemistry Co. Ltd. (Shanghai, China). All chemical solvents and reagents were of analytical grade and were used without additional purification. Deionized water was purified by a Milli-Q plus 185 equip from Millipore (Bedford, MA).2.2 Synthesis of BiOCl–HPWs
The BiOCl–HPWs were prepared through a one-step hydrothermal approach. In a typical synthesis, the reaction solution containing Bi(NO3)3·5H2O (0.97 g), polyvinylpyrrolidone (0.40 g), and 25 mL of ethylene glycol was sonicated for 10 min. Subsequently, 0.01 g HPW and 10 mL of saturated KCl solution were slowly added into the above mixture with stirring at room temperature for 10 min, and then the obtained solution was transferred into a 50 mL Teflon-lined stainless steel autoclave and hydrothermally heated at 160 °C for 3 h under vigorous stirring. After being naturally cooled, the mixture was centrifuged and washed several times with deionized water. Finally, the resulting solid powder was dried at 60 °C for 10 h and was denoted as BiOCl–HPW-1. According to this method, BiOCl–HPW-2, BiOCl–HPW-3, and BiOCl–HPW-4 were synthesized with different HPW content (0.02, 0.03, and 0.04 g), respectively.For comparison, a hydrothermal method was used to prepare BiOCl. The preparation of BiOCl was the same as the above method except for the use of HPW.
2.3 Characterization of BiOCl–HPWs
UV-Vis absorption spectra were recorded on a Cary 60 UV-vis spectrophotometer (Agilent Technologies) at room temperature. Fourier transform infrared spectroscopy (FTIR) was performed using a PerkinElmer FTIR spectrophotometer (PerkinElmer, USA) from 400 to 4000 cm−1. X-ray diffraction (XRD) patterns were recorded with monochromatic Cu Kα radiation (40 kV, 30 mA). Transmission electron microscopy (TEM) was conducted on a FEI Tecnai F-20 field emission HRTEM (Hillsboro, OR, USA) at a voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos XSAM 800 photoelectron spectrometer (Manchester, UK). Photoluminescence (PL) were performed on a FL7000 (Hitachi, Japan). N2 adsorption desorption isotherms was conducted on Belsorp-max (Belsorp-max II, Japan). The electrochemical impedance spectroscopy (EIS) were obtained on an electrochemical workstation (Chenhua, CHI-660D) with a three-electrode system where Ag/AgCl electrode as the reference, Pt wire as the counter and material-coated glassy carbon (GC) as the working electrode in 0.5 M Na2SO4 aqueous solution. The UV-vis diffuse reflectance spectra (DRS) were carried on Hitachi U-3010 spectrophotometer with BaSO4 as a reference. Bruker ESR A300 spectrometer was employed to record the electron spin resonance (ESR) signals under simulated light irradiation. Total organic carbon (TOC) test was conducted on a TOC-VCPH analyzer (Shimadzu, Japan).2.4 Photocatalytic activity of BiOCl–HPWs
The photocatalytic activities of BiOCl–HPW were evaluated by the decomposition of tetracycline (TC) and under simulated solar light irradiation. 10 mg of BiOCl–HPWs was dispersed in 50 mL TC with an initial concentration of 10 mg L−1 under magnetic stirring in the dark for 60 min. After achieving the adsorption–desorption equilibrium, photodegradation of TC was conducted under light irradiation using a PLS-SXE300UV Xe lamp (15 A, 300 W) as a light source. During tetracycline photodegradation, the reaction solutions were withdrawn at regular intervals for analysis using a UV-vis spectrophotometer in the range of 300–700 nm. All measurements were conducted in triplicate.In order to clarify the photocatalytic mechanism, free radical scavenge experiment was performed. Prior to addition of the photocatalyst, 1.0 mM p-benzoquinone (BQ), AO, and isopropanol (IPA) were added into the TC solution to investigate the effects of hydroxyl radicals (˙OH), superoxide radicals (˙O2−), and holes (h+) on TC photodegradation by BiOCl-HPWs, respectively. All experiments were conducted according to the photocatalytic activity experiment.
