
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe preparation of a difunctional porous β-tricalcium phosphate scaffold with excellent compressive strength and antibacterial properties†
Long Qin a,
Jiang Yia,
Lai Xuefeia,
Liao Lia,
Xie Kenana and
Xie Lu*b
a,
Jiang Yia,
Lai Xuefeia,
Liao Lia,
Xie Kenana and
Xie Lu*b
aSchool of Chemical Engineering, Sichuan University, Chengdu, 610065, China
bState Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases & Other Research Platforms, Dept. of Prosthodontic, West China Hospital of Stomatology, Sichuan University, China. E-mail: Xielu2017@163.com
First published on 30th July 2020
Abstract
Porous β-tricalcium phosphate (β-Ca3(PO4)2, β-TCP) scaffolds are widely applied in the field of bone tissue engineering due to their nontoxicity, degradability, biocompatibility, and osteoinductivity. However, poor compressive strength and a lack of antibacterial properties have hindered their clinical application. In order to address these disadvantages, graphene (G) and silver nanoparticles were introduced into β-TCP through a two-step method. In the synthesis process, G-β-TCP was prepared via an in situ synthesis method, and then silver nanoparticles and HAp particles were coated on the surface of the G-β-TCP scaffold in an orderly fashion using dopamine as a binder. From the results of characterization, when the content of graphene was 1 wt% of β-TCP, the G-β-TCP scaffold had the highest compression strength (127.25 MPa). And core–shell G-β-TCP-Ag-HAp not only had reduced cytotoxicity via the continuous release of Ag+, but it also achieved long-term antibacterial properties. Besides, the material still showed good cell activity and proliferation.
1 Introduction
Nowadays, biomaterials are introduced as promising substitutes for bone defect regeneration, including β-tricalcium phosphate (β-Ca3(PO4)2, β-TCP), calcium sulfate, and hydroxyapatite (HA). However, there are some limitation to these biomaterials. For instance, the absorption of calcium sulfate is too rapid for the formation of new bone in vivo. HA is hardly resorbed within the body, which restricts the formation of new bone and results in a poor local stability.1–4 β-TCP has good biocompatibility and osteoinduction, which is relatively balanced between absorption and new bone formation, and the degradation products can provide abundant Ca and P elements for the formation of new bone.5 However, the application of β-TCP in weight-bearing areas is hampered because its compression strength is poor with little brittleness and wear resistance. In the past years, much research has been to improve the compression strength of β-TCP porous scaffold in two ways. One approach is that a certain amount of polymer materials, such as poly L-lactic acid (PLLA), poly lactide–glycolide acid (PLGA), polyethylene, etc., is combined with the β-TCP porous scaffold.6–10 For example, Xiujuan Xu prepared a PLLA/β-TCP porous scaffold by combining PLLA with β-TCP in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to obtain a good compressive strength, and its degradation rate was effectively controlled by adjusting the ratio of PLLA to β-TCP.11 Yunqing Kang et al. prepared an interconnected porous β-TCP scaffold infiltrated with a thin layer of PLGA and the mechanical performance was improved. The infiltration of PLGA increased the compressive strength of β-TCP scaffolds from 2.90 to 4.19 MPa, bending strength from 1.46 to 2.41 MPa, and toughness from 0.17 to 1.44 MPa, which resulted from the combination of the systematic coating of struts, interpenetrating structural characteristics, and crack bridging.12 However, the compressive strength of these composites rarely met the demand of human cancellous bone (2–10 MPa). The other approach is that a certain amount of metal ions, metal oxides, metal salts, or other inorganic oxides are doped in the β-TCP scaffold to improve the compression property. Gary A. Fielding et al. fabricated macroporous TCP scaffolds doped with SiO2 (0.5 wt%)/ZnO (0.35 wt%) by using commercial 3D printing technology. The addition of dopants increased the densification of the scaffolds, showed up to a 250% increase in compressive strength when compared to that of the pure TCP scaffolds and the maximum compressive strength reached 10.21 ± 0.33 MPa. However, it is worth noting that the rate of dopants should be controlled because of their cytotoxicity. Kunio Ishikawa et al. proved that β-TCP doped with 10 wt% Zn showed obvious cytotoxicity.13 Amit Bandyopadhyay et al. prepared calcium phosphate ceramics doped with ZnO, and the microhardness increased up to 2.5 wt% of ZnO; however 3.5 wt% ZnO samples showed a low cell density, poor cell adhesion and some cell death.14 Therefore, it is necessary to hunt for more suitable materials to improve the mechanical property of β-TCP.
:2 to obtain a good compressive strength, and its degradation rate was effectively controlled by adjusting the ratio of PLLA to β-TCP.11 Yunqing Kang et al. prepared an interconnected porous β-TCP scaffold infiltrated with a thin layer of PLGA and the mechanical performance was improved. The infiltration of PLGA increased the compressive strength of β-TCP scaffolds from 2.90 to 4.19 MPa, bending strength from 1.46 to 2.41 MPa, and toughness from 0.17 to 1.44 MPa, which resulted from the combination of the systematic coating of struts, interpenetrating structural characteristics, and crack bridging.12 However, the compressive strength of these composites rarely met the demand of human cancellous bone (2–10 MPa). The other approach is that a certain amount of metal ions, metal oxides, metal salts, or other inorganic oxides are doped in the β-TCP scaffold to improve the compression property. Gary A. Fielding et al. fabricated macroporous TCP scaffolds doped with SiO2 (0.5 wt%)/ZnO (0.35 wt%) by using commercial 3D printing technology. The addition of dopants increased the densification of the scaffolds, showed up to a 250% increase in compressive strength when compared to that of the pure TCP scaffolds and the maximum compressive strength reached 10.21 ± 0.33 MPa. However, it is worth noting that the rate of dopants should be controlled because of their cytotoxicity. Kunio Ishikawa et al. proved that β-TCP doped with 10 wt% Zn showed obvious cytotoxicity.13 Amit Bandyopadhyay et al. prepared calcium phosphate ceramics doped with ZnO, and the microhardness increased up to 2.5 wt% of ZnO; however 3.5 wt% ZnO samples showed a low cell density, poor cell adhesion and some cell death.14 Therefore, it is necessary to hunt for more suitable materials to improve the mechanical property of β-TCP.
