
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of Cu(0) encapsulated nanocatalysts as superior catalytic systems in Cu-catalyzed organic transformations
Majid M. Heravi *,
Bahareh Heidari,
Vahideh Zadsirjan* and
Leila Mohammadi
*,
Bahareh Heidari,
Vahideh Zadsirjan* and
Leila Mohammadi
Department of Chemistry, School of Science, Alzahra University, P. O. Box 1993891176, Vanak, Tehran, Iran. E-mail: mmheravi@alzahra.ac.ir; mmh1331@yahoo.com; z_zadsirjan@yahoo.com; Fax: +98 21 88041344; Tel: +98 21 88044051
First published on 1st July 2020
Abstract
Recently, Cu nanoparticles (NPs) encapsulated into various materials as supports (e.g., zeolite, silica) have attracted much devotion due to their unique catalytic properties such as high catalytic activity, intensive reactivity and selectivity through highly protective properties. Nowadays, the superior catalytic activity of Cu-NPs, encapsulated onto zeolite, silica and different porous systems, is extensively investigated and now well-established. As a matter of fact, Cu-NPs are protected from deactivation by this kind of encapsulation. Thus, their exclusion proceeds smoothly, and their recyclability is significantly increased. Cu-NPs have been used as potential heterogeneous catalysts in different chemical transformations. In this review, we try to show the preparation and applications of Cu(0) encapsulated nanocatalysts in zeolite and silica as superior catalytic systems in Cu-catalyzed organic transformations. In addition, the catalytic activity of these encapsulated Cu-NPs in different important organic transformations (such as hydrogenation, oxidation and carbon–carbon bond formations) are compared with those of a variety of organic, inorganic and hybrid porous bearing a traded metal ion. Moreover, the results from the TGA/DTA analysis and optical properties of Cu-complexes are demonstrated. The inherited characteristic merits of the encapsulated Cu-NPs onto zeolite and silica, such as their low leaching, catalytic activity, reusability economic feasibility and originality are critically considered.
1. Introduction
The art of organic synthesis has overgrown a captivating transformation over the past few decades. The applications of various transition-metal species as effective catalysts in catalytic processes have permitted the rapid and effective synthesis of both simple and structurally complex molecules. This is achieved using commercially available or easily accessible starting materials under mild and green reaction conditions.1 The importance of transition-metal catalysis on the art of organic synthesis may be evaluated by the broad range and wide spectrum of molecules that have been synthesized. These molecules extend from simple ones, such as biaryls, to rather complex pharmaceutical intermediates, medication, and very complex structures.2 Transition-metal-catalyzed catalysis has been divided into two major classes. They are homogeneous and heterogeneous catalysis, and both have their own merits and drawbacks. This classification, especially in practice, has given researchers in both academia and industry an opportunity to expand their choices.2b,3 Homogeneous catalysis has been developed as an influential synthetic tool due to the well-definite nature of the catalysts. Thus, the catalytic processes can be studied from mechanistic and kinetic points of view. These developments have led the wide variety of commercial processes to use homogeneous catalysts as the chief catalytic species.4 In this regard, several industrial processes, such as, the important Ziegler–Natta polymerization, Wacker process, the Monsanto carbonylation process hydroformylation (Otto Roelen oxo process) and the SHOP process (Shell higher olefin process), use homogeneous catalysis.4c,d,5 The main drawback related to homogeneous catalysis is the problem of catalyst separation thus, an improvement strategy to circumvent this problem was found by anchoring the catalytic species to a solid support.6 A literature survey disclosed a plethora of reports over the past decade, underscoring the commanding nature of homogeneous catalysis in catalyzing such transformations.7 Transition metal complexes, having unique physio-chemical and structural properties, showed high catalytic activities under mild reaction conditions. Although they have been used as oxidizing agents of organic compounds by alchemists in the early age of chemistry as a science, they have been found to be useful as selective oxidation catalysts in the past five decades. Thus, transition metal complexes have attracted much interest from the chemical community.8 Cu(II), in particular, is well known as an appropriate and powerful oxidizing agent for various organic substrates. They show the merits of high selectivity. Thus, they can be considered as mild and compatible oxidizing agents, being found to exist in the 0, 1+, 2+, and 3+ oxidation states.9 The catalytic-selective oxidation reaction of aromatic compounds in the presence of heterogeneous and reusable catalysts under mild (nearby ambient) and green reaction conditions using “clean oxidants”, such as molecular-oxygen and hydrogen peroxide, has been an overgrowing field of investigation in recent years.10 The oxidation and hydroxylation reactions of aromatic compounds by O2 extensively take place in nature. In this regard, the homogeneous copper-(N-base)-O2 systems have been selected as a model for biological processes of Cu-bearing proteins, including tyrosinase and hemocyanin.11 For example, the copper–pyridine–O2 system, has been found as an active homogeneous catalyst for the efficient oxidative coupling of aromatic acetylenes,12 phenols13 and amines.14 In recent years, different homogeneous catalysts have been converted to their heterogeneous counterparts with the well-known advantages over homogeneous catalysts, such as the ease of separation from the reaction mixture, effective reusability for several times and being utilized in continuous flow processes.15 It is worth mentioning that heterogeneous catalysts often show limited activity and selectivity.16 Different methods for the heterogenization of homogeneous catalytic systems to render the catalyst and the product(s) are known. The homogeneous catalyst can be immobilized onto different solid supports.17 In general, immobilization comprises the interaction of two entities known as the biomaterial (as an immobilizer) and an immobilization agent. Various immobilization methods are expected to have a bright future in the field of pharmacy, medicine or industry. Biocatalyst re-usage will significantly decrease the costs. Immobilization in nanodelivery systems may soon dominate the field of medical sciences. The vast problem of civilization diseases is the difficulty in the administration of medication, either due to the size of the drug or the inaccessibility of the treated site. The method chosen for immobilization depends on the suitability of both entities to react, and it forms a stable and appropriate immobilized phase. Characteristically, the biomaterials employed as an immobilizer are an insoluble matrix, which is inactive with the neighboring and also its immobilized agent. An immobilized system is provided to attain its specific objective, for example, to achieve a supporting immobilization, to enhance agent reusability, purification, process control or as sequential immobilization either to enhance the rate of release or prolong it.18 Different immobilization methods were developed and reported.17a These techniques were classified into two main distinct physical and chemical immobilizations. This classification is based on its preparation method. Nevertheless, nowadays, immobilization can be achieved by combining multiple disparities of immobilizations.1.1. Supporting materials
Zeolites are also useful as catalysts, sorbents and solid supports. Their well-defined pore structure and variable acidity make them very active catalysts in the catalysis of a wide range of organic transformations.31 Zeolites are crystalline microporous aluminosilicates, which are made from corner-sharing SiO4- and AlO4-tetrahedra. These porous materials are generated in nature as a result of volcanic activity. Nowadays, a collection of zeolites can be easily prepared in the standard chemistry laboratory. To date, about 300 different kinds of zeolites have been prepared and fully characterized. The homogeneous Cu-pyridine system has been immobilized onto zeolites, converting it into a heterogeneous catalyst with the same active species. The intracrystalline cages or channel structures of zeolites were prepared. After full characterization, they were successfully employed as a heterogeneous catalyst in organic transformation.32
Moreover, the mechanically stable beaded gel resins can be used as support. Several metal catalysts have been immobilized onto resins, and polymers were used as heterogeneous catalysts in different chemical transformations. This led to the selective oxidation of alcohols into the corresponding carbonyl compounds,33 and the construction of different heterocyclic systems.34 Different metal nanoparticles (MNPs) can also be loaded to different supports to effectively act as heterogeneous catalysts.35 In such heterogeneous catalysts, the metal particle size and oxidation state are considered for selecting an appropriate oxidation catalyst. Furthermore, it is well-recognized that the reactivity and selectivity of monodisperse metal NPs can be improved by changes in their size and shape.36
Yuji and co-workers successfully fixed a Cu-pyridine derivative complex on silica. It was subjected to a reversible redox cycle between copper(II) and copper(I), and applied successfully as a catalyst for the oxidative coupling reaction of methylacetylene.12,37 The design and improvement of an eco-friendly catalytic method is of urgent significance. The immobilization of homogeneous metal complexes on solid supports (including alumina, silica and polymers) has been investigated widely in order to provide heterogeneous catalysts with the desirable structure to act effectively, and show other advantages expected from a heterogeneous catalysts.
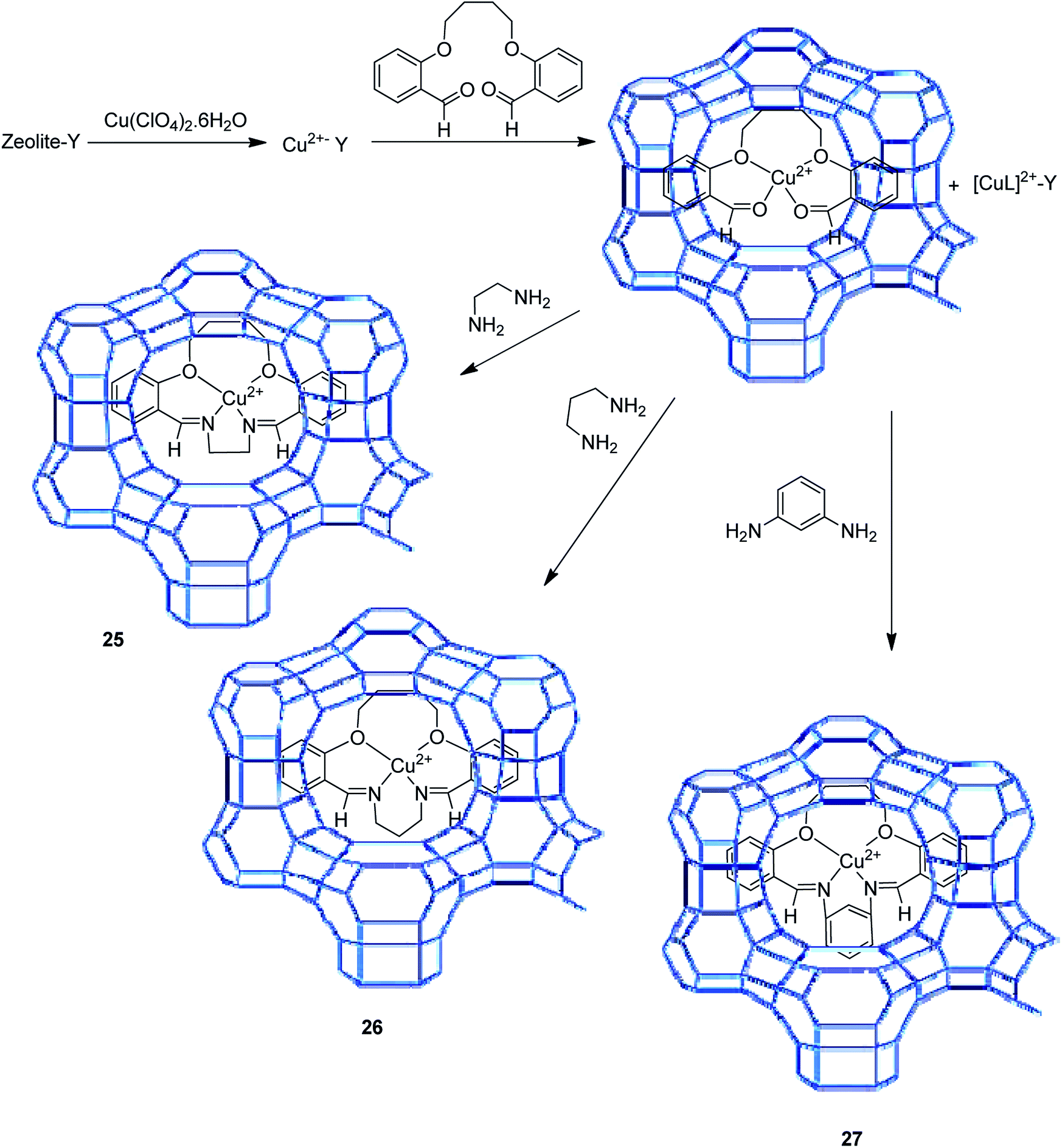
2. Applications of Cu(0) encapsulated nanocatalysts as superior catalytic systems in Cu-catalyzed organic transformations
2.1. Zeolite-encapsulated Cu NPs
Notably, the substitution of the aromatic hydrogen atoms in the salen ligand with electron-withdrawing groups (including –Br, –Cl, and NO2 groups) have two main influences: the retention and concentration of the Cu complex in the zeolite cavities are improved (due to the larger size of the groups), and the spectral properties of the encapsulated complex are also changed. Remarkably, the catalytic activity of the encapsulated copper salen in the decomposition of hydrogen peroxide, tert-butyl hydroperoxide (TBHP) and also the peroxidative oxidation of styrene, phenol and para-xylene were examined. For the oxidation of styrene under this condition, the major products benzaldehyde, phenyl acetaldehyde and styrene oxide were provided with the minor quantities of benzene-1,2-ethane diol and benzoic acid. In addition, hydrogen peroxide catalyzed the peroxidative oxidation of phenol (to hydroquinone and catechol), which is related linearly (major) to the catalytic efficacy of the encapsulated copper salens. The major products of oxidation of p-xylene are p-toluic acid, p-toluyalcohol and also p-tolualdehyde (Table 1).
| Catalyst | p-Xylene conversion (%) | TOF (h−1) | Products (wt%) | |||
|---|---|---|---|---|---|---|
| p-Toluic acid | p-Tolualdehyde | p-Toluyalcohol | 4-Carboxy benzyl alcohol, 4-carboxy benzaldehyde and traces of terephthalic acid | |||
| a Reaction conditions: p-xylene, catalyst, 0.5 g TBHP, 403 K, 18 hours. | ||||||
| CuSal-X | 45.1 | 182![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 285 285 |
46.3 | 29 | 13.5 | 11.1 |
| CuCl2Sal-X | 48.9 | 92200 |
37.8 | 33.3 | 14.7 | 14.1 |
| CuBr2Sal-X | 47.9 | 28829 |
39.6 | 35 | 13.3 | 11.9 |
| Cu(NO2)2Sal-X | 50.9 | 5238 | 38.7 | 29.6 | 19.4 | 12.2 |
The zeolite-Y encapsulated metal complex, [Cu(sal-dach)]-Y, was synthesized by the reaction of N,N-bis(salicylidene)cyclohexane-1,2-diamine (H2sal-dach) and Cu(II) exchanged zeolite-Y under reflux condition in methanol. Outstandingly, this encapsulated complex catalyzed the oxidation reaction of styrene, cyclohexane and cyclohexene using H2O2 (ref. 48) (Schemes 1 and 2). Under the optimal conditions, the oxidation reaction of styrene catalyzed by (1) gave 21.7% conversion with four products, including benzaldehyde (69.0%), styrene oxide (12.9%), phenyl acetaldehyde (12.0%) and benzoic acid (7.1%). Based on this method, the conversion of cyclohexene to cyclohexene oxide, 2-cyclohexene-1-one, 2-cyclohexene-1-ol, and cyclohexane-1,2-diol as major products with [Cu(sal-dach)]-Y was 18.1%. A maximum of 78.1% of cyclohexane with [Cu(sal-dach)]-Y was produced in which the selectivity of three main products follows the sequence: cyclohexanol > cyclohexane-1,2-diol > cyclohexanone.
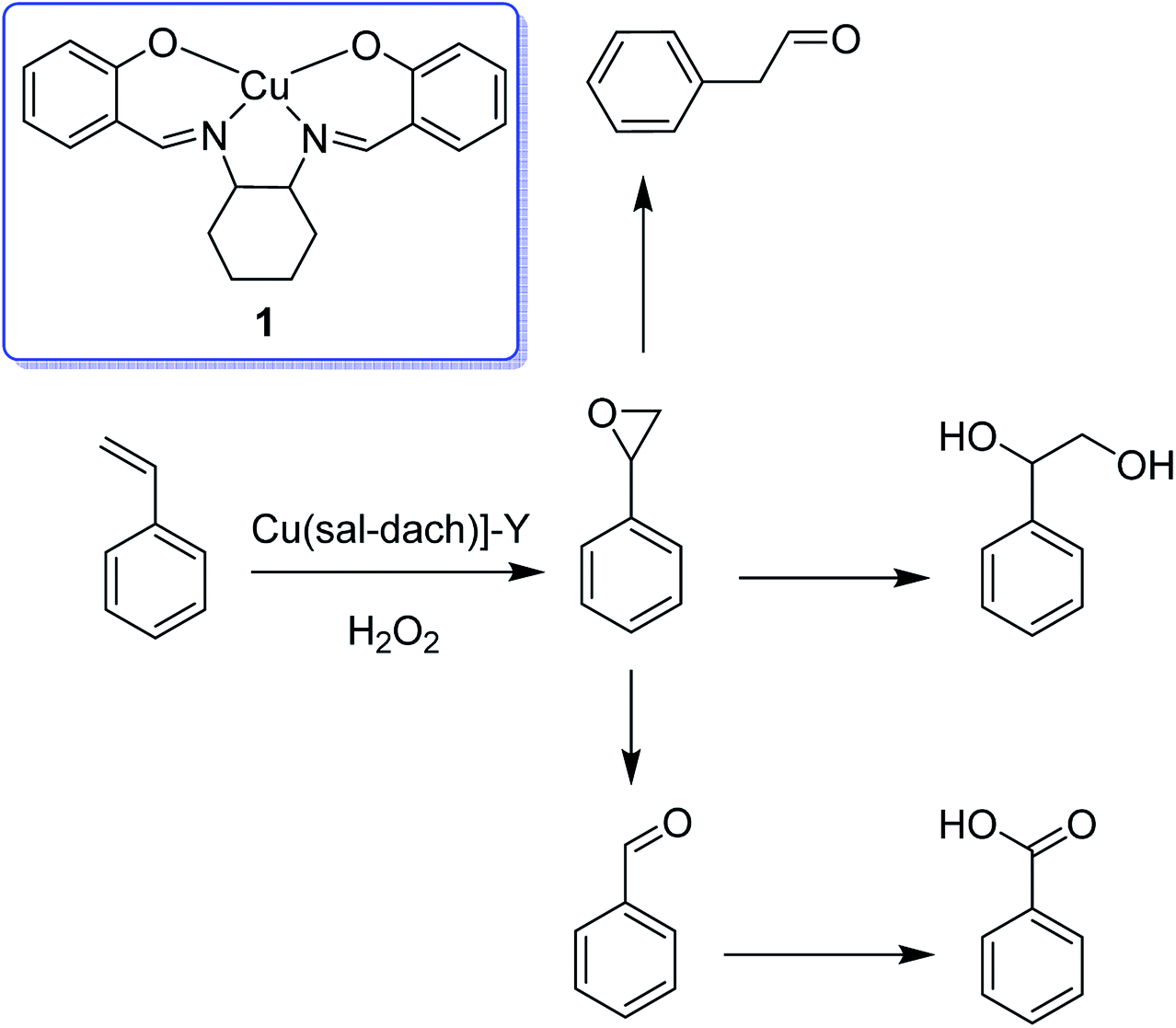 | ||
| Scheme 1 Oxidation reaction of styrene in the presence of [Cu(sal-dach)]-Y as an efficient catalyst. | ||
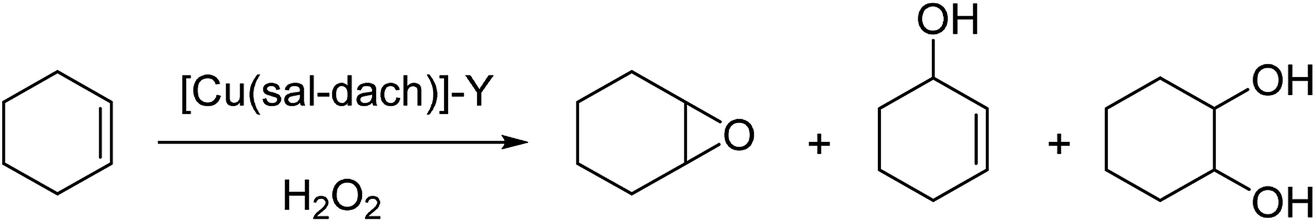 | ||
| Scheme 2 Oxidation reaction of cyclohexene in the presence of [Cu(sal-dach)]-Y as an efficient catalyst. | ||
The encapsulation of the copper(II) complex of monobasic bidentate NS donor ligand, 2-mercaptomethylbenzimidazole (Htmbmz) (2) in the cavity of zeolite-Y was achieved via flexible ligand method. [Cu(tmbmz)2]-Y was used as a catalyst in the oxidation of styrene, diphenyl sulfide and methyl phenyl sulfide.49 Under the optimized condition, the conversion of styrene in the presence of [Cu(tmbmz)2]-Y has poor efficiency (36.9%). It should be mentioned that four products (including benzaldehyde, styrene oxide, benzoic acid and phenylacetaldehyde) were produced. Outstandingly, this catalyst is very active in the oxidation of diphenyl sulfide and methyl phenyl sulfide (Scheme 3). The oxidation of diphenyl sulfide was achieved in the presence of at least hydrogen peroxide to diphenyl sulfide in a molar ratio of 3:1 to give 91.7% conversion in 7 hours. However, 94.3% conversion of methyl phenyl sulfide was obtained in 3 hours of contact time at a precursor to hydrogen peroxide molar ratio of 1:1.
