
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sol–gel synthesis to prepare size and shape-controlled mesoporous nanostructures of binary (II–VI) metal oxides†
Ryan Yarbrough,
Klinton Davis,
Sheeba Dawood and
Hemali Rathnayake *
*
Department of Nanoscience, Joint School of Nanoscience and Nanoengineering, University of North Carolina at Greensboro, Greensboro, NC 27401, USA. E-mail: hprathna@uncg.edu; Tel: +1-336-285-2860
First published on 6th April 2020
Abstract
A base-catalyzed sol–gel approach combined with a solvent-driven self-assembly process at low temperature is augmented to make manganese oxide (Mn3O4), copper oxide (CuO), and magnesium hydroxide (Mg(OH)2) nanostructures with size- and shape-controlled morphologies. Nanostructures of Mn3O4 with either hexagonal, irregular particle, or ribbon shape morphologies with an average diameter ranged from 100 to 200 nm have been prepared in four different solvent types. In all morphologies of Mn3O4, the experimental XRD patterns have indexed the nanocrystal unit cell structure to triclinic. The hexagonal nanoparticles of Mn3O4 exhibit high mesoporocity with a BET surface area of 91.68 m2 g−1 and BJH desorption average pore diameter of ∼28 nm. In the preparation of CuO nanostructures, highly nanoporous thin sheets have been produced in water and water/toluene solvent systems. The simulated XRD pattern matches the experimental XRD patterns of CuO nanostructures and indexes the nanocrystal unit cell structure to monoclinic. With the smallest desorption total pore volume of 0.09 cm3 g−1, CuO nanosheets have yielded the lowest BET surface area of 18.31 m2 g−1 and a BHJ desorption average pore diameter of ∼16 nm. The sol of magnesium hydroxide nanocrystals produces highly nanoporous hexagonal nanoplates in water and water/toluene solvent systems. The wide angle powder XRD patterns show well-defined Bragg's peaks, indexing to a hexagonal unit cell structure. The hexagonal plates show a significantly high BET surface area (72.31 m2 g−1), which is slightly lower than the surface area of Mn3O4 hexagonal nanoparticles. The non-template driven sol–gel synthesis process demonstrated herein provides a facile method to prepare highly mesoporous and nanoporous nanostructures of binary (II–IV) metal oxides and their hydroxide derivatives, enabling potential nanostructure platforms with high activities and selectivities for catalysis applications.
Introduction
Sol–gel chemistry, which transforms molecular precursors into polymeric oxide networks by hydrolysis and condensation, often offers a facile wet-chemical synthesis path to make size and shape-controlled nanostructures. Since the introduction of Stöber silica particles from base catalyzed hydrolysis and condensation of tetraethyl orthosilicate by Stöber et al.,1 the sol–gel method has been significantly advanced and implemented to make solid materials, such as inorganic oxides, organic–inorganic hybrids, and organosilica, with excellent electrical, optical, magnetic, thermal, and mechanical properties.2–4 The precursor to base concentration, solvent polarity, low temperature, additives type, and nucleation growth are few important factors that add merit to obtain morphology-controlled nanomaterials with high surface to volume ratio. As shape, size, and packing structure of the nanostructures play a crucial role for building next-generation devices and therapeutic materials, the sol–gel method is an excellent tool to fabricate metal oxide nanostructures, with size-dependent superior properties, particularly for optics and electronics. Up to date, a wide range of functional metal oxides by varying the metal centre has been synthesized using sol–gel syntheses at relatively low temperatures with better reproducibility.4–7 The sol–gel process, its-base catalyzed nucleation, nanocrystals ripening, and crystal growth process have been extensively studied to make homogenous, highly stoichiometric, and high-quality metal oxide morphologies, such as nanorods,8,9 nanoflakes,10 nanotubes,8,11 and nanofibers.12The sol–gel process has emerged as a versatile production technique for engineering materials and allows for preparing “one-pot” synthesis of porous microstructures from homogeneous sol of nanocrystals.4,13–15 Owing to its low operation temperature and aqueous-based fabrication process, the sol–gel method is considered as “soft chemistry” over more classical industrial techniques for glass and ceramic manufacturing, which require very high temperatures.4 The typical sol–gel synthesis is a two-step process, which involves two distinct phases: sol and gel: a sol is a colloidal suspension of homogeneous nanocrystals, and a gel is an interconnected network of solid phase particles that form a continuous entity throughout a liquid phase.16–18 With careful control of several reaction variables at either sol or gel stage, a wide range of accessible materials, such as fine powders, fibbers, thin films, xerogels, aerogels, and monoliths can be obtained.19 Depending on the monomer type (metal alkoxide or metal halide) and the reaction medium (aqueous–organic mixed or organic solvents alone), sol–gel process further divided into aqueous sol–gel routes17,19,20 and non-hydrolytic sol–gel pathways.21–23 However, these traditional aqueous sol–gel route and non-hydrolytic sol–gel process have their own limitations when it comes to the wide applicability of both processes for different types of metal precursors, in particularly for the preparation of transition metal oxide nanostructures. For example, with the particular exceptions of titanium and zirconium, alkoxide-transition metal precursors are not readily available. In hydrolytic sol–gel process, some metal alkoxides are expensive and still others are sensitive to moisture, heat, and light, making long term storage difficult. Further, some metal alkoxides are not commercially available or are difficult to obtain. In contrast, non-hydrolytic sol–gel method uses only organic ether-based solvents, thus precluding its versatility and applicability for large-scale environmentally benign production.
Overcoming drawbacks in aqueous and non-hydrolytic sol–gel routes, researchers have used more traditional approaches for the successful sol–gel preparation of transition metal oxide. This approach typically involves aqueous precipitation of the metal ion with a base that involves hydrolysis and condensation, yielding colloidal sols.24–26 The colloidal sol (seed crystals) of metal oxide nanocrystals can either be deposited onto a substrate to grow nanostructures or be continued through the polycondensation process to form gels. The gel can be used to form particles, xerogels, aerogels, glass, and ceramics, depending upon the final processing step involved.17 Owing to its versatility, scalability, and solution processability with ability to control the dimensionality at nanoscale via post processing methods, like hydrothermal, solvothermal, and low temperature solvent-driven shape-controlled crystal growth process and self-assembly, the modified sol–gel route is adaptable to prepare metal oxide nanostructures and hierarchical microstructures.
In our recent study, for the first time, we developed a facile sol–gel approach, which combines with a solvent polarity driven self-assembly and solvothermal crystal growth process to prepare shape-controlled ZnO nanostructures.5 We demonstrated that the difference in polarity and surface adhesion of an organic solvent controlled the nanocrystal growth where solvent molecules act as surfactants that adsorb onto surfaces of the growing crystallites.5 Thus, this base-catalyzed solution-based sol–gel approach serves as a versatile wet-chemical synthesis path to create shape-controlled metal oxide nanostructures by modulating the surface energy of nanocrystals via selective adhesion of solvent molecules (act as surfactants). Augmenting this novel sol–gel approach developed by our research group, herein, we demonstrate its wide applicability to make size and shaped-controlled highly crystalline nanostructures of manganese(II,III)oxide, copper oxide, and magnesium hydroxide from metal ion precursors of Mn2+, Cu2+, and Mg2+ at low temperature (<80 °C) using a series of oil–water solvent systems. There have been various synthesis methods developed to make these three metal oxides in the past where their properties and morphologies were extensively studied. For example; manganese oxide nanostructures with a wide variety of morphologies, such as hexagonal plates,27 cubes,28 nanorods,29 and spherical nanoparticles30 were synthesized by either hydrothermal techniques,27 thermal evaporation methods,28 or wet-chemical processes.29–31 The stoichiometries of the manganese oxide nanostructures reported up to date are Mn3O5, α-Mn2O3, Mn3O4, and MnO.27–31 Similarly, copper oxide nanostructures in the form of either Cu2O or CuO were prepared by sol–gel chemical syntheses,29–34 hydrothermal,33 solvothermal processes,35 and electrocatalytic methods36 to yield nanoparticles, rods, nanosheets, needles, and nanowires.29,31–36 From alkaline metal oxides nanomaterials, magnesium hydroxide has been successfully synthesized with four different morphologies; plates, needles, rods and tubes, via wet chemical synthesis processes,37–41 and hydrothermal methods.42–45 However, to the best of our knowledge, application of the sol–gel chemical process combined with colloidal nanocrystals self-assembly in different organic solvents to control the morphologies at nanoscale have not been demonstrated.