2.5 Recyclability of BiOCl–HPWs
BiOCl–HPW-3 was employed to evaluate the recyclability of BiOCl–HPWs by photodegradation of TC under simulated solar light irradiation. Ten milligrams of BiOCl–HPW-3 and 10 mg L−1 of TC underwent similar photocatalytic degradation processes, following which the used BiOCl–HPW-3 was collected by centrifugation; then, BiOCl–HPW-3 was washed and dried at 80 °C for 10 h for recyclability tests.3. Results and discussion
3.1 Structure of BiOCl–HPWs
X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) profiles were performed to investigate the structure of BiOCl–HPWs. As presented in Fig. 1a, all photocatalysts exhibit good crystallinity. For the as-prepared BiOCl, the diffraction peaks at 2θ = 11.95, 25.85, 32.80, 33.50, 36.50, 41.50, 46.64, 49.90, and 54.09 could be readily attributed to the (001), (101), (110), (102), (003), (112), (200), (113), and (211) crystal faces of BiOCl, respectively, (JCPDS Card No. 06-2049). The diffraction peaks of HPW resulting from the amorphous state of HPW cannot be observed, which is in accordance with previously reported results.35 All diffraction peaks of BiOCl–HPWs were similar to those of BiOCl, demonstrating the basic structure of BiOCl in BiOCl–HPWs. | ||
| Fig. 1 XRD patterns (a) of as-synthetised BiOCl and BiOCl–HPWs. XPS survey spectrum (b, inset: Bi 4f XPS spectrum), O 1s XPS spectrum (c) and W 4f XPS spectrum (d) of the as-prepared BiOCl–HPWs. | ||
To confirm the introduction of HPW, the Fourier transform infrared spectroscopy (FTIR) spectra of as-prepared BiOCl and BiOCl–HPWs are presented in Fig. S1 (ESI†). For the as-prepared BiOCl, the peak at approximately 527 cm−1 was assigned to the Bi–O stretching vibrations of BiOCl (Fig. S1a, ESI†). Distinct peaks at approximately 1650 cm−1 and 3400 cm−1 were observed which were attributed to the stretching vibration modes of –OH after the absorption of water molecules.36 For BiOCl–HPWs, peaks at 817 cm−1 and 980 cm−1 could be observed compared with the FT-IR spectra of as-prepared BiOCl (Fig. S1b, ESI†). These peaks are attributed to the typical vibration of W–Oc–W and W–O,35 indicating the existence of HPW in BiOCl-HPWs.
To further verify the fabrication of BiOCl–HPWs, XPS profiles of BiOCl–HPWs were analyzed. Fig. 1b shows the existence of Cl, O, Bi, and W atoms in BiOCl–HPWs. However, it is difficult to observe the peaks of P in the XPS profiles possibly because of the low amount of P in HPW. This observation agrees well with previously reported results.35 The high-resolution Bi 4f spectrum (inset of Fig. 1b) shows peaks at 159.1 eV and 164.4 eV corresponding to Bi 4f7/2 and Bi 4f5/2 of Bi3+, respectively.37 In Fig. S2 (ESI†), two peaks located at 197.9 eV and 199.1 eV are consistent with the Cl 2p3/2 and Cl 2p1/2 region belonging to Cl 2p peaks.36 Fig. 1c shows that two signals at 531.5 eV and 533.0 eV can be observed, which are attributed to Bi–O in BiOCl and W–O in the Keggin structure of HPW, respectively,38,39 which agreed well with the FT-IR results. Fig. 1d indicates that the XPS peaks of W 4f5/2 and W 4f7/2 in BiOCl–HPWs appear at 37.4 and 35.3 eV, respectively, which are characteristic of the W6+ state.40 These above results confirm the successful fabrication of BiOCl–HPWs. As shown in Fig. S3 (ESI†), compared with the as-prepared BiOCl, a small shift in peaks of Bi and Cl to the lower energy region indicates the coordination of BiOCl with HPW by chemical bonding in BiOCl–HPWs.41 This suggests HPW underwent self-assembly on the BiOCl surface through the hydrothermal method to formed BiOCl–HPW heterojunction.
3.2 Morphology of BiOCl–HPWs