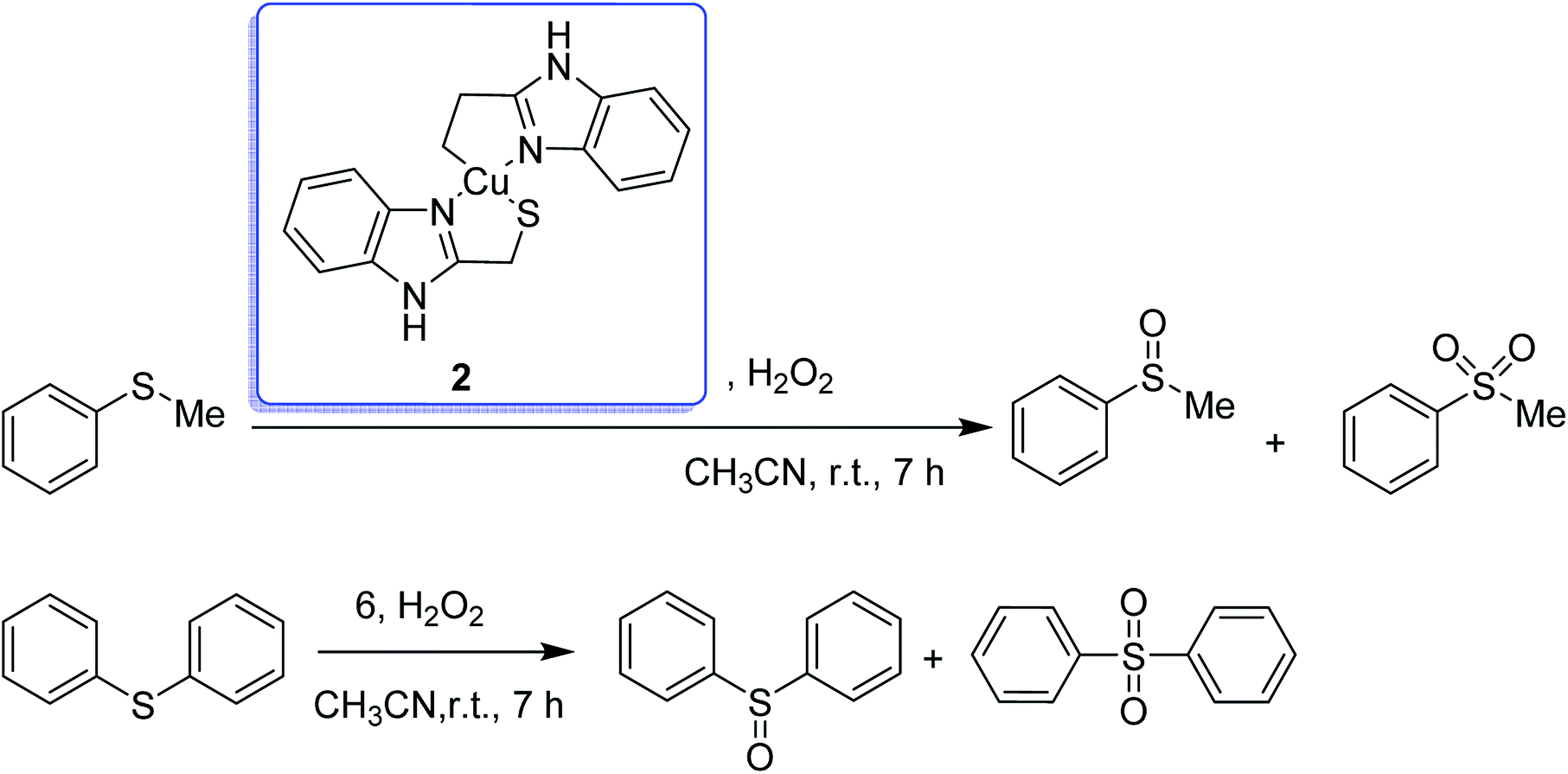 | ||
| Scheme 3 Oxidation products of methyl phenyl sulfide and diphenyl sulfide in the presence of [Cu(tmbmz)2]-Y. | ||
The tetraazamacrocyclic complex of copper(II) encapsulated into zeolite-Y [(CuMe4)2[14]tetraneN4]-Y (3) was synthesized and used as a heterogeneous catalyst for the aerobic oxidation of styrene (Scheme 4).50 This complex was constructed via a ship-in-a-bottle method in which the transition metal cations were ion-exchanged into zeolite-Y and reacted with ethylenediamine, and subsequently by acetylacetone.
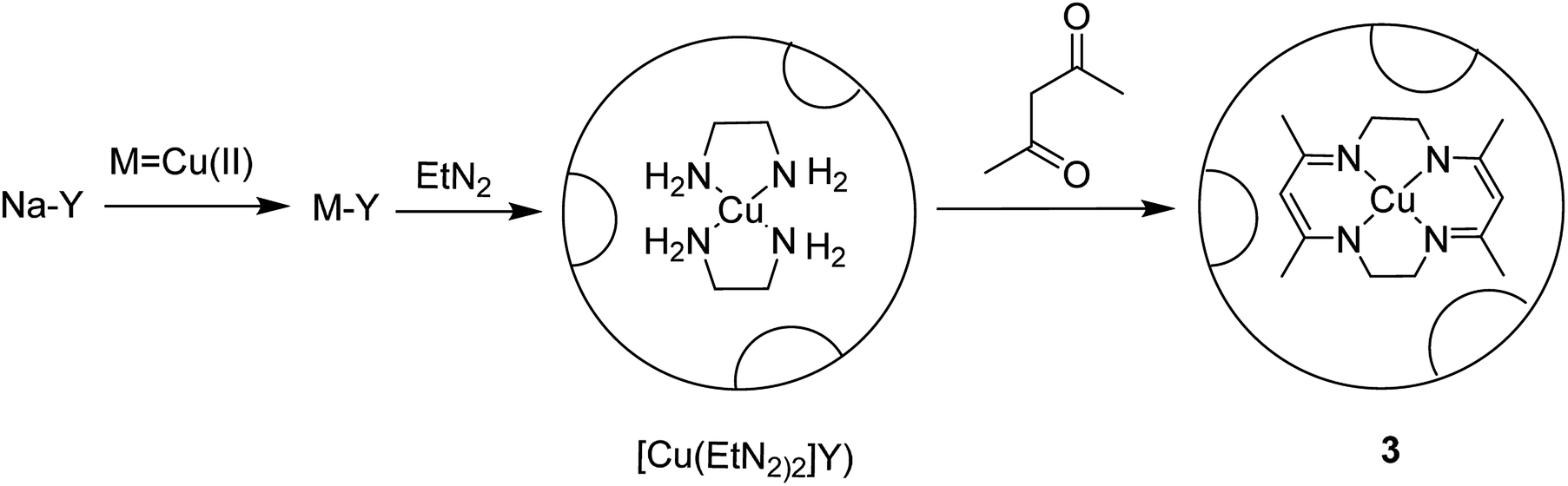 | ||
| Scheme 4 Synthesis of Y ([(CuMe4)2[14]tetraneN4]-Y) (3). | ||
In this method, to synthesize Na-Y, sodium hydroxide and NaAlO2 were dissolved in water and silica sol was gradually added to it at ambient temperature. The desired Na-Y product was obtained. For the synthesis of the Mn+-exchanged-Y zeolites, Na-Y was then added to a solution of [Cu(OAc)2] in water, and the mixture was refluxed at 90 °C. The formation of the [Cu(EtN2)2]-Y catalyst was adapted depending on the usual method, and ethylenediamine was added to the resulting Cu-Y in MeOH at 80 °C. Acetylacetone was added to a suspension of [Cu(EtN2)2]-Y in MeOH at 80 °C. Lastly, the desired Cu-encapsulated catalyst was successfully synthesized (Scheme 4).
[(CuMe4)2[14]tetraneN4]-Y (3) was used as heterogeneous catalyst for the aerobic oxidation of styrene. The recoverability and stability of [Cu{Me4(Et)2[14]tetraneN4}]-Y was explored. The results demonstrated that the catalyst could be effectively recycled three times without reducing the efficacy and selectivity.
In addition, to explore the heterogeneity of this catalytic method, a leaching examination was accomplished. For this purpose, the catalyst was eliminated using filtration at the reaction temperature (80 °C). Subsequently, the filtrated catalyst was known to transform styrene at only a very low rate. The quantity of Cu in the filtrate was known to be insignificant using ICP-AES. Therefore, the majority of the catalysis is actually heterogeneous.
It should be mentioned that, the homogeneous complex CuMe4(Et)2[14]tetraneN4 exhibited lower activity in comparison with the relevant heterogeneous complexes under the analogue conditions (Table 2).
| Catalyst | Cycle | Metal content (mmol g−1) | Styrene conversion (%) | TOF (h−1) | Product selectivity (mol%) styrene oxide, benzaldehyde, benzoic acid, phenyl acetaldehyde |
|---|---|---|---|---|---|
| [CuMe4[Et]2[14]tetraneN]-Y | 1 | 0.110 | 68 | 154.5 | 40.0, 54.5, 5.0 |
| [CuMe4[Et]2[14]tetraneN4] | 1 | 1.581 | 27 | 4.3 | 35.4, 39.3, 25.3 |
The encapsulation of copper(II) complexes of 8-quinolinol into the supercages of zeolite-Y (CuQn-Y) was achieved via the flexible ligand method.51 In this route, the transition metal cations were initially ion-exchanged into zeolite-Y and subsequently complexed by an 8-quinolinol ligand. Next, the catalytic activity of the metal-exchanged zeolites, metal complexes encapsulated into zeolite-Y and non-encapsulated homogeneous catalysts were examined in the aerobic oxidation of styrene. Strikingly, it was found that the encapsulated complexes constantly demonstrated greater activity in comparison with their corresponding non-encapsulated counterparts. Table 3 illustrates the results for the aerobic oxidation of styrene in the presence of various catalysts. It should be mentioned that except for Na-Y, all metal-exchanged zeolites and metal complexes either encapsulated or did not exhibit catalytic activity. This shows that the active sites were replaced metal ions and encapsulated metal complexes. Indeed, based on the elemental analysis, most of the replaced transition metals in MQn-Y are linked with 8-quinolinol, namely 88.9% for CuQ2-Y. This demonstrates that the performance of MQn-Y is mostly performed with the encapsulated metal complexes and not with the replaced metal ions. CuQ2-Y is more active than its corresponding metal exchanged zeolites and neat metal complexes. Remarkably, the encapsulated metal complexes show different advantages in comparison with their homogeneous equivalents. Notably, the catalytic activity of MQn-Y is greater in comparison with its corresponding nonencapsulated complexes (Table 3, entries 10 and 11), which is due to the site-isolation of the metal complexes.52
| Entry | Catalyst | M (mmol g−1) total | Styrene conversion (%) | TOF (h−1) | Product | Selectivity | (%) |
|---|---|---|---|---|---|---|---|
| Styrene oxide | Benzaldehyde | Benzoic acid, phenyl acetaldehyde, 1-phenylethane-1,2-diol | |||||
| 1 | Fe-Y | 0.204 | 60.6 | 74.3 | 30.1 | 61.7 | 8.2 |
| 2 | FeQ3 | 2.048 | 80.1 | 89.4 | 43.2 | 56.8 | 0 |
| 3 | FeQ3-Y | 0.224 | 83.4 | 93.1 | 24.5 | 58.2 | 17.3 |
| 4 | FeQ3-Y (2nd) | 0.217 | 74.2 | 85.5 | 37.4 | 60.7 | 1.9 |
| 5 | Fe-Y + FeQ3d | 0.518 | 66.5 | 74.7 | 45.1 | 51.5 | 3.4 |
| 6 | Co-Y | 0.080 | 63.3 | 197.8 | 33.2 | 58.5 | 8.3 |
| 7 | CoQ2 | 2.880 | 51.0 | 117.0 | 46.2 | 53.0 | 0.8 |
| 8 | CoQ2-Y | 0.109 | 56.7 | 130.0 | 39.2 | 59.6 | 1.2 |
| 9 | Cu-Y | 0.065 | 54.2 | 208.5 | 30.8 | 62.8 | 6.4 |
| 10 | CuQ2 | 2.840 | 61.8 | 441.4 | 40.4 | 59.6 | 0 |
| 11 | CuQ2-Y | 0.045 | 89.3 | 637.9 | 31.0 | 59.0 | 10.0 |
Two heterogeneous catalysts based on the copper(II) complexes bearing the 1,5-bis((E)-5-chloro-2-hydroxybenzylideneamino)-1H-imidazole-4-carbonitrile (H2-imida-salen) ligand (4) (Fig. 1) were considered as candidates in the styrene oxidation reaction.53 In this route, the non-encapsulated and encapsulated copper complexes were applied as catalysts in the oxidation of styrene using tert-butylhydroperoxide as the oxygen source and CH3CN as the solvent. Under the optimized reaction conditions, the catalysts exhibited good activity with greater selectivity to benzaldehyde. It is noteworthy that no Cu leaching was detected during the reaction cycle.
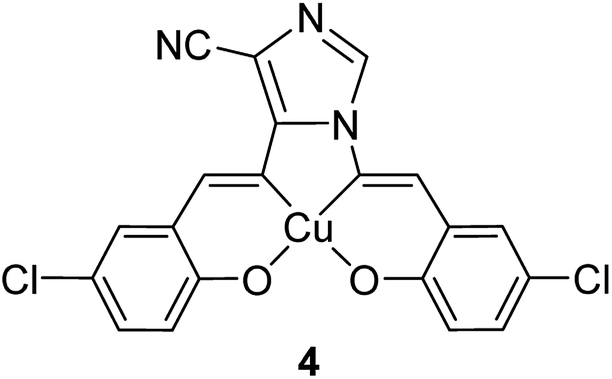 | ||
| Fig. 1 Molecular structure of complex 4. | ||
Interaction of the copper(II) exchanged zeolite-Y with the Schiff base produced from 2-aminomethylbenzimidazole (Hsal-ambmz) (5) and salicylaldehyde afforded the encapsulated copper(II) complex [Cu(sal ambmz)Cl]-Y.54 Under optimal reaction conditions, about 42% conversion of phenol was provided in the presence of this catalyst in which the selectivity of catechol changed in the order (73.9% and 56.7% conversion of styrene), and also the oxidation of styrene gave benzaldehyde, phenylacetaldehyde, styrene oxide, benzoic acid, and 1-phenylethane-1,2-diol as major products (Scheme 5).
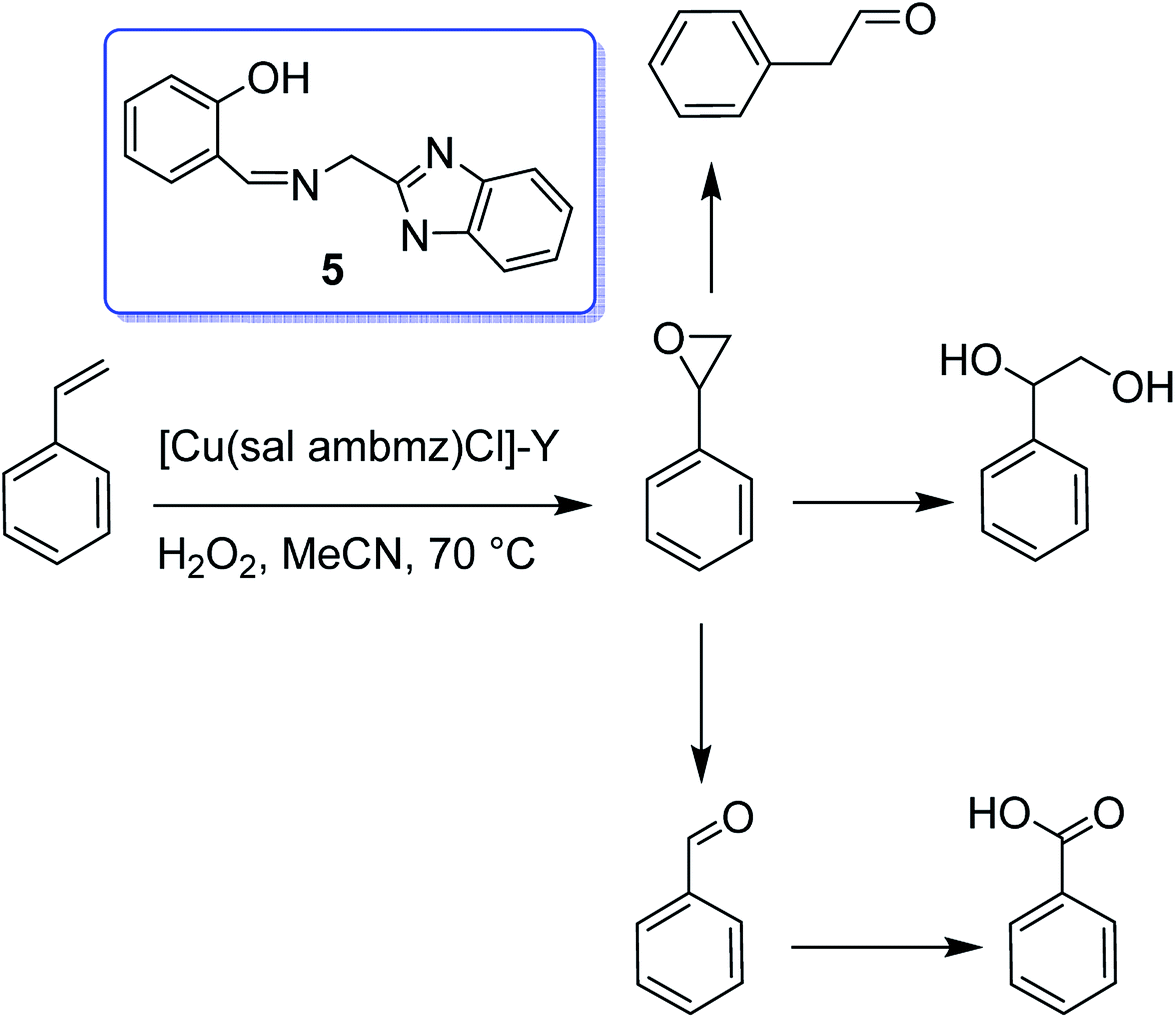 | ||
| Scheme 5 The oxidation reaction of styrene. | ||
The copper(II) complex of the Schiff base generated from the reaction of o-aminobenzyl alcohol and salicylaldehyde (H2sal-oaba) was encapsulated into the nanopores of zeolite-Y through the flexible ligand method.55 The TGA results for [Cu(sal-oaba)(H2O)]-Y demonstrated its decomposition in three stages. The exothermic elimination of only trapped H2O of ca. 4.7% happened up to 150 °C, although an exothermic weight loss of ca. 8.0% related to the elimination of intra-zeolite water happened in the temperature range from 150 °C to 350 °C. The encapsulated complex had one or more coordinated water molecules, and their elimination in this stage was also anticipated. In addition, the third stage included the slow exothermic weight loss of ca. 15.6% in a wider temperature range because of the decomposition of the metal complex. A low weight loss percentage in the higher temperature range is in agreement with the low percentage of metal content obtained for the encapsulated complexes.
Notably, the encapsulated complex [Cu(sal-oaba)(H2O)]-Y catalyzed the oxidation of cyclohexane and styrene using H2O2 as oxidant. Styrene in the presence of [Cu(sal-oaba)(H2O)]-Y (6) as catalyst gave five reaction products, including styrene oxide, benzaldehyde, phenylacetaldehyde, benzoic acid and 1-phenylethane-1,2-diol (Scheme 6). It is noteworthy that styrene oxide was produced only in poor yield, while the yield of benzaldehyde was the maximum. This catalyst, using tert-butylhydroperoxide, gave styrene oxide in the main yield. However, the overall conversion was low (10–30%). Using [Cu(sal-oaba)(H2O)]-Y with 45.8% conversion of cyclohexane, the selectivity of the two products follow the sequence: cyclohexanone (50.7%) > cyclohexanol (44.8%).