Graphene (G) is a two-dimensional (2D) crystal which attracted interest in recent years due to its specific surface area, high conductivity, superior compressive strength and good biocompatibility. Its theoretical tensile strength (150 GPa) and Young's modulus (1.0 TPa) make it viable as a functional material to improve the mechanical properties of the biomaterials. Gao et al. improved the mechanical properties of nano-58S bioactive glass with graphene, and significantly increased its compressive strength by 105%,15 and Mehdi M. et al. prepared calcium silicate ceramic composites reinforced with graphene nanoplatelets, which improved fracture toughness and brittleness index by ∼130% and ∼40%, respectively. The composites also had good biocompatibility and promoted cell viability and cell proliferation.16 Moreover, studies have revealed that graphene could induce directional differentiation of bone marrow stem cells to osteoblasts.17,18 Zengjie Fan et al. synthesized a graphene/hydroxyapatite (HA) nanorod composite by a one-pot hydrothermal strategy, and the introduced graphene effectively improved the hardness (242.06 ± 7.28 MPa) and Young's modulus (6.20 ± 1.67 GPa) of HA, and the composite containing 40 wt% HA showed a better biocompatibility and superior bone cellular proliferation.19 Ci et al. researched a diopside–graphene nanoplatelet composite, and the results showed that the compressive strength and fracture toughness of 1 wt% graphene nanoplatelets of the composite improved by 102% and 34%, respectively, compared with the diopside scaffold without graphene.20
For bone implants, infection of the implantation site may delay the healing, cause implant failure and repeated surgeries. To treat the infection, antibiotics are always the choice. However, with the increasing resistance of pathogens, can be formidable to cure infection owing to the overuse and abuse of antibiotics.21–23 Therefore, it is important to develop implants with local long term antibacterial properties. Silver has long been known for its broad-spectrum antibacterial property, which has been used as a dopant or surface modifier in bone tissue engineering scaffolds.24–28 The silver containing composites show an excellent antibacterial property when the amount of silver is suitable, otherwise they may exhibit cytotoxicity. In addition, the controlled release of Ag+ is the key to solve the problems of unabiding antibacterial efficacy and high cytotoxicity. For instance, N. Matsumoto et al. prepared AgZn-TCP and its antimicrobial activity and cytotoxicity were tested; the result showed the material exhibited a higher antimicrobial than that of Ag-TCP; however, AgZn-TCP containing 9.09 mol% Ag+ and 9.09 mol% Zn2+ had a low cytotoxicity. Sean Hoover et al. synthesized silver doped porous β-TCP at three levels: 0.5 wt%, 1 wt% and 2 wt% Ag2O; they claimed that silver can be released over a long period and the scaffolds showed no cytotoxicity.
In this study, a porous difunctional β-TCP scaffold was prepared by a two-step method. Firstly, the G-β-TCP was fabricated by an in situ synthesis method where G acts as a mechanical filler. Secondly, based on our previous research,29 silver nanoparticles and HA particles were orderly coated on the surface of G-β-TCP, using dopamine as a binder. To our best knowledge, this is first report of G-β-TCP-Ag-HAp with excellent compressive strength, antibacterial properties and good cell activity, and is expected to be a potential biomaterial for load-bearing applications.
2 Materials and methods
2.1 Materials
Calcium nitrate tetra hydrate (Ca(NO3)2·4H2O), diammonium hydrogen phosphate ((NH4)2HPO4), trihydroxymethyl aminomethane (Tris, C4H11NO3), hydrazine hydrate (N2H4·H2O, 80 vol%), silver nitrate (AgNO3), ammonia (NH4OH, 25–28 vol%), soluble starch ((C6H10O5)n), stearic acid (C18H36O2), polyvinyl alcohol (CH2CH(OH)n, PVA) and hydrochloric acid (30%, HCl) were purchased from Kelong Reagent Co., Ltd. (Chengdu, China). Dopamine (DA) was obtained from Aladdin Reagent Co., Ltd. (Shanghai, China) and graphene was purchased from Suzhou Tanfeng Graphene Technology Co., Ltd. (Suzhou, China). All other chemicals were of analytical reagent grade and were used as received without further purification. All aqueous solutions were prepared with de-ionized water (D.I. water).2.2 Preparation of the G-β-TCP scaffold