The morphologies of BiOCl–HPWs were characterized through SEM and TEM. As displayed in Fig. 2a, the BiOCl–HPWs are sheet-like structures, which are similar to the structure of BiOCl (Fig. S4a, ESI†). The as-prepared BiOCl–HPWs nanosheets have large and smooth surfaces, which are advantageous for the attachment of HPW. The TEM images (Fig. 2b) further confirmed the sheet-like structure of BiOCl–HPWs. High resolution TEM (HRTEM) imaging (inset of Fig. 2b) showed the prepared BiOCl–HPWs displayed a highly crystalline structure with a 0.27 nm lattice parameter, which was ascribed to the (110) plane of the BiOCl nanosheets.36 The lattice spaces of HPW on the surfaces of BiOCl nanosheets could not be observed, which match the XRD results of BiOCl–HPWs. However, the mapping images of P and W confirm the elemental composition and distribution in BiOCl–HPWs (Fig. 2f and h). Fig. 2c–h shows the same shape of the mapping images of Bi, P, and W, further suggesting the successful construction of BiOCl–HPW heterojunctions. Furthermore, we observed small particles distributed on the nanosheets (Fig. 2b), further demonstrating that HPW was decorated on the surface of BiOCl to form heterojunctions. | ||
| Fig. 2 The images of TEM (a), SEM (b, inset: HRTEM of BiOCl–HPW-3) and EDX elemental mappings (c–h) of BiOCl–HPW-3. | ||
3.3 Photocatalytic activity
Based on the photocatalytic activity of BiOCl, whether the prepared BiOCl–HPWs also possessing the photocatalytic activity on the photocatalytic degradation of TC under the simulated solar light was analyzed. Fig. 3a shows that the photocatalytic degradation capacity of BiOCl–HPWs is much higher than that of HPW and as-prepared BiOCl. This suggests that the introduced HPW has an important role in enhancing the photocatalytic degradation capacity of BiOCl–HPWs. To investigate the photocatalytic activity of as-prepared BiOCl, HPWs, and BiOCl–HPWs, the corresponding photocatalytic degradation kinetics were analyzed. Fig. S5 (ESI†) shows that the experimental data fit the first-order kinetic model with good linearity. As shown in Fig. 3b, the apparent rate constant (k) of BiOCl–HPW-3 (k = 0.0195 min−1) was approximately 5.3 and 13.0 times that of as-prepared BiOCl (k = 0.0037 min−1) and HPW (k = 0.0015 min−1), respectively. These results imply HPW has crucial roles in promoting the photocatalytic degradation efficiency of TC by BiOCl–HPWs. To further investigate the photocatalytic activity of BiOCl–HPWs prepared using different HPW contents, the k of BiOCl–HPWs were compared. As can be seen in Fig. 3b, the k of the respective BiOCl–HPWs increased with the incorporation of HPW. However, the k of BiOCl–HPWs decreased when HPW content became 0.04 g. The k of BiOCl–HPW-3, as a result, was higher when compared with that of BiOCl–HPW-1 (k = 0.0084 min−1), BiOCl–HPW-2 (k = 0.0106 min−1) and BiOCl–HPW-4 (k = 0.0133 min−1), which further confirms that HPW has crucial roles in promoting the photocatalytic performance of BiOCl–HPWs. Based on the above observations, BiOCl–HPW-3 was chosen to investigate the possible reason for the differences in photodegradation efficiencies of the photocatalysts in the subsequent TC degradation experiments.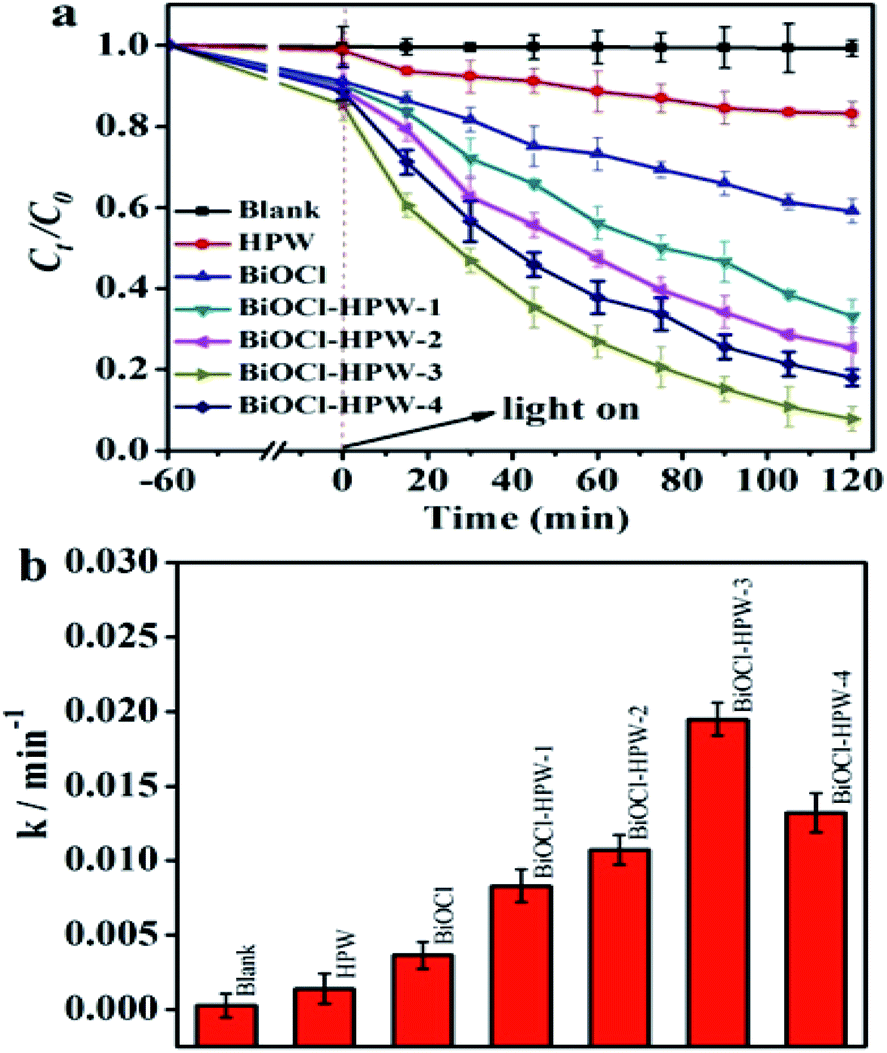 | ||
| Fig. 3 The photocatalytic efficiency (a) and the apparent rate constant (b) of BiOCl, HPW and BiOCl–HPW-3. The photo-catalyst concentration was 0.2 mg mL−1 and TC concentration was 10 mg L−1. | ||
To reveal the reasons for the enhanced photocatalytic degradation efficiency of TC by BiOCl–HPWs, the optical absorption properties of HPWs, as-prepared BiOCl, and BiOCl–HPWs were investigated by UV-vis diffuse reflectance spectra. As shown in Fig. 4a, the UV-vis spectra of BiOCl–HPWs were a combination of the spectra of HPW and as-prepared BiOCl. It is obvious that the absorption capacity of BiOCl is much lower than that of HPW. Additionally, an increasing red shift of BiOCl–HPWs to the longer wavelength region with increased HPW content was observed, suggesting that the modification of BiOCl with HPW greatly improves its absorption range and optical absorption capacity. Consequently, more e− and h+ pairs were easily produced after more light was adsorbed, thus resulting in the improved photocatalytic performances of BiOCl–HPWs.