In this work, a facile sol–gel chemical process followed by solvent-driven self-assembly process is conducted by varying the molar ratio of each metal precursor to the base at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1:10, and 1:15 in four different solvent systems (water, 70% ethanol, dimethyl formamide, and toluene). The effect of precursor to base concentration and solvent type on the morphologies and crystal packing is investigated to produce size and shape-controlled nanostructures of manganese oxide (Mn3O4), copper oxide (CuO), and magnesium hydroxide (Mg(OH)2) with high reproducibility in large-scale. All metal oxide nanostructures, prepared in this manner are characterized for crystallinity, solid-state packing, structural composition, and morphology. Combining powder XRD traces with simulated XRD patterns provide the respective crystal packing pattern and the chemical formula for each metal oxide form. In each case, individual oxide has produced somewhat different morphologies with respect to the solvent type, but in most cases, water/toluene system has yielded well-defined hexagonal nanoparticles for Mn3O4 and Mg(OH)2, whereas CuO has yielded irregular sheets in all four solvent systems. As the sol–gel approach follows seed crystals formation, then their self-assembly, followed by crystal growth, yielding void spaces among nanocrystals, nanostructures could possess mesoporous structures. Thus, the mesoporosity of these nanostructures are also evaluated. The nanostructures of manganese oxide and magnesium hydroxide exhibit highly mesoporous surfaces with considerably high surface area. Thus, the sol–gel route developed and demonstrated herein enables to make well-defined, highly mesoporous nanostructures of manganese oxide and magnesium hydroxide with high crystallinity.
:5, 1:10, and 1:15 in four different solvent systems (water, 70% ethanol, dimethyl formamide, and toluene). The effect of precursor to base concentration and solvent type on the morphologies and crystal packing is investigated to produce size and shape-controlled nanostructures of manganese oxide (Mn3O4), copper oxide (CuO), and magnesium hydroxide (Mg(OH)2) with high reproducibility in large-scale. All metal oxide nanostructures, prepared in this manner are characterized for crystallinity, solid-state packing, structural composition, and morphology. Combining powder XRD traces with simulated XRD patterns provide the respective crystal packing pattern and the chemical formula for each metal oxide form. In each case, individual oxide has produced somewhat different morphologies with respect to the solvent type, but in most cases, water/toluene system has yielded well-defined hexagonal nanoparticles for Mn3O4 and Mg(OH)2, whereas CuO has yielded irregular sheets in all four solvent systems. As the sol–gel approach follows seed crystals formation, then their self-assembly, followed by crystal growth, yielding void spaces among nanocrystals, nanostructures could possess mesoporous structures. Thus, the mesoporosity of these nanostructures are also evaluated. The nanostructures of manganese oxide and magnesium hydroxide exhibit highly mesoporous surfaces with considerably high surface area. Thus, the sol–gel route developed and demonstrated herein enables to make well-defined, highly mesoporous nanostructures of manganese oxide and magnesium hydroxide with high crystallinity.
Results and discussion
A base-catalyzed sol–gel approach combined with a solvent-driven self-assembly process was augmented to make manganese oxide, copper oxide, and magnesium hydroxide nanostructures with different morphologies. The nanocrystals self-assembly and crystal growth were performed at low operation temperature in four different oil/water solvent systems. The sol–gel approach developed here is a modified aqueous-based hydrolytic sol–gel route, using either metal halides or metal acetates as metal precursors. The morphology and crystal packing of each metal oxide nanostructures with respect to different solvent systems were investigated and results are discussed below. The chemistry of either metal oxide or metal hydroxide nanostructures formation is depicted in Scheme 1 along with sol–gel process for sol formation.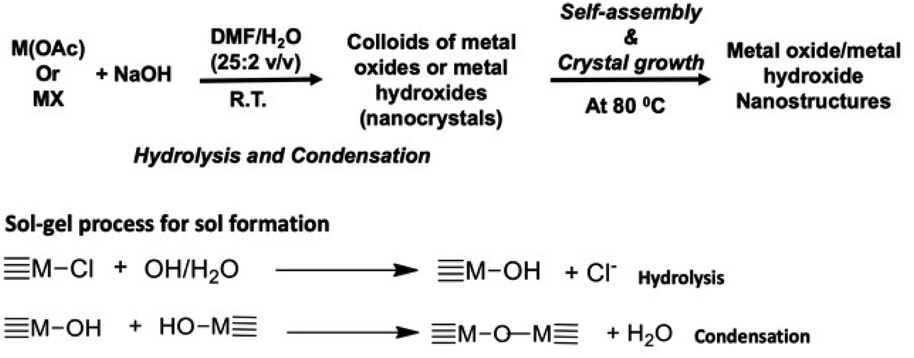 | ||
| Scheme 1 Sol–gel chemical process followed by self-assembly approach for the formation of either metal oxides or metal hydroxide nanostructures. | ||
Preparation, morphology, and crystal packing of manganese oxide nanostructures
Among different forms of manganese oxides, Mn3O4 is well-known as an active catalyst for the oxidation of methane and carbon monoxide,46 the selective reduction of nitrobenzene47 or the combustion of organic compounds,48 providing solution to control air pollution problems and organic waste. Additionally, Mn3O4 is used in lithium-ion batteries for electrode materials49,50 and in the production of ferrite soft magnetic materials.51,52 However, synthesis methods are limited to calcination,53–55 chemical bath deposition,56 sol–gel technique,57 coprecipitation,58 and hydrothermal/solvothermal synthesis.59–61 To the best of our knowledge, colloidal Mn3O4 nanoparticles were previously only prepared either from thermal decomposition of manganese acetylacetonate in oleylamine62 or ultrasonic-assisted synthesis at ambient temperature and pressure without any additional surfactant or template.63 The work described herein is one of a few demonstration where a reliable sol–gel-based wet-chemical synthesis method was developed to make shape and size-controlled Mn3O4 nanoparticles with high crystallinity and mesoporosity.Augmenting a previously developed sol–gel approach followed by solvothermal self-assembly process,5 manganese oxide nanostructures were prepared in four different solvent systems. The effect of the base concentration and solvent type on the morphology was evaluated. The reaction conditions, morphologies, and dimensions of respective nanostructures are summarized in Table 1. In a typical synthesis, manganese oxide nanomaterials in the oxide form of Mn3O4 were prepared by hydrolysis and condensation of anhydrous manganese(II)acetate (Mn(CH3COO)2) in the presence of sodium hydroxide as a base in a solution of de-ionized water/organic solvent mixture at ambient atmosphere. The formation of MnO2 by oxidizing Mn2+ to Mn4+ was not observed under the base-catalyzed low temperature reaction conditions. Because, the most stable possible forms of manganese ion intermediates formed under basic, low temperature conditions are Mn(II,III), the sol–gel reaction was favoured of yielding Mn3O4 complex. As demonstrated in the past, in order to form MnO2 from Mn2+, a strong highly acidic oxidizing agent is needed. For example, strong oxidizing HNO3 vapor was used to tune the oxidation state of Mn from 2+ in MnO to 4+ in MnO2.64 The formation of only Mn3O4 oxide form was also confirmed by powder XRD analysis and is discussed below. The effect of molar ratio on the precursor to NaOH and solvent type on the shape and size of nanoparticles were evaluated. After 24 hours reaction time at 80 °C, morphologies were analysed by scanning electron microscopy (SEM). The crystal packing was evaluated from powder XRD and selective area electron diffraction (SAED) analysis using X-ray powder diffractometer and transmission electron microscope (TEM) respectively.