At first, a certain amount of graphene (0.2 wt%, 0.5 wt%, 1 wt%, 1.5 wt%, and 2 wt% of β-TCP) was added into 200 mL of a Ca(NO3)2 aqueous solution (0.5 M) with ultrasonic dispersion and intense agitation. Meanwhile, (NH4)2HPO4 solution (0.5 M) was dropped into the mixed solution with a molar ratio of Ca/P 1.5 and the pH value was adjusted to 9–10 by NH3·H2O with intensive stirring for 2 h. Then, the precipitation powder was centrifuged, washed with D.I. water at least three times, dried through a vacuum oven at 100 °C for 24 h, and the dried powders were ground evenly and pressed into disc shapes (8 mm diameter, 2 mm thick) at 10 MPa for 3 min. Finally, the disc samples were transferred into a tube furnace and heated to 200 °C for 1 h with a heating rate of 5 °C min−1 before sintering at 1000 °C for 3 h. All the heating and cooling processes were carried out in a N2 flux atmosphere. The samples are referred to as x wt% G-β-TCP scaffold (x = 0.2, 0.5, 1, 1.5, and 2).2.3 Fabrication of G-β-TCP-Ag-HAp
After testing the compressive strength, we selected the best performing samples for the next experiment (in our work, 1 wt% G-β-TCP is the best one). Dopamine was dissolved in a Tris buffer (pH = 8.5, 10 mM Tris–HCl) solution at a concentration of 3.04 mg mL−1 with vigorous stirring at 25 °C for 2 h, and it is called the D-T solution. The G-β-TCP-Ag-HAp sample was prepared by the following steps, as shown in Fig. 1. Firstly, The G-β-TCP sample was immersed in 2 mL of D-T solution for 2 h, then washed with D.I. water at least three times and dried through vacuum freeze-drying for 24 h. The surface of the G-β-TCP was modified by dopamine. Secondly, the modified G-β-TCP was soaked in 2 mL of AgNO3 solution (2 mM) and reacted for 6 h in darkness, and then washed and dried by the previous method. In this process, Ag+ can be reduced to Ag nanoparticles (Ag-NPs) by dopamine self-polymerization, and the latter adhered to the surface of G-β-TCP uniformly, which is referred as G-β-TCP-Ag. Then, the G-β-TCP-Ag sample was coated by dopamine again in the same way. After that, the G-β-TCP-Ag sample was soaked in 200 mL of Ca(NO3)2 solution (0.5 M), rinsed with D.I. water three times after 6 h in the static state. Subsequently, the sample was immersed in 200 mL of (NH4)2HPO4 solution (0.3 M). The pH of the solution was adjusted to 10 by NH3·H2O. After reaction for 12 h at 60 °C, the final product was washed with D.I. water at least three times and dried through vacuum freeze-drying for 24 h, and referred to as G-β-TCP-Ag-HAp.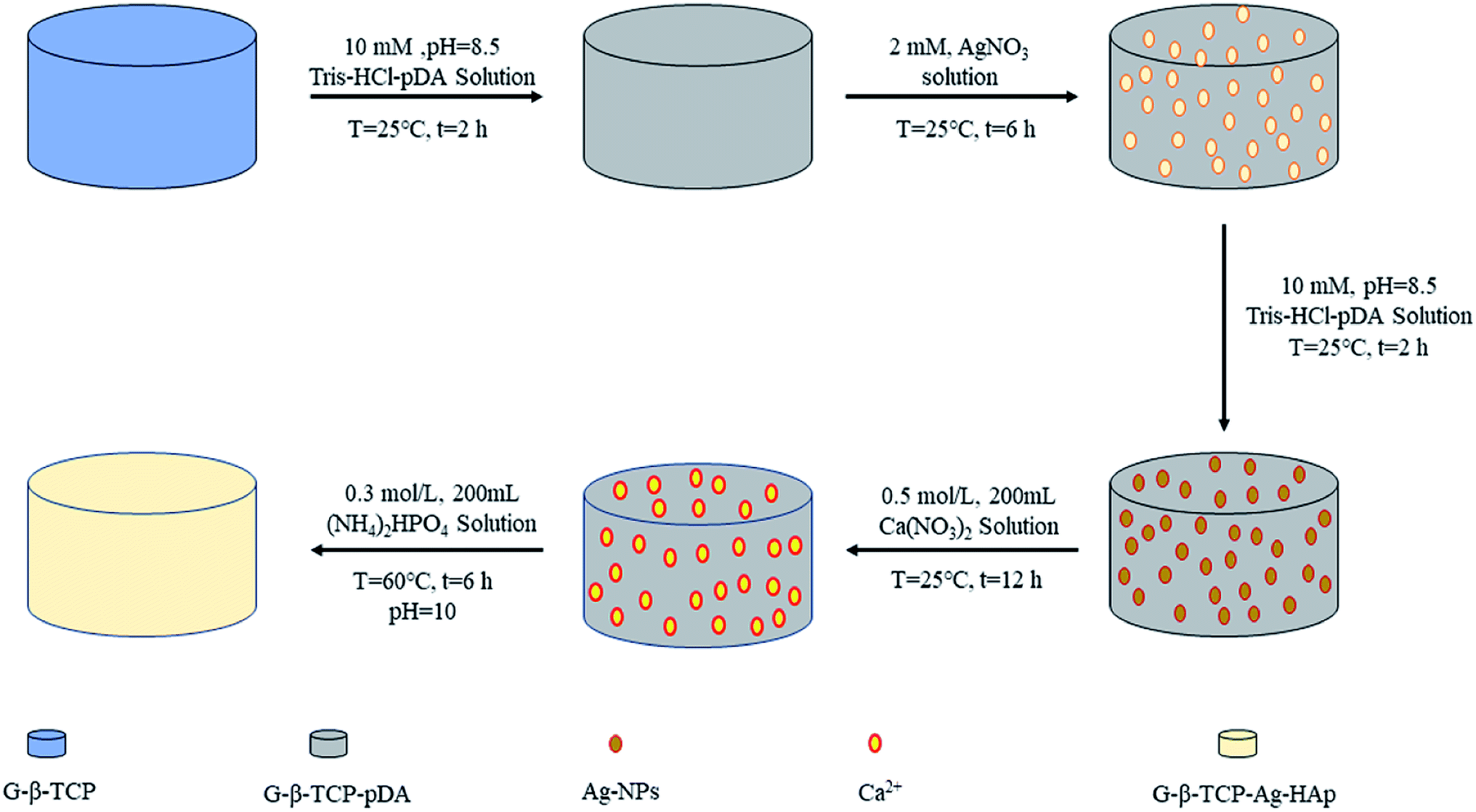 | ||
| Fig. 1 The preparation process diagram of G-β-TCP-Ag-HAp. | ||
2.4 Characterization
The phase composition of G-β-TCP was examined by X-ray diffraction analysis (XRD, PANalytical B.V., PANalytical) using Cu Kα radiation (λ = 0.154249 nm). The diffraction angle (2θ) range was between 10° and 70° with a scanning step size of 0.026°. The phase analysis was performed by the comparing sample diffraction pattern with the JCPDS standard XRD card.A field emission scanning electron microscope (FE-SEM, JSM-7500F, Japan) equipped with an energy dispersive X-ray spectroscope (EDS) was applied to analyze morphology and elemental composition of G-β-TCP, G-β-TCP-Ag and G-β-TCP-Ag-HAp. Transmission electron microscopy (TEM, TecnaiG2F20S-TWIN, Netherlands) was employed to detect the microstructure of G-β-TCP. All the powder samples were ultrasonically dispersed in ethanol to form dilute suspensions. Then, a drop of dilute suspension was placed onto a copper grid and the solvent was evaporated in air at ambient temperature before the TEM test.
Raman spectroscopy was performed on the graphene of the G-β-TCP by a laser confocal Raman spectrometer (Raman, HR800, France). The laser wavelength was set at 633 nm, the scanning range was between 500 cm−1 and 1800 cm−1, and the spectral resolution was 1.5 cm−1.
