
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 1,3-diaryl-spiro[azetidine-2,3′-indoline]-2′,4-diones via the Staudinger reaction: cis- or trans-diastereoselectivity with different addition modes†
Vadim Filatov a,
Maksim Kukushkina,
Juliana Kuznetsovaa,
Dmitry Skvortsovab,
Viktor Tafeenkoa,
Nikolay Zyka,
Alexander Majougaacd and
Elena Beloglazkina*a
a,
Maksim Kukushkina,
Juliana Kuznetsovaa,
Dmitry Skvortsovab,
Viktor Tafeenkoa,
Nikolay Zyka,
Alexander Majougaacd and
Elena Beloglazkina*a
aMoscow State University, Department of Chemistry, Leninskie gory 1-3, Moscow 119991, Russia. E-mail: bel@org.chem.msu.ru
bSkolkovo Institute of Science and Technology, 4 Alfred Nobel Street, Skolkovo, 143025, Russian Federation
cNational University of Science and Technology “MISiS”, Leninskiy pr. 4, Moscow 119991, Russia
dDmitry Mendeleev University of Chemical Technology of Russia, Miusskaya sq. 9, Moscow 125047, Russia
First published on 6th April 2020
Abstract
A new synthetic approach for realizing biologically relevant bis-aryl spiro[azetidine-2,3′-indoline]-2′,4-diones was developed based on Staudinger ketene–imine cycloaddition through the one-pot reaction of substituted acetic acids and Schiff bases in the presence of oxalyl chloride and an organic base. A series of [azetidine-2,3′-indoline]-2′,4-diones were synthesized using this method. For comparison, the same compounds were obtained using a known technique, where ketene is generated from pre-synthesized acyl chloride. It was shown that the use of oxalyl chloride for ketene generation in the one-pot reaction at room temperature allows for the reversal of the diastereoselectivity of spiro-lactam formation, unlike previously described procedures.
Introduction
The search for the most efficient and harmless way to treat uncontrollable cell proliferation via targeted therapy is currently a promising research area. The general idea of all targeted methods is to use pharmacological agents that are highly selective towards specific biomolecules in cancer cells that are necessary for them to proliferate.1 One such target is the MDM2 protein,2 which has great importance due to its function in p53-protein activity control.3–5 p53 is known as a “genome guardian”; its function is to check the integrity of DNA and to trigger the repair mechanisms in the presence of inconsistencies or to induce apoptosis if the DNA is deemed unrepairable.5–8 In this context, spiro-oxindoles are attractive molecules for synthesis because the different spiro-oxindoles display considerable anticancer action, which is apparently related to their ability to inhibit p53-MDM2 protein–protein interaction.9–15 Some spiro- and dispiro-oxindoles have been tested as anticancer drugs during preclinical or clinical studies.9,16–18 For example, the spiro-oxindoles SAR405838 and APG-115 (Fig. 1) are currently under clinical phase I investigation;9,16,17 our research group has recently proposed 2-thiohydantoin-dispiro-oxindones as inhibitors of the p53-MDM2 interaction, as one 2-thiohydantoin-dispiro-oxindone (Fig. 1) has now completed preclinical studies.10,19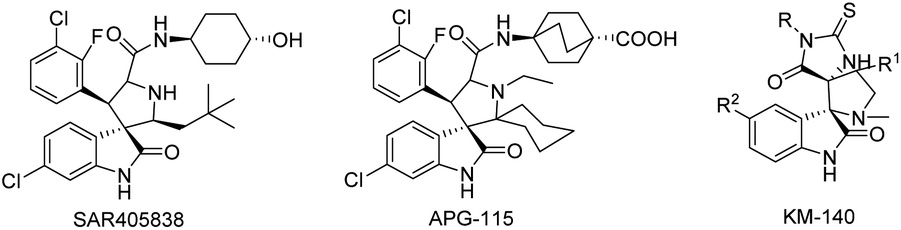 | ||
| Fig. 1 Spiro-oxindoles SAR405838 and APG-115,16,17 which are currently under clinical trials, and dispiro-oxindole KM-140 have successfully completed preclinical studies.10,19 | ||
Spirocyclicoxindolo-β-lactams are generally tested for antibacterial and antifungal activity but not for antiproliferative activity. We assumed that the spiro-conjugation of oxindole fragments with the four-membered ring of azetidin-2-one may prevent the fast biodegradation of potential p53-MDM2 inhibitors and provide at the same time conformational rigidity to synthesized molecules, which is necessary for effective binding to MDM2. A similar approach was previously proposed for the synthesis of combretastatin analogues.20
A number of alternative methods have been described in the literature for the formation of the β-lactam cycle including the Rh- or Ir-catalyzed reductive Mannich-type reaction,21,22 the Reformatsky reaction using Schiff bases and α-bromosubstituted esters derivatives,20,23 the direct regiospecific metal-catalyzed carbonilation of aziridines,24 the intramolecular nucleophilic cyclization of N-(p-hydroxyphenyl) cyanoacetamides,25 the intramolecular N-alkylation of amides,26 the Kinugasa reaction ([3 + 2] nitrones cycloaddition to acetylenide followed by acid-catalyzed rearrangement to β-lactam),27–29 the Pd-catalyzed carbonylation of allyl halides followed by the reaction with imines,30–33 the cyclization of β-amino acid esters,34–37 and Staudinger's ketene–imine cycloaddition.38–40 Staudinger synthesis is the most common method and can be carried out using various synthetic techniques with a well-established mechanism. The first stage of Staudinger reaction is a nucleophilic addition of imine to the ketene carbonyl carbon atom with the formation of a zwitterionic intermediate, which is cyclized to a four-membered ring at the second stage of the reaction with the formation, as a rule, of a mixture of isomeric cis- and trans-lactones with the predominance of one or the other diastereomer depending on the solvent, addition order of the reagents, the electronic effects of the substituents in ketene and imine, and the nucleophilicity of the base used for ketene generation.39,41,42 For the latter stage, two alternative mechanisms have been proposed43 (Scheme 1): intramolecular nucleophilic addition1 or electrocyclic conrotatory reaction.44 The Staudinger reaction was previously successfully used to synthesize spiro-β-lactams,45,46 but the diastereoselectivity of the reaction was not discussed in the cited articles.
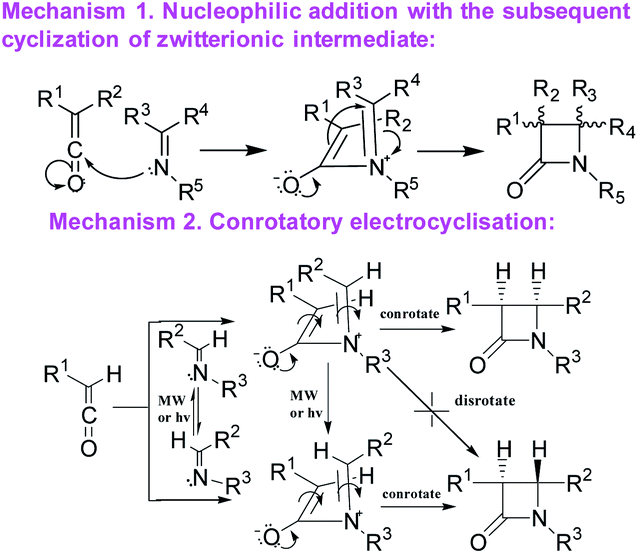 | ||
| Scheme 1 The possible mechanisms for the Staudinger reaction.3,43 | ||
Herein, we report a method for the synthesis of bis-aryl spiro[azetidine-2,3-indole]-2,4(1H)-diones via the Staudinger reaction with an in situ technique based on ketene generation from substituted phenyl acetic acid, oxalyl chloride and DIPEA. The proposed modification of the Staudinger reaction makes it possible to reverse the diastereoselectivity of β-lactam cycle formation, unlike previously described classic methods,45–47 and to shift the reaction stereoselectivity to the formation of diastereomers, which are considered as the most promising inhibitors of the p53-MDM2 protein–protein interaction according to molecular docking data.