 | ||
| Fig. 4 UV-vis absorbance spectra (a) and PL spectra (b) of BiOCl, HPW and BiOCl–HPW-3. Electrochemical impedance Nyquist plots (c) and photocurrent response (d) of BiOCl, HPW and BiOCl–HPW-3 heterojunction electrodes. | ||
In view of HPW regarding as an electron mediator, it was speculated that the separation of photogenerated carriers was promoted after the introduction of HPW. To confirm the above hypothesis, the photoluminescence (PL) spectra of BiOCl–HPW-3 were analyzed. Fig. 4b shows that the PL spectrum of BiOCl–HPW-3 agrees with that of the as-prepared BiOCl, which may be attributed to the defective emission of BiOCl. This observation is similar to a previously reported result.36 Under an excitation of 410 nm, the PL intensity of BiOCl–HPW-3 was lower than that of HPW and as-prepared BiOCl, which illustrates the separation of photogenerated e− and h+ pairs was promoted after the introduction of HPW.42 Therefore, it suggests that more photon participate in the photocatalytic reaction due to the decrease in recombination rate of photogenerated electron-hole pairs, resulting in an improved photocatalytic performance. However, as excess HPW was added, new photogenerated e− and h+ recombination centers were formed on the surface.9 Accordingly, the lower k (Fig. 1b) of BiOCl–HPW-4, compared with BiOCl–HPW-3, with the highest HPW content can also be explained reasonably.
To gain further insights into the promoted separation of photogenerated e− and h+ pairs of BiOCl–HPWs, the EIS Nyquist plots of HPW, as-prepared BiOCl, and BiOCl–HPW-3 were compared. Fig. 4c shows that the radii of the micro semicircle of BiOCl–HPW-3 are much smaller than those of as-prepared BiOCl and HPW, which indicates that the charge transfer resistance of BiOCl–HPW-3 is lowest.41 The lower electrical resistance can effectively facilitate the surface and interlayer electron transfer of BiOCl–HPWs, thus limiting the recombination of the charge carriers. Analogous, photocurrent response (Fig. 4d) of samples obviously display BiOCl–HPW-3 composites have a superior photocurrent response property than HPW and BiOCl, furtherly illustrate BiOCl–HPW-3 heterojunction facilitate the charge migration.
As is well-known, the activity of a catalyst depends on its surface area. Therefore, the surface areas of as-prepared BiOCl and BiOCl–HPW-3 were measured (Fig. S6, ESI†). Results show that the BET surface areas of as-prepared BiOCl and BiOCl–HPW-3 are 20.1 and 18.2 m2 g−1, respectively. The surface areas did decrease after the successful combination of BiOCl and HPW possibly because of the partially blocked pores by HPW.33 Indeed, after the introduction of HPW, the surface areas of BiOCl–HPW-3 decreased, while its photocatalytic activity was promoted, which suggests that the surface area has less impact on the photocatalytic activity of BiOCl–HPWs.
All of the above results clearly demonstrate that the enhanced photocatalytic activity of BiOCl–HPW-3 originates from the effective visible-light utilization, efficient photogenerated e− and h+ separation, and lower electrical resistance after the introduction of HPW.
3.4 Band structure
The photocatalytic activity is affected not only by the separation efficiency of photogenerated carriers but also by the band structure of the photocatalyst. Fig. 5a shows that the calculated valence band (VB) of BiOCl in heterojunction was 2.35 eV, which is higher than that reported for BiOCl (1.8 eV).43 Fig. 5b shows the VB of HPW is at 3.35 eV, which is more than that reported by Liu (3.28 eV).44 Theoretically, the oxidation ability of the photogenerated holes is promoted when the VB lies at a lower position. Accordingly, the photogenerated holes in the VB of BiOCl and HPW possess stronger oxidation ability. Besides, the oxidation ability of the photogenerated holes in the VB of HPW is higher than that of BiOCl in BiOCl–HPWs. As expected, the bandgap energy of BiOCl–HPWs (3.32 eV) is lower than that of BiOCl (3.55 eV) (Fig. 5c), resulting in the enhancement of the ability of harvesting visible light. | ||