| Molar ratio Mn+:base |
Solvent type/water | Morphology (dimension/nm) |
|---|---|---|
| a Number of moles of the metal precursor was kept constant and number of moles of the base was varied to maintain precursor to base concentration at 1:5, 1:10, and 1:15. |
||
| 1:5 |
Water | Hexagonal nanoparticles (130 ± 40) |
| 70% ethanol | Hexagonal nanoparticles (135 ± 32) | |
| DMF | Nanoribbons and nanoparticles (N/A) | |
| Toluene | Hexagonal nanoparticles (130 ± 40) | |
| 1:10 |
Water | Hexagonal nanoparticles (118 ± 50) |
| 70% ethanol | Hexagonal nanoparticles (135 ± 32) | |
| DMF | Nanoparticles (51 ± 21) | |
| Toluene | Hexagonal nanoparticles (117 ± 34) | |
| 1:15 |
Water | Hexagonal nanoparticles (73 ± 36) |
| 70% ethanol | Hexagonal nanoparticles (152 ± 50) | |
| DMF | Aggregated particles in gel form | |
| Toluene | Hexagonal nanoparticles (165 ± 53) | |
The effect of solvent type on the shape of the nanostructures was studied by performing a series of reactions in three different solvents (70% ethanol, dimethyl formamide, and toluene) mixed with de-ionized water at three different molar ratios (1:5, 1:10, and 1:15) of the precursor to base. These trials were performed according to the typical procedure described in the Experimental section with the organic solvent type being the only variable at each molar ratio. The morphologies formed with respect to each solvent type were compared with the nanomaterials, prepared in water alone. As summarized in Table 1, water, ethanol, and toluene yielded hexagonal nanoparticles whereas DMF produced irregular nanoparticles and, in some cases, a mixture of nanoparticles and nanoribbons. In three solvent systems; water, ethanol, and toluene, the average diameter of particles was ranged from 100–200 nm at all three different molar ratios, evidencing there is also no effect of the base concentration on the nanostructure's dimension. Although, we hypothesized that there may be significant changes in the morphologies as well as dimensionalities with respect to the solvent type and the different base concentrations, nanostructures exhibit no variations in shape and size with respect to the solvent type being water, ethanol, and toluene. This evidences that the formation of hexagonal nanoparticles in these three solvent systems is dominated by the crystal growth along particular preferred facets while hindering the directional growth along other facets. In order to investigate the nanocrystals self-assembly and their crystal growth for the formation of only hexagonal nanoparticles in all three solvent systems, we correlated morphologies of nanostructures with their crystal packing and is discussed later on.
Morphologies of nanostructures were analyzed from SEM and TEM after the samples were washed and re-dispersed in de-ionized water. As summarized in Table 1, Mn3O4 nanostructures with shape-controlled morphologies were obtained with respect to three different solvent systems and water. The SEM analysis of the final products resulted from different solvent systems exhibit two different distinct morphologies: hexagonal nanoparticles, and aggregated irregular shaped nanoparticles (see Fig. 1, S1 and S2†). The hexagonal nanoparticles with average diameter of 100 nm to 200 nm were resulted in water, ethanol, and toluene at 1:5 and 1:10 molar ratios of the precursor to the base whereas dimethyl formamide (DMF) mixture produced aggregated irregular shaped nanoparticles with average diameter ranged from 10 nm to 50 nm at all three different molar ratios. Although, hexagonal nanoparticles formed in water at 1:15 molar ratio, the reaction tends to exhibit rather aggregated nanoparticles trapped in a gel like structures. At 1:10 molar ratio of the precursor to the base in water/toluene solvent mixture, the hexagonal nanoparticles exhibit well-defined edges with rather uniform size distribution compared to the hexagonal nanoparticles formed in all other solvent systems and molar ratios. This may be due to rather low miscibility of toluene in water, yielding high concentration of non-solvated toluene molecules. Acting toluene molecules as shape-controllers, it could control the nanocrystal growth along the facets, yielding well-defined hexagonal nanoparticles compared to the particles formed in water and water/ethanol systems. However, in general, nanostructures produced in all four solvent systems show a wider size distribution at all three different base concentrations, but the particle size tends to decrease when the base concentration increases. For example, hexagonal nanoparticles formed in water show that diameter decreases with increasing the base concentration. At 1:5 molar ratio, particle's diameter is around 135 nm, whereas at 1:15 molar ratio, the average diameter was reduced to ∼73 nm. On the other hand, average particle size reduced from 170 nm to 150 nm for particles formed toluene when the precursor to base molar ratio increases from 1:5 to 1:10 and then the diameter increases at 1:15 molar ratio. The particles formed in ethanol do not show noticeable increase in the average diameter and maintained the average diameter around 135 nm, evidencing that regardless the increase in the base concentration, particle size was maintained at a narrow size distribution. Perhaps, this may be due to a higher solvation of the base in water/ethanol, yielding a homogeneous solution, where we believe that the nucleation rate maximizes at a specific base concentration, in our case at 1:5 molar ratio, and is independent at higher base concentrations. Thus, size distribution of seed crystals may homogeneous at all three base concentrations, while the growth of seed crystals governs by the surface energies of crystal facets, where solvent molecules act as surfactants to control the crystal growth, resulting in size-controlled nanostructures at all three base concentrations.
 | ||
| Fig. 1 SEM (top), TEM (down) and SAED pattern (inset) of manganese oxide nanostructures formed in each solvent system at the molar ratio of precursor to base 1:5. | ||
The TEM images further reveals the particles shape and size, confirming the hexagonal shape for nanoparticles formed in water, ethanol, and toluene. The SAED patterns confirm the high crystallinity of nanocrystals and in some cases reflects polycrystalline in nature where nanocrystals could arrange ordered and dis-ordered orientations in different solvent systems. Although, SEM images of nanoparticles formed in DMF show irregular shaped nanoparticles with aggregates, the SAED pattern shows uniform nanocrystal's diffraction pattern (a clearer ring pattern) compared to the hexagonal nanoparticles formed in water, ethanol, and toluene, and exhibit polycrystallinity.
The powder XRD results reveal the preferential crystal growth facets that modulate the crystal growth direction in the nanocrystal unit cell in each different solvent system. In our previous work, we showed that the polarity and chemical nature of the organic solvent in each reaction mixture act as a selective adhesion surfactant to facilitate the shape-controlled crystal growth.5,65,66 As studied in the past literature, the nanostructure morphology controls by surface energies of the facets. Depending on the polarity of solvent molecules, the solvent could adhere to the crystal facets, resulting in a different crystal growth pattern.65,66 Comparing the powder XRD results of Mn3O4 nanostructures (Fig. 2 and S3†), the intensities correspond to [101], [112], [013], and [211] were considered and compared for each solvent system to understand the preferential crystal growth direction and the morphology of the final nanostructure. The diffraction patterns obtained for all four different solvent systems exhibited [101] crystal plane reflection as a most dominant reflection, and [013] and [211] planes were visible for the hexagonal nanoparticles formed in water, ethanol, and toluene. The diffraction for [112] plane was not noticeable in hexagonal nanoparticles prepared in these three solvents, further convincing the crystal growth process to yield shape controlled hexagonal nanoparticles. However, we observed a clear intensity differences in these three reflection planes for the nanostructures formed in DMF. The reflection planes of [112], [013], and [211] were clearly noticeable and [112] plane was well-resolved. Thus, the difference in the diffraction pattern for the nanostructures formed in DMF supports the formation of spherical nanoparticles instead of hexagonal nanoparticles.
 | ||
| Fig. 2 Left – Powder XRD traces (coloured) along with respective simulated XRD patterns (black colour) for manganese oxide nanostructures formed in each different solvent system at the molar ratio of precursor to base 1:5; right – respective nanocrystals unit cell crystal structure (triclinic unit cell, COD ID: 1514121) acquired from the Crystallographic Open Data base (COD). | ||
Comparing the experimental powder XRD pattern with the simulated XRD pattern, acquired from screening the crystallographic open data base for crystal structures of different forms of manganese oxides, the chemical formula of the oxide form was identified. The simulated XRD pattern was acquired by screening respective crystal structures for three forms of manganese oxides; MnO, MnO2, and Mn3O4 from the crystallographic open data base. The simulated XRD pattern matched the experimental XRD pattern, confirming the crystal structure to Mn3O4, and indexed to triclinic unit cell structure, having calculated lattice constants of a = 5.76 Å, b = 5.76 Å, and c = 9.43 Å.