The compressive strengths of two G-β-TCP samples were tested by a microcomputer control electron universal testing machine (CMT4104, Shenzhen New Sansi Material Testing Co., Ltd.). The test results (elastic modulus and maximum load) were analyzed and processed by computer software. During the test, the loading speed was set as 0.05 mm min−1. And the basic physical properties, such as density and open porosity, were measured by the Archimedes method.30,31 The open porosity of G-β-TCP was measured as following. The weight of the dry sample was recorded as G1. The weight of the beaker and ethyl alcohol was recorded as m1. The samples were immersed in ethyl alcohol under a vacuum environment for 2 h, then suspended in the ethyl alcohol. The weight of the beaker, ethyl alcohol and suspended samples was recorded as m2. The weight of the beaker, ethyl alcohol and samples which sank to the bottom was recorded as m3. The open porosity (P) was calculated as follows: P = (m3 − m1 − G1)/(m2 − m1). The graphene that remained after sintering was tested and analyzed.
During the sintering process, graphene usually overflows from the sample and is carried away by the N2 flux, so a certain amount of graphene will be lost. For analyzing the graphene that remained after sintering, we separately took the ten samples (1 g) of different components (0.2 wt%, 0.5 wt%, 1 wt%, 1.5 wt%, and 2 wt% of β-TCP) and put them in a 0.05 M hydrochloric acid solution in order to dissolve the tricalcium phosphate. Then, the graphene remaining in the solution was centrifuged, dried, and weighed.
2.5 Silver ion release
The disc samples of G-β-TCP-Ag and G-β-TCP-Ag-HAp were submerged in 15 mL of phosphate buffer solution (PBS, pH = 7.4). The above mixture was incubated in an orbital shaker at 120 rpm at 37 °C. At predetermined times (0, 1, 2, 3, 4, 5, 6 and 7 days), 3 mL of the supernatant was collected, and then an equal amount of fresh PBS was refilled accordingly. The collected solution was analyzed by using inductively coupled plasma-atomic emission spectrometry (ICP-AES, iCAP Q, Thermo Fisher).2.6 Antibacterial tests
Escherichia coli (E. coli, ATCC8739) and Staphylococcus aureus (S. aureus, ATCC6538) were employed to evaluate the antibacterial properties of the materials. The bacteria were cultivated at 37 °C in liquid Luria Bertani (LB) medium with shaking at 150 rpm for 24 h. 100 μL of the E. coli or S. aureus suspension with a concentration of 106–107 CFU mL−1 were coated uniformly onto the agar LB medium. And then the sterilized disc samples of G-β-TCP, G-β-TCP-Ag and G-β-TCP-Ag-HAp were placed onto the foregoing agar LB medium in order to successfully evaluate the antibacterial properties of materials and release-ability of Ag+. After incubation for 24 h at 37 °C, bacterial growth inhibition zones were observed, and then the sizes of the bacterial growth inhibition zones were evaluated.100 μL of an E. coli suspension or S. aureus suspension with a concentration of 106–107 CFU mL−1 and 10 mL of liquid LB medium were added into each sterilized test tube. Three parallel samples of each group were used to evaluate the bacterial growth kinetics. Then, the sterilized disc samples of G-β-TCP, G-β-TCP-Ag and G-β-TCP-Ag-HAp were further introduced to the forgoing suspension. They were incubated at 37 °C in an orbital shaker for 24 h. The optical density value of the bacterial solution at 600 nm (OD600) was examined by microplate reader (SAF-680T, Shanghai) at various time periods (0, 3, 6, 9, 12 and 15 h). The bacterial solution was serially diluted 10−5-fold after reaction with materials for 24 h. In order to visually observe the effect of materials on the bacteria, 100 μL of the diluted bacterial suspension was coated uniformly onto agar LB mediums. The number of bacterial colonies was recorded after culturing for 24 h.
2.7 Cell viability
In vitro adhesion and growth of MG63 were performed using the cell counting kit-8 assay (CCK-8, KeyGEN Biotech Co., Ltd, China), and the density of cell suspension (500 μL) for seeding was about 2 × 104 cells mL−1. At every desired time point (1 d, 3 d, 5 d), the supernatant medium was discarded, then 500 μL of fresh medium, which consisted of DMEM (10 vol% CCK-8), was added into the well. After two hours of incubation, the absorbance of the supernatant was tested at 450 nm (OD450) using a microplate reader (SAF-6801, BAJIU Corporation, Shanghai, China). With respect to the control group, no cell was contained in the incubation medium.2.8 Statistical analysis
All the data are expressed as mean ± standard deviation (SD). Statistically significant differences (p) among groups were measured using the two-way analysis of variance and Tukey's multiple comparison tests. A value of p < 0.05 was considered significant.3 Results and discussion
3.1 The characterization of G-β-TCP
The crystalline structures of the G-β-TCP samples were investigated by using XRD.From Fig. 2, it can be seen that the XRD patterns of the as-obtained materials are very similar, and can be assigned to β-TCP (JCPDS card no. 09-0169). And after different amounts of graphene were added to the synthesis of β-TCP, characteristic graphene peaks were not detected by XRD, indicating the small amount of graphene was outside the scope of test and the addition of graphene had no effect on the stability of β-TCP. In particular, the XRD peak strength of the materials calcined at 1000 °C is significantly stronger than that of the materials calcined at 800 °C, so in the follow-up experiment, we chose a sintering temperature 1000 °C.