| Fig. 5 VB-XPS patterns of BiOCl (a) and HPW (b). The plots of (αhν)2 vs. photon energy (hν) for the band gap energies of as-prepared BiOCl, HPW and BiOCl–HPWs (c). | ||
3.5 The performance for the removal of TC
It is well known that the removal of TC includes two processes: the TC are first adsorbed on the surface of catalyst, followed by photocatalytic degradation. As shown in Fig. 3a, the removal of TC in the presence of different catalysts displayed similar trends during both adsorption and photocatalytic degradation. During both adsorption, the best adsorption capacity and the adsorption ability of BiOCl–HPW-3 was observed after achieving the adsorption–desorption equilibrium (Fig. S7, ESI†), which effectively not only strengthens the contact between TC and BiOCl–HPW-3 but also increased the utilization rate of the active species.45 Consequently, the best adsorption of BiOCl–HPW-3 facilitates subsequent photocatalytic degradation. As for the process of photocatalytic degradation, the TC degradation efficiency by BiOCl and HPW is relatively low with only 40.9% and 27.0%, respectively. However, the removal efficiency of TC by BiOCl–HPWs is remarkably higher than that of BiOCl and HPW. The highest TC degradation efficiency of BiOCl–HPW-3 reaches 92.2% after 120 min simulated solar light irradiation (Fig. 3a), which is similar to the result of Chen46 and much higher than that in the reported literature.47,48To evaluate TC mineralization by BiOCl, HPW, and BiOCl–HPW-3, TOC measurements were conducted, and the results are shown in Fig. 6a. As shown in Fig. 6a, only 21.1% and 6.1% TOC were removed in the presence of BiOCl and HPW after illumination for 120 min, respectively. Meanwhile, the TOC removal efficiency of TC by BiOCl–HPW-3 is higher than that of BiOCl and HPW, indicating that the efficiency of photocatalytic performance of BiOCl is greatly enhanced after combination with HPW. However, the mineralization rate of BiOCl–HPW-3, compared with its photodegradation efficiency, is much lower, which is resulted from the production of intermediate materials during photoreaction.46
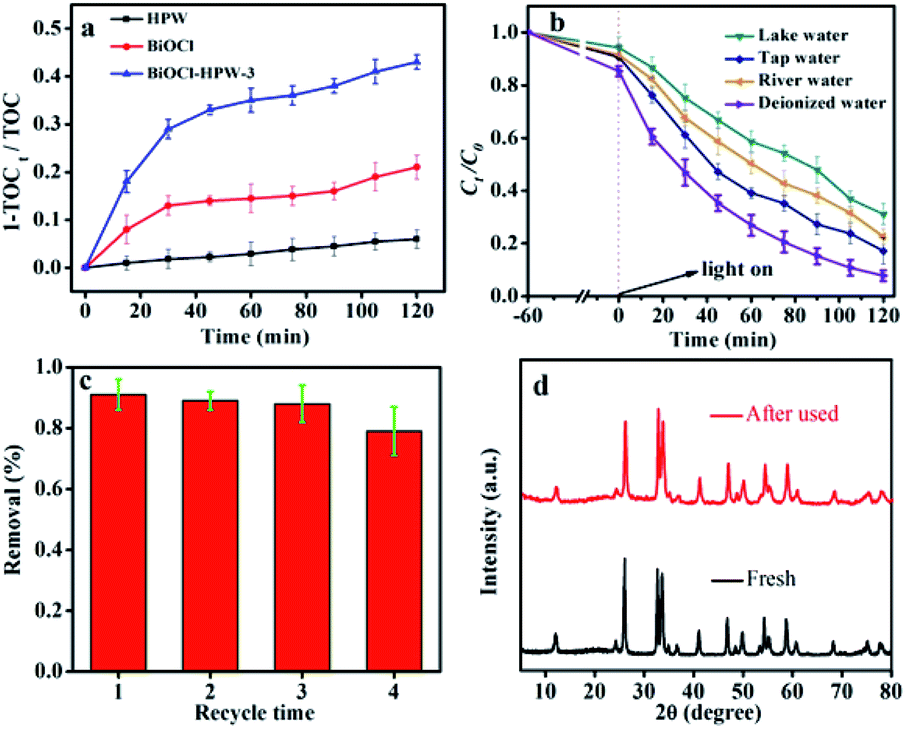 | ||
| Fig. 6 TOC removal of TC in the presence of BiOCl, HPW and BiOCl–HPW-3 within 120 min illumination (a), removal of TC in the presence of BiOCl–HPW-3 in real wastewater (b), the recyclability test (c) and XRD patterns (d) of BiOCl–HPW-3 (photocatalyst concentration was 0.2 mg mL−1 and TC concentration was 10 mg L−1). | ||
For evaluating the practical application of BiOCl–HPW-3 in complex samples, BiOCl–HPW-3 was applied for the photocatalytic degradation of TC in tap water, river water and lake water, respectively. Three different types of wastewater were spiked with TC standard solutions with concentrations of 10 mg L−1. As shown in Fig. 6b, the degradation efficiency of TC over BiOCl–HPW-3 in tap water, river water and lake water were 82.9%, 77.6% and 68.1%, respectively. A decrease in photocatalytic degradation efficiency of TC in three water samples was observed, which is attributed to the competition between TC and the interfering components in water on active sites and active species. Exciting, the photocatalytic degradation of TC in real water samples maintain a relatively high efficiency, indicating that the reliability and feasibility of BiOCl–HPW-3 in the photocatalytic degradation of TC in complex samples.
For further evaluating the practicality of photocatalysts, recyclability and stability of BiOCl–HPW-3 were performed. Fig. 6c shows that the catalytic stability of BiOCl–HPW-3 does not significantly decrease after four cycling runs. Besides, all diffraction peaks in the XRD patterns of BiOCl–HPW-3 before and after four recycles were well preserved (Fig. 6d). These results illustrate that BiOCl–HPW-3 has good stability.