Preparation, morphology, and crystal packing of copper oxide nanostructures
Copper oxide has been studied extensively in literature, owing to its p-type semiconducting character with narrow and indirect energy band gap of ∼1.2 eV. With its additional intriguing properties, such as environmental benignity, chemical stability, high catalytic activity, electrochemical activity, antimicrobial activity, and abundance, there have been synthetic methods developed to make diverse morphologies at nanoscale, including nanowires,67,68 nanoflowers,69–71 nanorods,33,72,73 nanoplatelets,74 nanotubes,75 nanobelts,67 nanoparticles,33,73 nanoleaves,72,75 and many more.68,74 The primary method for synthesizing nanoscale copper oxide is using a sol–gel method29,31–34 and has utilized to make both Cu2O and CuO nanoparticles with a diameter of 31 nm and 32 nm respectively.30 Other nanoparticles were produced using wet synthesis methods under nitrogen and high temperature, producing nanoparticles with a diameter ranging from 3.6 nm to 10.7 nm.32 Jisen et al. produced CuO nanorods with a length of about 200 nm.33 Among other methods, electrocatalytic methods have been used to produce microspheres, nanosheets, and nanowires.36 However, there is no extensive studies conducted on sol–gel synthesis with solvent-driven self-assembly process to make size and shape-controlled CuO nanostructures.Similar to the preparation of manganese oxide nanostructures, experimental parameters, along with the effect of solvent type on the nanostructures' morphologies were studied and are summarized in Table 2. The effect of solvent type on the shape of the nanostructures was studied by performing a series of trial reactions following the same procedure developed herein. The synthesis was conducted in the same four different solvent systems. In a typical procedure, copper(II)acetate was subjected to base-catalyzed hydrolysis and condensation under ambient atmosphere. After 24 hours reaction time, the morphologies were analyzed from SEM and crystal packing was evaluated from powder XRD and selective area diffraction analysis.
| Molar ratio Mn+:base |
Solvent type/water | Morphology (dimension/nm) |
|---|---|---|
| a Number of moles of the metal precursor was kept constant and number of moles of the base was varied to maintain precursor to base concentration at 1:5, 1:10, and 1:15. |
||
| 1:5 |
Water | Thin sheets (length 1498 ± 291; width 400 ± 10) |
| 70% ethanol | Thin sheets (length 145 ± 62) with rods like structures | |
| DMF | Rice like particles (28 ± 6) | |
| Toluene | Thin sheets (length 1079 ± 453; width 370 ± 10) | |
| 1:10 |
Water | Thin sheets (length 1168 ± 242; width 400 ± 10) |
| 70% ethanol | Rods (length 119 ± 55) | |
| DMF | Ribbons (length 94 ± 31) | |
| Toluene | Rods (length 273 ± 111) with sheets | |
| 1:15 |
Water | Thin sheets (length 767 ± 169) |
| 70% ethanol | Irregular structures | |
| DMF | Rods (length 129 ± 54) | |
| Toluene | Thin sheets (length 667 ± 284) | |
As summarized in Table 2 and depicted in Fig. 3, water, ethanol, DMF, and toluene yielded nanostructures with different morphologies. In all three different base concentrations, water produced thin sheets like structures. In ethanol, thin sheets were produced at the lowest base concentration, while small well-defined nanorods and irregular aggregates were produced at 1:10 and 1:15 molar ratio of precursor to the base (Fig. S4 and S5†).
 | ||
| Fig. 3 SEM (top), TEM (down) and SAED pattern (inset) of copper oxide nanostructures formed in each solvent system at the molar ratio of precursor to base 1:5. | ||
The nanostructures formed in toluene were also thin sheets at 1:5 and 1:15 molar ratio of the precursor to the base, but rods were produced at the precursor to the base concentration of 1:10. The reactions performed in DMF produced nanoparticles, ribbons, and rods with particles at each base concentration respectively. A series of trial reactions, conducted by maintaining the same experimental parameters at three different base concentrations, evidenced that nanorods formation is strictly controlled by both solvent type and the base concentration. The experimental conditions that produced well-defined nanorods in ethanol at 1:10 precursor to the base, could use as a facile, environmentally benign sol–gel synthesis path to produce dimensional controlled CuO nanorods. Nanostructures produced under all other experimental parameters have exhibited no shape and size-controlled morphologies by either solvent type or at different base concentrations. These results evidence that crystal growth is independent from the polarity of the solvent type for water and toluene, but morphology was tailored by the solvent polarity for ethanol and DMF.
The powder XRD traces of CuO nanostructures (Fig. 4 and S6†) exhibit two distinct well-defined peaks that correspond to the Bragg's reflection planes of [001] and [312] with a low intensity shoulder peak for the [110] reflection plane. The diffraction patterns obtained for all four different solvent systems at all three different molar ratios of the precursor to the base exhibited [001] and [312] crystal planes as most dominant reflections with the exception of additional peaks for the nanostructures produced in DMF at 1:10 molar ratio. The difference in the XRD powder pattern with additional XRD peaks very well supports the ribbons like morphology formed in DMF at 1:10 molar ratio.
 | ||
| Fig. 4 Left – Powder XRD traces (coloured) along with respective simulated XRD patterns (black colour) for CuO nanostructures formed in each solvent system at 1:5 molar ratio of precursor to base; right – respective CuO unit cell crystal structure (monoclinic unit cell, COD ID: 4105682). | ||
The SAED patterns obtained for the nanostructures formed in water at all three molar ratios exhibit well-resolved aligned dot patterns reflecting single crystalline in nature whereas the SAED patterns of nanostructures produced in ethanol and DMF show ring patterns with somewhat unorganized dot patterns. Although the morphology of nanostructures formed in toluene are similar to the nanostructures formed in water, the SAED pattern of the nanostructures formed in toluene reflects aligned dot patterns with much larger distance between lines. The simulated XRD pattern acquired from the crystallographic open data base, matched the experimental XRD patterns and indexed the nanocrystal unit cell structure to monoclinic (COD ID: 4105682) with calculated lattice constants of a = 4.7 Å, b = 3.4 Å, and c = 5.2 Å.
Preparation, morphology, and crystal packing of magnesium hydroxide nanostructures
Magnesium hydroxide (Mg(OH)2) is widely used as the flame-retardant filler in composite materials40,76–79 as well as a precursor for the preparation of magnesium oxide.80 Owing to its properties, which include: (1) its ability to undergo endothermic dehydration in fire conditions, and (2) its ionic constituents consist of a relatively small number of electrons with stable oxidation states, magnesium oxide (MgO) exhibits less well-defined surface defect structures such as low coordinated ions and/or vacancies on the surface. Recent research advancements have utilized MgO as one of the preferred targets of surface structural analysis and catalytic research.81–83 Additionally, Mg(OH)2 and its oxide form have been widely used in toxic waste remediation, or as additives in refractory, paint, and superconductor products, and steel manufacturing as anti-corrosive coating materials.84–86The synthesis methods for the preparation of Mg(OH)2 with different morphologies have been demonstrated in the past.40,42,78,80,87–89 Most of these syntheses based on hydrothermal reactions of pure magnesium or magnesium oxide powder in an autoclave at higher temperature using longer reaction time and low reactant concentration.42,78,80 Although a few wet chemical synthesis methods were demonstrated to make nanoparticles,40,42 they are limited to one or two morphologies, relatively low yields, and economically disadvantages. A simple, versatile, and environmentally benign method to make specifically shape and size-controlled magnesium hydroxide nanoparticles is still in the exploratory stage. In particular, developing a sol–gel-based synthesis method combined with a solvent driven self-assembly process developed in this work is the first demonstration of making shape and size-controlled Mg(OH)2 hexagonal nanoplates.
The sol–gel synthesis developed to make nanostructures of Mn3O4 and CuO was applied to make Mg(OH)2 nanostructures in four different solvent systems in the presence of water. The effect of size and shape with respect to the solvent type as well as the precursor to the base concentration were evaluated. In a typical procedure, the sol–gel synthesis was performed by stirring the solution mixture at room temperature followed by gradually heating to 80 °C and let it stand at 80 °C for 24 h. The nanostructures prepared in this manner were characterized by powder XRD and morphologies were visualized from SEM and TEM. The crystallinity, packing pattern, and crystal unit cell structure of self-assembled nanostructures were also evaluated by SAED analysis and comparing the simulated XRD patterns with the experimental powder XRD patterns. The unit cell crystal structure and its cell parameters were acquired from the Open Crystallography Data Base.