Results and discussion
Given the well-known fact that even different optical isomers of the same substance can provide completely different biological activity, with one isomer being acure while the other is toxic,7 we determined which enantiomer can become a possible inhibitor of the targeted MDM2 molecules among the potentially obtained diastereomers. For this, the molecular docking of stereoisomeric 1,3-bis(4-chlorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-diones optimized with a MMFF94 procedure with AutoDock Vina48 in the active site of MDM2 shaped by a complimentary fragment of p53 was carried out. We found that the best binding mode is observed for 2R,3R enantiomer and that in this binding mode, one of the lateral aryl fragments is placed into the hydrophobic pocket of MDM2 (Fig. 2), which corresponds to an affinity energy of −7.7 kcal mol−1. The second “good” mode is one where an indolin-2-one fragment is placed into the said pocket albeit in a slightly shifted manner (Fig. 2a). The affinity energy here is −7.7 kcal mol−1 as well. On the other hand, the 2R,3S (Fig. 2b) and 2S,3R (Fig. 2c) isomers of spiro-oxindoles did match the desired binding mode with affinity energies of −7.8 kcal mol−1 and −8.7 kcal mol−1, respectively. The 2S,3S enantiomer (Fig. 2d) is disallowed from being placed in the cavity of MDM2 in an advantageous way, which turned out to be the furthest from the desired binding mode, with an affinity energy of −6.9 kcal mol−1, corresponding to its best position. Thus, only two out of four enantiomers of 1,3-diaryl-spiro[azetidine-2,3′-indoline]-2′,4-dione demonstrate binding modes appropriate for the potential inhibition of p53-MDM2 interaction, and these isomers are compounds with a trans arrangement of six-membered aromatic rings of the starting phenyl acetic acid and isatin.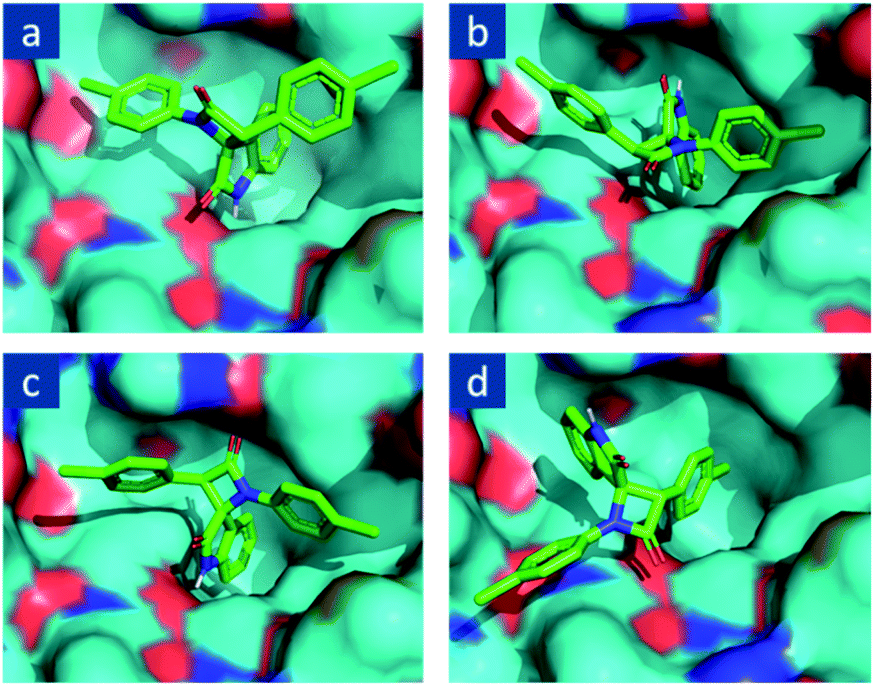 | ||
| Fig. 2 Molecular docking of 1,3-bis(4-chlorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione stereoisomers on MDM2 active site: (a) (2R,3R), (b) (2R,3S), (c) (2S,3R), (d) (2S,3S). | ||
The synthesis of target spiro-β-lactams was started with the preparation of the initial Schiff bases using the method described in (ref. 48) by the condensation of isatin with amines in refluxing ethyl alcohol in the presence of acetic acid (Table 1). Initial imines 1–21 were obtained in good yields as mixtures of E/Z isomers with an E-isomer predominance in accordance with the NMR 1H spectra and literature data.49 The resulting mixtures were introduced into further reactions without separation of the individual geometric isomers. It may be noted that the imine formation reactions did not occur with aliphatic amines and in the presence of anilines with strong electron donor/electron withdrawing substituents (4-NO2, 4-OH, 3,4-(OMe)2). Moreover, the imine formation reaction is also difficult in the presence of substituents in the ortho position of the starting aniline. Although 2-fluoroaniline reacts with isatin to give Schiff base 1, the isolated yield of imine 1 (36%) is significantly lower than that in other cases, and the reaction of 2-Cl and 2-Br-substituted anilines did not yield the desired Schiff bases.
|
|||
|---|---|---|---|
| Entry (compound #)a | R1 | R2 | Yield, %b [Z/E]c |
| a Reaction conditions: isatin (1.0 eq.), aniline (1.2 eq.), AcOH (2 drops) in 96% EtOH, reflux for 0.5–3 h.b Isolated yield.c Z/E ratios were determined based on 1H NMR data. | |||
| 1 | H | 2-F | 36 [1/2] |
| 2 | H | 3-F | 60 [1/4] |
| 3 | H | 4-F | 56 [1/2] |
| 4 | H | 3-Cl | 52 [1/2] |
| 5 | H | 4-Cl | 83 [1/2] |
| 6 | H | 3-Br | 51 [1/2] |
| 7 | H | 4-Br | 89 [1/3] |
| 8 | H | 3-I | 96 [1/3] |
| 9 | H | 3-Cl, 4-F | 89 [2/5] |
| 10 | H | 4-OH | 92 [1/5] |
| 11 | H | 4-OMe | 93 [1/4] |
| 12 | H | 4-OEt | 87 [1/5] |
| 13 | H | 3-Me | 84[1/3] |
| 14 | H | 4-Me | 93[1/4] |
| 15 | Cl | 4-F | 63 [1/2] |
| 16 | Cl | 4-OMe | 88 [1/4] |
| 17 | Cl | 3-Me | 85 [1/3] |
| 18 | Cl | 4-Me | 91 [1/3] |
| 19 | Br | 4-F | 73 [3/5] |
| 20 | Br | 4-OMe | 79 [1/4] |
| 21 | Br | 3-Me | 82 [2/5] |
Imines 1–21 were then used for Staudinger's imine–ketene cycloaddition. There are various methods of β-lactam synthesis involving the Staudinger reaction;23,39,42 these methods differ in the solvent used and the route for ketene generation (photolysis or thermolysis of α-diazoketones,44 the dehydrohalogenation of halides of substituted acetic acids20,38), and the use of different one-pot techniques for ketene generation (phosphonitrile chloride, SOCl2, or a DMSO–Ac2O complex40,50,51). First, we tested the known literature method used for the synthesis of target spiro-β-lactams with the trans arrangement of aromatic rings in the 2 and 3 positions of the four-membered cycle. It was found that isatin imines 1, 3, 5, 7, 9, 10, 12 interact in the presence of Et3N with 4-chlorophenyl acetyl chloride pre-generated from acid by addition of oxalyl chloride/DMF. This experimental technique allows one to create desired the spiroconjugation in a similar way to that reported previously45 (Scheme 2). Refluxing of the imines 1, 3, 5, 7, 9, 10, 12 with acyl chloride and Et3N in DMF gives pairs of diastereomers, which were then easily separated by column chromatography. However, in most of these reactions, the diastereomer ratio was close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, with a predominance of the undesired cis (2R*,3R*) isomers azetidinones 22b–28b (Table 2).
:2, with a predominance of the undesired cis (2R*,3R*) isomers azetidinones 22b–28b (Table 2).
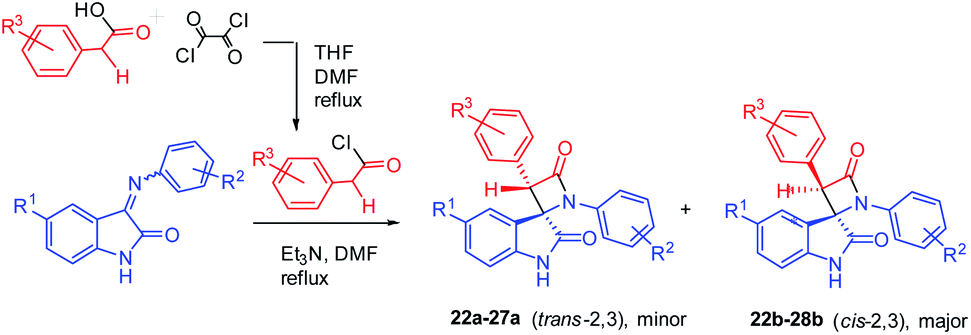 | ||
| Scheme 2 Staudinger synthesis of bis-aryl spiro[azetidine-2,3′-indoline]-2′,4-diones using pre-synthesized acyl chloride. | ||
| Compound # | R1 | R2 | R3 | a/b | Yield,% |
|---|---|---|---|---|---|
| a Reaction conditions: Schiff base (1.0 eq.), 4-chlorophenylacetyl chloride (1.5 eq.), Et3N (3.0 eq.), DMF, reflux for 6–9 h. | |||||
| 22a,b | H | 4F | 4-Cl | 1/2 | 64 |
| 23a,b | H | 4-Cl | 4-Cl | 1/2 | 53 |
| 24a,b | H | 4-Br | 4-Cl | 1/2 | 49 |
| 25a,b | H | 4-OH | 4-Cl | 1/2 | 62 |
| 26a,b | H | 4-OEt | 4-Cl | 1/5 | 57 |
| 27a,b | H | 3-Cl, 4-F | 4-Cl | 2/5 | 55 |
| 28b | H | 2-F | 4-Cl | — | 44 |
Thus, the diastereoselectivity of the literature method was unsatisfactory for the selective preparation of trans-isomeric 2,3- diaryl-spiro[azetidine-2,3′-indoline]-2′,4-diones. To find a convenient way to change the diastereoselectivity, we examined the possibility of using ketene–imine Staudinger cycloaddition in more mild conditions by avoiding the use of moisture-sensitive acyl chlorides. Some methods described in earlier publications40,50,51 seemed attractive, but poor solubility of the reagents or difficulties with the final work-up made them unacceptable in our case.
Starting from the idea of acid activation by strengthening the electron-withdrawing character of the carboxylic group, we guessed that oxalyl chloride, which we previously used for formation of acyl halide from acid, can be used in a one-pot process as an acid activator in a fashion similar to that described in the aforementioned publications. Using this novel one-pot technique of β-lactam synthesis (Scheme 3) by activation of α-substituted acetic acids with oxalyl chloride in a reaction mixture at room temperature, we have successfully synthesized a series of target spiro-oxindoles 22–43 with a predominance or exceptional formation of the target trans-diastereomers 22a–43a (Table 3).
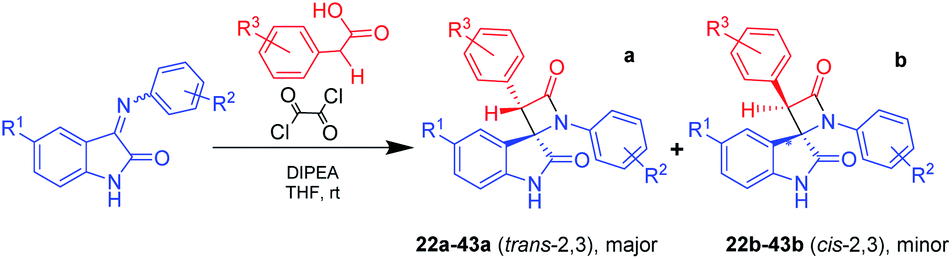 | ||
| Scheme 3 Staudinger synthesis of bis-aryl spiro[azetidine-2,3′-indoline]-2′,4-diones without using presynthesized acyl chloride. | ||
| Compound # | R1 | R2 | R3 | a/b | Yields,% |
|---|---|---|---|---|---|
| 22a,b | H | 4-F | 4-Cl | 2/1 | 65 |
| 23a,b | H | 4-Cl | 4-Cl | 1.2/1 | 71 |
| 24a,b | H | 4-Br | 4-Cl | 1.5/1 | 59 |
| 25a,b | H | 4-OH | 4-Cl | — | 0 |
| 26a,b | H | 4-OEt | 4-Cl | 3.4/1 | 75 |
| 27a,b | H | 3-Cl, 4-F | 4-Cl | 5/1 | 41 |
| 28b | H | 2-F | 4-Cl | — | 32 |
| 29a,b | H | 3-F | 4-Cl | 1.5/1 | 45 |
| 30b | H | 3-Cl | 4-Cl | — | 25 |
| 31a,b | H | 3-Br | 4-Cl | 1.4/1 | 47 |
| 32a,b | H | 3-I | 4-Cl | 1.3/1 | 80 |
| 33a,b | H | 4-OMe | 4-Cl | 4.5/1 | 78 |
| 34a | H | 3-Me | 4-Cl | — | 65 |
| 35a | H | 4-Me | 4-Cl | — | 73 |
| 36a | Cl | 4-F | 4-Cl | — | 27 |
| 37a | Cl | 4-OMe | 4-Cl | — | 48 |
| 38a | Cl | 3-Me | 4-Cl | — | 58 |
| 39a | Cl | 4-Me | 4-Cl | — | 54 |
| 40a | Br | 4-F | 4-Cl | — | 51 |
| 41a | Br | 4-OMe | 4-Cl | — | 74 |
| 42a,b | Br | 3-Me | 4-Cl | 4/1 | 84 |
| 43a | H | 4-OMe | 4-Br | -/1 | 75 |
All reactions were carried out at room temperature. For the base, we used diisopropylethylamine (DIPEA); however, the use of other similar tertiary amines, such as TEA, does not affect the reaction course significantly.