 | ||
| Fig. 2 XRD patterns of β-TCP and x wt% G-β-TCP. | ||
Fig. 3 exhibits the Raman spectra of graphene and G-β-TCP with different amounts of graphene. The typical G band (1593 cm−1) and D band (1355 cm−1) of graphene are shown in Fig. 3. Note that the structure of graphene remains intact after combining with β-TCP, and the characteristic peaks between 954 cm−1 and 985 cm−1 and at 1041 cm−1 are mainly attributed to the stretching vibration of the PO4 group in β-TCP, and the weak peak of PO3 group appears at around 735 cm−1, which may be due to a small amount of calcium pyrophosphate.32
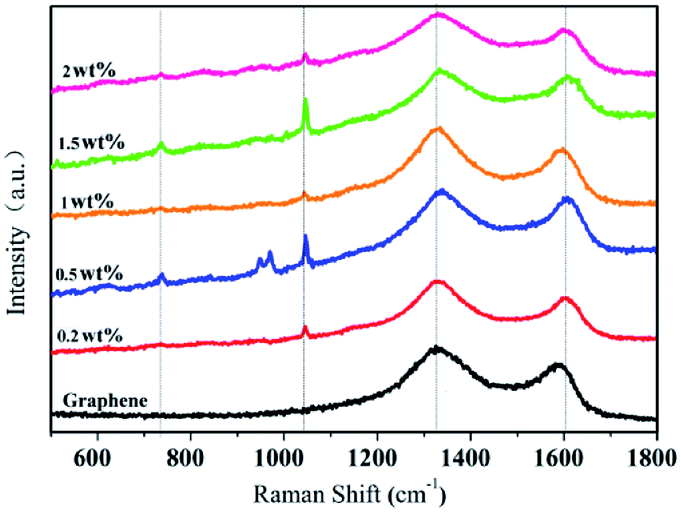 | ||
| Fig. 3 Raman spectra of graphene and x wt% G-β-TCP. | ||
The microstructure of the prepared G-β-TCP was performed by TEM analysis as shown in Fig. 4. It shows that graphene has a flake structure, and the β-TCP particles are needle-like or rod-like about 100 nm in length. In particular, when the amount of graphene is 1 wt%, graphene and the β-TCP were compounded uniformly. These results confirm that G-β-TCP was synthesized successfully.
 | ||
| Fig. 4 TEM images of the prepared materials: pure graphene (a), and 0.2 wt% (b), 0.5 wt% (c), 1 wt% (d), 1.5 wt% (e), and 2 wt% G-β-TCP (f). | ||
3.2 Basic physical properties
It can be seen from Fig. 5 that the surface morphology of the material, with different qualities of graphene, is obviously transformed. After sintering, the diameter and thickness of the specimens reduce to about 7.8 mm and 1.7 mm, respectively. By weighing, the density of the samples is concluded as in Fig. 6, which also delineates the open porosity of the prepared materials. | ||
| Fig. 5 Micrographs of the prepared materials: pure β-TCP (a), 0.2 wt% graphene-β-TCP (b), 0.5 wt% graphene-β-TCP (c), 1 wt% graphene-β-TCP (d), 1.5 wt% graphene-β-TCP (e), and 2 wt% graphene-β-TCP (f). | ||
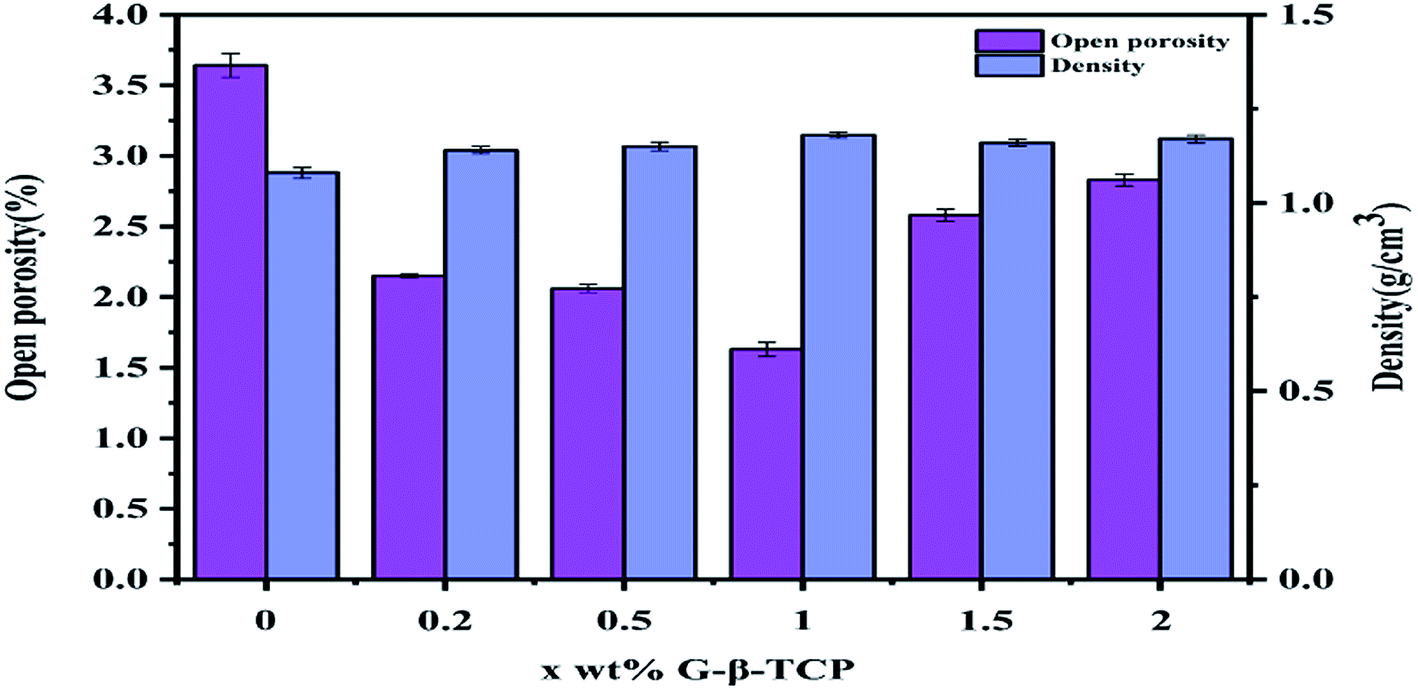 | ||
| Fig. 6 The open porosity and density properties of x wt% G-β-TCP. | ||
Finally, we get the remaining amount of graphene shown in Fig. 7. And from Fig. 7, with the increase of graphene, the loss during the sintering process was increased.