All observations further indicate the great potential applications of BiOCl–HPW-3 in wastewater purification.
3.6 Possible photocatalytic mechanism
Generally, photocatalysis is controlled by ˙O2−, ˙OH, and h+ radicals. As shown in Fig. 7, no signals of ˙O2− and ˙OH observed illustrated that no photodegradation occurred in the dark. As expected, four characteristic peaks of DMPO-˙OH were detected after 5 min of simulated solar light irradiation, indicating that ˙OH is produced during the photocatalytic degradation process. On the other hand, the fluorescence intensity of peak at 424 nm (characteristic peak of TA-OH) gradually increases with the increase of reaction time (Fig. S8, ESI†), which further confirmed the result of ESR signals of the DMPO-˙OH. Similarly, six characteristic signals of DMPO-˙O2− were clearly observed under the same conditions. In addition, the intensity of BiOCl–HPW-3 triggering DMPO-˙OH and DMPO-˙O2− was all higher than that of BiOCl triggering DMPO-˙OH and DMPO-˙O2−, further confirming the superior photodegradation activity of BiOCl–HPWs to that of BiOCl. | ||
| Fig. 7 ESR signals of the DMPO-˙OH (a) and DMPO-˙O2− (b) for BiOCl–HPW-3 in the dark and under 300 W Xe lamp irradiation. | ||
To identify the key active species responsible for the degradation, the scavenger regents (BQ for ˙O2−, AO for h+, and IPA for ˙OH) were analyzed. As indicated in Fig. S9 (ESI†), photodegradation is greatly suppressed by addition of IPA, illustrating ˙OH was the most active species for the degradation of TC. Meanwhile, h+ has the lowest effect on TC degradation, indicating h+ may tend to generate more ˙OH not to degrade TC. From Fig. 7a and b, the intensity of ˙OH signals was stronger than that of ˙O2−, indicating that the content of ˙OH was higher than that of ˙O2− in the photodegradation reaction and ˙OH was of a critical species, which is consistent with the result of the scavenger regent experiments.
Based on the above experimental results of band gap energy, the main reactive species, and the position of valence band and conduction band, the possible photocatalytic mechanism of TC photo-degradation by BiOCl–HPW heterojunctions was discussed. First, a conventional type-II heterojunction was proposed to explain the reaction mechanism of the BiOCl–HPW. If the transfer path of the photogenerated electrons and holes of BiOCl–HPW followed the type-II, then, under light illumination, the photogenerated e− on the conduction band (CB) of BiOCl (CB = −1.2 eV) would transfer to and concentrated on the CB of HPWs (CB = 0.24 eV), as shown in Fig. 8 (left). However, the concentrated photogenerated electrons (e−) on the CB of HPWs could not reduce the absorpted O2 to yield ˙O2− radicals due to the more positive potential than O2/˙O2− (−0.33 eV), which was inconsistent with the results of radical species trapping. These indicated that the photocatalytic mechanism of BiOCl–HPW could not be described by the charge transfer of type-II heterojunction, which is contrary to the results of the active species trapping experiments and ESR spectra. Therefore, direct Z-scheme mechanism is indirectly supported by the above result. As illustrated in Fig. 8 (right), under simulated solar light irradiation, e− remain in the CB of BiOCl and h+ in the VB of HPW. The VB potential of HPW (3.35 eV) is lower than the H2O/˙OH position (2.72 eV vs. NHE), and thus, h+ oxidize H2O to generate ˙OH for the oxidization of TC, which is in agreement with the above experimental results. Meanwhile, the remaining e− in the CB of BiOCl reduced O2 to produce ˙O2− for further oxidization of TC.