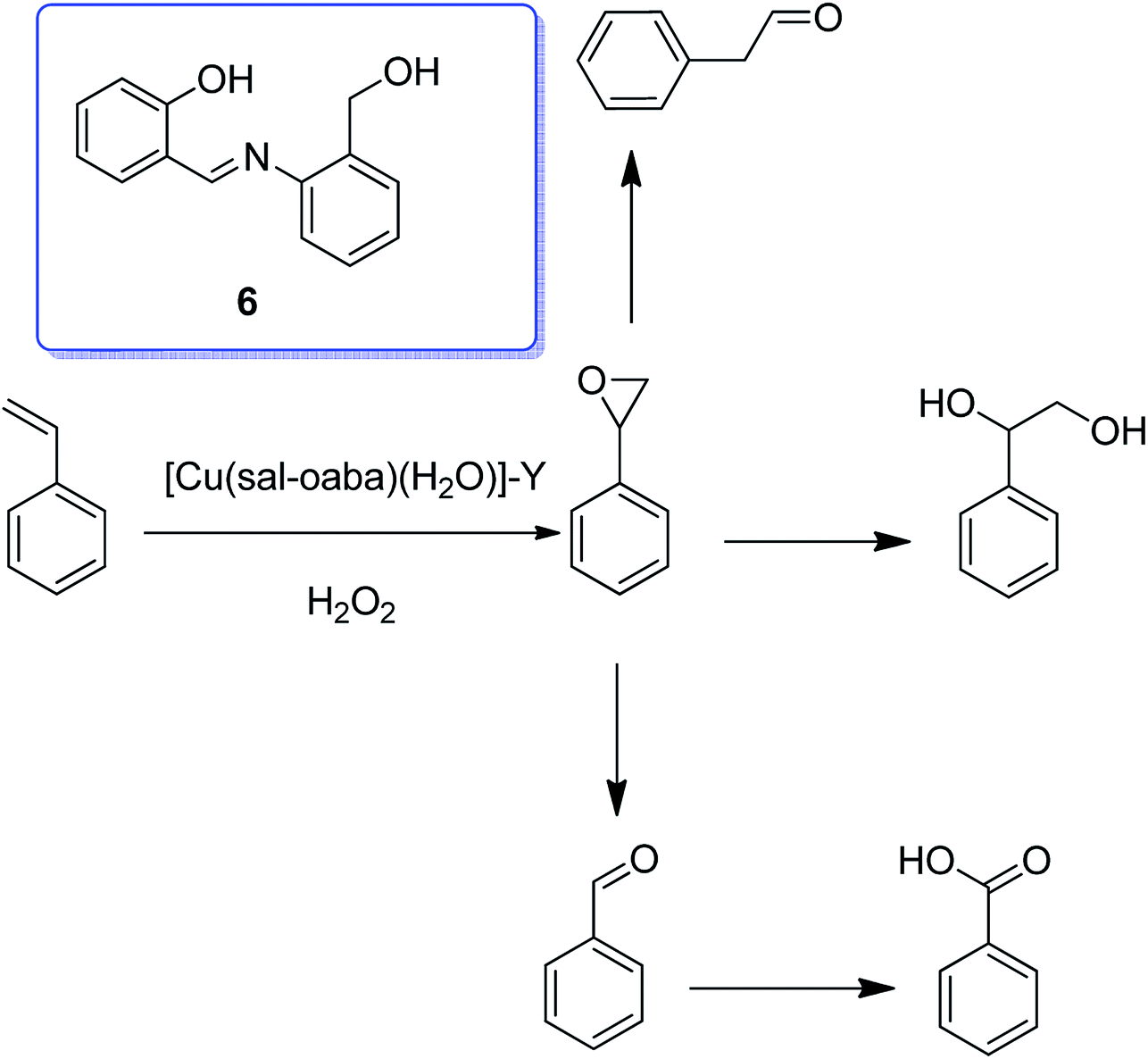 | ||
| Scheme 6 Oxidation reaction of styrene in the presence of [Cu(sal-oaba)(H2O)]-Y using hydrogen peroxide. | ||
In 2016, Gayathri and co-workers reported the formation of a copper complex of 2-(2′-hydroxyphenyl)benzimidazole (ohpbmzl) (7) encapsulated into the cavity of zeolite-Y (8) via the flexible ligand method (Scheme 7).56 The catalytic activity of the non-encapsulated and encapsulated Cu-Y complex was explored in the oxidation reaction of phenol and styrene. Hydroquinone, catechol, and parabenzoquinone were the only major products obtained in the oxidation reaction of phenol and parabenzoquinone that were identified amongst the products using zeolites with moderately larger quantities of encapsulated CuCll4Pc complexes (with 0.27 and 0.28 wt% of Cu, respectively). The encapsulated complex showed greater catalytic activity and selectivity in comparison with the non-encapsulated complex. Notably, the catalytic activity of the catalysts in the phenol oxidation reaction surveyed the order: Cu(opbmzl)2-Y (86.7%) > Cu(opbmzl)2 (70.6%) > Cu-Y (64.3%) > Na-Y (22.6%). However, the catalytic activity of the catalysts in their selectivity towards catechol was in the order: Cu(opbmzl)2-Y (66.8%) > Cu-Y (51.2%) > Cu(opbmzl)2 (48.2%) > Na-Y (18.6%). Na-Y resulted in a lower phenol conversion (22.6%), exhibiting the importance of a transition metal in the zeolite cavity for greater catalytic activity. As a result, the reactions were accomplished without the plausibility of free radicals, and the probable mechanistic pathways based on the intermediate complexes and the Cu-oxygen species were proposed with some uncertainty (Schemes 8 and 9).
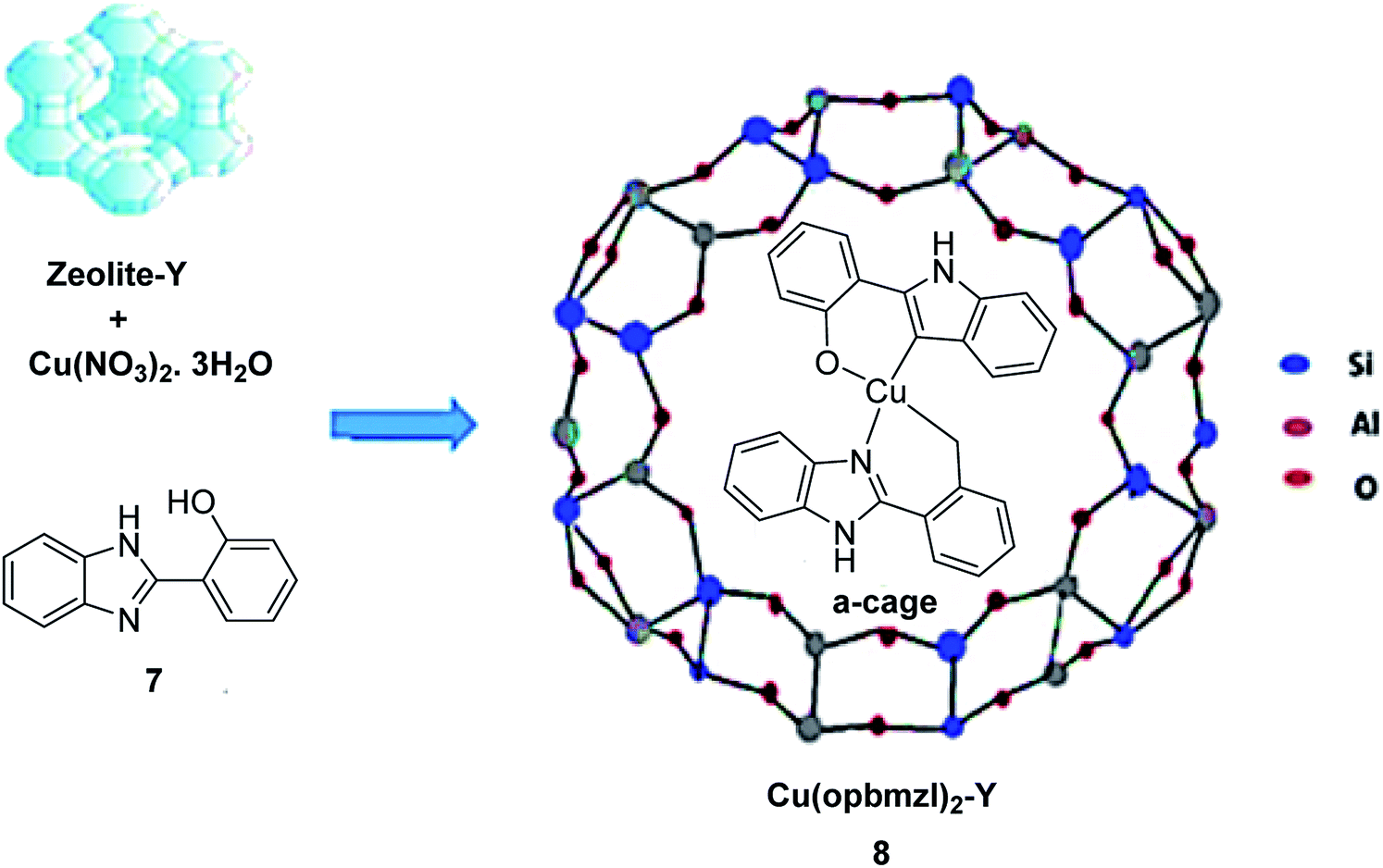 | ||
| Scheme 7 Schematic illustration of the preparative pathway for Cu(opbmzl)2-Y 8 via flexible ligand method. Reprinted from (ref. 56) with permission of the Royal Society of Chemistry. | ||
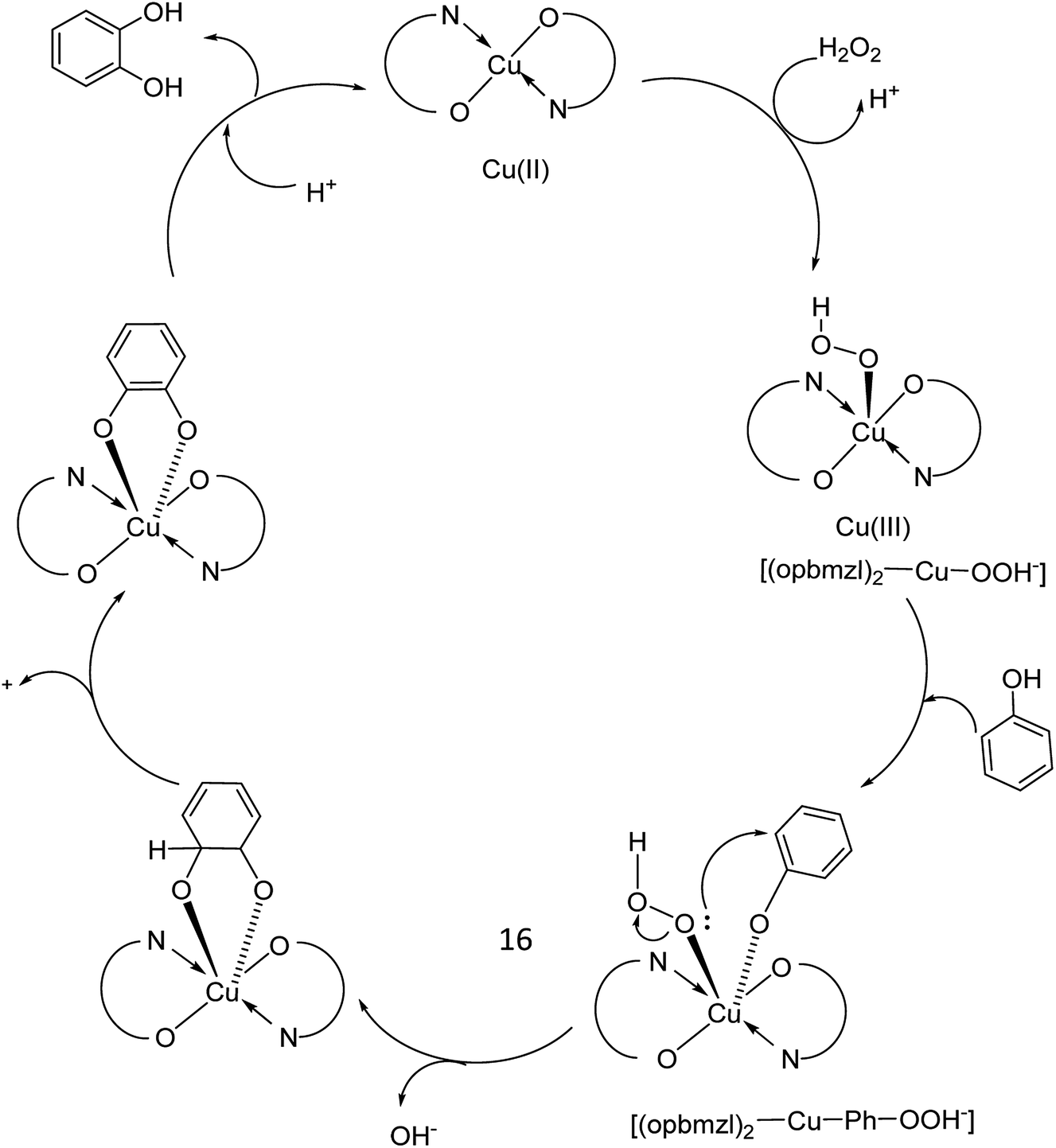 | ||
| Scheme 8 Suggested probable mechanistic route for the oxidation of phenol. | ||
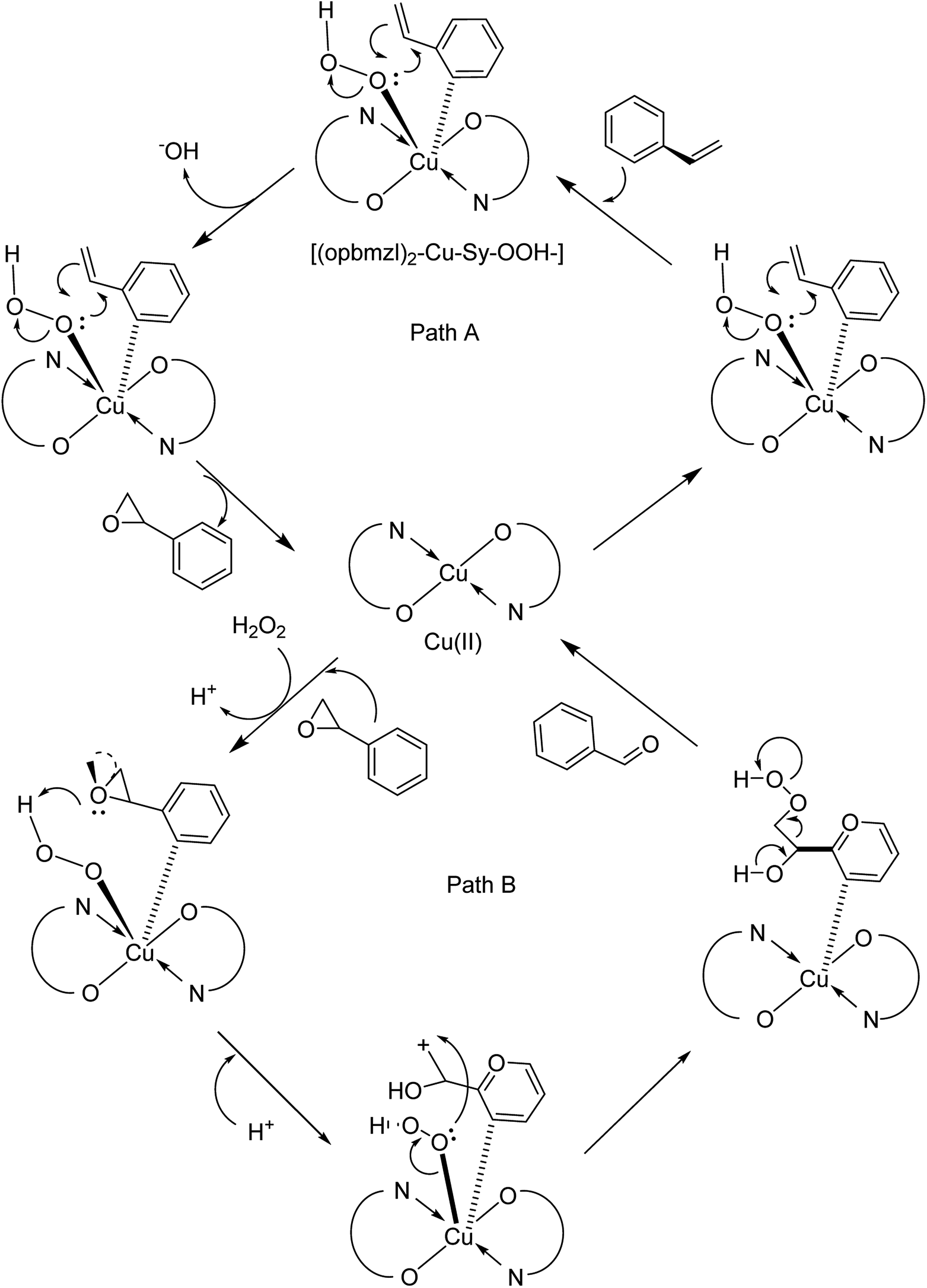 | ||
| Scheme 9 Suggested probable mechanistic route for the oxidation of styrene. | ||
The effect of various factors on the catalytic activity of Cu(opbmzl)2-Y was investigated by changing the phenol, solvents, temperature, catalyst dosage and H2O2 molar ratio. Initially, the reactions were examined with H2O2 and TBHP under the particular conditions. H2O2 was also selected as the desirable oxidant to attain the optimal conditions. Next, the influence of the temperature on the phenol oxidation was explored by repeating the reaction in the temperature range from 303 K to 343 K. It was noteworthy that this group proposed that the phenol conversion was very susceptible to temperature changes. The most promising temperature at which the reactant molecules would have the threshold energy to overcome the barrier to provide products was known to be 333 K at a reaction time of 6 hours. In addition, the solvent can either stabilize or destabilize the transition state. It changes the concentration and distribution of the intermediates generated throughout the reaction progress at the active site of the catalyst, and affects the reaction rate by dissolving the reactants and intermediates in the solution. Moreover, phenol oxidation was performed in several solvents such as MeOH, EtOH, MeCN, MeCOMe and tetrahydrofuran (THF). As a result, MeCN (4 mL) was selected as the best solvent.
It should be mentioned that AAS analysis demonstrated no sign of metal leaching into the reaction mixture. In addition, no further conversion was detected upon the elimination of the catalyst from the reaction mixture or extending the reaction time (up to 7 hours for the oxidation of styrene and 6 hours for the oxidation of phenol). Notably, this result is very important in highlighting the heterogeneous catalyst as a candidate for industrial usage.
Maurya et al. in 2003 reported the encapsulation of transition metal complexes of N,N′-bis(salicylidene) propane-1,3-diamine (H2salpn) (9) into zeolite-Y (Fig. 2). The catalytic activity of this catalyst was examined in the oxidation reaction of phenol.57 In this pathway, N,N′-bis(salicylidene)propane-1,3-diamine copper(II), [Cu(salpn)] was encapsulated in the supercages of zeolite-Y.58 The catalytic activity of [Cu(salpn)]-Y was examined in the oxidation of phenol to a mixture of hydroquinone and catechol using H2O2 as an oxidant. Based on the optimal reaction conditions, the selectivity towards catechol and hydroquinone formation was about 80 and 20%, respectively. Elevating the temperature from 50 to 80 °C in acetonitrile increased the percentage of hydroquinone formation from 12 to 27%. In addition, an analogous pattern was followed with CCl4 as a solvent in which the hydroquinone selectivity increased from 20 to 32% on elevating the temperature from 50 to 80 °C. It is noteworthy that ethanol served quite differently as a solvent in which the selectivity towards catechol and hydroquinone construction was 85 and 15%, respectively.