According to the LCMS and 1H NMR data, compounds 34b–41b and 43b were obtained in trace amounts but were not isolated in pure form, so in Table 3, only the diastereomers 34a–41a and 43a are indicated. Unlike all the other reactions, in the case of the spiro-compounds 28 and 30, we were able to isolate only cis diastereomers 28b, 30b. Note also that, unlike the classical Staudinger method, we could not manage to obtain the 4-hydroxy-substituted derivative 25 using an acyl chloride-free technique.
To increase the yield of the final product, an optimization of the reaction conditions was carried out using Schiff base 5 and 4-chlorophenyl acetic acid as a model system. We varied the solvent, temperature, ratio of starting acid, DIPEA and oxalyl chloride as well as the order of component addition. The results obtained from optimization are shown in Table 4.
| Entry | Solvent | T, °C | Acid, mol | DIPEA, mol | Diastereomers ratio (23a/23b) | Yield, % |
|---|---|---|---|---|---|---|
| 1 | THF | 20 | 1 | 3.00 | 1:1 |
56 |
| 2 | THF | 20 | 1.5 | 3.00 | 1.2:1 |
73 |
| 3 | THF | 20 | 1.5 | 6.00 | 1:1 |
58 |
| 4 | THF | 0 | 1.5 | 6.00 | 1:1 |
60 |
| 5 | DCM | 20 | 1.5 | 3.00 | 1:1 |
4 |
| 6 | DCM | 0 | 1.5 | 3.00 | 1.2:1 |
9 |
| 7 | Acetone | 20 | 1.5 | 3.00 | 1:1 |
26 |
| 8 | Acetone | 0 | 1.5 | 3.00 | — | n/a |
| 9 | Et2O | 20 | 1.5 | 3.00 | — | n/a |
After optimization, it was discovered that the rate of the reaction, yield and presence of byproducts depends greatly on the solubility of the Schiff bases in the reaction solvent as well as the ratio of imine and acid. It was emphasized that THF dissolves the components of the reaction mixture much better than other tested solvents, which may explain why the best results were obtained using such a solvent. A decrease in the temperature of most solvents aside from THF leads to a decline in the solubility, and, as a consequence, a decline in yields. To our surprise, the choice of solvent did not noticeably affect the diastereoselectivity of the model reaction. The excess of acid proved necessary to lower the byproduct yields. It may be assumed that the oxalyl chloride used to convert starting acid into ketene may interact with the Schiff base as well as DIPEA, but, in the presence of an excess of acid, the probability of the target interaction becomes greater. However, the addition of too much excess acid negatively affected the yield, supposedly due to a drop in the solubility of the imine. In addition, the simultaneous mixing of all components as well as the excessive addition of oxalyl chloride resulted in a decline in the product yield and an increase in the colored byproduct content.
According to the results shown in Table 3, the ratio of the obtained diastereomers depends on the electron-donating ability of the substituent connected with the imine nitrogen. In general, the greater the donating character of the substituent in the phenyl ring of the isatin imine, the greater the diastereoselectivity shifted towards the formation of trans-spiro-oxindolo-β-lactams. The overall diastereomer yields in these cases were higher as well.
This can be explained by the assumption that Schiff bases with more electron-donating substituents have more electron density on the imine nitrogen, leading to increased nucleophilicity. Since the first stage of the Staudinger reaction is known to be a nucleophilic attack of the lone electron pair of the imine nitrogen by the electron-deficit carbonyl of ketene, this explanation seems plausible. A similar trend is observed when moving from nonsubstituted isatin to 5-chloroisatin. The yield of 5-chloroxindole spiro-β-lactams tended to be lower. On the contrary 5-bromoisatin derivatives have been obtained with generally better yields which seems to be related to better solubility of 5-bromo substituted Schiff bases.
To check whether the reaction proceeds with the formation of acyl halide in the reaction mixture or without intermediate formation, we tried to carry out the target reaction by mixing all components (Schiff base, 4-chlorophenylacetic acid and oxalyl chloride) except for the base (DIPEA). In this case, Staudinger ketene–imine cycloaddition did not occur, supposedly due to the absence of the ketene in the reaction mixture. The same result occurred when mixing imine, acid and DIPEA without oxalyl chloride. Additionally, we tried to carefully mix together oxalyl chloride and DIPEA and to add the resulting brown mixture to the remaining components. In this case, the formation of the target spiro-β-lactam was also not observed. Hence, contrary to the reaction with DMF, the oxalyl chloride/DIPEA mixture does not yield an active chlorinating agent.
Thus, for the key stage of the Staudinger synthesis without using pre-synthesized acyl chloride, we used the addition of oxalyl chloride to acetic acid with the generation of mixed anhydride (Scheme 4). Acid chloride apparently does not form in the reaction mixture.
 | ||
| Scheme 4 The proposed mechanism for acid activation and ketene formation. | ||
The proposed method of the Staudinger ketene–imine cycloaddition at room temperature without acyl chloride allows one to reverse the diastereoselectivity of the formation of spiro-β-lactams compared to the classical high-temperature method for generating ketene in the Staudinger reaction (see Tables 2 and 3). It should also be noted that oxalyl chloride converts acid into ketene during the reaction without formation of acyl halide, which was proven by a series of trials wherein we mixed 4-chlorophenyl acetic acid with oxalyl chloride and tertiary amine but with no DMF present.
In summary, the two synthetic techniques used in this work, as summarized in Scheme 5, complement each other, making it possible to selectively obtain cis or trans diastereomers of the target bis-aryl spiro[azetidine-2,3′-indoline]-2′,4-diones.
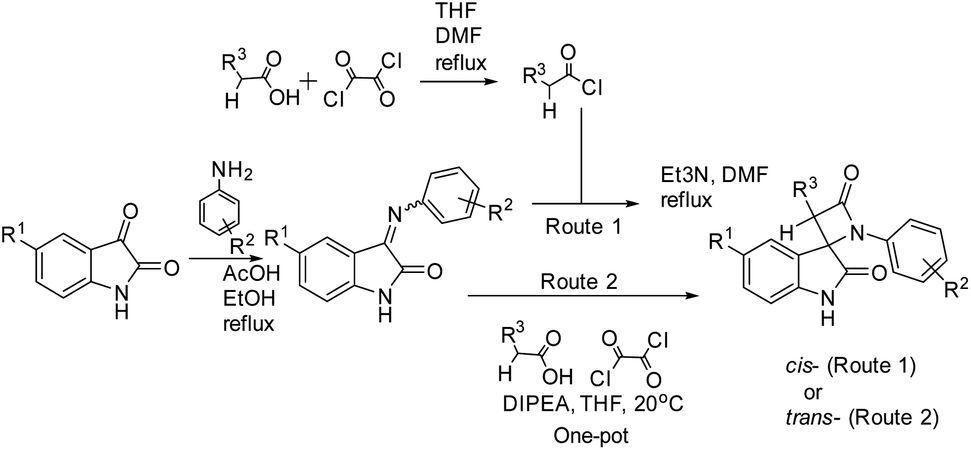 | ||
| Scheme 5 Generalization of two approaches for spirocyclic oxindolo-β-lactams synthesis by Staudinger ketene imine-cycloaddition applied in the present work (predominantly formed diastereomers are indicated). | ||
To confirm the structures of the synthesized spiro-oxindolo-β-lactams as well as correlate the 1H NMR data with the specific diastereomers, the structures of five obtained compounds (22a, 22b, 25a, 28b, 29b) were determined using X-ray diffraction analysis. The molecular structures of these compounds are shown in Fig. 3; the selected bond lengths and angles together with the numbering scheme used are all summarized in Table 5.