 | ||
| Fig. 7 The remaining amount of graphene. | ||
3.3 Compressive strength of the G-β-TCP scaffold
In general, the compressive strength of human cortical bone is in the range of 88 MPa to 164 MPa.33 When the compressive strength of synthetic bone repair materials is in this range, it is an ideal replacement material for bone repair. Changgu Lee et al. measured the monolayer graphene elastic properties and intrinsic strength and the result showed that it yields second- and third-order elastic stiffnesses of 340 N m−1 and −690 N m−1, respectively. The breaking strength is 42 N m−1, which represents the intrinsic strength of a defect-free sheet.34 Hence, the effects of the quality of the graphene in the composites investigated on compressive strength were investigated. Fig. 8 shows the stress–strain curve of two specimens, and Table 1 shows the compressive strength parameters of the corresponding materials. According to Fig. 8 and Table 1, when the content of graphene is between 0 and 1 wt%, the maximum compressive strength (MCS) and maximum load (ML) of the material increased gradually with the mass fraction of graphene. When the content of graphene is 1 wt%, the value of the two aforementioned quantities of these samples are the largest, 127.25 MPa and 6111.86 N (sample A), and 124.72 MPa and 6082.65 N (sample B), respectively, and the MCS is about 8 times that of the pure β-TCP scaffold, suggesting that the introduction of graphene can enhance the compressive strength of β-TCP. The reason is as follows: β-TCP particles are wrapped by graphene more evenly with increasing content of graphene from 0 to 1 wt%, which can improve the bonding force between graphene and the matrix and thus inhibit the crack growth. On the one hand, the graphene may be controlled as a nucleus for the crystallization of β-TCP crystals,35 which in turn may restrain the growth of β-TCP nanoparticles so that the fracture toughness of the composites is significantly increased. It is reported that the fine grain strengthening mechanism could be a possible cause for the improved mechanical properties of the composites.36 According to Fig. 4, the nucleus is a rod-like or needle-like crystal, which plays key roles in the interface and crack bridging and improves the compressive strength. On the other hand, graphene nanosheets have a high specific surface area enabling an increased contact area with the matrix, which is the major toughening mechanism that resists crack propagation.37 And, due to its high Young's modulus and flexibility, graphene may be located around the grain boundaries and aligned with the grain shape,38 which is expected to enhance the mechanical interlocking and adhesion strength within the matrix.35 When the graphene content is between 1 wt% and 2 wt%, the dispersion of graphene becomes difficult, and it is easy to cause agglomeration. And the aggregates can be peeled off from the composite matrix or crack itself, reducing the interface bonding strength between graphene and the matrix, which decreases the fracture toughness, and further disturbs the compressive strength of the specimens.36 From Fig. 8, the MCS and ML values of the material gradually decrease as the graphene content increases. According to the literature, the enhancement of compressive strength is mainly due to crack bridges, graphene pull-out, crack deflection and crack tip shielding. Firstly, when graphene bridges two crack surfaces, it hampers the relative displacement between the opposite crack surfaces by alleviating the stress required for further crack propagation. Secondly, the pull-out of graphene from the matrix requires more energy dissipation due to the high toughness and large surface area of graphene. Thirdly, when the crack propagation encounters graphene, the crack plane is not perpendicular to the axis of stress and the deflection process generates a tortuous path. The crack propagation can be slowed down by additional resistance from the interfacial friction between graphene and β-TCP, which allows much more energy dissipation. Crack deflection may be ascribed to the large specific surface area of graphene. Lastly, the crack tip is restricted in the vicinity of graphene due to the insufficient energy required for interface debonding.15 | ||
| Fig. 8 Stress–strain curves of specimens. | ||
| Sample | Maximum load/N (A/B) | Maximum compressive strength/MPa (A/B) | Maximum load deformation/% (A/B) | Compressive modulus/MPa (A/B) |
|---|---|---|---|---|
| β-TCP | 769.41/749.13 | 16.06/14.36 | 0.083/0.081 | 0.0289/0.0208 |
| 0.2 wt% G-β-TCP | 3897.34/3888.81 | 82.41/82.12 | 0.293/0.281 | 0.0375/0.0321 |
| 0.5 wt% G-β-TCP | 5030.51/5006.31 | 105.55/105.28 | 0.133/0.130 | 0.1143/0.0947 |
| 1 wt% G-β-TCP | 6111.86/6082.65 | 127.25/124.72 | 0.095/0.093 | 0.201/0.1768 |
| 1.5 wt% G-β-TCP | 3078.40/3007.42 | 65.09/58.28 | 0.123/0.121 | 0.0806/0.0509 |
| 2 wt% G-β-TCP | 728.65/711.73 | 15.61/14.58 | 0.066/0.065 | 0.0334/0.0233 |
3.4 Morphological observations
The microstructure of the surface-modified G-β-TCP porous scaffold was observed by SEM, as shown in Fig. 9. There are nano-sized spherical particles on the surface of G-β-TCP, and a small amount of grains on its surface, which indicates that dopamine reduces and adheres Ag+ on the surface of the scaffold (Fig. 9(a)). When G-β-TCP-Ag was modified by pDA, the nano-sized spherical particles become more ambiguous, and it is difficult to see the crystal grains on the G-β-TCP surface, indicating that pDA was successfully coated on the surface (Fig. 9(b)). After the reaction of G-β-TCP-Ag-pDA with Ca(NO3)2 and (NH4)2HPO4, some pompon-like amorphous apatite particles were formed on the surface, as shown in Fig. 9(c). To further confirm the chemical composition of G-β-TCP-Ag-HAp, EDS was employed to analyze the prepared sample. Ag, Ca, and P elements were detected (Fig. 9(d)), which means that Ag-NPs and apatite were formed G-β-TCP-Ag-HAp on the surface. | ||
| Fig. 9 SEM images of G-β-TCP-Ag (a), G-β-TCP-Ag-pDA (b), and G-β-TCP-Ag-HAp (c), and the EDS spectrum of G-β-TCP-Ag-HAp (d). | ||
To further observe the microstructure of the surface-modified G-β-TCP porous scaffold, it was characterized by transmission electron microscopy (TEM), as shown in Fig. 10. After the G-β-TCP porous scaffold reacted with dopamine and AgNO3, Ag-NPs with a particle size of about 15 nm were distributed on the surface. The SAED pattern consisted of several diffraction rings and some diffraction spots, indicating that the scaffold belongs to polycrystalline (Fig. 10(a)). There is obvious amorphous apatite on the surface of the material in Fig. 10(b), which is consistent with the Fig. 9(c) test results.