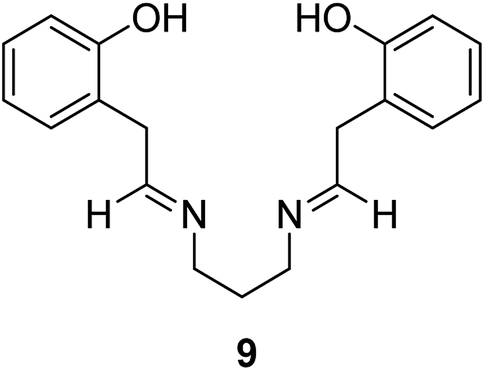 | ||
| Fig. 2 The formation of transition metal complexes of (H2salpn) 9. | ||
It is noteworthy to mention that phenol was successfully oxidized using the heterogeneous catalysts based on Fe(II) or Cu(II) pyrrolylazine (10) complexes that were encapsulated into the NaY zeolite (Scheme 10).59 Notably, the encapsulated metal pyrrolylazine complexes in the NaY zeolite increased the superior conversion of phenol into catechol (Table 4).
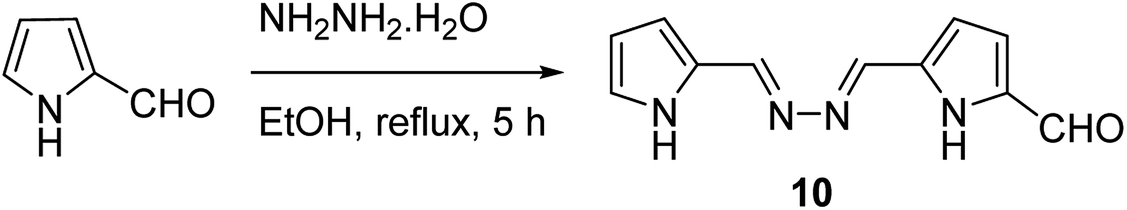 | ||
| Scheme 10 Synthesis of the pyrrolyl-azin derivative. | ||
| Entry | Run | Catalyst | Tb (h) | %Cc | CAT | Others | ||
|---|---|---|---|---|---|---|---|---|
| %S | %Y | %Sd | %Ye | |||||
| a Reaction conditions: PhOH, 1:3 PhOH:tBuOOH, heterogeneous catalyst.b Reaction time at which the PhOH conversion starts to become constant.c Reaction runs under homogeneous conditions.d Equivalent 100 mg of Fe2L@Y.e Equivalent 100 mg of Fe2L@YB. |
||||||||
| 1 | 1 | NaY | 48 | 4 | — | — | — | — |
| 2 | 1 | Cu2L@YB | 6 | 86 | 61 | 53 | 39 | 33 |
| 3 | 2 | 24 | 67 | 100 | 67 | 0 | 0 | |
| 4 | 1 | Cu2L@YA | 24 | 34 | 50 | 17 | 50 | 17 |
| 5 | 2 | <24 | 38 | 100 | 29 | 0 | 0 | |
Crystalline copper(II) Schiff base complexes of [CuL1NO3]n 11 and [CuL2Cl] 12 (where HL1 = 1-[(3-dimethylaminopropylimino)-methyl]-naphthalen-2-ol and HL2 = 3-[(3-dimethylamino-2,2-dimethyl-propylimino)-methyl]-naphthalen-2-ol) were constructed in 2018 by Mukhopadhyay et al. (Scheme 11).60
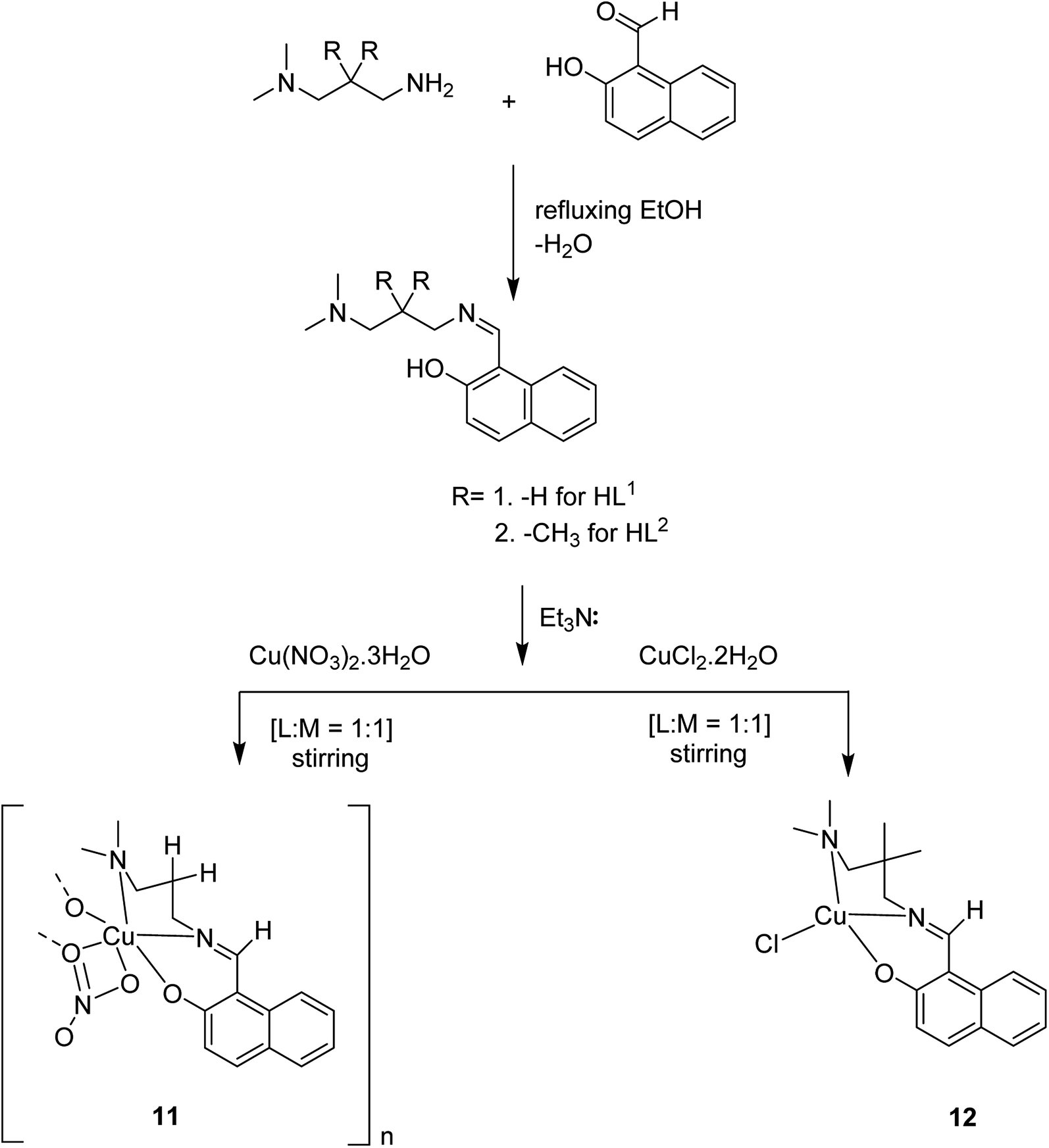 | ||
| Scheme 11 Construction of Schiff-base ligands HL1 and HL2, and their Cu(II) complexes (11 and 12). | ||
Various solvents comprising diethyl ether, acetonitrile and methanol were used to purify the resultant products to eliminate the unexpected species or the species, which were adsorbed on the zeolite surfaces. The formation of the host–guest complexes into the zeolite supercages was further confirmed via the colour of the target product. Finally, anhydrous [CuL1NO3@Y or CuL2Cl@Y] (13) was provided as a blue-violet powder (Scheme 12).
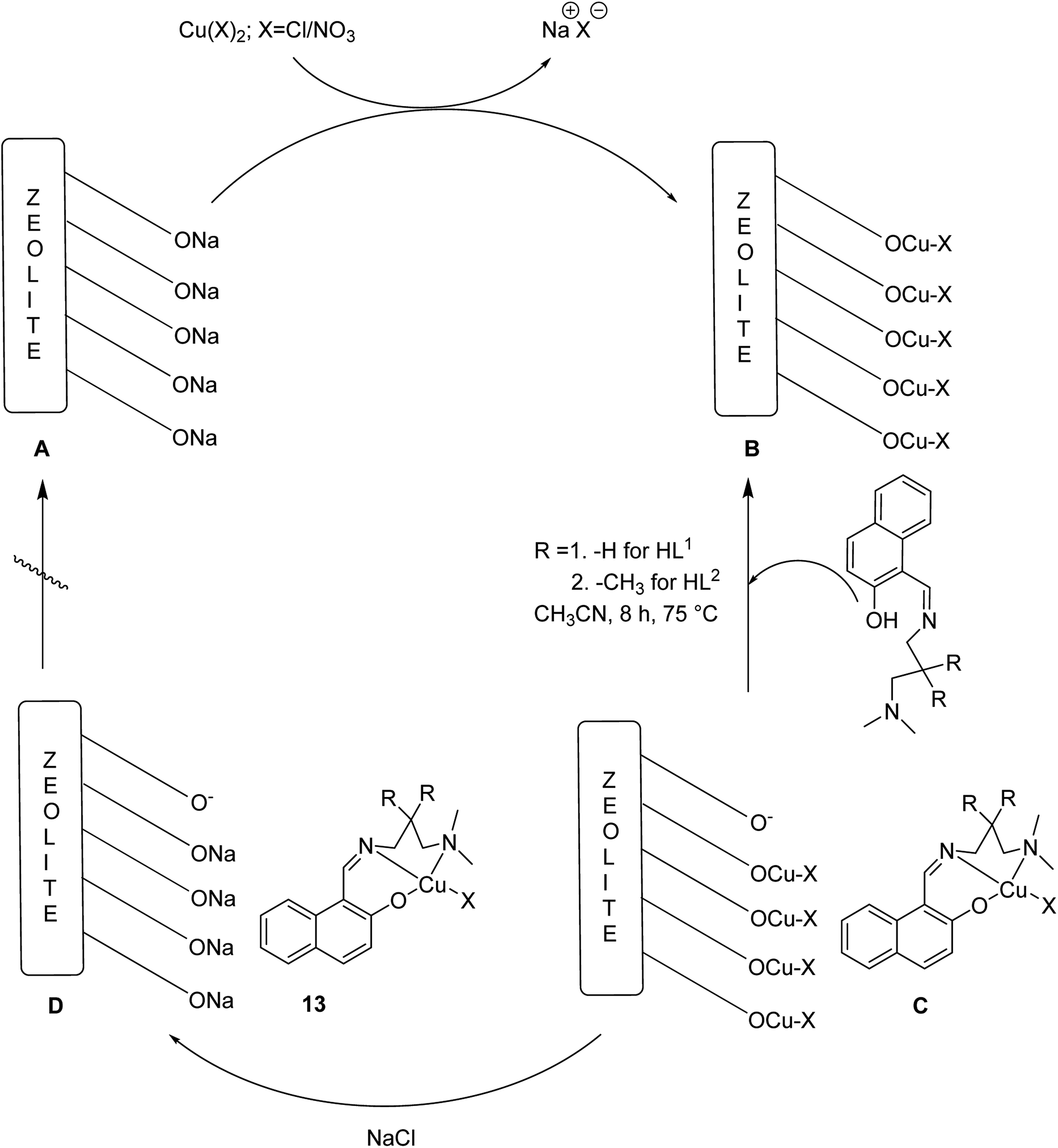 | ||
| Scheme 12 Synthesis of heterogeneous catalysts via metal (M = Cu) incorporation followed by immobilization of the Schiff base ligands into the zeolite, NaY. | ||
Thermogravimetric analysis (TGA) was used to attain information about the thermal stability of the neat and metal-exchanged zeolite NaY, and also the encapsulated complexes under static nitrogen atmosphere. Both pure and metal exchanged zeolite presented only distinct degradation at 192 °C, due to the loss of peripheral –OH moieties or loss of physically and chemically adsorbed water. Moreover, it exhibited minor mass reduction until 400 °C, which is apparent from the differential thermal analysis (DTG) profiles. The zeolite-encapsulated complexes exhibited additional decomposition stages because of the loss of ligand molecules, and the relevant temperature range extended up to ∼500 °C. This demonstrates the improvement in the thermal stability of the complexes after encapsulation in the zeolite framework. A slight mass loss (∼4%, nCuL2Cl@Y) obviously indicates the presence of slight quantities of metal complex in the supercages of zeolite, which is related to the low metal concentration estimated by ICP-MS analysis.
Notably, this method exhibits the advantageous of heterogeneous catalytic reactions for the stability of metal complexes in the supercages of zeolite-Y and the selective oxidation reaction of phenol, styrene, and cyclohexene using H2O2. The catalytic activity of the zeolite entrapped Cu(II) complexes were compared with their homogeneous analogues as well. Catalytic hydroxylation of phenol gave two major products, including hydroquinone and catechol; however, it can be further oxidized to para-benzoquinone (Scheme 13).
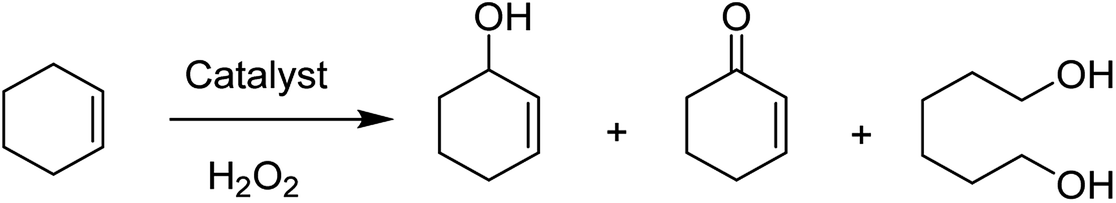 | ||
| Scheme 13 Different oxidation products of cyclohexene. | ||
Table 5 displays the percentage conversion of the oxidation products of cyclohexene using various catalysts and keeping other parameters fixed.
| Entry | Catalysts | Conversion (%) | Selectivity | Of (%) | Product | TOF (min−1) |
|---|---|---|---|---|---|---|
| Cyol | Cyone | Other | ||||
| 1 | Blank | 1.91 | — | 100 | — | — |
| 2 | [CuL1NO3]n | 44.42 | 23.68 | 51.57 | 24.75 | — |
| 3 | CuL2Cl | 35.24 | 28.63 | 65.32 | 6.05 | — |
| 4 | NaY | 9.84 | 29.47 | 70.42 | — | — |
| 5 | Cu(II)@Y(NO3) | 18.83 | 20.02 | 62.34 | 17.64 | 73.1 |
| 6 | Cu(II)@Y(Cl) | 15.84 | 22.09 | 69.93 | 8.28 | 70.6 |
| 7 | CuL1NO3@Y | 62.69 | 19.12 | 69.03 | 11.85 | 322.9 |
| 8 | CuL2Cl@Y | 58.67 | 18.70 | 72.83 | 8.47 | 384.2 |
Scheme 14 displays the suggested possible mechanism for the oxidation of phenol in the presence of homogeneous complexes (by [CuL1NO3]n; X = –NO3 and R = –H and [CuL2Cl]; X = –Cl and R = –CH3) using H2O2 in which complex 13 gives the potent oxidative metal-hydroperoxide species.61
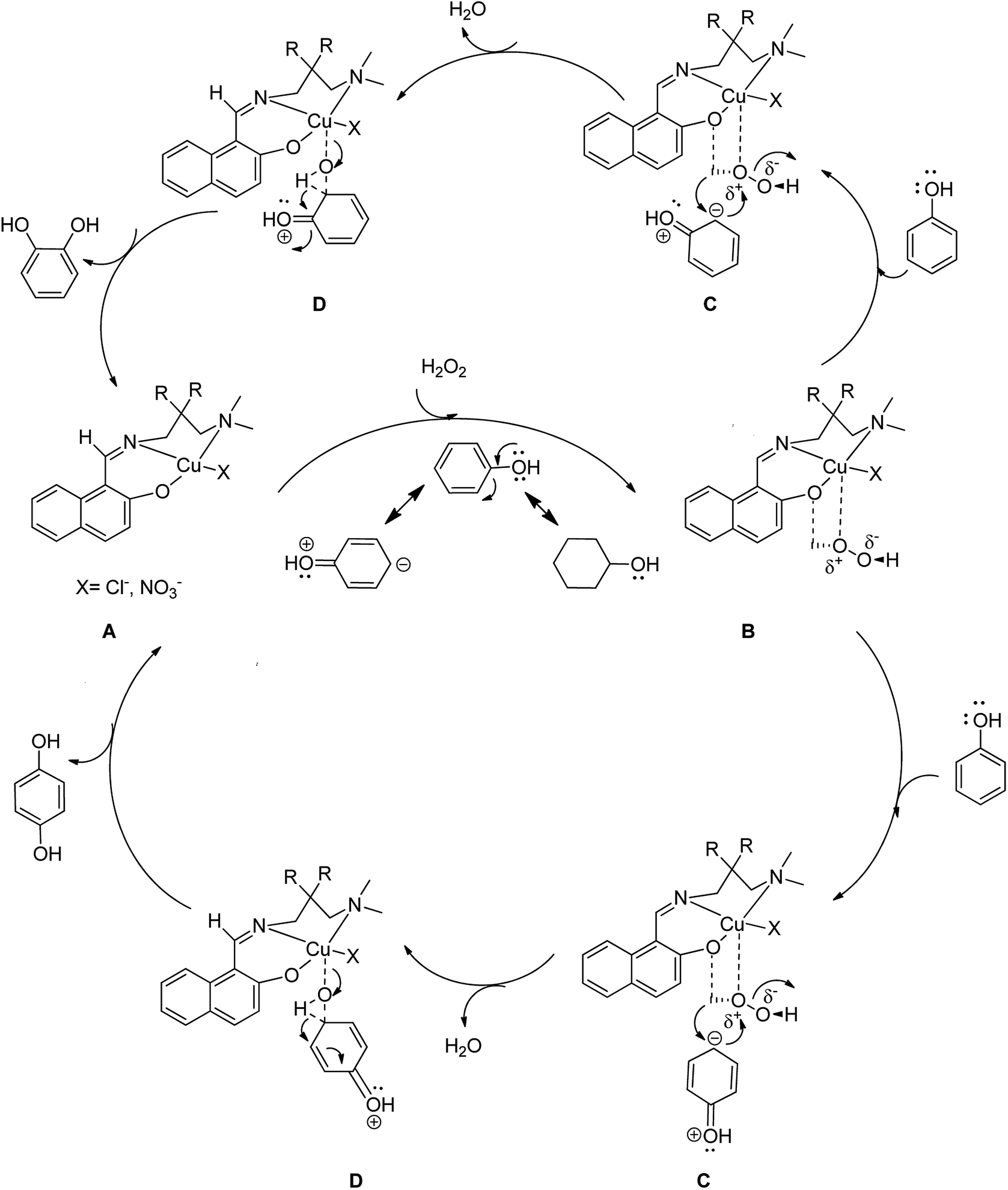 | ||
| Scheme 14 Probable catalytic cycle for the phenol hydroxylation reaction to form catechol or/and hydroquinone as the major products. | ||
The catalytic oxidation of styrene provided benzaldehyde, 1-phenylethane-1,2-diol, acetophenone and 2-hydroxy-1-phenylethanone (Scheme 15).
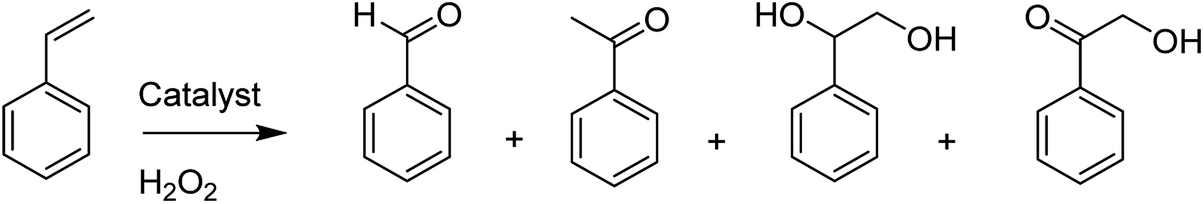 | ||
| Scheme 15 Various products in the oxidation of styrene. | ||
Mobinikhaledi and co-workers in 2014 reported the encapsulation of copper(II) complexes of methyl-2-(2-hydroxybenzylideneamino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (HL1) and 2-ethyl-4-methyl 5-(2-hydroxybenzylideneamino)-3-methylthiophene-2,4-dicarboxylate (HL2) (Fig. 3) into the supercages of zeolite NaY via the flexible ligand method.62 The neat and encapsulated complexes were used as catalysts in the oxidation reaction of phenol and benzyl alcohol. Remarkably, the encapsulated complexes were both more stable and reactive catalysts in comparison with the desired free complexes. It should be mentioned that the complex of HL2 in both free and encapsulated modes illustrated greater activity in comparison to the complex of HL1. This is related to the electron-withdrawing groups that decrease the electron density of the copper(II) centers, and so they increase their Lewis acidity.