The morphologies of the nanostructures along with their dimensionalities with respect to the experimental optimization parameters are summarized in Table 3. Mg(OH)2 nanostructures with two distinct shape-controlled morphologies were obtained with respect to all four different solvent systems at all three molar ratios. The hexagonal nanoplates, with an average diameter ranged from 30 nm to 170 nm were produced in water, DMF, and toluene at all three molar ratios of the precursor to the base. The solvent mixture of ethanol/water produced irregular shaped nanoparticles with average diameter ranged from 25 nm to 70 nm at all three different molar ratios. The average diameter of hexagonal plates produced in water exhibited similar size range (70 ± 30) at 1:5 and 1:10 molar ratios whereas the average particle diameter was increased to above 120 nm at the highest base concentration. At 1:5 molar ratio, hexagonal nanoplates obtained in toluene were the smallest size range and with the increase of base concentration, the average particle size tends to increase. The average diameter of hexagonal plates resulted from DMF at the lowest base concentration was rather smaller compared to the nanoplates produced at 1:10 molar ratio.
| Molar ratio Mn+:base |
Solvent type/water | Morphology (dimension/nm) |
|---|---|---|
| a Number of moles of the metal precursor was kept constant and number of moles of the base was varied to maintain precursor to base concentration at 1:5, 1:10, and 1:15. |
||
| 1:5 |
Water | Hexagonal plates (78 ± 39) |
| 70% ethanol | Nanoparticles (48 ± 22) | |
| DMF | Hexagonal plates (53 ± 13) | |
| Toluene | Hexagonal plates (45 ± 15) | |
| 1:10 |
Water | Hexagonal plates (71 ± 25) |
| 70% ethanol | Nanoparticles (44 ± 17) | |
| DMF | Hexagonal plates (136 ± 31) | |
| Toluene | Hexagonal plates (73 ± 49) | |
| 1:15 |
Water | Hexagonal plates (97 ± 24) |
| 70% ethanol | Nanoparticles (53 ± 13) | |
| DMF | Hexagonal plates (69 ± 20) | |
| Toluene | Hexagonal plates (67 ± 26) | |
Morphologies of nanostructures, analyzed from SEM and TEM after washing and re-dispersing in de-ionized water, are depicted in Fig. 5, S7 and S8.† With the base concentration increases, the shape of nanostructures formed in water tends to exhibit rather well-defined hexagonal nanoplates. However, at 1:10 molar ratio of the precursor to the base in water/toluene solvent mixture, the hexagonal nanoparticles exhibit well-defined edges with rather uniform size distribution compared to the hexagonal nanoplates formed in water and DMF systems. As we observed the similar effect in the formation of well-defined Mn3O4 hexagonal nanoparticles formed in water/toluene, toluene molecules act as surfactants to control the size and shape. However, in most of all cases, nanostructures show a wide size distribution at all three different base concentrations. At all three base concentrations, ethanol produced irregular nanoparticles while maintaining the particle diameter in the same size range. Perhaps, this may be due to the high solubility of the base in water/ethanol system as we observed in the previous two metal oxides nanoparticles formation. The TEM images further reveals the particles shape, confirming the hexagonal shape for nanoparticles formed in water, DMF, and toluene, and irregular nanoparticles formed in ethanol.
 | ||
| Fig. 5 SEM (top), TEM (down) and SAED pattern (inset) of Mg(OH)2 nanostructures formed in each solvent system at the molar ratio of precursor to base 1:5. | ||
The SAED patterns (Fig. 5-inset) confirm the crystallinity of hexagonal nanoplates with crystalline ring structure, evidencing self-assembled nanocrystals to form hexagonal nanoplates. The Fig. 6 represents the comparison TEM images along with their SAED pattern for hexagonal nanostructures formed in water and water/toluene mixture. With the increase in the base concentration, the nanostructures formed in water and water/toluene mixture show high intense diffraction ring patterns, confirming high crystalline nature of the nanostructures with the trend of ordered self-assembly of nanocrystals to form hexagonal nanoplates. For example, hexagonal nanoplates formed in water at 1:10 and 1:15 molar ratios show high intensity diffraction ring pattern. The SAED diffraction patterned obtained for the nanostructures formed in toluene/water mixture at 1:10 molar ratio, shows clear evidence of nanocrystals self-assembly to yield hexagonal nanoplates. The diffraction ring pattern of the hexagonal nanoplates formed in water/toluene mixture at 1:10 molar ratio also provides clear evidence for the hexagonal crystallographic characteristics of brucite as identified in the past literature.42
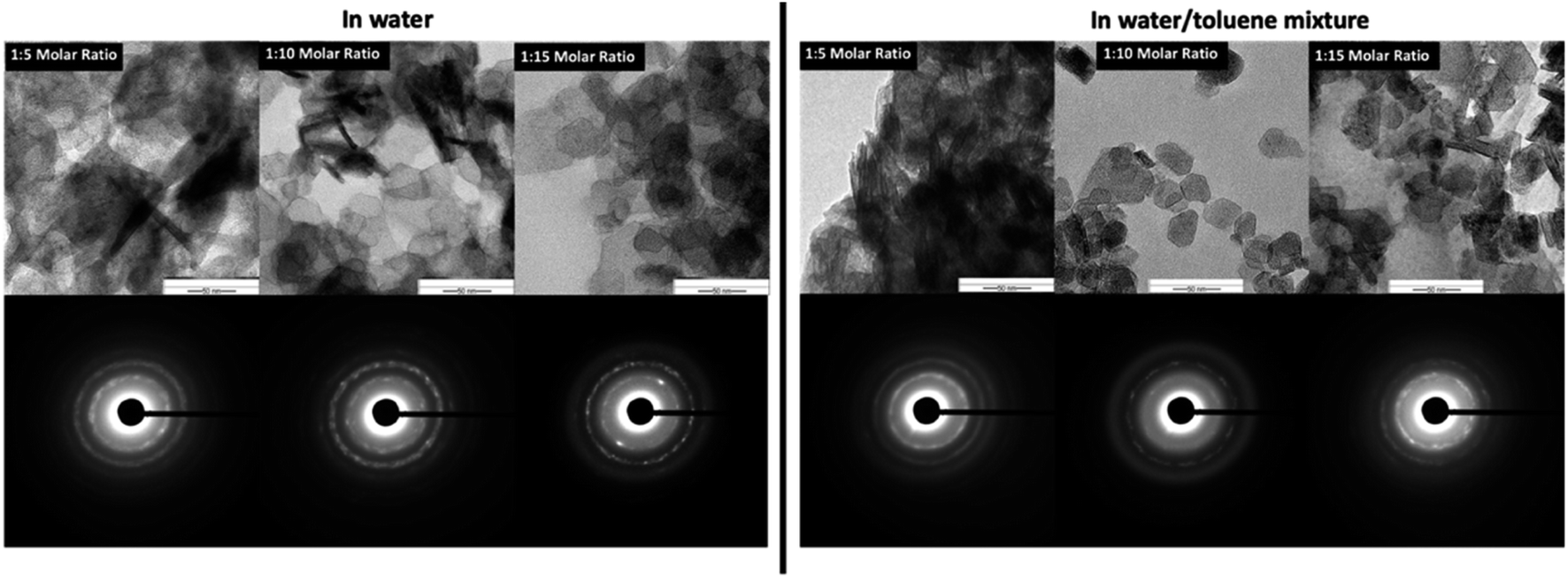 | ||
| Fig. 6 The comparison TEM Images and SAED patterns of hexagonal nanoplates produced in water and water/toluene mixture at three different base concentrations. | ||
The wide angle powder XRD traces of Mg(OH)2 nanostructures (Fig. 7 and S9†) exhibit three distinct well-defined peaks that correspond to the Bragg's reflection planes of [001], [101], and [102] with a low intensity shoulder peak for the [110] reflection plane. The diffraction patterns obtained for all four different solvent systems at all three different molar ratios of the precursor to the base exhibited these three most prominent Bragg's reflections. However, the intensity of [102] reflection plane for the nanostructures produced in ethanol is weak and the peak tends to be broader compare to that of for other solvents. This supports the formation of irregular nanoparticles in ethanol whereas formation of hexagonal nanoplates in all other three solvents. The simulated XRD pattern acquired from the crystallographic open data base (COD ID: 1000054), matched the experimental XRD patterns and indexed the nanocrystal unit cell structure to hexagonal with the lattice constants a = 3.1 Å, b = 3.1 Å, and c = 4.8 Å, in agreement with the previously reported data.42
 | ||
| Fig. 7 Left – Powder XRD traces (coloured) along with respective simulated XRD patterns (black colour) for Mg(OH)2 nanostructures formed in each solvent system at 1:5 molar ratio of precursor to base; right – respective Mg(OH)2 unit cell crystal structure (hexagonal structure, COD ID: 1000054) acquired from the Crystallographic Open Data base (COD). | ||
Nanoporosity, BET surface area, and BHJ pore size distribution analysis
As revealed from the TEM images of Mn3O4 hexagonal nanoparticles, CuO nanosheets, and Mg(OH)2 hexagonal nanoplates, the sol–gel synthesis method developed herein has produced mesoporous structures. The formation of mesoporous structures is unique to our synthesis approach, as it follows sol formation (nanocrystals) followed by self-assembly of nanocrystals in ordered manner prior to the nanocrystal growth. During this process, nanocrystals arrange into a particular order, directed by surface energies of crystal facets, leaving void spaces among nanocrystals. Although the reaction conditions are consistent in the preparation of all three types of oxides/hydroxides, the nanocrystals arrangement and their growth govern by the surface energies of crystal facets. Thus, the final nanostructure, with different crystal unit cell and crystal packing, exhibits mesoporosity, which processes variations in the pore volume. For example, most of the nanostructures formed in water and water/toluene show highly mesoporous morphologies (Fig. 8 – insets). In particularly, hexagonal nanoparticles of Mn3O4 formed in water at 1:10 molar ratio of the precursor to the base exhibit a wormlike mesoporous structure, having large, noticeable void spaces among nanocrystals in high density (Fig. 8(a) – inset). The thin sheets of CuO nanostructures prepared under same conditions exhibit uniformly distributed spherical nanopores but void spaces are smaller and evenly distributed (Fig. 8(b) – inset). In Mg(OH)2 nanostructures, hexagonal nanoplates prepared in toluene at 1:10 molar ratio of the precursor to the base also exhibit dense wormlike mesoporous surface with noticeable void spaces among nanocrystals (Fig. 8(c) – inset) and comparable to the mesoporous hexagonal nanoparticles of Mn3O4 formed in water and water/toluene.