 | ||
| Fig. 10 TEM images of samples: G-β-TCP-Ag (a) and G-β-TCP-Ag-HAp (b); the insets show the selected area electron diffraction patterns of the corresponding samples. | ||
3.5 Ag+ release assessment
The release concentration of Ag+ determines the antibacterial properties and cytotoxicity, and an effective control of the Ag+ release not only reduces cytotoxicity but also achieves long-lasting antibacterial properties. As shown in Fig. S1,† on the first day, the rate of Ag+ release in G-β-TCP-Ag-HAp is only 50.89% of that of G-β-TCP-Ag. And in the next four days, the curve of the latter becomes sharply flat, and its slope decreases significantly. The oxidation of Ag-NPs and hydrolysis of silver oxide (AgO) play key roles in the release of Ag+.39 The oxidized Ag-NPs result in a rapid release of large amounts of Ag+ within a short time. Owing to the pDA and the HAp coating, specimens can effectively help the Ag-NPs avoid exposure to the dissolved oxygen in the PBS solution, so only a small amount of Ag-NPs is oxidized. Therefore, the rate of release of Ag+ from G-β-TCP-Ag-HAp is slower throughout the release process.3.6 Antibacterial assessment
As shown in Fig. 11, because of the excellent antibacterial effect of silver ions, more significant inhibition zones can be clearly observed both around G-β-TCP-Ag and G-β-TCP-Ag-HAp compared with G-β-TCP, indicating that not only do all the materials have good antibacterial properties, but also, it is worth noting, that the inhibitory ring around G-β-TCP-Ag is significantly larger than that around G-β-TCP-Ag-HAp both against S. aureus and E. coli, which further indicates that G-β-TCP-Ag-HAp can effectively control the Ag+ release area. | ||
| Fig. 11 Growth inhibition zones of G-β-TCP, G-β-TCP-Ag, and G-β-TCP-Ag-HAp against S. aureus (a) and E. coli (b). | ||
From Fig. S2,† it can be seen that G-β-TCP had no significant effect on the growth of S. aureus and E. coli. But G-β-TCP-Ag and G-β-TCP-Ag-HAp significantly inhibited the growth of S. aureus and E. coli, and a small amount of Ag+ with excellent antibacterial properties, which may be attributed to the dual antibacterial action of polydopamine and Ag+.
To further observe the effect of the material on bacterial growth, the bacterial suspension after 24 hours of incubation was evenly spread on agar LB medium, as shown in Fig. 12. Both G-β-TCP-Ag and G-β-TCP-Ag-HAp can kill more than 99.99% of S. aureus and E. coli, indicating that G-β-TCP-Ag-HAp has excellent antibacterial properties, and because G-β-TCP-Ag-HAp can effectively control the release of Ag+, G-β-TCP-Ag-HAp is beneficial for achieving long-lasting antibacterial properties.
 | ||
| Fig. 12 Photos showing the growth of E. coli (a–c) and S. aureus (d–f) on LB agar plates after 24 hours of incubation. The bacteria reacted with G-β-TCP (a and d), G-β-TCP-Ag (b and e), and G-β-TCP-Ag-HAp (c and f) in a liquid LB medium for 24 hours before testing. | ||
3.7 Cytotoxicity assessment
MG63 cells were seeded on G-β-TCP, G-β-TCP-Ag and G-β-TCP-Ag-HAp porous scaffolds, and cultured in DMEM medium for 1, 3, and 5 days, and cytotoxicity was measured by the CCK-8 kit. The cells proliferated significantly on both G-β-TCP and G-β-TCP-Ag-HAp, while the cells on G-β-TCP-Ag did not proliferate significantly (Fig. 13). On the one hand, the exposed Ag-NPs on G-β-TCP-Ag is directly in contact with oxygen, releasing a large amount of Ag+. It inhibits cell proliferation and even kills cells; on the other hand, bare Ag-NPs on graphene-β-TCP-Ag reacted directly with cells to exhibit a stronger cytotoxicity. It is worth noting that the proliferation of cells on G-β-TCP-Ag-HAp is much higher than that on G-β-TCP, indicating G-β-TCP-Ag-HAp has good cell activity. This may be because the synergistic action of polydopamine and pompon-like HAp increases the cellular activity of the material. | ||
| Fig. 13 Optical density values illustrating MG63 cell proliferation on G-β-TCP, G-β-TCP-Ag, and G-β-TCP-Ag-HAp scaffolds after culturing for 1, 3, and 5 days. Each value is the mean ± standard error from three observations. * represents p < 0.05 between groups on the same day. | ||
4 Conclusions
In summary, we provide an effective way to prepare G-β-TCP-Ag-HAp, with excellent compressive strength, antibacterial properties, and good cell activity. The results revealed that the introduction of 1 wt% graphene to β-TCP could markedly enhance the compressive strength of the material, and Ag NPs and HAp particles were successfully coated on the surface of G-β-TCP using dopamine as a binder and reducing agent. From the controlled release of Ag+, the bactericide rate of G-β-TCP-Ag-HAp was increased to 99.99% after culturing for 24 h. The material also had excellent cell activity and can significantly promote cell proliferation due to the action of dopamine and HAp. Therefore, the difunctional porous β-TCP (G-β-TCP-Ag-HAp) scaffold is a promising alternative material for regenerative bone repair.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by The Applied Basic Research Program of Science and Technology Department of Sichuan Province in 2019 (2019YJ0141).References
- A. M. Ng, K. K. Tan, M. Y. Phang, O. Aziyati, G. H. Tan, M. R. Isa, B. S. Aminuddin, M. Naseem, O. Fauziah and B. H. Ruszymah, J. Biomed. Mater. Res., Part A, 2010, 85A, 301–312 CrossRef PubMed.
- D. Y. Suh, S. D. Boden, J. Louis-Ugbo, M. Mayr, H. Murakami, H. S. Kim, A. Minamide and W. C. Hutton, Spine, 2002, 27, 353–360 CrossRef PubMed.
- H. S. Sandhu and S. D. Boden, Spine, 1995, 20, 124S CrossRef PubMed.
- A. Elghannam, Expert Rev. Med. Devices, 2005, 2, 87–101 CrossRef PubMed.
- J. Wang, W. Chen, Y. Li, S. Fan, J. Weng and X. Zhang, Biomaterials, 1998, 19, 1387–1392 CrossRef CAS PubMed.