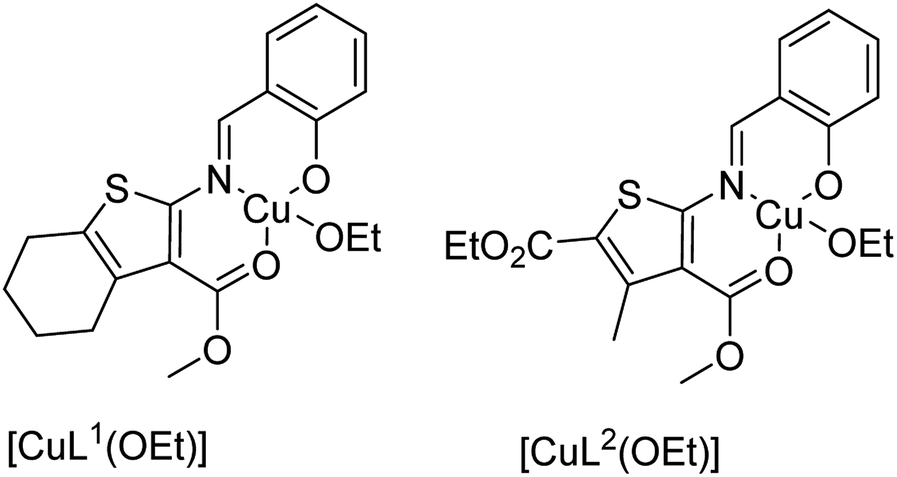 | ||
| Fig. 3 Proposed structure for free and encapsulated complexes [CuL1(OEt)] and [CuL2(OEt)]. | ||
The catalytic activity of CuAc-Y and CuClAc-Y, CuAc and CuC1Ac in the oxidation of phenol to o-benzoquinone was examined (Table 6). Some features are mentioned in which the hydroxylation reaction does not occur in the absence of any catalyst. Both CuClAc-Y and CuAc-Y selectively oxidize phenol to o-benzoquinone. In addition, the turnover frequency for oxidation is remarkably increased upon encapsulation of the complexes in zeolite-Y for both CuC1Ac and CuAc. Finally, the particular catalytic activity of CuClAc-Y is greater than that of CuAc-Y at analogous levels of phenol conversion.
| Catalyst | Phenol conv. (mol%) | BZQ (mol%) | Others (mol%) | TOF (h−1) |
|---|---|---|---|---|
| a TON = turnover frequency (number of molecules of phenol converted per atom of Cu per hour). Reaction condition: pH = 6.5, 298 K, 19 hours. | ||||
| CuAc “neat” | 5.1 | 4.9 | 0.2 | 3.6 |
| CuAc-Y | 11.5 | 11.5 | — | 60.9 |
| CuClAc “neat” | 1.0 | 1.0 | — | 0.9 |
| CuClAc-Y | 12.0 | 11.9 | 0.1 | 227.1 |
The reaction between excess N,N′-bis(salicylidene)diethylenetriamine (H2saldien) (14) and copper(II) exchanged zeolite-Y at ca. 90 °C resulted in the encapsulation of the ligand in the supercages of zeolite. This was followed by the complexation with the metal ion (15) (Scheme 16).57 The encapsulated complex was used as the significant catalyst for the decomposition of hydrogen peroxide, and for the oxidation reaction of phenol to a mixture of hydroquinone and catechol using H2O2 as an oxidant. Based on the optimal condition in the presence of Cu(saldien)]-Y as the catalyst and using hydrogen peroxide in MeCN, the oxidation reaction of phenol at 80 °C gave the maximum conversion of phenol. Based on these conditions, the catalytic activity of Cu(saldien)]-Y is 45% upon 24 hours.
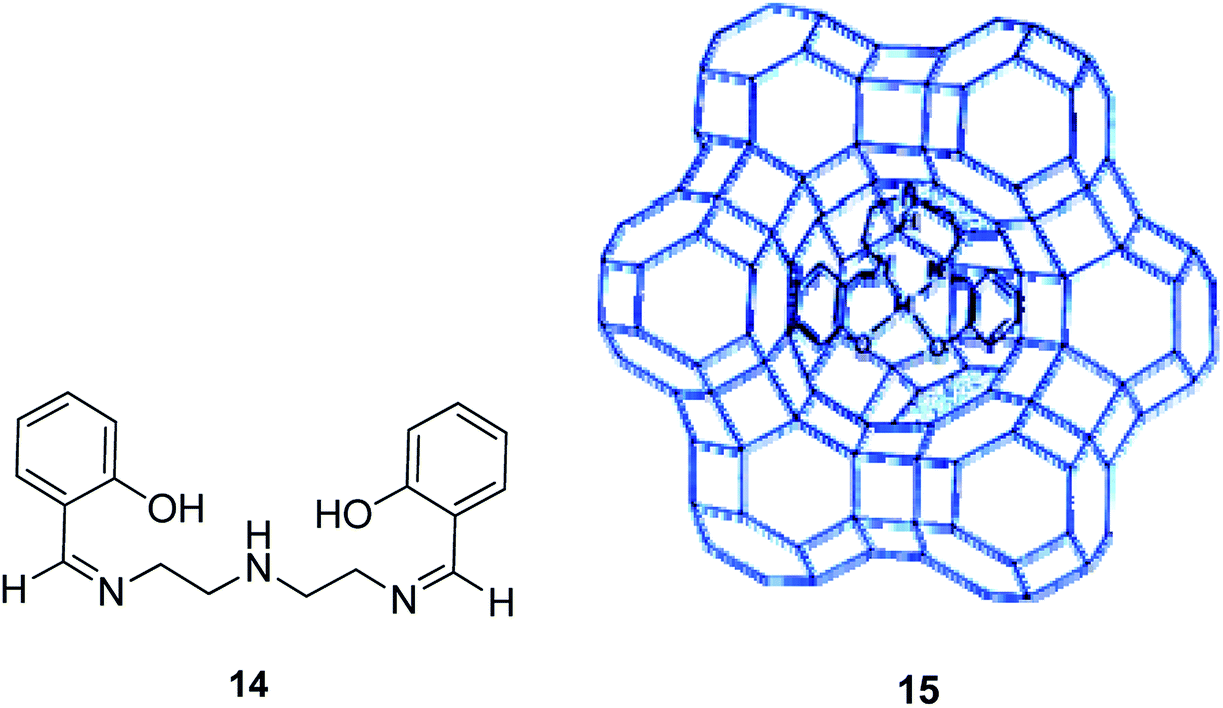 | ||
| Scheme 16 Suggested structure of [M(saldien)]-Y (M = Cu(II), Ni(II) and Zn(II)). Reprinted from (ref. 57) with permission of the Royal Society of Chemistry. | ||
The selective oxidation reaction with O2 and hydrogen peroxide of n-hexane, phenols and naphthalene in the presence of Cu complexes was demonstrated in 1996 by Raja and co-workers.63 The copper complexes included copper acetate (Cu(CH3COO)2), copper phthalocyanine (C32H16CuN8), copper nitrophthalocyanine (C32H16CuN9O2) and copper chlorophthalocyanine (C32H17ClCuN8), and were encapsulated in molecular sieves (X, Y, MCM-22 and VPI-5). The oxidation of phenols to hydroquinone and catechol was achieved with high yields in the presence of Cu-phthalocyanines encapsulated into the zeolites Na-X or Na-Y.
Zeolite NaX or Na-Y with changed loadings of Cu(NO2)4Pc or CuCl14Pc were provided successfully. The oxidation of phenol using hydrogen peroxide in the presence of CuPc-based catalysts was achieved (Table 7). Notably, both Cu(NO2)4Pc and CuCl14Pc are active catalysts for the oxidation reaction of phenol to catechol/hydroquinone using H2O2 as the oxidant. Table 7 demonstrates the effect of the Cu content (and, therefore the concentration of the Cu complex) in the zeolite on the rate of oxidation of phenol and in the product distribution. Herein, the noticeable aspects are as follows: (i) the catalytic activity (turnover number) of the Cu ions is greater in the encapsulated state in comparison with the “neat” complex; (ii) the Cl complex is also more potent than the NO2 complex; (iii) the TON does not change considerably with Cu content, suggesting that the Cu complexes are well separated from each other (the conversion of phenol, obviously, improves with the concentration of the Cu complex into the zeolite); (iv) parabenzoquinone hydroquinone and catechol are the only main products generated in the oxidation of phenol. It should be mentioned that the quantity of ‘tar’ products is low (less than 1%). Notably, the resultant product was almost colourless. In addition, parabenzoquinone is detected between the products having zeolites with comparatively larger quantities of encapsulated CuCl14Pc complexes (having 0.27 and 0.28 wt% copper, respectively).
| Catalyst | Cu (wt%) | TON (moles of phenol converted per mol of Cu in the catalyst) | Products p-benzoquinone catechol hydroquinone |
|---|---|---|---|
| a Reaction condition: catalyst, phenol/H2O:(3:1), 3 hours, 353 K. |
|||
| CuCl14Pc | — | 2.1 | 2.0, 59.5, 38.5 |
| Cu(NO2)4Pc | — | 1.21 | 12.5, 0, 87.5 |
| CuCl14Pc-Na-Y | 0.11 | 7.95 | 0, 65.8, 34.2 |
| CuCl14Pc-Na-Y | 0.17 | 8.27 | 0, 63.1, 36.9 |
| CuCl14Pc-Na-Y | 0.26 | 6.77 | 0, 50.9, 49.1 |
| CuCl14Pc-Na-Y | 0.27 | 7.35 | 6.1, 49.1, 44.8 |
| CuCl14Pc-Na-X | 0.14 | 8.25 | 0, 62.8, 37.2 |
| CuCl14Pc-Na-X | 0.28 | 7.36 | 11.2, 46.4, 34.4 |
| Cu(NO2)4Pc-Na-Y | 0.09 | 7.40 | 0, 0, 100 |
| Cu(NO2)4Pc-Na-Y | 0.16 | 3.95 | 0, 0, 100 |
| Cu(NO2)4Pc-Na-X | 0.11 | 4.25 | 0, 0, 100 |
| Cu(NO2)4Pc-Na-X | 0.14 | 3.95 | 0, 0, 100 |
The copper(II)-pyridine complex was encapsulated into the supercage of the Y zeolite. The complex was simply reduced to Cu(I) at 200 °C under vacuum and re-oxidized to Cu(II) upon exposure to molecular oxygen in its reduced state. The oxidative coupling reaction of 2,6-dimethylphenol was selected as a model reaction.12 This reaction was explored in the presence of homogeneous copper-amine catalysts in solution using O2.13 Two corresponding products, diphenoquinone (DPQ) and polyphenylene oxide (PPO) were prepared through C–C and C–O coupling reactions, respectively (Scheme 17).64
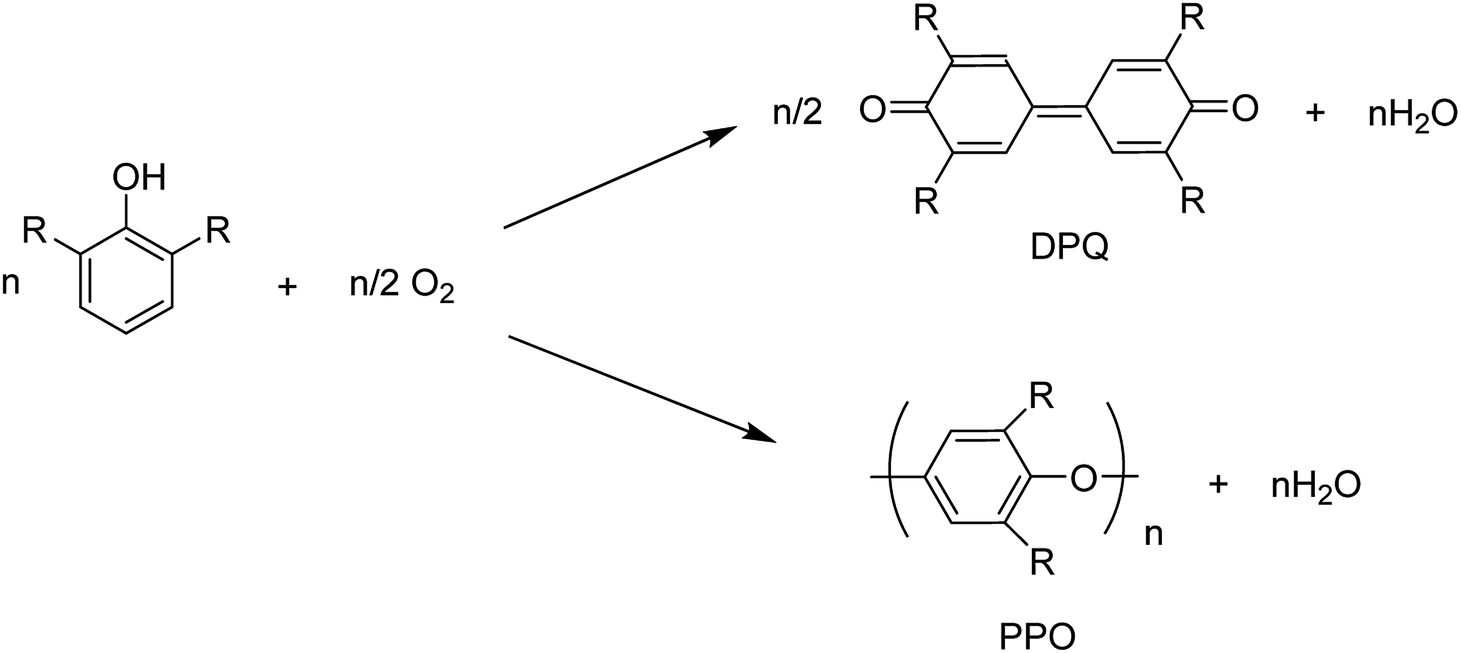 | ||
| Scheme 17 The oxidative coupling reaction of 2,6-dimethylphenol using homogeneous Cu-amine catalysts. | ||
The results demonstrate that Cu–Py–NaY affords extraordinary selectivity to PPO in comparison with CuCl+Py, in spite of its low catalytic activity. Although various probable reaction mechanisms were proposed, Oyama and co-workers (1987)65 and Waters (1971)66 recommended in their homogeneous investigations that PPO or DPQ is produced via different intermediates (Scheme 18). Initially, the dissociation of phenol into a phenolate anion occurs using bases. Next, PPO is generated via an intermediate ArO° radical or DPQ via an ArO+ cation, in which a one- or two-electron transfer occurs from the phenolate anion to Cu(II) ion, respectively. To provide the catalytic reaction, the reduced copper(I) ions should be reoxidized to Cu(II) ions with O2. It was found that Cu–Py–NaY favors the C–O coupling since the catalytically active species for it is separated in the zeolite supercage, and must subsequently admit only one electron from a phenolate anion.
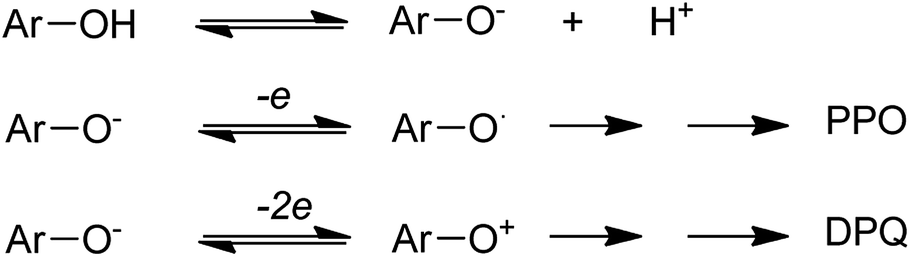 | ||
| Scheme 18 Dissociation of phenol to the phenolate anion. | ||
Salavati-Niasari and co-workers in 2008 reported the encapsulation of [M(H4C6N6S2)] (H6C6N6S2 = 1,2,5,6,8,11-hexaazacyclododeca-7,12-dithione-2,4,8,10-tetraene) (16) into the nanocavity of zeolite-Y via the flexible ligand method (Scheme 19).67
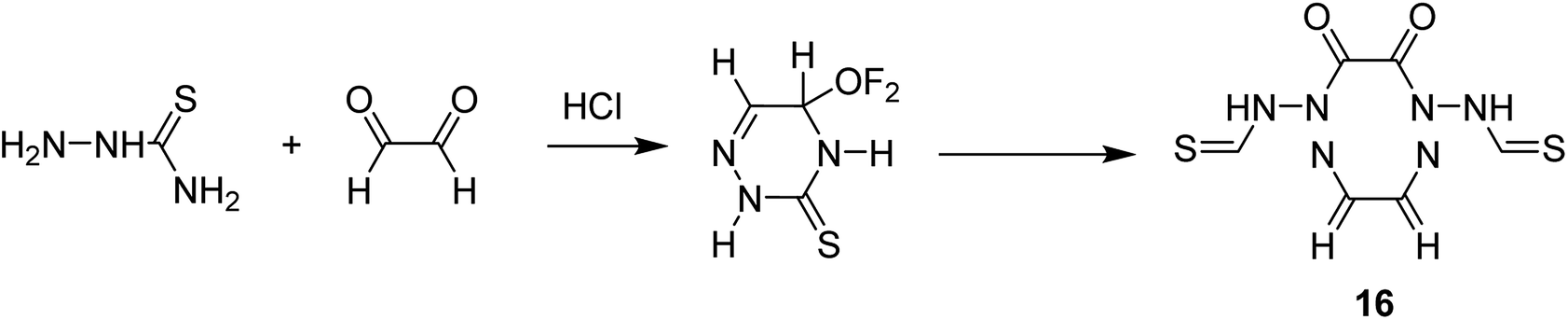 | ||
| Scheme 19 Preparation of 16. | ||
Based on this method, an ethanolic solution of thiosemicarbazide in the presence of HCl was added to an ethanolic solution of glyoxal. Then, an ethanolic solution of CuCl2 was added to an ethanolic suspension of the intermediate ligand, 6-ethoxy-4-thio-2,3,5-triazine to provide 1,2,5,6,8,11-hexaazacyclododeca-7,12-dithione-2,4,8,10-tetraene. Next, a hot ethanolic solution of relevant metal acetate was added to a hot ethanolic suspension of the 12-membered macrocyclic ligand under reflux to form the desired complex. M(II)-NaY was synthesized using the ion-exchange approach. Then, NaY was added to the solution of manganese acetate in water. It is noteworthy that the encapsulated complex was produced via FLM. For this purpose, M(II)-NaY and H6C6N6S2 = 1,2,5,6,8,11-hexaazacyclododeca-7,12-dithione-2,4,8,10-tetraene were mixed and heated at 250 °C. The melted ligand served as the solvent and as reactant. The resulting material was isolated using MeOH until the complex was free from the unreacted H6C6N6S2. The resulting zeolites were isolated with N,N-dimethylformamide (DMF) and with EtOH to eliminate excess unreacted diamine and any M(II) complexes adsorbed onto the external surface of the zeolite crystallites.