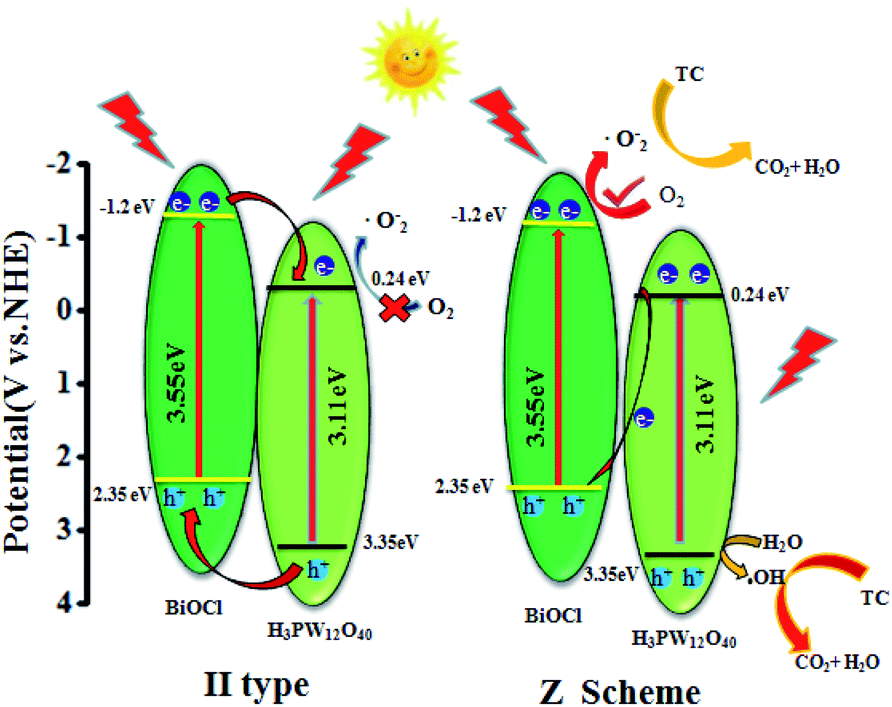 | ||
| Fig. 8 Schematic diagram of direct Z-scheme pathway for charge carrier separation. | ||
4. Conclusions
In summary, we have prepared a direct Z-scheme heterojunction photocatalyst BiOCl–HPWs by a facile hydrothermal synthesis method. Results demonstrate that BiOCl–HPWs prepared using 0.03 g HPW displayed superior photocatalytic ability for RhB and TC degradation under simulated solar light irradiation. The excellent photocatalytic ability was probably caused by the effective visible-light utilization, efficient photogenerated e− and h+ separation, as well as lower electrical resistance after the introduction of HPW with the Keggin unit. The well-matched energy band structures as well as ˙OH and ˙O2− mediated degradation suggest the photodegradation by BiOCl–HPW following a direct Z-scheme-dictated charge carrier transformation mechanism, which enhanced the photocatalytic activity of the BiOCl–HPWs. The photocatalytic activity of BiOCl–HPWs only slightly changed even after they were reused up to the fourth cycle. Consequently, BiOCl–HPWs as a Z-scheme heterojunction photocatalyst showed great potential for photocatalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (No. 21665001), and BAGUI Scholar Program.References
- K. Kümmerer, Chemosphere, 2009, 75, 417–434 CrossRef PubMed.
- C. I. Brinzila, N. Monteiro, M. J. Pacheco, L. Ciriaco, I. Siminiceanu and A. Lopes, Environ. Sci. Pollut. Res. Int., 2014, 21, 8457–8465 CrossRef PubMed.
- H. Shi, X. Xiao, L. Zeng, Q. Zhang, J. Nan and L. Wang, J. Nanosci. Nanotechnol., 2014, 14, 6934–6940 CrossRef PubMed.
- S. Sharma, V. Dutta, P. Singh, P. Raizada, A. Rahmani-Sani, A. Hosseini-Bandegharaei and V. K. Thakur, J. Cleaner Prod., 2019, 228, 755–769 CrossRef CAS.
- D. Yue, J. Lei, Y. Peng, J. Li and X. Du, Fuel, 2018, 226, 148–155 CrossRef CAS.
- S. Chan, T. Wu, J. Juan and C. Teh, J. Chem. Technol. Biotechnol., 2011, 86, 1130–1158 CrossRef CAS.
- H. Sun, S. Liu, S. Liu and S. Wang, Appl. Catal., B, 2014, 146, 162–168 CrossRef CAS.
- J. Zhang, S. Wageh, A. Al-Ghamdi and J. Yu, Appl. Catal., B, 2016, 192, 101–107 CrossRef CAS.
- P. Meng, H. Heng, Y. Sun, J. Huang, J. Yang and X. Liu, Appl. Catal., B, 2018, 226, 487–498 CrossRef CAS.
- S. Garg, M. Yadav, A. Chandra and K. Hernadi, J. Nanosci. Nanotechnol., 2019, 19, 280–294 CrossRef CAS.
- S. Cao, P. Zhou and J. Yu, Chin. J. Catal., 2014, 35, 989–1007 CrossRef.
- Y. Yang, C. Zhang, D. Huang, G. Zeng, J. Huang, C. Lai, C. Zhou, W. Wang, H. Guo, W. Xue and R. Deng, Appl. Catal., B, 2019, 245, 87–99 CrossRef CAS.
- K. Zhang, C. Liu, F. Huang, C. Zheng and W. Wang, Appl. Catal., B, 2006, 68, 125–129 CrossRef CAS.
- R. S. Yuan, C. Lin, B. C. Wu and X. Z. Fu, Eur. J. Inorg. Chem., 2009, 24, 3537–3540 CrossRef.
- H. Feng, Z. Xu, L. Wang, Y. Yu, D. R. G. Mitchell, D. Cui, X. Xu, J. Shi, T. Sannomia, Y. Du, W. Hao and S. Dou, ACS Appl. Mater. Interfaces, 2015, 7, 27592–27596 CrossRef CAS PubMed.
- L. Ye, L. Zan, L. Tian, T. Peng and J. Zhang, Chem. Commun., 2011, 47, 6951–6953 RSC.
- R. Jitta, R. Gundeboina, N. Veldurthi, R. Guje and V. Muga, J. Chem. Technol. Biotechnol., 2015, 24, 1937–1948 CrossRef.
- J. Bolobajev, M. Kasle, K. Kreek, M. Kulp, M. Koel and A. Goib, Chem. Eng. J., 2019, 357, 120–128 CrossRef CAS.
- H. Che, G. Che, P. Zhou, C. Liu and H. Dong, J. Colloid Interface Sci., 2019, 546, 262–275 CrossRef CAS.