- F. G. Torres, S. N. Nazhat, S. H. S. M. Fadzullah, V. Maquet and A. R. Boccaccini, Compos. Sci. Technol., 2007, 67, 1139–1147 CrossRef CAS.
- P. Fabbri, V. Cannillo, A. Sola, A. Dorigato and F. Chiellini, Compos. Sci. Technol., 2010, 70, 1869–1878 CrossRef CAS.
- M. Peroglio, L. Gremillard, J. Chevalier, L. Chazeau, C. Gauthier and T. Hamaide, J. Eur. Ceram. Soc., 2007, 27, 2679–2685 CrossRef CAS.
- J. Zhao, X. Lu, K. Duan, L. Y. Guo, S. B. Zhou and J. Weng, Colloids Surf., B, 2009, 74, 159–166 CrossRef CAS PubMed.
- X. Miao, D. M. Tan, L. Jian, X. Yin and R. Crawford, Acta Biomater., 2008, 4, 638–645 CrossRef CAS PubMed.
- M. H. Huang, Q. H. Chen, L. Lei, D. C. Wang and T. T. Yan, Adv. Mater. Res., 2013, 712–715, 415–419 CAS.
- Y. Kang, A. Scully, D. A. Young, S. Kim, H. Tsao, M. Sen and Y. Yang, Eur. Polym. J., 2011, 47, 1569–1577 CrossRef CAS PubMed.
- I. Kunio, Y. Miyamoto, T. Toh, T. Yuasa, A. Ito, M. Nagayama and K. Suzuki, Key Eng. Mater., 2001, 192–195, 785–788 Search PubMed.
- G. A. Fielding, A. Bandyopadhyay and S. Bose, Dent. Mater., 2012, 28, 113 CrossRef CAS PubMed.
- C. Gao, T. Liu, C. Shuai and S. Peng, Sci. Rep., 2014, 4, 4712 CrossRef PubMed.
- M. Mehdi, M. Ehsan, S. S. S. Farid, B. Saeid, M. Mohammad, L. S. Tahan, M. H. S. Cornelis, K. N. Adib, Z. Keivan and O. N. A. Abu, PLoS One, 2014, 9, e106802 CrossRef PubMed.
- J. Kim, Y. R. Kim, Y. Kim, K. T. Lim, H. Seonwoo, S. Park, S. P. Cho, B. H. Hong, P. H. Choung and T. D. Chung, J. Mater. Chem. B, 2013, 1, 933–938 RSC.
- J. Lu, Y. S. He, C. Chi, W. Yi, Q. Ling, L. Dan and D. Zou, Adv. Funct. Mater., 2013, 23, 3494–3502 CrossRef CAS.
- Z. J. Fan, J. Q. Wang, Z. F. Wang, H. Q. Ran, Y. Li, L. Y. Niu, P. W. Gong, B. Liu and S. R. Yang, Carbon, 2014, 66, 407–416 CrossRef CAS.
- C. Shuai, T. Liu, C. Gao, P. Feng and T. Xiao, J. Alloys Compd., 2016, 655, 86–92 CrossRef CAS.
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- D. I. Andersson and D. Hughes, Nat. Rev. Microbiol., 2010, 8, 260–271 CrossRef CAS PubMed.
- M. J. Schwaber, T. De-Medina and Y. Carmeli, Nat. Rev. Microbiol., 2004, 2, 979–983 CrossRef CAS PubMed.
- S. Hoover, S. Tarafder, A. Bandyopadhyay and S. Bose, Mater. Sci. Eng., C, 2017, 79, 763–769 CrossRef CAS PubMed.
- H. Fan, Y. Hu, L. Qin, X. Li, H. Wu and R. Lv, J. Biomed. Mater. Res., Part A, 2010, 77A, 785–794 CrossRef PubMed.
- A. Shivaram, S. Bose and A. Bandyopadhyay, Acta Biomater., 2017, 58, 550–560 CrossRef CAS PubMed.
- N. Matsumoto, K. Sato, K. Yoshida, K. Hashimoto and Y. Toda, Acta Biomater., 2009, 5, 3157–3164 CrossRef CAS PubMed.
- L. Shen, C. Fan, J. Fang, Z. Li, K. Dai and J. Lu, Int. J. Appl. Ceram. Technol., 2013, 12, 1–7 Search PubMed.
- K. Chen, K. Xie, L. Qin, L. Deng, Z. Fu, H. Xiao and X. Lu, RSC Adv., 2017, 7, 29368–29377 RSC.
- Y. Tian, T. Lu, F. He, Y. Xu, H. Shi, X. Shi, F. Zuo, S. Wu and J. Ye, Colloids Surf., B, 2018, 318–327 CrossRef CAS PubMed.
- S. Yang, S. Zhao and G. Li, Lab. Sci., 2009, 71–72 CAS.
- Q. Chen, S. P. Li, J. P. He, N. T. Yu, Q. X. Zheng and Y. H. Yan, Acta Biochim. Biophys. Sin., 1999, 31, 409–414 CAS.
- V. P. Orlovskii, V. S. Komlev and S. M. Barinov, Inorg. Mater., 2002, 38, 973–984 CrossRef CAS.
- J. Hone, Science, 2008, 321, 385–388 CrossRef PubMed.
- L. Ming, Y. Wang, L. Qian, Q. Li and S. Wei, J. Mater. Chem. B, 2012, 1, 475–484 Search PubMed.
- Y. Liu, J. Huang and H. Li, J. Mater. Chem. B, 2013, 1(13), 1826–1834 RSC.
- L. Zhang, W. Liu, C. Yue, T. Zhang, P. Li, Z. Xing and Y. Chen, Carbon, 2013, 61, 105–115 CrossRef CAS.
- Y. Zhao, K. N. Sun, W. L. Wang, Y. X. Wang, X. L. Sun, Y. J. Liang, X. N. Sun and P. F. Chui, Ceram. Int., 2013, 39, 7627–7634 CrossRef CAS.
- A. D. Dwivedi, S. P. Dubey, M. Sillanpaa, Y.-N. Kwon, C. Lee and R. S. Varma, Coord. Chem. Rev., 2015, 287, 64–78 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02388d |
| This journal is © The Royal Society of Chemistry 2020 |