The activity of this catalyst was examined in the oxidation of cyclohexene. This complex exhibited significant activity in the oxidation of cyclohexene to 2-cyclohexene-2-ol, 2-cyclohexene-1-one, and 1-(tert-butylperoxy)-2-cyclohexene, where tert-butylhydroperoxide was provided as the product with the maximum yield (Table 8).
| Catalyst | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|
| 2-Cyclohexene-1-one | 2-Cyclohexene-1-ol | 1-(Tert-butylproxy)-2-cyclohexene | ||
| [Cu(H4C6N6S2)]-NaY | 40.1 | 65.5 | 22.0 | 12.5 |
Iron and copper(II)salen complexes were spontaneously encapsulated into zeolite-Y via the flexible ligand method. Notably, this complex demonstrated considerable greater activity in comparison with the neat Fe(salen) and Cu(salen) in the oxidation reaction of cyclohexane with H2O2.68 It should be mentioned that the presence of a synergetic influence is the reason for this high activity. This synergetic influence may originate from the interaction of the adjacent moderately coordinated Fe(salen) and Cu(salen) complex molecules and/or the formation of the dinuclear salen complexes via the lattice oxygen of the zeolitic host. This reaction was achieved more rapidly on (Cu–Fe) (salen)/Y than on Cu(salen)/Y or Fe(salen)/Y. The optimal molar ratio of Cu2+ to Fe3+ in the resulting material is 1.93:1. This reaction was accomplished in the presence of a catalyst in CH3CN with H2O2 at 60 °C. These catalysts exhibited considerably greater activity in comparison with the zeolite Y-encapsulated iron salen and zeolite Y-encapsulated copper(II) salen for the oxidation of cyclohexane with hydrogen peroxide (Table 9).
| Catalyst | Amounts of Cu2+ and Fe3+ cations (mmol) | Cu2+/Fe3+ (mol mol−1) | TON | Selectivity (%) | |
|---|---|---|---|---|---|
| Cyclohexanol | Cyclohexanone | ||||
| a Reaction conditions: the designed amount of catalyst, 10 mL of CH3CN, 18.5 mmol of substrate, 19.5 mmol of H2O2 (30% in aqueous solution), 60 °C, 2 hours. | |||||
| Cu(salen)/Y + Fe(salen)/Y | 0.013 | 2.80 | 89 | 50.5 | 49.5 |
| Cu(salen)/Y + Fe(salen)/Y | 0.013 | 1.93 | 85 | 56.8 | 43.2 |
| Cu(salen)/Y + Fe(salen)/Y | 0.013 | 0.6 | 70 | 55.9 | 44.1 |
| Cu(salen)/Y + Fe(salen)/Y | 0.013 | 0.21 | 62 | 57.3 | 42.7 |
The Cu(II) complex having a pentadentate Schiff-base ligand, N,N-bis(salicylidene)-2,6-pyridinediaminato, and H2[sal-2,6-py], was encapsulated into the nanocavity of zeolite-Y (17) (Scheme 20).69 The results demonstrated that the encapsulated Cu(II) complexes in the nanodimensional pores of zeolite-Y were different from those of the free complexes. These differences can arise from distortions provided by steric effects due to the presence of sodium cations, or from interactions with the zeolite matrix. The host–guest nanocomposite materials [M(sal-2,6-py)]-NaY catalyzed the oxidation reaction of cyclohexene using TBHP.
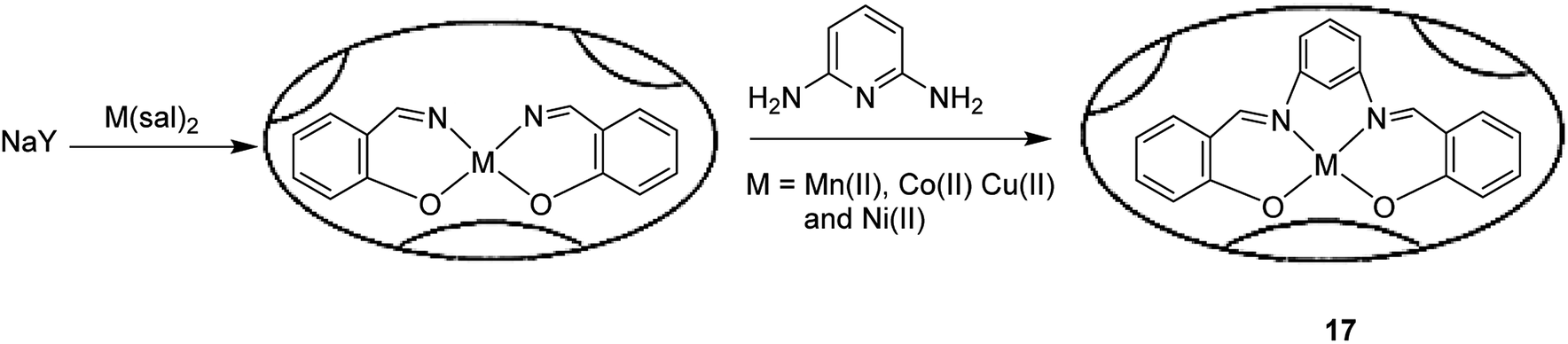 | ||
| Scheme 20 The encapsulation of transition metal complexes in the supercages of a NaY zeolite. | ||
As shown in Table 10, the activity of these catalysts in cyclohexene oxidation using different solvents decreases in the order: CH2Cl2 > CH3Cl > MeOH > CH3CN. Based on this method, only allylic oxidation occurred with the formation of 2-cyclohexene-1-ol, 2-cyclohexene-1-one, and 1-(tert-butylperoxy)-2-cyclohexene and 2-cyclohexene-1-one (as the major product).
Cu(II) complexes having tetrahydro-salophen (salophen = N,N-bis-(salycilidene)-1,2-phenylenediamine; H2[H4salophen] = 2-(2-[(2 hydroxybenzyl)amino] anilinomethyl)phenol) (18) was encapsulated into the nanopores of zeolite-Y, [M(H4salophen)]-NaY (19), via the flexible ligand method.70 For the formation of the ligand, 1,2-phenylenediamine and salicylaldehyde were used. This method resulted in the encapsulation of Mn(II), Co(II), Ni(II) and Cu(II) complexes of salophen and tetrahydro-salophen ligands in the nanopores of zeolite (Scheme 21). The catalytic activity using host–guest nanocomposite complexes in the oxidation of cyclohexane was examined. This catalytic complex exhibited excellent activity in the oxidation of cyclohexane under mild conditions (Table 11). Therefore, zeolite-encapsulated systems increased the catalytic potential, particularly in comparison with the activity for limited oxidation and stability.
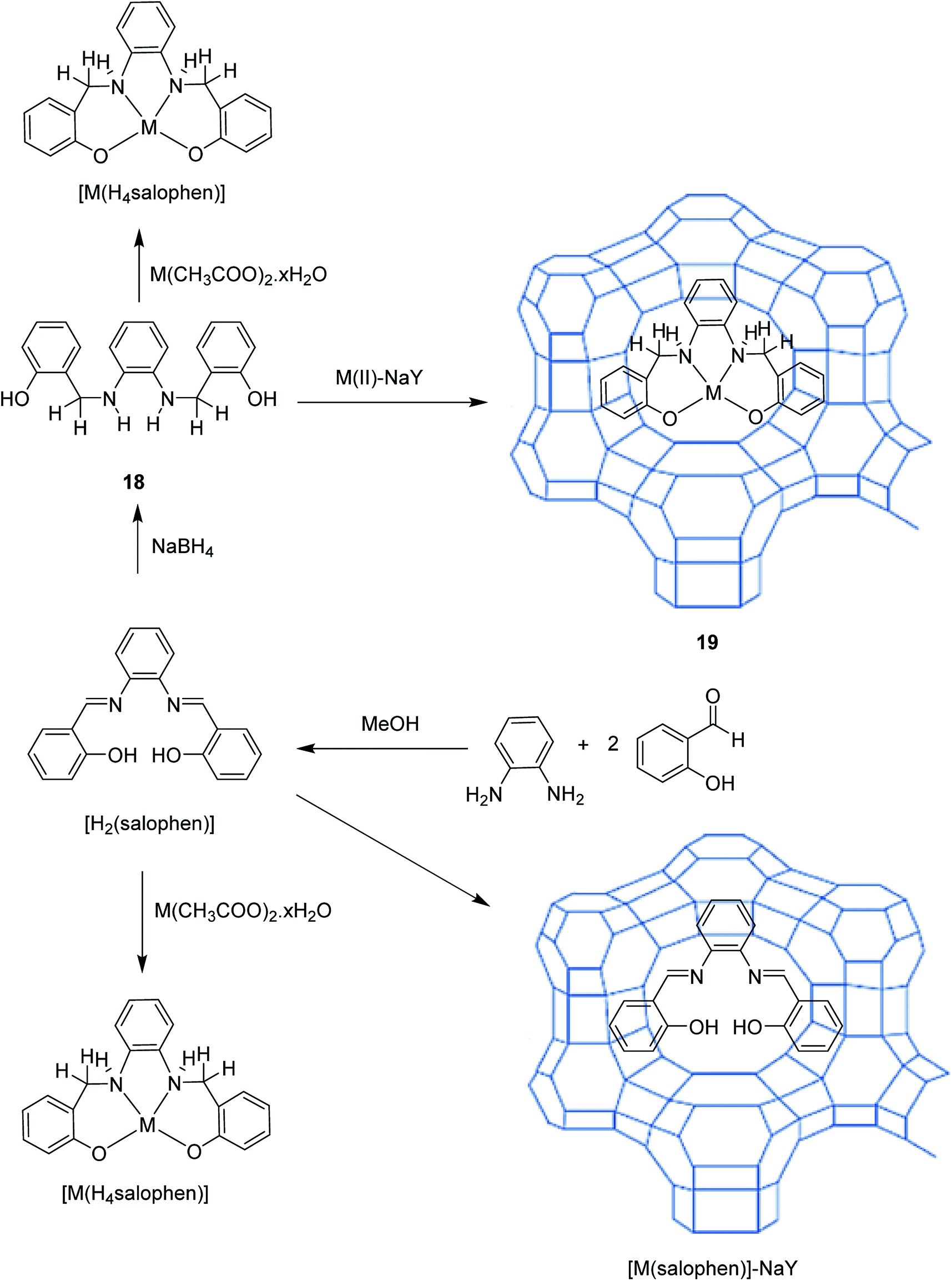 | ||
| Scheme 21 Encapsulation of Mn(II), Co(II), Ni(II) and Cu(II) complexes of salophen nanopores of zeolite. Reprinted from (ref. 70) with permission of the Royal Society of Chemistry. | ||
| Catalyst | Conversion (%) | Selectivity (%) | |
|---|---|---|---|
| Cyclohexanol | Cyclohexanone | ||
| a Reaction condition: time (2 hours), cyclohexane (10 mmol), catalyst (1.02 × 10−5 mol), H2O2 (20 mmol), CH3CN (5 mL).b A similar procedure as system was carried out with 0.5 × 10−5 mol catalyst instead of 1.02 × 10−5.c A similar procedure as system was carried out with 2.04 × 10−5 mol catalyst.d A similar procedure as system was carried out with 4.08 × 10−5 mol catalyst. | |||
| [Cu(salphen)]a | 43.2 | 36.4 | 63.6 |
| [Cu(H4salphen)]a | 48.7 | 42.3 | 57.7 |
| [Cu(H4salphen)]b | 26.8 | 60.9 | 39.1 |
| [Cu(H4salphen)]c | 42.1 | 38.9 | 61.1 |
| [Cu(H4salphen)]d | 18.6 | 31.4 | 68.6 |
Cu(II) complexes bearing octahydro-Schiff base (H4-N4O4) = 2,7,13,18-tetramethyl-3,6,14,17-tetraazatricyclo-[17.3.1.1]-tetracosa-1(23),2,6,8(24),9,11,13,17,19,21-decaene-9,11,20,22-tetraol, H4([H]8-N4O4) = 2,7,13,18-tetramethyl-3,6,14,17-tetraazatricyclo-[17.13.1.1.]-tetracosa-1(23),8(24),9,11,19,21-hexane-9,11,20,22-tetraol, were encapsulated into the nanopores of zeolite-Y, [M([H]8-N4O4)]@NaY, via the flexible ligand method (Scheme 22). The zeolite-encapsulated octahydro-Schiff base copper(II) complex, [Cu([H]8-N4O4)]@NaY, demonstrated high activity in the cyclohexane oxidation.71 It is noteworthy that the catalytic activity of these complexes are influenced by the steric environment of the active site and by their geometry. These complexes are also more active than the relevant neat complexes, [Cu([H]8-N4O4)]. Remarkably, the encapsulation of the Schiff base complexes found as the most widely explored in the investigation arena of ship-in-a-bottle materials in this kind of complex has a flexible conformation with different geometries, namely stepped configurations, planar, and umbrella-type. As a result, they could provide various active site environments for several oxidations. However, the amount of the encapsulated Schiff base complex is very limited due to the existence of a comparatively severe C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. On the other hand, the hydrogenation reaction of CN to C–N would increase the nitrogen basicity and make the conformation of the complex more flexible, leading to the more readily accessible coordination to the metal centers in a folded method. Hence, this results in the inclusion of more transition metal complexes or active sites without rigid blockage of the channels in the zeolitic matrix. Moreover, the transition metal tetrahydro-salophen complex (M-[H4]salophen) is considered to be more active than [Cu-salophen] in the homogeneous oxidation reaction of cyclohexane.70 This resulted in [M-([H4] salophen)]-Y as an active heterogeneous catalyst for the oxidation reaction of this type of organic substrates. As a result, this group demonstrated the formation of a wide range of [M([H]8-N4O4)]-Y catalysts and the investigation of catalytic activities in the oxidation of cyclohexane.
N bond. On the other hand, the hydrogenation reaction of CN to C–N would increase the nitrogen basicity and make the conformation of the complex more flexible, leading to the more readily accessible coordination to the metal centers in a folded method. Hence, this results in the inclusion of more transition metal complexes or active sites without rigid blockage of the channels in the zeolitic matrix. Moreover, the transition metal tetrahydro-salophen complex (M-[H4]salophen) is considered to be more active than [Cu-salophen] in the homogeneous oxidation reaction of cyclohexane.70 This resulted in [M-([H4] salophen)]-Y as an active heterogeneous catalyst for the oxidation reaction of this type of organic substrates. As a result, this group demonstrated the formation of a wide range of [M([H]8-N4O4)]-Y catalysts and the investigation of catalytic activities in the oxidation of cyclohexane.
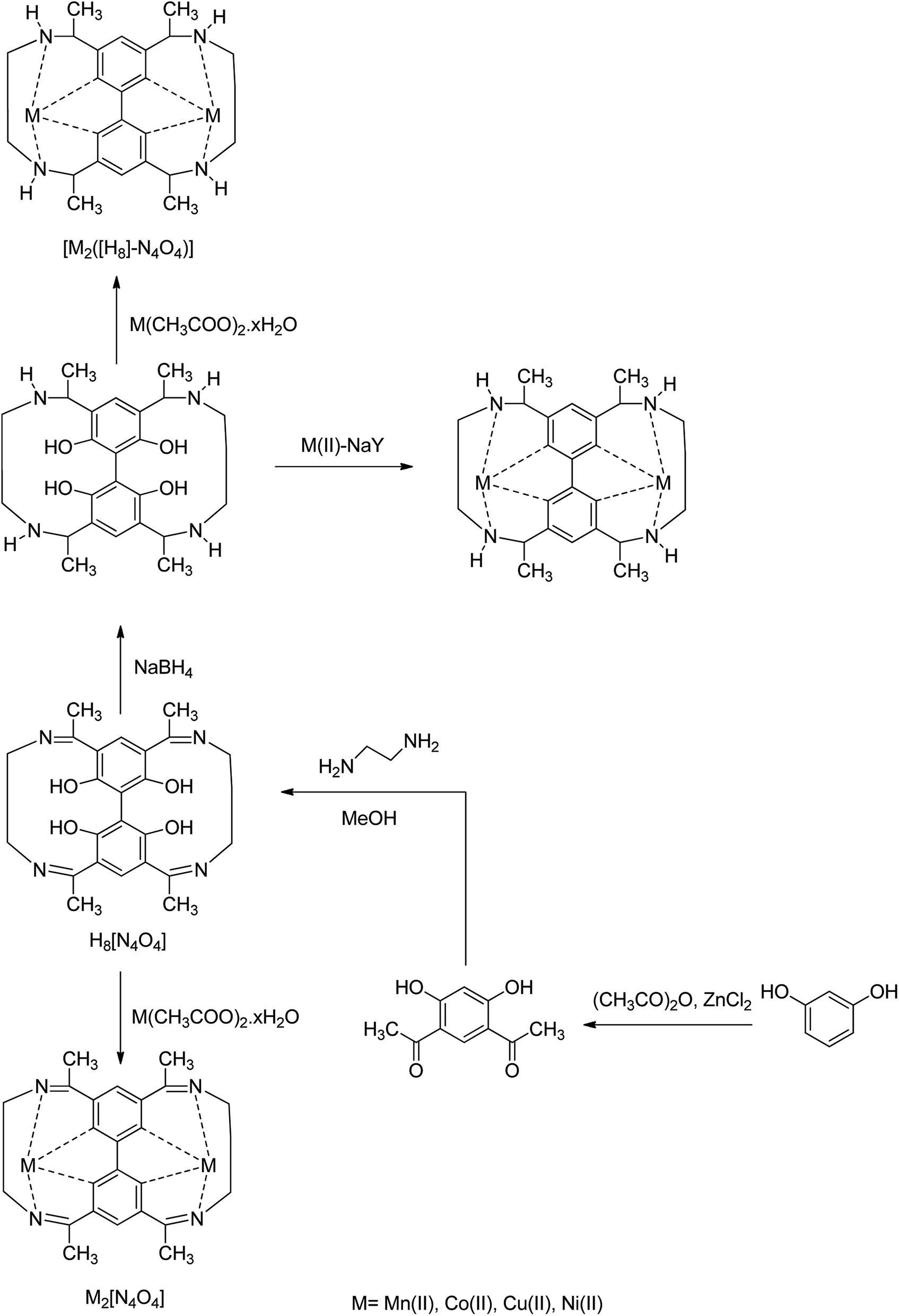 | ||
| Scheme 22 Schematic diagram exhibiting the construction of host–guest nanocomposites. | ||
The Cu(II) ion with the 1-(2-pyridylazo)-2-naphthol (PAN) ligand was encapsulated into supercages of the Y zeolite (host) via the flexible ligand method. The catalytic activity of this catalyst was examined in liquid phase in the cyclohexanol and cyclohexene oxidations using tert-butylhydroperoxide as the oxidizing agent at 40 °C and 60 °C, respectively.72 Cyclohexene is appropriate for both allylic oxidation and epoxidation. To attain proof for or against a radical mechanism and to evaluate the catalyst selectivity, the oxygenation reaction of cyclohexene was achieved. The resultant products were identified as 2-cyclohexene-1-one, 2-cyclohexene-1-ol and 1-tert-butylperoxy-2-cyclohexene.73 The results for the oxidation of cyclohexene and cyclohexanol in the presence of this heterogeneous catalyst, [Cu(PAN)]@Y, are shown in Tables 12 and 13, respectively.
| Catalyst | Conversion (%) | Selectivity (%) | TON | ||
|---|---|---|---|---|---|
| 2-Cyclohexene-1-ol | 2-Cyclohexene-1-one | 1-Tert-butylperoxy-2-cyclohexene | |||
| [Cu(PAN)Cl] | 26 | — | 32 | 68 | 83.4 |
| [Cu(PAN)]@Y | 28 | — | 11 | 89 | 56.7 |
| Catalyst | Time (h) | Conversion (%) | Yield (%) | TON |
|---|---|---|---|---|
| [Cu(PAN)]@Y | 3 | 46 | 46 | 2722 |
The reaction between CuCl2 and (Z)-2-(1-(2-(1H-benzo[d]imidazol-2-yl)ethylimino)ethyl) phenol(Hhap-aebmz) (20) that was generated from 2-aminoethylbenzimidazole (aebmz) and o-hydroxyacetophenone (Hhap) gave [Cu(hap-aebmz)Cl]. Next, [Cu(hap-aebmz)Cl] was encapsulated in the nanocavity of zeolite-Y through various physico-chemical approaches.74 The encapsulated system was used as an efficient catalyst for the oxidation of cyclohexene and phenol using H2O2 (Schemes 23 and 24). With the approximately quantitative oxidation of cyclohexene, the selectivity of the oxidation products follows the order: 2-cyclohexene-1-ol (44%) > 2-cyclohexene-1-one (40%) > cyclohexeneoxide (12%) > cyclohexane-1,2-diol (4%). Obviously, the best result for the oxidation of phenol was obtained in the presence of [Cu(II)(hap-aebmz)Cl]-Y, hydrogen peroxide at 80 °C. [Cu(hap-aebmz)Cl]-Y exhibited a conversion of 65.7% with selectivity of the main products, catechol (66.1%) and hydroquinone (32.9%).