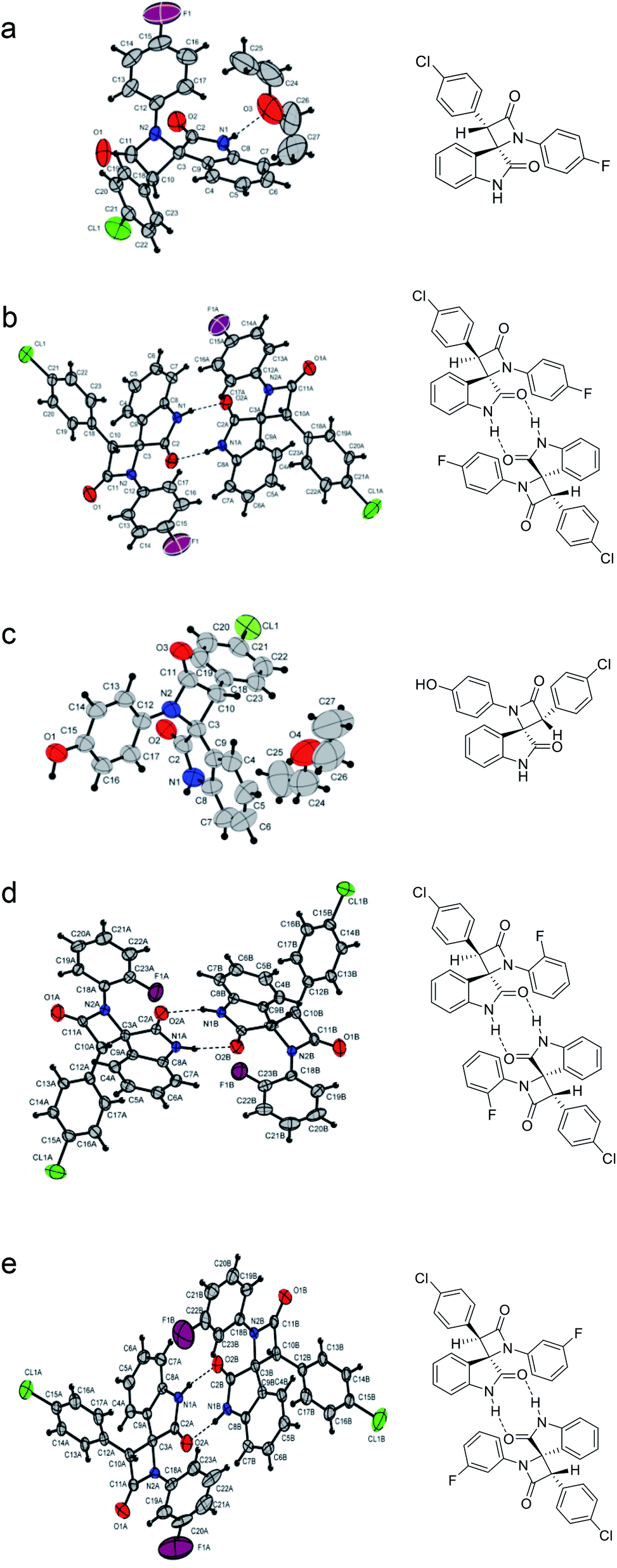 | ||
| Fig. 3 Molecular structures spiro-oxindolo-β-lactams: (a) compound 22a (CCDC 1963622); (b) compound 22b (CCDC 1963623); (c) compound 25a (CCDC 1963617); (d) compound 28b (CCDC 1963625); (e) compound 29b (CCDC 1963624). | ||
|
|||||
|---|---|---|---|---|---|
| Bond/angle | 22a | 22b | 25a | 28b | 29b |
| C2–C3 | 1.541 | 1.540 | 1.539 | 1.534 | 1.524 |
| C3–C9 | 1.494 | 1.489 | 1.495 | 1.495 | 1.501 |
| C3–N2 | 1.468 | 1.458 | 1.480 | 1.476 | 1.475 |
| C3–C10 | 1.582 | 1.609 | 1.583 | 1.592 | 1.579 |
| C10–C11 | 1.525 | 1.527 | 1.522 | 1.523 | 1.53 |
| N2–C11 | 1.363 | 1.378 | 1.353 | 1.384 | 1.371 |
| N2–C12 | 1.404 | 1.407 | 1.417 | 1.415 | 1.415 |
| C10–C18 | 1.493 | 1.493 | 1.493 | 1.479 | 1.495 |
| C2–C3–C9 | 103.31 | 103.50 | 103.31 | 103.3 | 103.7 |
| C2–C3–N2 | 114.04 | 118.04 | 115.27 | 117.7 | 117.9 |
| C9–C3–C10 | 117.74 | 118.88 | 118.00 | 118.3 | 118.4 |
| C3–N2–C11 | 95.48 | 96.05 | 95.16 | 95.1 | 94.7 |
| C3–C10–C11 | 84.84 | 84.46 | 84.77 | 85.3 | 85.4 |
| N2–C3–C10 | 86.68 | 86.54 | 86.45 | 86.5 | 86.3 |
| N2–C11–C10 | 92.81 | 92.75 | 93.63 | 92.7 | 92.9 |
| C18–C10–H | 113.21 | 111.12 | 110.07 | 109.9 | 110.9 |

By analyzing the X-ray data, we note that the compounds 22a and 25a are enantiomeric pairs of (2R,3S)- and (2S,3R)- spiro[azetidine-2,3′-indoline]-2′,4-diones that crystallized in a monoclinic centrosymmetric P21/c group. This correlates with the characteristic signals observed in the 1H NMR spectra at ∼10.9 ppm (NH of oxindole) and ∼5.35 ppm (γ-CH of azetidin-2-one ring). Compounds 22b, 28b, and 29b are enantiomeric pairs of (2R,3R)- and (2S,3S)-3-(4-chlorophenyl)-1-(4-aryl)spiro[azetidine-2,3′-indoline]-2′,4-diones. These pairs crystallize in a noncentrosymmetric P41 group with a pseudo center of symmetry. These structures correlate with the characteristic signals observed in the 1H NMR spectra at ∼11.1 ppm (NH of oxindole) and ∼5.1 ppm (γ-CH of azetidin-2-one ring).
For all studied compounds, the bond lengths and bond angles are found to be weakly dependent on the substituents in the aryl fragments. The planes of the spiro-fused heterocycles are nearly perpendicular to each other. The nitrogen atom of the azetidine fragment has an approximately planar trigonal environment, and the aryl substituent is almost coplanar with the four-membered ring.
To explain the change in the diastereoselectivity of the acyl chloride-free Staudinger synthesis at room temperature compared with the high-temperature technique described in the literature, we employed quantum chemical calculations. Geometry optimization for the compounds, vibrational frequency analysis, transition state (TS) searches of the studied transformations and intrinsic reaction coordinate calculations (IRC) were carried out using PRIRODA-04 software52 with the Perdew–Burke–Ernzerhof (PBE)53 functional in combination with a 3ζ basis set for the model reaction of compound 23 synthesis. We assumed that the structure of the final product depends on the organization of Staudinger's zwitterionic intermediate, which in turn can be formed from a Z- or E-Schiff base by attachment to a ketene carbonyl either on the side of the hydrogen atom or phenyl ring (Fig. 4).
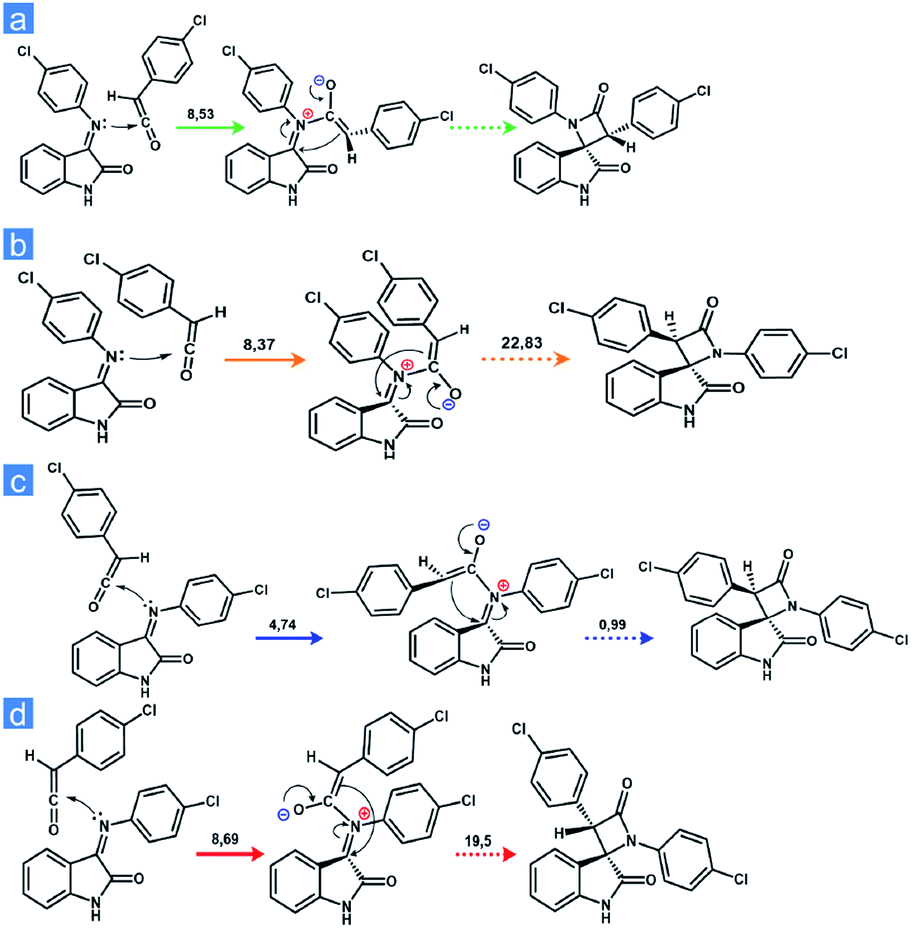 | ||
| Fig. 4 Schiff base attacking a ketene carbonyl: (a) E-imine attacks from the hydrogen side; (b) E-imine attacks from the phenyl side; (c) Z-imine attacks from the hydrogen side; (d) Z-imine attacks from the phenyl side. | ||
It is obvious that ketene attack from the side of the phenyl ring will have a greater activation barrier compared with attack from the hydrogen side, but it is important to determine how large this energy gap is, and, moreover, what the difference between the E- and Z-imines is. We found that the 2R*,3S*-isomer of 1,3-bis(4-chlorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (trans-product) is obtained by the cyclization of the intermediate formed from Z-imine attacking ketene from the phenyl side as well as E-imine attacking ketene from the hydrogen side. In contrast, the 2R*,3R*-isomer (cis-product) is obtained by the cyclization of the intermediate formed from the E-imine attacking ketene from the phenyl side as well as the Z-imine attacking ketene from the hydrogen side. For the E-imine, a zwitterionic Staudinger intermediate leading to a 2R*,3S* product is formed with 0.11 kcal mol−1 lower activation energy compared to that leading to the 2R*,3R* configuration, whereas the next stage of the process (cyclization) proceeds without an activation barrier, contrary to that of 2R*,3R* precursor, which is cyclized with an activation energy of approximately 22.8 kcal mol−1.
For the Z-imine, the calculations show a completely opposite result; the generation of the 2R*,3S* product proceeds with a higher activation barrier for both the formation of the intermediate (approximately 4 kcal mol−1 higher) and its cyclization into a four-membered β-lactam cycle (approximately 18.5 kcal mol−1 higher). Thus, the results of the quantum-chemical calculations indicate the predominant formation of a diastereomer with the a 2R*,3S* configuration only if the initial mixture of isomeric Schiff bases is enriched with the E-isomer.