 | ||
| Fig. 8 Nitrogen adsorption–desorption isotherms along with respective TEM images (in-set) for nanostructures of: (a) Mn3O4 prepared in water, (b) CuO prepared in water, and (c) Mg(OH)2 prepared in toluene by maintaining the molar ratio of the precursor to the base at 1:10. | ||
In order to evaluate the surface porosity distribution and the surface area of each nanostructure type, the nanoporosity, pore volume distribution, and surface area analysis were conducted utilizing Barret–Joyner–Halenda (BJH) and Barrett–Emmett–Teller (BET) analyses. The representative BET surface area results, BHJ pore volume, and average pore diameters are summarized in Table 4 and their BET adsorption–desorption linear isotherm plots are depicted in Fig. 8. The corresponding BJH pore size distribution curves for respective two metal oxides and the metal hydroxide are shown in Fig. S10.† As depicted in Fig. 8(a), the N2 isotherm of the Mn3O4 hexagonal nanoparticles shows a type III isotherm with a very narrow type H1 hysteresis loop. The shape of the type III isotherm with H1 loop evidences that agglomerates of nanocrystals of self-assembled hexagonal particles arranged in a rather uniform way with cylindrical pore geometry.89 The average pore diameter, as calculated by the BJH method from the desorption branch of the isotherm is 27.9 nm with the smallest nanopore size of 1.7 nm and the largest mesopores diameter of 48.5 nm. With the highest pore volume of 0.45 cm3 g−1 (see Fig. S11†), the BET surface area of Mn3O4 nanoparticles is 91.68 m2 g−1, which is the highest BET surface area compared to BET surface area of CuO and Mg(OH)2 nanostructures. The CuO nanosheets also produced the type III isotherm with similar H1 hysteresis behaviour (Fig. 8(b)), indicating distribution of cylindrical pore geometry. However, average pore diameter is smaller compared to the average pore diameter of Mn3O4 hexagonal nanoparticles. The pore diameter distribution of CuO ranges from nanopore diameter of 1.8 nm to mesopore diameter of 49.7 nm with average pore diameter of 14.7 nm. Since CuO nanosheets resulted the lowest pore volume of 0.098 cm3 g−1, nanosheets also yielded the lowest BET surface area of 18.31 m2 g−1. The hexagonal plates of Mg(OH)2 nanostructures made in toluene at 1:10 molar ratio of the precursor to the base also exhibit the type III isotherm but having the type H3 hysteresis loop.89 The H3 loop does not exhibit any limiting adsorption at high P/P°, evidencing aggregates of plate-like particles giving rise to slit-shaped pores. The porous Mg(OH)2 plates has a higher BET surface area of 72.31 m2 g−1, which is slightly lower than the surface area of Mn3O4 particles. The pore size distribution, as calculated by the BJH method from the desorption branch of the nitrogen isotherm, reveals that this material contains small mesopores with a pore size of 1.8 nm and large mesopores with a pore size of 39.4 nm. The resulting average pore diameter was found to be 14.88 nm.
:10 molar ratio of the precursor to the base
| BET analysis | Mn3O4 1:10 in water |
CuO 1:10 in water |
Mg(OH)2 1:10 in toluene |
|---|---|---|---|
| BET surface area (m2 g−1) | 91.68 | 18.31 | 72.31 |
| Single point desorption total pore volume (cm3 g−1) | 0.45 | 0.09 | 0.29 |
| BJH desorption average pore diameter (nm) | 27.92 | 14.68 | 12.62 |
| BJH adsorption average pore diameter (nm) | 28.84 | 16.73 | 14.88 |
Overall, the variation in desorption pore volume and BET surface area of Mn3O4, CuO, and Mg(OH)2 nanostructures show direct correlation to the sol–gel synthesis method, where nanocrystals self-assembled, creating void spaces among nanocrystals, and the crystal growth is governed by surface energies of crystal facets to yield mesoporous structures. Moreover, the high mesoporosity with a combination of independently controlled, well-connected smaller and larger mesopore distribution, observed, specially, in Mn3O4 and Mg(OH)2 nanostructures could result higher activities and better controlled selectivity in catalysis reactions, providing potential nanostructure platforms for catalysis applications.
Experimental
Materials
N,N-Dimethylformamide (anhydrous, 99.8%) was obtained from Sigma-Aldrich chemicals. Ethanol (Reagent Alcohol, 140 Proof), toluene, magnesium(II)chloride hexahydrate, manganese(II)acetate (anhydrous), and sodium hydroxide pellets were obtained from Fisher Scientific. Copper(II) acetate monohydrate was obtained from Alfa Aesar. All chemicals were used as received.Characterization
Scanning Electron Microscopy (SEM) was performed using Zeiss Auriga FIBFESEM and a Hitachi S-4800 FESEM. The typical imaging accelerating voltage was 3 kV with a working distance of 4 mm and 5 kV and a working distance of 5 mm respectively unless otherwise specified. EDS analysis was done using the Zeiss Auriga FIBFESEM with the Working Distance set to 6 mm. Transmission Electron Microscopy (TEM) was performed using Zeiss Libra 120 TEM. Powder X-ray Diffraction (XRD) was performed using the Agilent Gemini XRD; a Mo Kα X-ray source was used operating with a voltage and current of 40 kV and 40 mA respectively. Nitrogen adsorption measurements were performed at 77 K, using an automatic volumetric adsorption equipment obtained from Micromeritics utilizing Barrett–Emmett–Teller (BET) calculations for surface area and BJH calculations for pore size distribution for the desorption branch of the isotherm.The typical procedure for the preparation of metal oxide nanostructures
Reactions were run in 250 mL round bottom flasks in an oil bath under ambient atmosphere. Each metal oxide nanomaterial was synthesized in four different solvents: de-ionized H2O, 70% EtOH, DMF and toluene. A metal precursor was prepared using one of the following for each nanomaterial: magnesium chloride (1.20 g, 13.0 mmol), manganese acetate (1.20 g, 6.90 mmol) and copper acetate (1.2 g, 9.80 mmol). In a typical procedure, a solvent (160 mL) was added to the flask. A metal precursor was dissolved in de-ionized water (10.0 mL) and added to the reaction flask. Sodium hydroxide (NaOH) pellets were dissolved in de-ionized water (10 mL) and was added dropwise at a rate of 10.0 mL min−1 to the reaction flask and stirred for another 5 min. The molar ratio of the precursor to NaOH was maintained at 1:5, 1:10 and 1:15. After stirring 5 min at room temperature, the stir-bar was removed, and the flask was put in a non-heated oil bath at room temperature. Before heating the bath boiling stones were added to the flask to prevent bumping. The flask was then gradually heated to 80 °C and left for 24 hours. Then the reaction flask was allowed to cool down to room temperature over a period of several hours. The colloidal solution was transferred into centrifuge tubes and centrifuged at a rate of 10000 rpm for 10 minutes, decanted the liquid, and washed the product with de-ionized water. The washing step was repeated several times and the final product collected in this manner was freeze dried at 0.140 mBar at −50 °C for 24 hours. The dried powder was analyzed using SEM, TEM, and XRD. All SEM samples were prepared by sonicating the nanomaterial powder in de-ionized water and then drop casting the solution onto a silicon wafer. All SEM samples are sputtered coated using 5 nm Gold/Palladium to ground the sample. The TEM samples were prepared by dropping the sample onto a TEM grid coated with Formvar and left to air dry before imaging. The powder XRD was performed by suspending the powder on a glass fibber using vacuum grease.