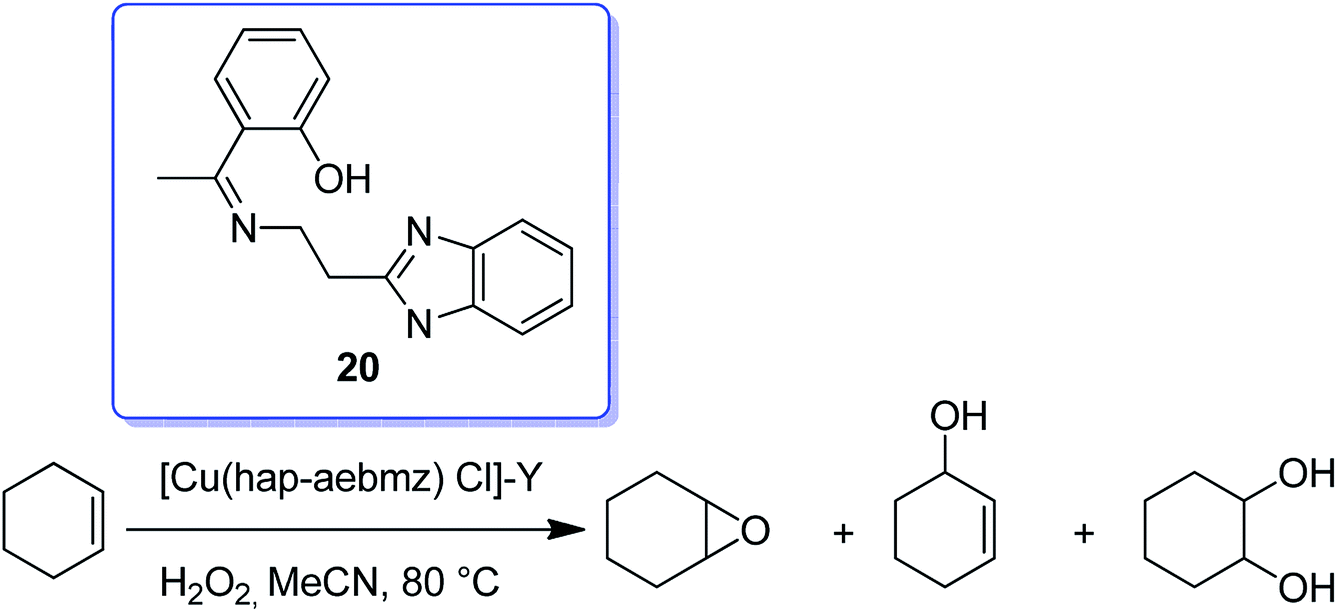 | ||
| Scheme 23 Cyclohexene oxidation reaction in the presence of [Cu(hap-aebmz)Cl]. | ||
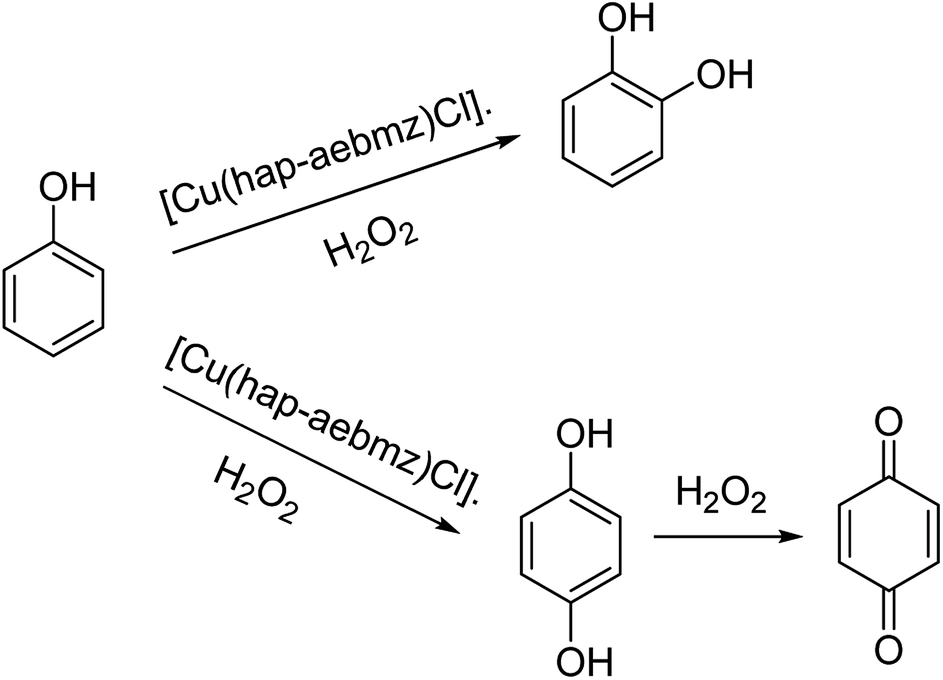 | ||
| Scheme 24 Oxidation of phenol using [Cu(hap-aebmz)Cl]. | ||
The copper(II) complex of a Schiff base ligand (H2L) (21) generated from o-phenylenediamine and pyrrolecarbaldehyde was encapsulated in the Y-zeolite matrix (22) (Scheme 25).75 In this approach, for the synthesis of the ligand, pyrrolecarbaldehyde was added to an ethanolic solution of o-phenylenediamine. The reaction mixture was then heated to reflux. Then, for the formation of the copper(II) complex (CuL), the (H2L) ligand was dissolved in EtOH and an ethanolic solution of Cu(CH3COO)2·H2O was added to this solution under reflux. Next, the Na-Y zeolite was suspended in water containing Cu(NO3)2 and Cu-NaY (Scheme 25, I). For the immobilization of CuL in Cu-NaY, a quantity of Cu-NaY and MeCN solution of H2L were mixed at 100 °C. The resultant material was isolated until the complex was free from the unreacted ligand. The non-complexed metal ions present in the zeolite were eliminated with an aqueous sodium chloride solution (Scheme 25). Finally, the desired encapsulated copper(II) catalyst was efficiently obtained.
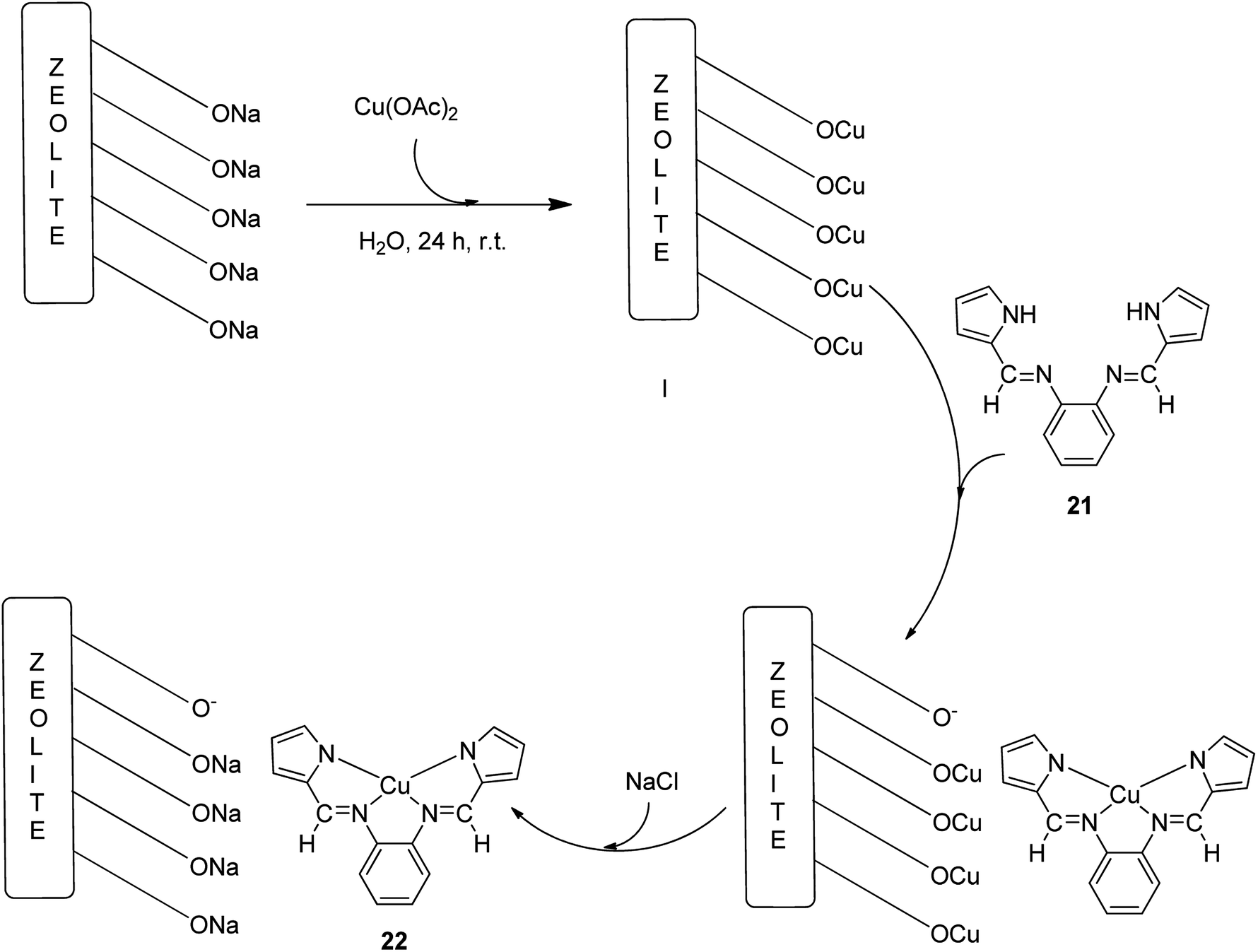 | ||
| Scheme 25 Encapsulation of Cu(II) into NaY (metal exchanged Y-zeolite). | ||
The encapsulated copper(II) catalyst is an active catalyst for the oxidation of cyclohexene and cyclooctene using H2O2 as an oxidant. Notably, catalyst 22 gave 81% conversion of cyclohexene along with two main oxidation products, including 2-cyclohexenone and 2-cyclohexene-1-ol. In addition, 87% conversion of cyclooctene with 46% selectivity for epoxide construction was attained. Remarkably, the calcination of the resultant copper(II)-incorporated zeolite was prevented from arresting the migration of copper(II) ions from the vicinity of the supercage (Scheme 25).
The oxidation reaction of cyclooctene in the presence of 22 as a catalyst using hydrogen peroxide and tert-butyl hydroperoxide (TBHP) afforded cyclooctene oxide as the major product, along with a minor quantity of the alcohol product. Two different oxidants containing H2O2 and tert-butylhydroperoxide were used for the oxidation of cyclooctene. The hexenyl radical reacted with LCu(III)-OH to give 2-cyclohexene-1-ol demonstrated greater conversion and selectivity for epoxide formation. Then, 2-cyclohexene-1-ol reacted with another LCu(III)-OH to afford 2-cyclohexenone (Scheme 26).
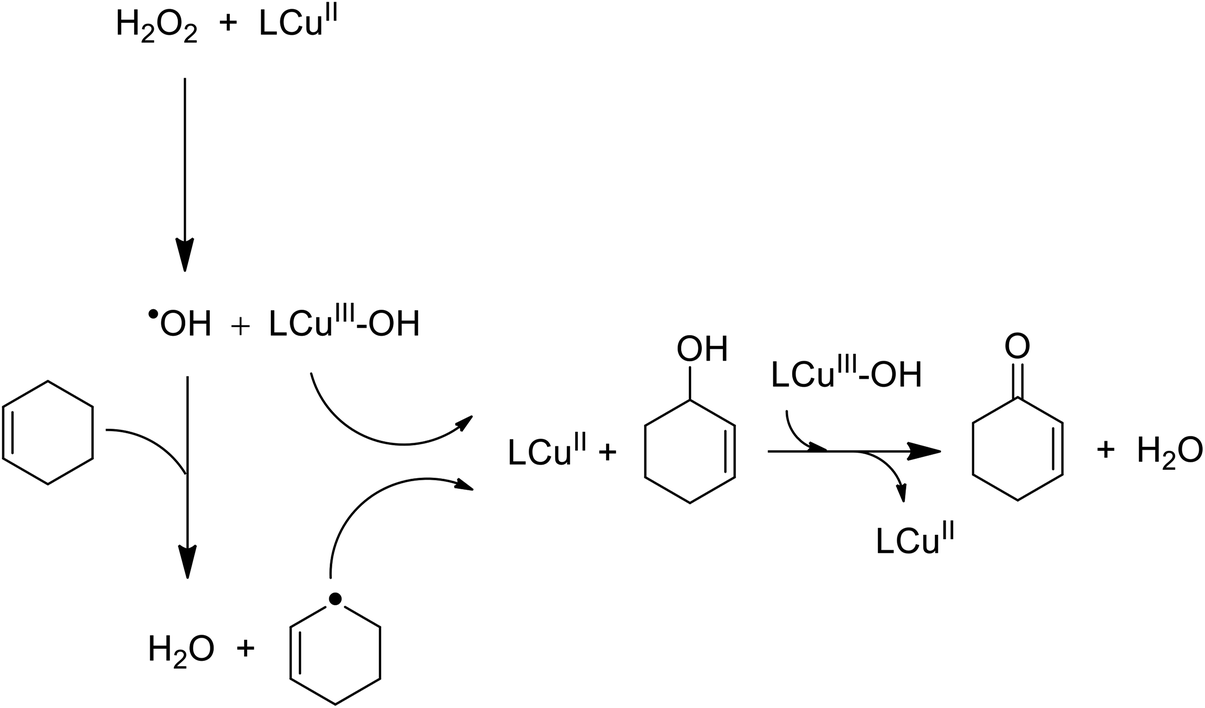 | ||
| Scheme 26 Oxidation reaction of cyclooctene. | ||
Hacpy-oap (23), an ONN donor, was used to form [CuII(acpy-oap)Cl]. The encapsulation into the supercages of zeolite-Y afforded [CuII(acpy-oap)Cl]-Y (Scheme 27). The resultant encapsulated complex is active for the oxidation reaction of cyclohexene and styrene.76 The oxidation reaction of styrene gave 60.2% conversion with two major products, including styrene oxide (78.2%) and benzaldehyde (18.2%). In addition, the oxidation reaction of cyclohexene was accomplished to attain equilibrium with 61.3% conversion and selectivity: cyclohexane-1,2-diol (77.7%) > 2-cyclohexene-1-ol (11.1%) > cyclohexene epoxide (4.7%) > 2-cyclohexene-1-one (4.6%) (Scheme 28). In addition, the neat analogue [Cu(II)(acpy-oap)Cl] is active. However, it exhibits less conversion in comparison with the encapsulated complexes. The oxidation reaction of cyclohexene was accomplished using [(HOO)-CuIII(acpy-oap)Cl], formation, which was demonstrated with electronic absorption spectroscopy. The oxidation of styrene in the presence of [CuII(acpy-oap)Cl]-Y, was performed using 70% tert-butylhydroperoxide as an oxidant to afford benzaldehyde and styrene oxide, along with only minor amounts of unrecognized products.
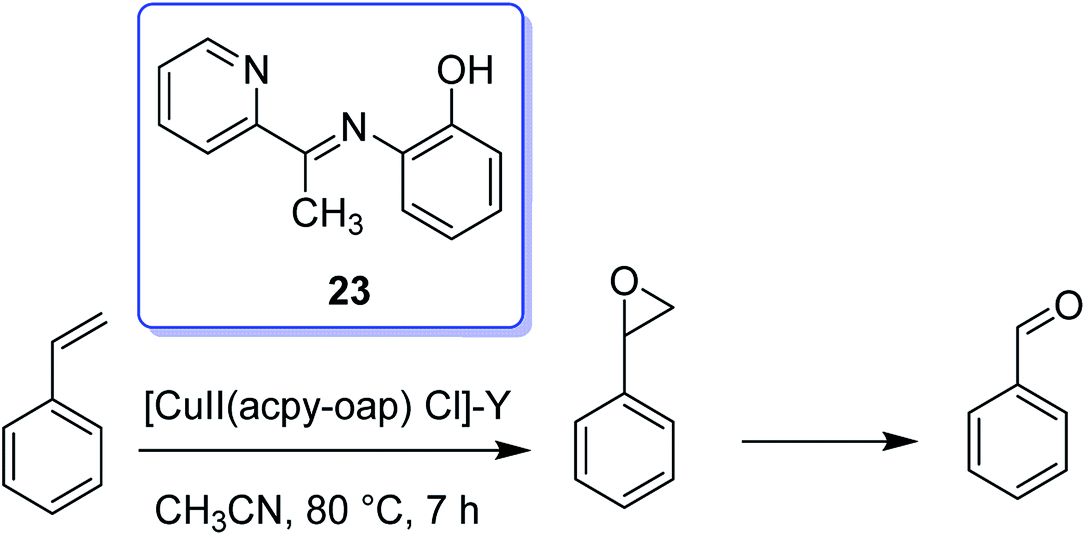 | ||
| Scheme 27 The oxidation of styrene in the presence of [Cu(II)(acpyoap)Cl]-Y. | ||
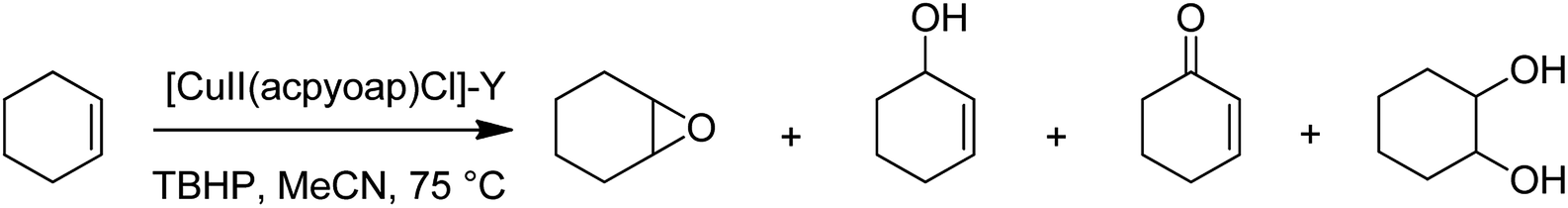 | ||
| Scheme 28 Oxidation of cyclohexene in the presence of [Cu(II)(acpyoap)Cl]-Y. | ||
The copper(II) complex (CuL) (24), a Schiff base ligand provided from the reaction of 3,4-diaminotoluene and 2-hydroxy-naphthaldehyde, was encapsulated into the nanocavity of Y-zeolite (CuL–NaY) (Scheme 29).77 Based on this method, 2-hydroxy-naphtaldehyde was added to an ethanolic solution of 4-methyl-o-phenylenediamine. The reaction mixture was heated to reflux (Scheme 29, II). The ligand (H2L) was dissolved in EtOH and an ethanol solution of Cu(CH3COO)2·H2O was added to this solution under reflux conditions (Scheme 29, III). Next, the Na-Y zeolite was suspended in water containing Cu(NO3)2, and Cu-NaY was provided after 24 h (Scheme 29, I). A quantity of Cu-NaY and MeCN solution of H2L were then mixed and heated at 85 °C for 7 hours. In an oil bath, the resulting material was isolated until the complex was free from the unreacted ligand. Finally, the non-complexed metal ions in the zeolite were eliminated by exchange with aqueous sodium chloride solution (Scheme 29, IV).
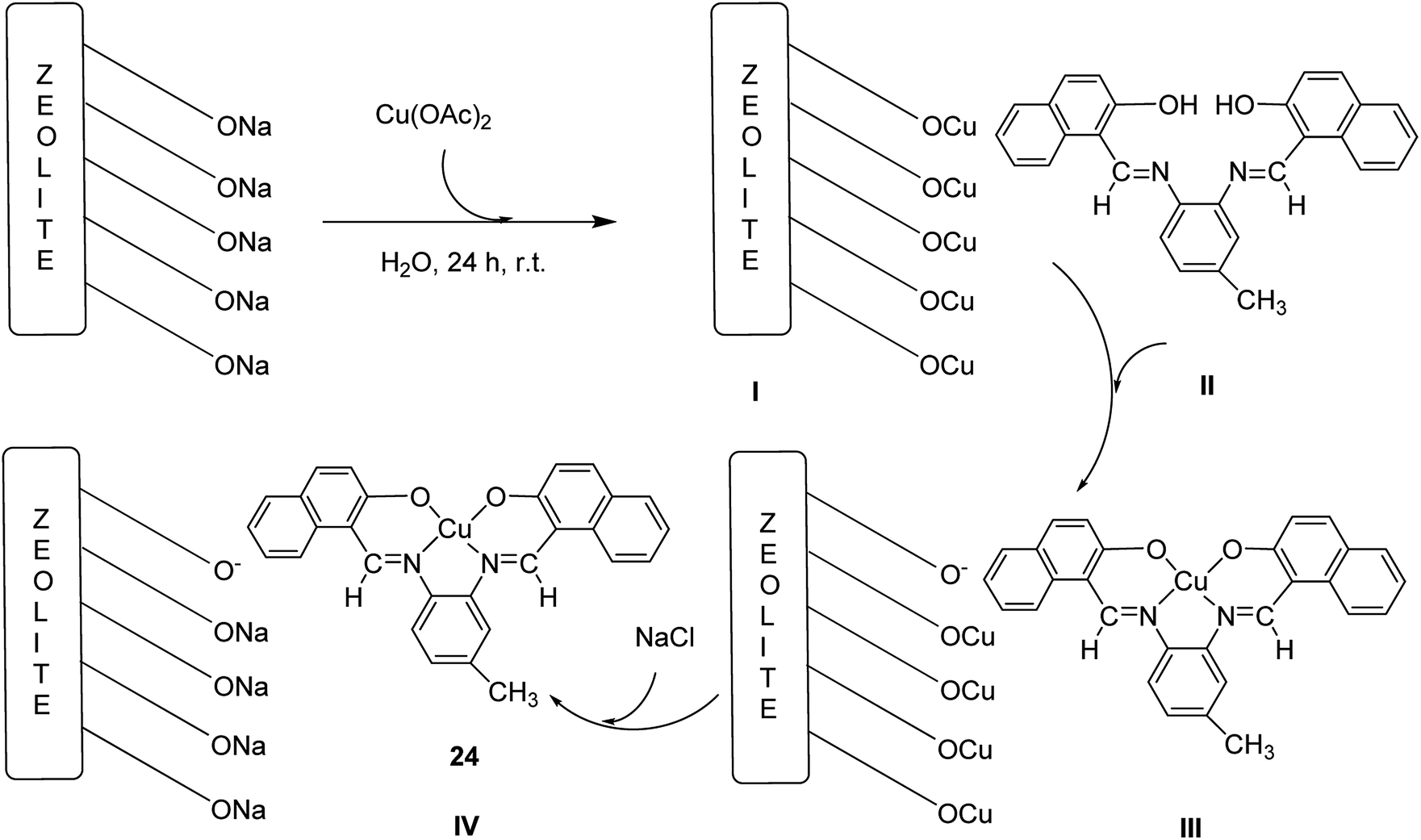 | ||
| Scheme 29 The formation of Cu-NaY 24. | ||
The oxidation of olefins in the presence of CuL and CuL–NaY as an efficient catalyst using tert-butylhydroperoxide as an oxidant was achieved to provide the desired products in moderate yields. The oxidation of cyclooctene, methyl styrene, styrene and cyclohexene in the presence of CuL gave 83%, 84%, 81% and 56% conversion, respectively. In addition, the oxidation of cyclooctene, styrene, α-methyl styrene and cyclohexene in the presence of CuL–NaY gave 100%, 84%, 79% and 45% conversion, respectively.