Summarizing everything up to this point we have obtained some qualitative explanation for presence of diastereoselectivity in formation of our spiro-β-lactams. Yet according to calculations diastereomeric ratio should correlate with ratio of E/Z Schiff base in mixture, which doesn't seem to occur.
So further computational research is necessary in order to verify whether isomerization of Schiff base or more significantly isomerization of zwitterionic intermediate takes place. This could have explained dependency of diastereoselectivity on temperature as well as polarity of solvent.
To verify the correctness of the assumptions made on the basis of molecular docking in terms of the preference of trans-2,3-diaryl-spiro[azetidine-2,3′-indoline]-2′,4-diones as p53-MDM2 interaction inhibitors, we carried out preliminary in vitro cytotoxicity tests for compounds 23a,b and 27a,b. Cytotoxicity was evaluated using the standard MTT method.54 For testing, we selected the cell cultures of prostate cancer LNCap, PC3 and colorectal cancer HCT(p53+), HCT(p53−), both of which express p53 protein (LNCap and HCT (p53+)) and do not express PC3 and HCT116 (p53−). The results are shown in Table 6.
| No. | IC50 (μM) | |||
|---|---|---|---|---|
| LNCap | PC3 | HCTwt (P53+) | HCT−/− (P53−) | |
| 23a | 14.3 ± 4.5 | 27.6 ± 3.0 | 81.8 ± 16.0 | 152.1 ± 26.6 |
| 23b | 37.0 ± 8.1 | >50 M | 141.6 ± 40.1 | >50 M |
| 27a | 13.1 ± 5.8 | >50 M | 29.6 ± 16.9 | >50 M |
| 27b | 17.2 ± 3.3 | >50 M | 127.7 ± 14.3 | >50 M |
According to the data presented in Table 6, it is seen that all studied compounds are more cytotoxic to cell lines expressing p53 protein than to lines not expressing it. Moreover, the cytotoxicity of compounds 23a and 27a on p53-positive cell lines LNCap and HCT (p53+) is higher than for the corresponding isomeric compounds 23b and 27b. Such results are consistent with the molecular docking data for the higher affinity of trans-isomeric spiro[azetidine-2,3′-indoline]-2′,4-diones towards the MDM2 protein, and, accordingly, their higher anticancer activity associated with the inhibition of p53-MDM2 interaction.
Experimental
General techniques
All solvents used were purified and dehydrated using methods described in (ref. 55). All starting reagents were purchased from Sigma-Aldrich. Reactions were checked by TLC analysis using silica plates with a fluorescent indicator (254 nm) and visualized with a UV lamp. 1H and 13C NMR spectra were recorded on a Bruker Avance spectrometer (400 MHz for 1H, 100 MHz for 13C) in DMSO-d6. Chemical shifts are reported in parts per million relative to TMS. High resolution mass spectra (HRMS) were recorded on an Orbitrap Elite (Thermo Scientific) mass spectrometer with electrospray ionization (ESI) and orbital trap. To inject solutions with a concentration of 0.1 to 9 μg mL−1 (in 1% formic acid in acetonitrile), direct injection into the ion source using a syringe pump (5 μL min−1) was used. The spray voltage was ±3.5 kV, the temperature of the capillary was 275 °C.General procedure for preparation of Schiff base 1–21
Aromatic amine (1.1 mmol) was added into boiling solution of isatin (1 mmol) in 25 mL of absolute ethanol containing a few drops of glacial acetic acid. The reaction mixture was refluxed for 3 hours. Completion was checked by TLC. After the cooling of the reaction mixture to room temperature it was filtered under reduced pressure. Precipitate was washed with cold EtOH (2 × 5 mL) then dried. The spectral characteristics of the obtained compounds correspond to the literature data:56 (compounds 1, 2),57 (compounds 3–4, 6, 7, 19),58 (compounds 4–9, 11, 13, 14),59 (compound 10),60 (compound 10),61 (compound 12),62 (compound 15),63 (compounds 16, 18),64 (compound 17),65 (compounds 18, 20, 21).:ethyl acetate (35%) mixture. The products were recrystallized from diethyl ether giving a white solid.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(4-fluorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (22a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.88 (s, 1H), 7.67 (d, J = 7.34 Hz, 1H), 7.36–7.42 (m, 3H), 7.15–7.22 (m, 4H), 7.12 (t, J = 7.58 Hz, 1H), 7.02–7.07 (m, 2H), 7.00 (d, J = 7.70 Hz, 1H), 5.35 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.52, 164.06, 141.93, 133.04, 132.87, 131.05, 130.33, 130.23, 128.48, 124.78, 123.99, 122.91, 118.10, 118.02, 116.67, 116.44, 111.07, 66.29, 64.63. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 393.0801, found: 393.0792.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(4-fluorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (22b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.14 (s, 1H), 7.34 (d, J = 8.31 Hz, 2H), 7.21–7.25 (m, 3H), 7.17–7.21 (m, 2H), 7.05 (dd, J = 9.11, J = 4.71 Hz, 2H), 6.95 (d, J = 7.82 Hz, 1H), 6.71–6.74 (m, 2H), 5.08 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.72, 163.81, 142.53, 132.87, 131.45, 130.92, 130.77, 130.68, 128.70, 128.25, 125.50, 122.04, 120.99, 118.13, 116.70, 116.47, 111.02, 65.55, 63.69. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 393.0801, found: 393.0798.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(4-chlorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (23a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.91 (s, 1H), 7.67 (d, J = 7.39 Hz, 1H), 7.42–7.38 (m, 5H), 7.16 (d, J = 8.37 Hz, 2H), 7.12 (t, J= 7.52 Hz, 1H), 7.04–6.99 (m, 3H), 5.37 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.27, 164.14, 141.83, 135.27, 132.80, 130.95, 130.14, 130.10, 129.59, 128.38, 128.18, 124.70, 123.81, 110.98, 66.13, 64.91, 64.62, 15.16. HRMS (ESI) Calc for C22H15Cl2N2O2 [M + H+]: 409.0505, found: 409.0496.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(4-chlorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (23b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.16 (s, 1H), 7.40 (d, J = 8.80 Hz, 2H), 7.32 (d, J = 8.44 Hz, 2H), 7.18–7.25 (m, 3H), 7.02 (d, J = 8.80 Hz, 2H), 6.95 (d, J = 7.76 Hz, 1H), 6.67–6.75 (m, 2H), 5.09 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.48, 174.36, 163.92, 142.26, 135.27, 132.80, 130.86, 130.69, 130.50, 129.64, 128.60, 128.41, 125.44, 121.94, 120.76, 117.78, 110.90, 65.37, 63.65. HRMS (ESI) Calc for C22H15Cl2N2O2 [M + H+]: 409.0505, found: 409.0496.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(4-bromophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (24a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.91 (s, 1H), 7.67 (d, J = 7.39 Hz, 1H), 7.53 (d, J = 8.76 Hz, 2H), 7.35–7.43 (m, 3H), 7.16 (d, J = 8.38 Hz, 2H), 7.12 (t, J = 7.58 Hz, 1H), 7.00 (d, J = 7.99 Hz, 1H), 6.97 (d, J = 8.76 Hz, 2H), 5.37 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.26, 164.17, 141.81, 135.66, 132.79, 132.47, 130.96, 130.11, 130.09, 128.39, 124.71, 123.78, 122.79, 118.00, 116.20, 110.98, 66.11, 64.62. HRMS (ESI) Calc for C22H15BrClN2O2 [M + H+]: 453.0000, found: 452.9988.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(4-bromophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (24b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.16 (s, 1H), 7.53 (d, J = 8.68 Hz, 2H), 7.29–7.36 (m, 2H), 7.18–7.26 (m, 3H), 6.97 (t, J = 9.18 Hz, 3H), 6.67–6.77 (m, 2H), 5.10 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.43, 163.90, 142.40, 135.63, 132.77, 132.48, 130.81, 130.64, 130.45, 128.56, 125.39, 121.89, 120.76, 118.07, 116.42, 110.91, 65.34, 63.65. HRMS (ESI) Calc for C22H14BrClN2O2 [M + H+]: 453.0000, found: 452.9989.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(4-hydroxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (25a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.78 (s, 1H), 9.43 (s, 1H), 7.62 (d, J = 7.34 Hz, 1H), 7.33–7.41 (m, 3H), 7.16 (d, J = 8.44 Hz, 2H), 7.10 (t, J = 7.58 Hz, 1H), 6.97 (d, J = 7.82 Hz, 1H), 6.88 (d, J = 8.80 Hz, 2H), 6.68 (d, J = 8.80 Hz, 2H), 5.24 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.79, 163.41, 154.39, 141.86, 132.63, 130.71, 130.66, 130.20, 128.47, 128.32, 124.57, 124.50, 122.65, 118.24, 115.86, 110.80, 66.17, 64.33. HRMS (ESI) Calc for C22H16ClN2O3 [M + H+]: 391.0844, found: 391.0834.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(4-hydroxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (25b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.04 (s, 1H), 9.46 (s, 1H), 7.33 (d, J = 8.31 Hz, 2H), 7.21 (d, J = 8.44 Hz, 3H), 6.92 (d, J = 7.83 Hz, 1H), 6.88 (d, J = 8.80 Hz, 2H), 6.65–6.74 (m, 4H), 4.97 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 175.03, 163.17, 154.54, 142.46, 132.63, 131.02, 130.65, 130.58, 128.56, 128.46, 125.34, 121.80, 121.48, 118.30, 115.86, 110.75, 65.44, 63.27. HRMS (ESI) Calc for C22H16ClN2O3 [M + H+]: 391.0844, found: 391.0838.