- H. Wang, B. Zhang, F. Zhao and B. Zeng, ACS Appl. Mater. Interfaces, 2018, 10, 35281–35288 CrossRef CAS.
- W. Lin, X. Yu, Y. Shen, H. Chen, Y. Zhu, Y. Zhang and H. Meng, J. Nanopart. Res., 2017, 19, 56–67 CrossRef.
- Y. Bai, P. Wang, J. Liu and X. Liu, RSC Adv., 2014, 4, 19456–19461 RSC.
- F. Deng, Q. Zhang, L. Yang, X. Luo, A. Wang, S. Luo and D. Dionysiou, Appl. Catal., B, 2018, 238, 61–69 CrossRef CAS.
- R. Jiang, G. Lu, Z. Yan, D. Wu, R. Zhou and X. Bao, Chem. Eng. J., 2019, 374, 79–90 CrossRef CAS.
- M. Liang, T. Borjigin, Y. Zhang, H. Liu, B. Liu and H. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 34123–34131 CrossRef CAS PubMed.
- W. Zhu, D. Han, L. Niu, T. Wu and H. Guan, Int. J. Hydrogen Energy, 2016, 41, 14713–14720 CrossRef CAS.
- R. Wang, K. Lu, Z. Tang and Y. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- T. Bao, L. Song and S. Zhang, Chem. Eng. J., 2018, 351, 189–194 CrossRef CAS.
- A. Hiskia, M. Ecke, A. Troupis, A. Kokorakis, H. Hennig and E. Papaconstantinou, Environ. Sci. Technol., 2001, 35, 2358–2364 CrossRef CAS PubMed.
- G. Wang, Y. Shen and X. Wu, Eur. J. Org. Chem., 2008, 25, 4367–4371 CrossRef.
- A. Jha, A. Garade, S. Mirajkar and C. Rode, Ind. Eng. Chem. Res., 2012, 51, 3916–3922 CrossRef CAS.
- S. Wang and G. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- M. Betiha, H. Hassan, E. El-Sharkawy, A. Al-Sabagh, M. Menoufy and H. Abdelmoniem, Appl. Catal., B, 2016, 182, 15–25 CrossRef CAS.
- X. Zeng, X. Xiao, J. Chen and H. Wang, Appl. Catal., B, 2019, 248, 573–586 CrossRef CAS.
- L. Li, L. Li, T. Sun, X. Yu, L. Long, X. Lei and J. Yan, J. Solid State Chem., 2019, 274, 152–161 CrossRef CAS.
- D. Wu, X. Wang, H. Wang, F. Wang, D. Wang, Z. Gao, X. Wang, F. Xu and K. Jiang, J. Colloid Interface Sci., 2019, 533, 539–547 CrossRef CAS.
- X. Zhang, X. Wang, L. Wang, W. Wang, L. Long, W. Li and H. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 7766–7772 CrossRef CAS PubMed.
- F. Gao, D. Zeng, Q. Huang, S. Tian and C. Xie, Phys. Chem. Chem. Phys., 2012, 14, 10572–10578 RSC.
- J. Liu, S. Xie, Z. Geng, K. Huang, L. Fan, W. Zhou, L. Qiu, D. Gao, L. Ji, L. Duan, L. Lu, W. Li, S. Bai, Z. Liu, W. Chen, S. Feng and S. Zhang, Nano Lett., 2016, 16, 6568–6575 CrossRef CAS PubMed.
- P. Meng, H. Heng, Y. Sun, J. Huang, J. Yang and X. Liu, Appl. Catal., B, 2018, 226, 487–498 CrossRef CAS.
- W. Yang, B. Ma, W. Wang, Y. Wen, D. Zeng and B. Shan, Phys. Chem. Chem. Phys., 2013, 15, 19387–19394 RSC.
- K. Li, L. Yan, Z. Zeng, S. Luo, X. Luo, X. Liu, H. Guo and Y. Guo, Appl. Catal., B, 2014, 156, 141–152 CrossRef.
- C. Liu, J. Zhou, J. Su and L. Guo, Appl. Catal., B, 2019, 241, 506–513 CrossRef CAS.
- J. Meng, X. Wang, X. Yang, A. Hu, Y. Guo and Y. Yang, Appl. Catal., B, 2019, 251, 168–180 CrossRef CAS.
- X. Li, J. Wang, A. Rykov, V. Sharma, H. Wei, C. Jin, X. Liu, M. Li, S. Yu, C. Sun and D. Dionysiou, Catal. Sci. Technol., 2015, 5, 504–514 RSC.
- S. Chen, D. Huang, G. Zeng, X. Gong, W. Xue, J. Li, Y. Yang, C. Zhou, Z. Li, X. Yan, T. Li and Q. Zhang, Chem. Eng. J., 2019, 370, 1087–1100 CrossRef CAS.
- Y. Liu, J. Kong, J. Yuan, W. Zhao, X. Zhu, C. Sun and J. Xie, Chem. Eng. J., 2018, 331, 242–254 CrossRef CAS.
- L. Rimoldi, A. Giordana, G. Cerrato, E. Falletta and D. Meroni, Catal. Today, 2019, 328, 210–215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02396e |
| This journal is © The Royal Society of Chemistry 2020 |