Conclusions
In summary, a versatile sol–gel synthesis, combined with low-temperature solvothermal process allows us to make highly crystalline, mesoporous binary (II–VI) metal oxide nanostructures of Mn3O4 and CuO and metal hydroxide nanostructures of Mg(OH)2 with controlled-morphologies. The hexagonal nanoparticles of Mn3O4 with an average diameter ranged from 100 to 200 nm were yielded in water, water/ethanol, and water/toluene solvent mixtures and evidenced that there was no solvent polarity effect on the crystal growth with respect to each organic solvent. The SAED patterns of Mn3O4 nanostructures confirmed the high crystallinity of self-assembled nanocrystals and in some cases reflects polycrystalline in nature where nanocrystals could arrange ordered and dis-ordered orientations in different solvent systems. The experimental powder XRD patterns indexed the nanocrystal unit cell structure to triclinic with unit cell parameters of a = b = 5.76 Å, and c = 9.43 Å. The hexagonal nanoparticles with highly mesoporous morphologies produced in water at 1:10 molar ratio of the precursor to the base exhibited the highest BET surface area of 91.68 m2 g−1 and BJH desorption average pore diameter of ∼28 nm. Augmenting the same sol–gel process, the CuO nanostructures of nanoporous thin sheets, nanoparticles, rods, and ribbons were prepared in different solvent systems. The simulated XRD pattern confirmed that the experimental XRD patterns of all types of CuO nanostructures were indexed the nanocrystal unit cell structure to monoclinic. The mesoporosity of nanosheets, prepared in water at 1:10 molar ratio of the precursor to the base, yielded the smallest desorption total pore volume of 0.09 cm3 g−1, with the lowest BET surface area of 18.31 m2 g−1 and the BHJ desorption average pore diameter of ∼16 nm. The sol of magnesium hydroxide nanocrystals self-assembled to produce highly nanoporous hexagonal nanoplates in water and water/toluene solvent systems. The wide angel powder XRD patterns showed well-defined Bragg's peaks, indexing to hexagonal unit cell structure. The hexagonal nanoplates produced in water/toluene system show rather well-defined edges with worm-like porous morphologies, which were very similar to mesoporous Mn3O4 hexagonal nanoparticles produced in water and water/toluene. These hexagonal plates exhibited a high BET surface area of 72.31 m2 g−1, which is slightly lower than the surface area of Mn3O4 hexagonal nanoparticles.
In overall, we demonstrated that the sol–gel approach developed herein is rather advantageous for the preparation of highly mesoporous binary and ternary metal oxides nanostructures, in terms of scalability, processability, and reliability compared to other wet-chemical and hydrothermal fabrication processes, that utilize high temperatures and highly toxic hydrazine-based solvents and additives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is performed at the Joint School of Nanoscience and Nanoengineering, a member of the Southeastern Nanotechnology Infrastructure Corridor (SENIC) and National Nanotechnology Coordinated Infrastructure (NNCI), supported by the NSF (Grant ECCS-1542174). Financial support for this work is provided, in part, from the Joint School of Nanoscience and Nanoengineering and the Office of Research, University of North Carolina at Greensboro. The authors also gratefully acknowledge Dr Kyle Nowlin for SEM and TEM support, and Mr Nafeezuddin Mohammad for BET analysis.References
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- H. Rathnayake, J. Binion, A. McKee, D. J. Scardino and N. I. Hammer, Nanoscale, 2012, 4, 4631 RSC.
- H. Rathnayake, N. Wright, A. Patel, J. Binion, L. E. McNamara, D. J. Scardino and N. I. Hammer, Nanoscale, 2013, 5, 3212 RSC.
- R. Sui and P. Charpentier, Chem. Rev., 2012, 112, 3057–3082 CrossRef CAS PubMed.
- N. Baccile, F. Babonneau, B. Thomas and T. Coradin, J. Mater. Chem., 2009, 19, 8537–8559 RSC.
- Z. Zhang, J. Liu, J. Gu, L. Su and L. Cheng, Energy Environ. Sci., 2014, 7, 2535–2558 RSC.
- G. Oskam, J. Sol-Gel Sci. Technol., 2006, 37, 161–164 CrossRef CAS.
- M. Zhang, Y. Bando and K. Wada, J. Mater. Sci. Lett., 2001, 20, 167–170 CrossRef CAS.
- K. Woo, H. J. Lee, J.-P. Ahn and Y. S. Park, Adv. Mater., 2003, 15, 1761–1764 CrossRef CAS.
- M. Kashif, S. M. Usman Ali, M. E. Ali, H. I. Abdulgafour, U. Hashim, M. Willander and Z. Hassan, Phys. Status Solidi A, 2012, 209, 143–147 CrossRef CAS.
- J. Li, Y. Wu, M. Yang, Y. Yuan, W. Yin, Q. Peng, Y. Li and X. He, J. Am. Ceram. Soc., 2017, 100, 5460–5470 CrossRef CAS.
- S. Maensiri, W. Nuansing, J. Klinkaewnarong, P. Laokul and J. Khemprasit, J. Colloid Interface Sci., 2006, 297, 578–583 CrossRef CAS PubMed.
- M. A. Cauqui and J. M. Rodríguez-Izquierdo, J. Non-Cryst. Solids, 1992, 147–148, 724–738 CrossRef CAS.
- D. A. Ward and E. I. Ko, Ind. Eng. Chem. Res., 1995, 34, 421–433 CrossRef CAS.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- J. Zha and H. Roggendorf, Adv. Mater., 1991, 3, 522 CrossRef.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- The Sol-Gel handbook: synthesis, characterization, and applications, ed. D. Levy and M. Zayat, Wiley-VCH, Weinheim, 2015 Search PubMed.
- R. Roy, Science, 1987, 238, 1664–1669 CrossRef CAS PubMed.
- V. G. Kessler, J. Sol-Gel Sci. Technol., 2009, 51, 264–271 CrossRef CAS.
- A. Vioux and D. Leclercq, Heterog. Chem. Rev., 1996, 3, 65–73 CrossRef CAS.
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- C. B. Tay, S. J. Chua and K. P. Loh, J. Cryst. Growth, 2009, 311, 1278–1284 CrossRef CAS.
- K. Kim, K. Utashiro, Y. Abe and M. Kawamura, Materials, 2014, 7, 2522–2533 CrossRef PubMed.
- T.-Z. Ren, Z.-Y. Yuan, W. Hu and X. Zou, Microporous Mesoporous Mater., 2008, 112, 467–473 CrossRef CAS.
- J. Du, Y. Gao, L. Chai, G. Zou, Y. Li and Y. Qian, Nanotechnology, 2006, 17, 4923–4928 CrossRef CAS.
- A. Vázquez-Olmos, R. Redón, G. Rodríguez-Gattorno, M. Esther Mata-Zamora, F. Morales-Leal, A. L. Fernández-Osorio and J. M. Saniger, J. Colloid Interface Sci., 2005, 291, 175–180 CrossRef PubMed.
- N. Topnani, S. Kushwaha and T. Athar, Int. J. Green Nanotechnol., 2010, 1, M67–M73 CrossRef.
- T. Ozkaya, A. Baykal, H. Kavas, Y. Köseoğlu and M. S. Toprak, Phys. B, 2008, 403, 3760–3764 CrossRef CAS.
- M. Yin, C.-K. Wu, Y. Lou, C. Burda, J. T. Koberstein, Y. Zhu and S. O'Brien, J. Am. Chem. Soc., 2005, 127, 9506–9511 CrossRef CAS PubMed.
- W. Jisen, Y. Jinkai, S. Jinquan and B. Ying, Mater. Des., 2004, 25, 625–629 CrossRef.
- Z. N. Kayani, M. Umer, S. Riaz and S. Naseem, J. Electron. Mater., 2015, 44, 3704–3709 CrossRef CAS.
- A. Ananth, S. Dharaneedharan, M.-S. Heo and Y. S. Mok, Chem. Eng. J., 2015, 262, 179–188 CrossRef CAS.
- X. Liu, S. Cui, Z. Sun and P. Du, Electrochim. Acta, 2015, 160, 202–208 CrossRef CAS.
- J. Lv, L. Qiu and B. Qu, J. Cryst. Growth, 2004, 267, 676–684 CrossRef.