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(4-ethoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (26a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.82 (br. s., 1H), 7.64 (d, J = 7.39 Hz, 1H), 7.35–7.41 (m, 3H), 7.18 (d, J = 8.30 Hz, 2H), 7.10 (t, J = 7.50 Hz, 1H), 6.94–7.01 (m, 3H), 6.87 (d, J = 8.91 Hz, 2H), 5.28 (s, 1H), 3.92 (q, J = 6.90 Hz, 2H), 1.26 (t, J = 6.93 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.65, 163.52, 155.30, 141.86, 132.63, 130.73, 130.52, 130.13, 129.70, 128.29, 124.55, 124.33, 122.65, 117.88, 115.21, 110.79, 66.12, 64.37, 63.21, 14.52. HRMS (ESI) Calc for C24H20ClN2O3 [M + H+]: 419.1157, found: 419.1147.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(4-ethoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (26b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.09 (s, 1H), 7.33 (d, J = 8.31 Hz, 2H), 7.17–7.25 (m, 3H), 6.92–6.99 (m, 3H), 6.87 (d, J = 8.93 Hz, 2H), 6.68–6.74 (m, 2H), 5.01 (s, 1H), 3.92 (q, J = 6.95 Hz, 2H), 1.26 (t, J = 6.94 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.91, 163.31, 155.45, 142.47, 132.65, 130.91, 130.66, 129.70, 128.56, 125.36, 121.83, 121.33, 117.93, 115.23, 110.78, 65.38, 63.33, 63.23, 14.56. HRMS (ESI) Calc for C24H20ClN2O3 [M + H+]: 419.1157, found: 419.1150.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(3-chloro-4-fluorophenyl)spiro [azetidine-2,3′-indoline]-2′,4-dione (27a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.94 (s, 1H), 7.70 (d, J = 7.52 Hz, 1H), 7.34–7.43 (m, 5H), 7.09–7.18 (m, 3H), 7.00 (d, J = 7.76 Hz, 1H), 6.72 (dd, J = 7.27, 4.46 Hz, 1H), 5.41 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 171.90, 171.78, 170.12, 164.05, 152.33, 141.63, 141.47, 133.23, 132.63, 130.93, 129.87, 129.73, 128.20, 124.67, 123.17, 122.68, 120.21, 120.02, 117.97, 115.74, 110.75, 66.12, 64.52. HRMS (ESI) Calc for C22H14Cl2FN2O2 [M + H+]: 427.0411, found: 427.0402.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(3-chloro-4-fluorophenyl)spiro [azetidine-2,3′-indoline]-2′,4-dione (27b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.19 (s, 1H), 7.38–7.44 (m, 2H), 7.34 (d, J = 8.44 Hz, 2H), 7.21–7.28 (m, 3H), 6.97 (d, J = 7.76 Hz, 1H), 6.69–6.80 (m, 3H), 5.14 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.31, 164.02, 152.68, 142.48, 132.86, 131.02, 130.74, 130.36, 128.60, 125.58, 122.02, 120.46, 120.26, 118.46, 118.30, 118.08, 116.10, 110.95, 65.62, 63.86. HRMS (ESI) Calc for C22H14Cl2FN2O2 [M + H+]: 427.0411, found: 427.0404.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(2-fluorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (28b). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.88 (s, 1H), 7.87 (td, J = 7.58, 1.45 Hz, 1H), 7.33 (d, J = 8.45 Hz, 2H), 7.15–7.25 (m, 5H), 7.12 (td, J = 7.63, 1.10 Hz, 1H), 6.83 (d, J = 7.69 Hz, 1H), 6.69–6.74 (m, 1H), 6.61–6.68 (m, 1H), 5.14 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 175.53, 164.75, 153.40, 150.94, 142.71, 132.73, 130.96, 130.51, 130.39, 128.61, 127.22, 127.15, 125.28, 124.88, 123.20, 123.08, 122.72, 122.30, 121.46, 116.81, 116.61, 110.42, 67.73, 64.41. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 393.0801, found: 393.0794.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(3-fluorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (29a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.93 (s, 1H), 7.70 (d, J = 7.40 Hz, 1H), 7.40 (d, J = 8.56 Hz, 3H), 7.32–7.38 (m, 1H), 7.11–7.18 (m, 3H), 7.02 (d, J = 7.70 Hz, 1H), 6.92–6.99 (m, 2H), 6.59 – 6.66 (m, 1H), 5.41 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.26, 164.45, 141.86, 137.85, 132.88, 131.81, 131.71, 131.11, 130.15, 130.08, 128.47, 124.85, 123.77, 122.90, 111.60, 111.05, 103.70, 103.44, 66.29, 64.56. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 393.0801, found: 393.0790.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(3-fluorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (29b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.19 (s, 1H), 7.31–7.40 (m, 3H), 7.20–7.27 (m, 3H), 6.93–7.02 (m, 3H), 6.69–6.78 (m, 2H), 6.63 (dd, J = 8.10, 1.19 Hz, 1H), 5.13 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.40, 164.16, 163.43, 161.00, 142.44, 137.78, 132.84, 131.78, 131.69, 130.93, 130.71, 130.41, 128.62, 125.50, 121.98, 120.74, 111.65, 111.62, 111.54, 111.33, 110.94, 103.83, 103.57, 65.51, 63.61. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 393.0801, found: 393.0790.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(3-chlorophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (30b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.20 (s, 1H), 7.31–7.37 (m, 3H), 7.23–7.28 (m, 4H), 7.19 (td, J = 8.07, 1.10 Hz, 1H), 6.97 (d, J = 7.82 Hz, 1H), 6.69–6.79 (m, 3H), 5.13 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.41, 164.20, 142.43, 137.57, 133.82, 132.86, 130.97, 130.75, 130.40, 128.62, 125.55, 124.50, 122.01, 120.68, 116.27, 114.18, 110.94, 65.46, 63.66. HRMS (ESI) Calc for C22H15Cl2N2O2 [M + H+]: 409.0505, found: 409.0498.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(3-bromophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (31a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.91 (s, 1H), 7.67 (d, J = 7.27 Hz, 1H), 7.53 (d, J = 8.86 Hz, 2H), 7.37–7.43 (m, 3H), 7.16 (d, J = 8.44 Hz, 2H), 7.12 (td, J = 7.55, 0.67 Hz, 1H), 7.00 (d, J = 7.70 Hz, 1H), 6.96 (d, J = 8.86 Hz, 2H), 5.37 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.28, 164.20, 150.16, 141.82, 135.67, 132.80, 132.51, 130.99, 130.14, 130.12, 128.42, 124.75, 123.79, 122.82, 118.00, 116.22, 111.01, 66.11, 64.62. HRMS (ESI) Calc for C22H15BrClN2O2 [M + H+]: 453.0000, found: 452.9981.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(3-bromophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (31b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.20 (s, 1H), 7.42 (s, 1H), 7.32–7.36 (m, 2H), 7.30 (d, J = 6.42 Hz, 1H), 7.20–7.27 (m, 4H), 6.97 (d, J = 7.76 Hz, 1H), 6.70–6.79 (m, 3H), 5.13 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.41, 164.18, 142.43, 137.68, 132.85, 131.77, 130.96, 130.75, 130.40, 128.61, 127.37, 125.56, 122.20, 120.67, 119.17, 114.48, 110.91, 65.43, 63.65. HRMS (ESI) Calc for C22H15BrClN2O2 [M + H+]: 453.0000, found: 452.9984.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(3-iodophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (32a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.94 (s, 1H), 7.69 (d, J = 7.34 Hz, 1H), 7.59 (s, 1H), 7.46 (d, J = 7.82 Hz, 1H), 7.40 (d, J = 8.50 Hz, 3H), 7.16 (d, J = 8.56 Hz, 2H), 7.07–7.13 (m, 2H), 7.01 (d, J = 7.76 Hz, 1H), 6.76 (d, J = 8.31 Hz, 1H), 5.39 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.26, 164.35, 141.81, 137.59, 132.99, 132.83, 131.62, 131.07, 130.12, 128.43, 124.88, 124.84, 123.75, 122.86, 114.74, 110.94, 95.39, 66.07, 64.47. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 500.9861, found: 500.9848.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(3-iodophenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (32b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.20 (s, 1H), 7.64 (br. s., 1H), 7.46 (d, J = 7.64 Hz, 1H), 7.34 (d, J = 8.01 Hz, 2H), 7.19–7.28 (m, 3H), 7.09 (t, J = 7.92 Hz, 1H), 6.97 (d, J = 7.64 Hz, 1H), 6.69–6.78 (m, 3H), 5.11 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.46, 164.10, 161.77, 153.07, 142.42, 137.55, 133.20, 132.83, 131.59, 130.74, 130.44, 128.60, 125.54, 125.06, 121.97, 114.76, 110.87, 95.39, 65.32, 63.58. HRMS (ESI) Calc for C22H15ClFN2O2 [M + H+]: 500.9861, found: 500.9845.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(4-methoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (33a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.83 (s, 1H), 7.64 (d, J = 7.40 Hz, 1H), 7.35–7.40 (m, 3H), 7.18 (d, J = 8.13 Hz, 2H), 7.10 (t, J = 7.52 Hz, 1H), 6.96–7.00 (m, 3H), 6.86–6.90 (m, 2H), 5.28 (s, 1H), 3.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.71, 163.57, 156.07, 141.92, 132.70, 130.79, 130.56, 130.19, 129.86, 128.35, 124.62, 124.36, 122.70, 117.89, 114.76, 110.85, 66.15, 64.40, 55.28. HRMS (ESI) Calc for C23H18ClN2O3[M + H+]: 405.1000, found: 405.0997.
(2R*,3R*)-3-(4-Chlorophenyl)-1-(4-methoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (33b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.10 (s, 1H), 7.34 (d, J = 8.50 Hz, 2H), 7.23 (d, J = 8.50 Hz, 2H), 7.18–7.21 (m, 1H), 6.87–7.00 (m, 5H), 6.69–6.73 (m, 2H), 5.01 (s, 1H), 3.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.89, 163.29, 156.17, 142.48, 132.66, 131.32, 130.88, 130.62, 129.80, 128.55, 125.34, 121.81, 121.31, 117.93, 114.73, 110.76, 68.25, 65.38, 55.25. HRMS (ESI) Calc for C23H18ClN2O3 [M + H+]: 405.1000, found: 405.1000.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(m-tolyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (34a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.87 (s, 1H), 7.64 (d, J = 7.34 Hz, 1H), 7.36–7.42 (m, 3H), 7.14–7.19 (m, 3H), 7.08–7.14 (m, 2H), 7.00 (d, J = 7.76 Hz, 1H), 6.90 (d, J = 7.58 Hz, 1H), 6.58 (d, J = 8.25 Hz, 1H), 5.32 (s, 1H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.19, 163.85, 141.37, 138.74, 136.32, 132.48, 130.59, 129.92, 129.11, 128.13, 124.89, 124.38, 122.50, 116.81, 112.49, 110.55, 64.00, 59.53, 20.89. HRMS (ESI) Calc for C23H18ClN2O2 [M + H+]: 389.1051, found: 389.1046.
(2R*,3S*)-3-(4-Chlorophenyl)-1-(p-tolyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (35a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.86 (s, 1H), 7.63 (d, J = 7.40 Hz, 1H), 7.37–7.41 (m, 3H), 7.17 (d, J = 7.89 Hz, 2H), 7.10 (d, J = 7.76 Hz, 3H), 7.00 (d, J = 7.70 Hz, 1H), 6.93 (d, J = 7.82 Hz, 2H), 5.31 (s, 1H), 2.19 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.68, 163.91, 141.87, 134.18, 133.75, 132.77, 131.42, 130.87, 130.49, 130.23, 129.92, 128.42, 128.21, 124.64, 124.36, 122.78, 116.19, 110.93, 66.02, 64.38, 20.47. HRMS (ESI) Calc for C23H17ClN2O2 [M + H+]: 389.1051, found: 389.1045.
(2R*,3S*)-5′-Chloro-3-(4-chlorophenyl)-1-(4-fluorophenyl)spiro [azetidine-2,3′-indoline]-2′,4-dione (36a). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.01 (s, 1H), 7.87 (d, J = 1.98 Hz, 1H), 7.45 (dd, J = 8.38, 2.13 Hz, 1H), 7.39 (d, J = 8.45 Hz, 2H), 7.15–7.25 (m, 4H), 6.98–7.09 (m, 3H), 5.41 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.05, 163.69, 159.77, 157.37, 140.62, 132.90, 130.84, 130.28, 130.02, 128.60, 128.41, 126.87, 125.92, 125.12, 118.05, 116.67, 116.44, 112.47, 66.10, 64.65. HRMS (ESI) Calc for C22H14Cl2FN2O2 [M + H+]: 427.0411, found: 427.0404.
(2R*,3S*)-5′-Chloro-3-(4-chlorophenyl)-1-(4-methoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (37a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.97 (s, 1H), 7.84 (s, 1H), 7.44 (dd, J = 8.30, 1.29 Hz, 1H), 7.39 (d, J = 8.07 Hz, 2H), 7.20 (d, J = 8.30 Hz, 2H), 6.95–7.02 (m, 3H), 6.91 (d, J = 8.83 Hz, 2H), 5.36 (s, 1H), 3.68 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.45, 163.31, 156.12, 140.79, 132.79, 130.68, 130.29, 129.66, 128.36, 126.77, 126.35, 125.01, 117.88, 114.84, 112.36, 66.04, 64.41, 55.27.HRMS (ESI) Calc for C23H17Cl2N2O3[M + H+]: 439.0611, found: 439.0601.
(2R*,3S*)-5′-Chloro-3-(4-chlorophenyl)-1-(m-tolyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (38a). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.00 (s, 1H), 7.84 (d, J = 2.14 Hz, 1H), 7.45 (dd, J = 8.38, 2.20 Hz, 1H), 7.39 (d, J = 8.50 Hz, 2H), 7.15–7.21 (m, 3H), 7.09 (s, 1H), 7.01 (d, J = 8.31 Hz, 1H), 6.92 (d, J = 7.52 Hz, 1H), 6.56 (dd, J = 8.01, 1.71 Hz, 1H), 5.38 (s, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.30, 163.82, 140.68, 139.08, 136.43, 132.84, 130.28, 130.11, 129.45, 128.39, 126.78, 126.32, 125.24, 125.01, 117.05, 112.64, 112.38, 65.86, 64.31, 21.14. HRMS (ESI) Calc for C23H17Cl2N2O2 [M + H+]: 423.0662, found: 423.0657.
(2R*,3S*)-5′-Chloro-3-(4-chlorophenyl)-1-(p-tolyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (39a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.98 (s, 1H), 7.82 (s, 1H), 7.44 (dd, J = 8.34, 1.94 Hz, 1H), 7.39 (d, J = 8.38 Hz, 2H), 7.19 (d, J = 8.30 Hz, 2H), 7.12 (d, J = 8.22 Hz, 2H), 7.01 (d, J = 8.30 Hz, 1H), 6.92 (d, J = 8.30 Hz, 2H), 5.37 (s, 1H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.33, 163.53, 140.71, 133.97, 133.75, 132.79, 130.65, 130.25, 130.17, 129.90, 128.34, 126.74, 126.31, 124.94, 116.10, 112.35, 65.85, 64.36, 20.40. HRMS (ESI) Calc for C23H17Cl2N2O2 [M + H+]: 423.0662, found: 423.0654.
(2R*,3S*)-5′-Bromo-3-(4-chlorophenyl)-1-(4-fluorophenyl)spiro [azetidine-2,3′-indoline]-2′,4-dione (40a). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.03 (s, 1H), 7.99 (d, J = 1.83 Hz, 1H), 7.58 (dd, J = 8.31, 2.02 Hz, 1H), 7.36–7.42 (m, 2H), 7.17–7.24 (m, 4H), 7.02–7.09 (m, 2H), 6.97 (d, J = 8.31 Hz, 1H), 5.41 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.07, 163.69, 159.78, 157.38, 141.22, 133.69, 132.91, 130.28, 130.05, 128.42, 127.79, 126.33, 118.04, 117.96, 116.69, 116.46, 114.58, 112.98, 66.05, 64.67. HRMS (ESI) Calc for C22H14BrClFN2O2 [M + H+]: 470.9906, found: 470.9898.
(2R*,3S*)-5′-Bromo-3-(4-chlorophenyl)-1-(4-methoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (41a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.98 (s, 1H), 7.94 (s, 1H), 7.57 (dd, J = 8.31, 1.96 Hz, 1H), 7.39 (d, J = 8.44 Hz, 2H), 7.19 (d, J = 8.44 Hz, 2H), 6.88–6.99 (m, 5H), 5.36 (s, 1H), 3.67 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 171.95, 163.04, 155.87, 140.95, 140.79, 133.28, 132.54, 130.04, 129.41, 128.11, 127.42, 126.41, 117.60, 114.60, 114.23, 112.54, 65.70, 64.17, 55.03. HRMS (ESI) Calc for C23H17BrClN2O3 [M + H+]: 483.0106, found: 483.0100.
(2R*,3S*)-5′-Bromo-3-(4-chlorophenyl)-1-(m-tolyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (42a). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.01 (br. s., 1H), 7.95 (d, J = 1.59 Hz, 1H), 7.57 (dd, J = 8.31, 1.65 Hz, 1H), 7.39 (d, J = 8.38 Hz, 2H), 7.14–7.21 (m, 3H), 7.09 (s, 1H), 6.97 (d, J = 8.31 Hz, 1H), 6.93 (d, J = 7.52 Hz, 1H), 6.55 (d, J = 7.95 Hz, 1H), 5.38 (s, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.17, 163.81, 141.10, 139.09, 136.43, 133.57, 132.84, 130.28, 130.12, 129.46, 128.39, 127.68, 126.68, 125.24, 117.05, 114.49, 112.84, 112.62, 64.95, 64.32, 21.15. HRMS (ESI) Calc for C23H17BrClN2O2 [M + H+]: 467.0156, found: 467.0148.
(2R*,3R*)-5′-Bromo-3-(4-chlorophenyl)-1-(m-tolyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (42b). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.27 (s, 1H), 7.34–7.42 (m, 3H), 7.27 (d, J = 8.44 Hz, 2H), 7.13–7.22 (m, 2H), 6.87–6.97 (m, 3H), 6.54 (d, J = 8.62 Hz, 1H), 5.08 (s, 1H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 174.31, 163.44, 141.61, 139.12, 136.42, 133.37, 132.99, 130.82, 130.45, 129.46, 128.58, 128.10, 125.47, 123.65, 117.23, 113.54, 112.66 (d, J = 3.13 Hz), 65.05, 63.63, 21.13. HRMS (ESI) Calc for C23H17BrClN2O2 [M + H+]: 467.0156, found: 467.0148.
(2R*,3S*)-3-(4-Bromophenyl)-1-(4-methoxyphenyl)spiro[azetidine-2,3′-indoline]-2′,4-dione (43a). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.84 (s, 1H), 7.64 (d, J = 7.34 Hz, 1H), 7.52 (d, J = 8.38 Hz, 2H), 7.38 (t, J = 7.37 Hz, 1H), 7.07–7.14 (m, 3H), 6.95–7.01 (m, 3H), 6.89 (d, J = 9.05 Hz, 2H), 5.27 (s, 1H), 3.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 172.68, 163.50, 156.04, 141.89, 131.26, 130.98, 130.46, 129.82, 124.63, 124.32, 122.70, 121.28, 117.87, 114.76, 110.84, 66.06, 64.39, 55.27. HRMS (ESI) Calc for C23H18BrN2O3 [M + H+]: 449.0495, found: 449.0477.
Conclusions
In this work, the possibility of using ketene–imine Staudinger cyclization for synthesis of 2,3-diaryl-spiro[azetidine-2,3′-indoline]-2′,4-diones with varying diastereoselectivity was demonstrated. Carrying out the reaction of isatin imines with acyl chlorides in the presence of Et3N under heating can yield cis-isomeric spiro-β-lactams as the main reaction products, and the reactions of isatin imines with substituted phenylacetic acids in the presence of oxalyl chloride and DIPEA at room temperature mainly yields trans isomeric spiro-β-lactams. The latter reaction proceeds, apparently, without the formation of acyl chloride in the reaction mixture and leads to the formation of products that demonstrate reasonable cytotoxic activity in an MTT test, which may be associated with inhibition of the p53-MDM2 protein–protein interaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Russian Foundation for Basic Research (Projects 19-03-00201 and 19-33-90282) for the financial support of this work. This work in part of NMR and X-ray study was supported by the M. V. Lomonosov Moscow State University Program of Development.References
- A.-M. Tsimberidou, Cancer Chemother. Pharmacol., 2015, 76, 1113–1132 CrossRef CAS PubMed.
- M. F. Shaikh, W. F. Morano, J. Lee, E. Gleeson, B. D. Babcock, J. Michl, E. Sarafraz-Yazdi, M. R. Pincus and W. B. Bowne, Ann. Clin. Lab. Sci., 2016, 46, 627–634 CAS.
- U. M. Moll and O. Petrenko, Mol. Cancer Res., 2003, 1, 1001–1008 CAS.
- R. Honda, H. Tanaka and H. Yasuda, FEBS Lett., 1997, 420, 25–27 CrossRef CAS PubMed.