- D. An, L. Wang, Y. Zheng, S. Guan, X. Gao, Y. Tian, H. Zhang, Z. Wang and Y. Liu, Colloids Surf., A, 2009, 348, 9–13 CrossRef CAS.
- D. Chen, L. Zhu, H. Zhang, K. Xu and M. Chen, Mater. Chem. Phys., 2008, 109, 224–229 CrossRef CAS.
- C. Henrist, J.-P. Mathieu, C. Vogels, A. Rulmont and R. Cloots, J. Cryst. Growth, 2003, 249, 321–330 CrossRef CAS.
- R. Giorgi, C. Bozzi, L. Dei, C. Gabbiani, B. W. Ninham and P. Baglioni, Langmuir, 2005, 21, 8495–8501 CrossRef CAS PubMed.
- J. C. Yu, A. Xu, L. Zhang, R. Song and L. Wu, J. Phys. Chem. B, 2004, 108, 64–70 CrossRef CAS.
- C. Y. Tai, C.-T. Tai, M.-H. Chang and H.-S. Liu, Ind. Eng. Chem. Res., 2007, 46, 5536–5541 CrossRef CAS.
- G. Song, S. Ma, G. Tang and X. Wang, Colloids Surf., A, 2010, 364, 99–104 CrossRef CAS.
- O. Baidukova and E. V. Skorb, Ultrason. Sonochem., 2016, 31, 423–428 CrossRef CAS PubMed.
- E. d Stobbe, B. A. De Boer and J. W. Geus, Catal. Today, 1999, 47, 161–167 CrossRef.
- E. J. Grootendorst, Y. Verbeek and V. Ponec, J. Catal., 1995, 157, 706–712 CrossRef CAS.
- M. Baldi, E. Finocchio, F. Milella and G. Busca, Appl. Catal., B, 1998, 16, 43–51 CrossRef CAS.
- L. Sánchez, J. Farcy, J.-P. Pereira-Ramos, L. Hernán, J. Morales and J. L. Tirado, J. Mater. Chem., 1996, 6, 37–39 RSC.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- V. V. Pankov, M. Pernet, P. Germi and P. Mollard, J. Magn. Magn. Mater., 1993, 120, 69–72 CrossRef CAS.
- R. Ramachandran, J. Mater. Sci.: Mater. Electron., 2002, 13, 257–262 CrossRef CAS.
- J. C. Southard and G. E. Moore, J. Am. Chem. Soc., 1942, 64, 1769–1770 CrossRef CAS.
- C. H. Shomate, J. Am. Chem. Soc., 1943, 65, 785–790 CrossRef CAS.
- I. Ursu, R. Alexandrescu, I. N. Mihailescu, I. Morjan, V. Jianu and C. Popescu, J. Phys. B: At. Mol. Phys., 1986, 19, L825 CrossRef CAS.
- W. S. Kijlstra, J. C. M. L. Daamen, J. M. van de Graaf, B. van der Linden, E. K. Poels and A. Bliek, Appl. Catal., B, 1996, 7, 337–357 CrossRef CAS.
- E. Mendelovici and A. Sagarzazu, Thermochim. Acta, 1988, 133, 93–100 CrossRef CAS.
- E. Finocchio and G. Busca, Catal. Today, 2001, 70, 213–225 CrossRef CAS.
- G. Demazeau, J. Mater. Chem., 1999, 9, 15–18 RSC.
- Y. C. Zhang, T. Qiao and X. Ya Hu, J. Solid State Chem., 2004, 177, 4093–4097 CrossRef CAS.
- Z. Yang, Y. Zhang, W. Zhang, X. Wang, Y. Qian, X. Wen and S. Yang, J. Solid State Chem., 2006, 179, 679–684 CrossRef CAS.
- W. S. Seo, H. H. Jo, K. Lee, B. Kim, S. J. Oh and J. T. Park, Angew. Chem., Int. Ed., 2004, 43, 1115–1117 CrossRef CAS PubMed.
- S. Lei, K. Tang, Z. Fang and H. Zheng, Cryst. Growth Des., 2006, 6, 1757–1760 CrossRef CAS.
- H. Antoni, W. Xia, J. Masa, W. Schuhmann and M. Muhler, Phys. Chem. Chem. Phys., 2017, 19, 18434–18442 RSC.
- J. Zhang, F. Huang and Z. Lin, Nanoscale, 2010, 2, 18–34 RSC.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 1188–1191 CrossRef CAS PubMed.
- G. H. Du and G. Van Tendeloo, Chem. Phys. Lett., 2004, 393, 64–69 CrossRef CAS.
- L. S. Huang, S. G. Yang, T. Li, B. X. Gu, Y. W. Du, Y. N. Lu and S. Z. Shi, J. Cryst. Growth, 2004, 260, 130–135 CrossRef CAS.
- K. Mageshwari and R. Sathyamoorthy, J. Mater. Sci. Technol., 2013, 29, 909–914 CrossRef CAS.
- D. P. Dubal, G. S. Gund, C. D. Lokhande and R. Holze, Mater. Res. Bull., 2013, 48, 923–928 CrossRef CAS.
- J. Y. Xiang, J. P. Tu, L. Zhang, Y. Zhou, X. L. Wang and S. J. Shi, J. Power Sources, 2010, 195, 313–319 CrossRef CAS.
- M. Cao, C. Hu, Y. Wang, Y. Guo, C. Guo and E. Wang, Chem. Commun., 2003, 1884 RSC.
- W.-T. Yao, S.-H. Yu, Y. Zhou, J. Jiang, Q.-S. Wu, L. Zhang and J. Jiang, J. Phys. Chem. B, 2005, 109, 14011–14016 CrossRef CAS PubMed.
- R. A. Zarate, F. Hevia, S. Fuentes, V. M. Fuenzalida and A. Zúñiga, J. Solid State Chem., 2007, 180, 1464–1469 CrossRef CAS.
- A. Bello, D. Dodoo-Arhin, K. Makgopa, M. Fabiane and N. Manyala, Am. J. Mater. Sci., 2014, 4, 64–73 Search PubMed.
- P. A. van Aken and F. Langenhorst, Eur. J. Mineral., 2001, 13, 329–341 CrossRef CAS.
- Y. Ding, G. Zhang, H. Wu, B. Hai, L. Wang and Y. Qian, Chem. Mater., 2001, 13, 435–440 CrossRef CAS.
- S. Utamapanya, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1991, 3, 175–181 CrossRef CAS.
- A. Durin-France, L. Ferry, J.-M. Lopez Cuesta and A. Crespy, Polym. Int., 2000, 49, 1101–1105 CrossRef CAS.
- Y. Li, M. Sui, Y. Ding, G. Zhang, J. Zhuang and C. Wang, Adv. Mater., 2000, 12, 818–821 CrossRef CAS.
- O. Diwald and E. Knözinger, J. Phys. Chem. B, 2002, 106, 3495–3502 CrossRef CAS.
- M. Anpo, Y. Yamada, Y. Kubokawa, S. Coluccia, A. Zecchina and M. Che, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 751–764 RSC.
- D. M. Murphy, R. D. Farley, I. J. Purnell, C. C. Rowlands, A. R. Yacob, M. C. Paganini and E. Giamello, J. Phys. Chem. B, 1999, 103, 1944–1953 CrossRef CAS.
- R. Richards, R. S. Mulukutla, I. Mishakov, V. Chesnokov, A. Volodin, V. Zaikovski, N. Sun and K. J. Klabunde, Scr. Mater., 2001, 44, 1663–1666 CrossRef CAS.
- S. Rajagopalan, O. Koper, S. Decker and K. J. Klabunde, Chem.–Eur. J., 2002, 8, 2602–2607 CrossRef CAS.
- B. M. Choudary, R. S. Mulukutla and K. J. Klabunde, J. Am. Chem. Soc., 2003, 125, 2020–2021 CrossRef CAS PubMed.
- R. C. Xie and B. J. Qu, CN Pat., CN1359853A, 2002.
- M. Alexandre, G. Beyer, C. Henrist, R. Cloots, A. Rulmont, R. Jérôme and P. Dubois, Macromol. Rapid Commun., 2001, 22, 643–646 CrossRef CAS.
- M. E. Spahr, P. Bitterli and R. Nesper, Angew. Chem., Int. Ed., 1998, 37, 1263 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SEM and TEM images, XRD powder diffraction traces and BET analysis data. See DOI: 10.1039/d0ra01778g |
| This journal is © The Royal Society of Chemistry 2020 |