- F. Rodier, J. Campisi and D. Bhaumik, Nucleic Acids Res., 2007, 35, 7475–7484 CrossRef CAS PubMed.
- A. Efeyan and M. Serrano, Cell Cycle, 2007, 6, 1006–1010 CrossRef CAS PubMed.
- T. Ozaki and A. Nakagawara, Cancers (Basel), 2011, 3, 994–1013 CrossRef CAS PubMed.
- D. Liu and Y. Xu, Antioxid. Redox Signaling, 2011, 15, 1669–1678 CrossRef CAS PubMed.
- M. Wade and G. M. Wahl, Mol. Cancer Res., 2009, 7, 1–11 CrossRef CAS PubMed.
- Y. A. Ivanenkov, S. V. Vasilevski, E. K. Beloglazkina, M. E. Kukushkin, A. E. Machulkin, M. S. Veselov, N. V. Chufarova, E. S. Chernyagina, A. S. Vanzcool, N. V. Zyk, D. A. Skvortsov, A. A. Khutornenko, A. L. Rusanov, A. G. Tonevitsky, O. A. Dontsova and A. G. Majouga, Bioorg. Med. Chem. Lett., 2015, 25, 404–409 CrossRef CAS PubMed.
- L. Shu, Z. Li, C. Gu and D. Fishlock, Org. Process Res. Dev., 2013, 17, 247–256 CrossRef CAS.
- Z. Zhang, X. J. Chu, J. J. Liu, Q. Ding, J. Zhang, D. Bartkovitz, N. Jiang, P. Karnachi, S. S. So and C. Tovar, ACS Med. Chem. Lett., 2014, 5, 124–127 CrossRef CAS PubMed.
- A. Gollner, D. Rudolph, H. Arnhof, M. Bauer, S. M. Blake, G. Boehmelt, X. L. Cockroft, G. Dahmann, P. Ettmayer and T. Gerstberger, J. Med. Chem., 2016, 59, 10147–10162 CrossRef CAS PubMed.
- Y. Zhao, L. Liu, W. Sun, J. Lu, D. McEachern, X. Li, S. Yu, D. Bernard, P. Ochsenbein and V. Ferey, J. Am. Chem. Soc., 2013, 135, 7223–7234 CrossRef CAS PubMed.
- A. Beloglazkina, D. Skvortsov, V. Tafeenko, A. Majouga, N. Zyk and E. Beloglazkina, Russ. Chem. Bull., 2018, 67, 562–569 CrossRef CAS.
- C. Sheng and G. I. Georg, Targeting Protein–Protein Interactions by Small Molecules, Springer Singapore, 2018. DOI:10.1007/978-981-13-0773-7.
- B. Yu, Y.-C. Zheng, X.-J. Shi, P.-P. Qi and H.-M. Liu, Anticancer Agents Med. Chem., 2016, 16, 1315–1324 CrossRef CAS PubMed.
- H. Chen, D. Luo, L. Zhang, X. Lin, Q. Luo, H. Yi, J. Wang, X. Yan, B. Li and Y. Chen, Oncotarget, 2017, 8, 43008–43022 Search PubMed.
- Y. A. Ivanenkov and A. G. Majouga, RU2629750C2, April 9, 2015, https://yandex.ru/patents/doc/RU2629750C2_20170901.
- A. M. Malebari, L. M. Greene, S. M. F. D. Nathwani, N. M. O'Boyle, S. Wang, B. Twamley, D. M. Zisterer and M. J. Meegan, Eur. J. Med. Chem., 2017, 130, 261–285 CrossRef CAS PubMed.
- M. Isoda, K. Sato, Y. Kunugi, S. Tokonishi, A. Tarui, M. Omote, H. Minami and A. Ando, Beilstein J. Org. Chem., 2016, 12, 1608–1615 CrossRef CAS PubMed.
- J. A. Townes, M. A. Evans, J. Queffelec, S. J. Taylor and J. P. Morken, Org. Lett., 2002, 4, 2537–2540 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Carr, L. M. Greene, O. Bergin, S. M. Nathwani, T. McCabe, D. G. Lloyd, D. M. Zisterer and M. J. Meegan, J. Med. Chem., 2010, 53, 8569–8584 CrossRef PubMed.
- H. Alper, F. Urso and D. J. H. Smith, J. Am. Chem. Soc., 1983, 105, 6737–6738 CrossRef CAS.
- H. Abdellaoui and J. Xu, Tetrahedron, 2014, 70, 4323–4330 CrossRef CAS.
- S. J. Balkrishna, B. S. Bhakuni and S. Kumar, Tetrahedron, 2011, 67, 9565–9575 CrossRef CAS.
- M. Kinugasa and S. Hashimoto, J. Chem. Soc. Chem. Commun., 1972, 466–467 RSC.
- R. Pal and A. A. Basak, Chem. Commun., 2006, 16, 2992–2994 RSC.
- D. R. Banerjee, R. Biswas, A. K. Das and A. Basak, Eur. J. Med. Chem., 2015, 100, 223–234 CrossRef CAS PubMed.
- L. Troisi, L. De Vitis, C. Granito, T. Pilati and E. Pindinelli, Tetrahedron, 2004, 60, 6895–6900 CrossRef CAS.
- C. S. Cho, L. H. Jiang and S. Shim, Synth. Commun., 1999, 29, 2695–2703 CrossRef CAS.
- P. Xie, B. Qian, H. Huang and C. Xia, Tetrahedron Lett., 2012, 53, 1613–1616 CrossRef CAS.
- S. J. Zhang, W. W. Sun, P. Cao, X. P. Dong, J. K. Liu and B. Wu, J. Org. Chem., 2016, 81, 956–968 CrossRef CAS PubMed.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010, pp. 521–524 Search PubMed.
- J. L. Vicario, D. Badía and L. Carrillo, Org. Lett., 2001, 3, 773–776 CrossRef CAS PubMed.
- M. Ohno, S. Kobayashi, T. Iimori, Y. F. Wang and T. Izawa, J. Am. Chem. Soc., 1981, 103, 2405–2406 CrossRef CAS.
- A. Iza, J. L. Vicario, L. Carrillo and D. Badía, Synthesis, 2006, 4065, 4065–4074 Search PubMed.
- Z. Wang, N. Chen and J. Xu, Tetrahedron, 2011, 67, 9690–9699 CrossRef CAS.
- Z. Yang, S. Li, Z. Zhang and J. Xu, Org. Biomol. Chem., 2014, 12, 9822–9830 RSC.
- M. A. Zarei, Tetrahedron Lett., 2014, 55, 5354–5357 CrossRef CAS.
- L. Jiao, Y. Liang and J. Xu, J. Am. Chem. Soc., 2006, 128, 6060–6069 CrossRef CAS PubMed.
- Y. Wang, Y. Liang, L. Jiao, D. M. Du and J. Xu, J. Org. Chem., 2006, 71, 6983–6990 CrossRef CAS PubMed.
- F. P. Cossio, A. Arrieta and M. A. Sierra, Acc. Chem. Res., 2008, 41, 925–936 CrossRef CAS PubMed.
- Y. Liang, L. Jiao, S. Zhang and J. Xu, J. Org. Chem., 2005, 70, 334–337 CrossRef CAS PubMed.
- R. J. Shah, N. R. Modi, M. J. Patel, L. J. Patel, B. F. Chauhan and M. M. Patel, Med. Chem. Res., 2011, 20, 587–594 CrossRef CAS.
- J. W. Skiles and D. McNeil, Tetrahedron Lett., 1990, 31, 7277–7280 CrossRef CAS.
- J. Mc. Conathy and M. J. Owens, Prim. Care Companion J. Clin. Psychiatry, 2003, 5, 70–73 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 32, 455–461 Search PubMed.
- M. Verma, S. N. Pandeya, K. N. Singh and J. P. Stables, Acta Pharm., 2004, 54, 49–56 CAS.
- M. Zarei, Tetrahedron, 2013, 69, 6620–6626 CrossRef CAS.
- A. Jarrahpour and M. Zarei, Tetrahedron Lett., 2009, 50, 1568–1570 CrossRef CAS.
- D. N. Laikov and Y. A. Ustynyuk, Russ. Chem. Bull., 2005, 54, 820–826, DOI:10.1007/s11172-005-0329-x.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- L. F. Tietze, T. Eicher, Reaktionen Und Synthesen Im Organisch-chemischenPraktikum Und Forschungslaboratorium; 1991. DOI:10.1002/3527601716.
- J. Y. Ma, Y. C. Quan, H. G. Jin, X. H. Zhen, X. W. Zhang and L. P. Guan, Chem. Biol. Drug Des., 2016, 87, 342–351 CrossRef CAS PubMed.
- M. V. Diurno, O. Mazzoni, E. Piscopo and A. Bolognese, Farmaco, 1993, 48, 435–441 CAS.
- K. Mohammed Khan, U. Rasool Mughal, N. Ambreen, N. Hasan Rama, F. Naz, S. Perveen and M. Iqbal Choudhary, Lett. Drug Des. Discovery, 2010, 7, 716–720 CrossRef.
- C. J. A. Ribeiro, R. C. Nunes, J. D. Amaral, L. M. Gonçalves, C. M. P. Rodrigues, R. Moreira and M. M. M. Santos, Eur. J. Med. Chem., 2017, 140, 494–509 CrossRef CAS PubMed.
- Y. Meng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, E66, o1055 CrossRef PubMed.
- Y. M. S. A. Al-Kahraman, G. S. Singh and M. Yasinzai, Lett. Drug Des. Discovery, 2011, 8, 491–495 CrossRef CAS.
- A. Dandia, H. Sachdeva and R. Devi, J. Chem. Res., Synop., 2000, 6, 272–275 CrossRef.
- A. González, J. Quirante, J. Nieto, M. R. Almeida, M. J. Saraiva, A. Planas, G. Arsequell and G. Valencia, Bioorg. Med. Chem. Lett., 2009, 19, 5270–5273 CrossRef PubMed.
- S. K. Sridhar, M. Saravanan and A. Ramesh, Eur. J. Med. Chem., 2001, 36, 615–625 CrossRef CAS PubMed.
- S. K. Sridhar and A. Ramesh, Biol. Pharm. Bull., 2001, 24, 1149–1152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR and HRMS data for synthesised compounds. CCDC 1963617, 1963622–1963625. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02374d |
| This journal is © The Royal Society of Chemistry 2020 |