The copper(II) complexes of 16- and 17-membered diazadioxamacrocycles, [L1]; 3,4,11,12-dibenzo-1,14-diaza-5,10-dioxacyclohexadecane-1,13-diene, [L2]; 3,4,11,12-dibenzo-1,14-diaza-5,10-dioxacycloheptadecane-1,13-diene, and [L3]; 3,4,11,12,15,16,17-tribenzo-1,14-diaza-5,10-dioxacycloheptadecane-1,13-diene, were encapsulated into the nanopores of zeolite-Y and synthesized through the template condensation reaction of diamine and 1,4-bis(2-carboxyaldehydephenoxy)butane [L′]; Cu[L1]2+-Y, Cu[L2]2+-Y and Cu[L3]2+-Y (Scheme 30).78 The neat and encapsulated complexes were examined in the oxidation of cyclooctene with tert-butyl hydroperoxide as the oxidant using different solvents. The supported Cu[L1]2+-Y showed a moderate 81.9% selectivity for epoxidation with 84.2% conversion.
 | ||
| Scheme 30 Encapsulation of Cu(II) complex nanoparticles of 16- and 17-membered diazadioxamacrocycles in nanopores of zeolite-Y. Reprinted from (ref. 78) with permission of the Royal Society of Chemistry. | ||
A copper(II) C2-symmetric bis(oxazoline), CuBox (28), was shown in two forms of marketable Y-zeolite: a sodium form (NaY) and an ultrastable form (NaUSY). CuBox was prepared via first partially replacing the sodium cations of both zeolites for copper(II), and then under reflux with the resultant precursors using a solution of bis(oxazoline) (Box) (29) (Fig. 4). Two different loadings were provided for each form of zeolite.79 These complexes were all active in the cyclopropanation of styrene with ethyldiazoacetate at room temperature, and were diastereoselective toward trans cyclopropanes. Therefore, the CuBox encapsulated into NaUSY is a more active catalyst in the cyclopropanation reaction of styrene at ambient temperature compared with NaY as the support. In addition, it was demonstrated that the catalytic activity increases with the amount of Box encapsulated. However, NaUSY showed greater activity in spite of the lower Box content, which can be identified with a stronger interaction of the zeolite and Box as a result of dealumination reactions. The CuBox@Y1, CuBox@Y2, CuBox@USY1 and CuBox@USY2 complexes were examined as heterogeneous catalysts in the cyclopropanation reaction of styrene and ethyldiazoacetate at ambient temperature (Scheme 31). These complexes served as significant heterogeneous catalysts in the cyclopropanation of styrene, and a side-product from the ethyldiazoacetate dimerization, diethyl fumarate. In addition, the materials were diastereoselective toward the trans cyclopropanes and the trans for 3 hours of reaction. The Box involving CuBox@Y2 and CuBox@USY2 complexes exhibited enantioselectivity (ee) for both trans and cis cyclopropane isomers: 8% (trans) and 6% (cis) for NaY and 5% (trans) and 4% (cis) for USY. It is noteworthy that in the case of CuBox@USY2, the enantioselectivity is dependent on the reaction time and is reduced with time. Therefore, the cis and trans cyclopropane ees are very poor in comparison with those that result once the reaction is performed in homogeneous cases based on the similar reaction conditions as the heterogeneous ones.
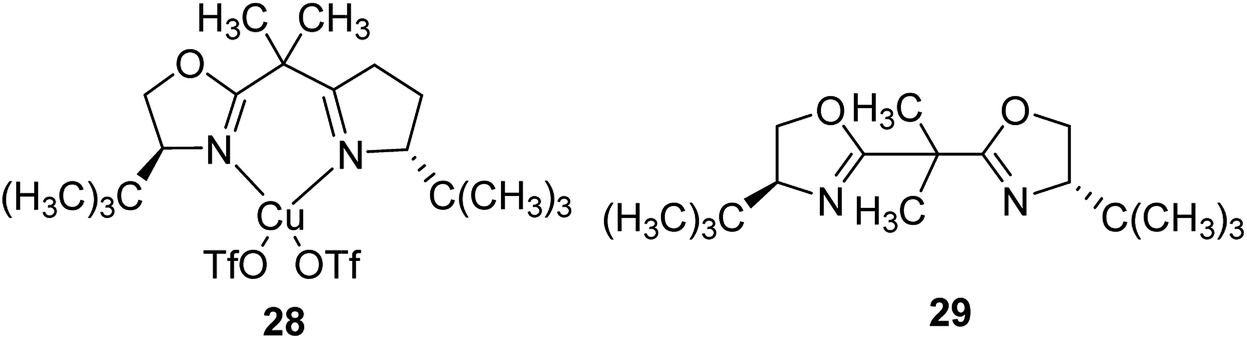 | ||
| Fig. 4 Structure of the CuBox (28) and Box ligand (29). | ||
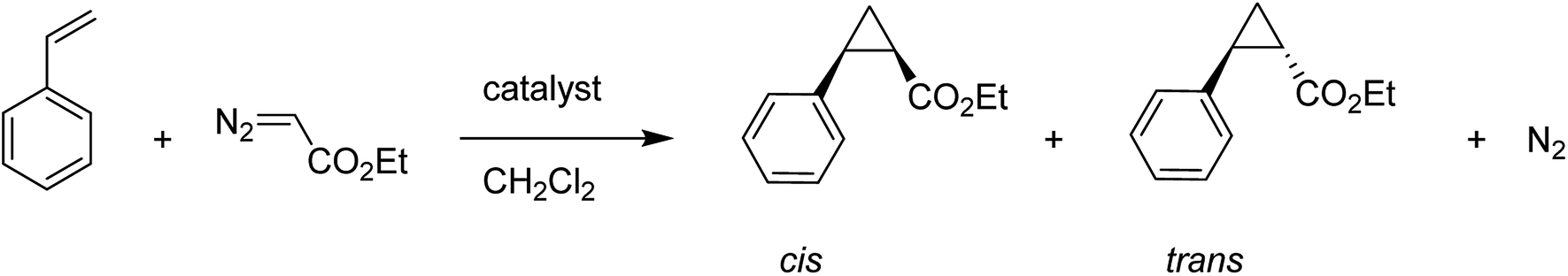 | ||
| Scheme 31 Cyclopropanation reaction of styrene with ethyldiazoacetate. | ||
The Cu(II) complex of dimethylglyoxime (dmgH2) 30 and N,N-ethylenebis(7-methylsalicylideneamine) (Me2salen) 31 were encapsulated in situ into the Y-zeolite using the reaction of ion-exchanged metal ions through the flexible ligand method, which had diffused into the cavities (Fig. 5).80 Data analysis exhibited the formation of complexes in the pores without influencing the zeolite framework structure, the absence of any unnecessary species and the geometry of the encapsulated complexes. The catalytic activity for hydrogen peroxide decomposition and the oxidation reaction of benzyl alcohol and ethylbenzene using zeolite complexes were examined. Of significance, the zeolite complexes were stable to be recycled and were appropriate to be employed as partial oxidation catalysts. The activity increases on elevating the reaction temperature from 323 to 343 K. The results demonstrated that YCu(dmgH)2 experienced a higher degree of deactivation (53% loss in activity), while YCuMe2salen exhibited improved poison-tolerance (Table 14). It is widely supposed that the stereochemical environment of the active site includes an extreme effect on the mobility of the molecules at the reaction center and therefore, on the poison-tolerance. Moderately superior activity of YCu(dmgH)2 in comparison with YCuMe2salen could be related to the increased mobility of the reactants at the active sites. The encapsulation of cobalt(II), nickel(II) and copper(II) complexes of dmgH2 and Me2salen ligands was accomplished into the Y-zeolite via a flexible ligand method. The ligands, that are flexible enough to diffuse via the zeolite channels, react with the pre-exchanged metal ions in the supercage to yield the encapsulated complexes.
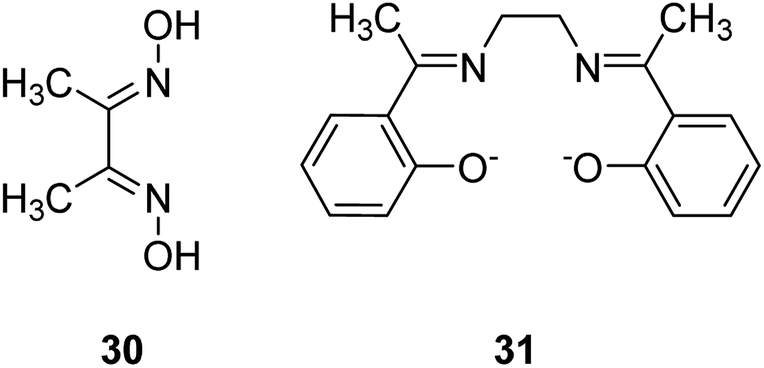 | ||
| Fig. 5 Structure of compounds 30 and 31. | ||
| Sample | Substrate | Oxidant | Temperature (K) | Percent conversion (wt%) | TOF |
|---|---|---|---|---|---|
| YCu(dmgH)2 | Benzyl alcohol | H2O2 | 323 | 30.0 | 90 |
| 333 | 40.8 | 123 | |||
| 343 | 52.6 | 158 | |||
| Ethylbenzene | H2O2 | 323 | 24.0 | 72 | |
| 333 | 33.6 | 101 | |||
| 343 | 46.3 | 139 | |||
| Benzyl alcohol | O2 | 323 | 18.6 | 56 | |
| Ethylbenzene | O2 | 323 | 16.8 | 51 | |
| Benzyl alcohol (poisoned) | H2O2 | 323 | 14.2 | 43 | |
| YCuMe2salen | Benzyl alcohol | H2O2 | 323 | 28.3 | 84 |
| 333 | 36.8 | 109 | |||
| 343 | 47.7 | 141 | |||
| Ethylbenzene | H2O2 | 323 | 23.1 | 68 | |
| 333 | 29.4 | 87 | |||
| 343 | 39.2 | 116 | |||
| Benzyl alcohol | O2 | 323 | 16.8 | 50 | |
| Ethylbenzene | O2 | 323 | 11.1 | 33 | |
| Benzyl alcohol (poisoned) | H2O2 | 323 | 20.5 | 61 |
To mimic the structure and function of the enzyme, metal-imidazolesalen complexes were provided in situ in the cavities of the Y zeolite. Zeolite-encapsulated Cu salen II demonstrated appropriate catalytic activity in the selective oxidation reaction of benzyl alcohol. In particular, the efficacy and selectivity of the zeozyme were compared to those of the free copper-salen II complex in solution. The resultant zeozyme concurrently contains high activity and homogeneous catalysts. The copper(II), complex of 2-methylimidazole (2-MeImzlH) was encapsulated into the supercages of zeolite-Y ([Cu(2-MeImzlH)]-Y) 32 (Scheme 32).81 Oxidation of benzyl alcohol in the presence of these encapsulated complexes gave only benzaldehyde as the desired product. In addition, the oxidation of phenol was accomplished in the presence of [Cu(2-MeImzlH)]-Y as a significant catalyst using H2O2 as an oxidant in acetonitrile. The two products hydroquinone and catechol were identified in the reaction mixture with a mass balance of 95%.
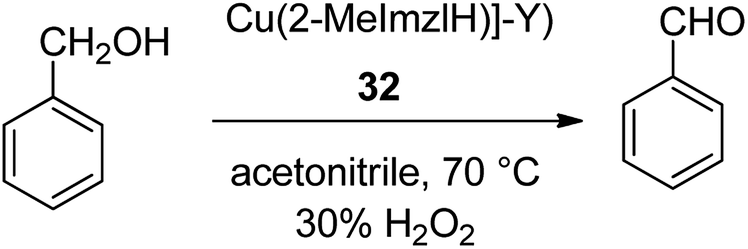 | ||
| Scheme 32 Oxidation reaction of benzyl alcohol in the presence of catalyst 32. | ||
The copper(II) complex of 4-((E)-(4-(1Hbenzo[d]imidazol-2-yl)phenyl)diazenyl)-2-((E)-(4-hydroxyphenylimino)methyl)phenol (HL) 33 was encapsulated into zeolite NaY through the flexible ligand method (Scheme 33).82 The neat and encapsulated complexes were examined in the oxidation of benzyl alcohol and the aldol condensation reaction. Notably, the encapsulated complex was more stable and active than the relevant neat complex.
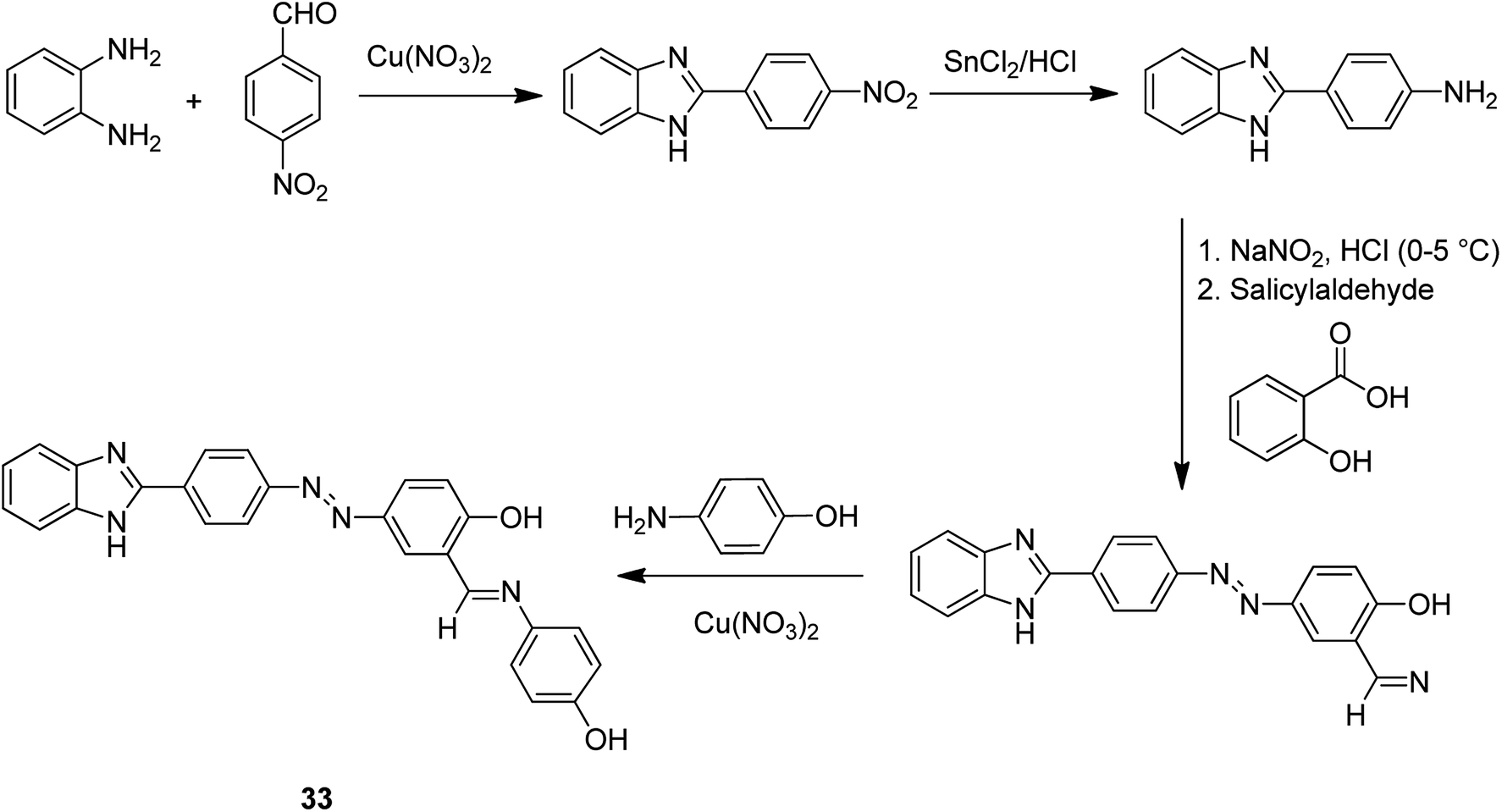 | ||
| Scheme 33 Common method for the formation of complex 33. | ||
This reaction was demonstrated to be very dependent on the effect of the solvent. The results exhibited that once the reaction was performed in solvents having coordinative capability like EtOH and THF, the yield of the reaction exhibited an extreme reduction. This is possibly related to the strong coordination of the solvent via oxygen atoms to acidic sites of the catalyst. In addition, the polarity of CH2Cl2 is greater than CHCl3. The mechanism of the reaction possibly also includes a polarized transition state. Consequently, it is not unreasonable that CH2Cl2, in comparison to CHCl3, improved the yield and stability of the transition state. Thus, CH2Cl2 was found to be the optimal solvent in this reaction.
It is noteworthy that the leaching from the supercages of zeolite was not detected during the reactions with this encapsulated complex.
The activity of this type of catalyst was examined in the benzyl alcohol oxidation. The corresponding products of the benzyl alcohol oxidation are shown in Scheme 34. Zeolite NaY, Cu(II)/Y, the encapsulated complex and free complex were applied and compared in this approach.
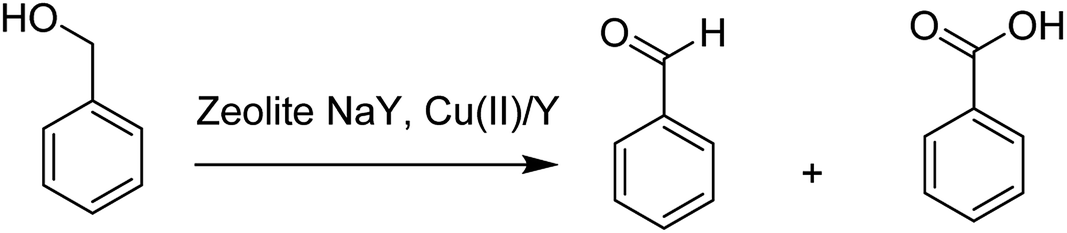 | ||
| Scheme 34 The oxidation reaction of benzyl alcohol in the presence of zeolite NaY and Cu(II)/Y. | ||
Moreover, the catalytic activity of the resultant complexes was explored in the acid-catalyzed aldol condensation. The reaction of benzaldehyde and cyclohexanone was achieved in the presence of zeolite NaY/Cu(II)/Y via aldol reaction (Scheme 35). Remarkably, the reactivity of the aldol reaction was intensely affected by the nature of the solvent, and the nature and quantity of catalyst.
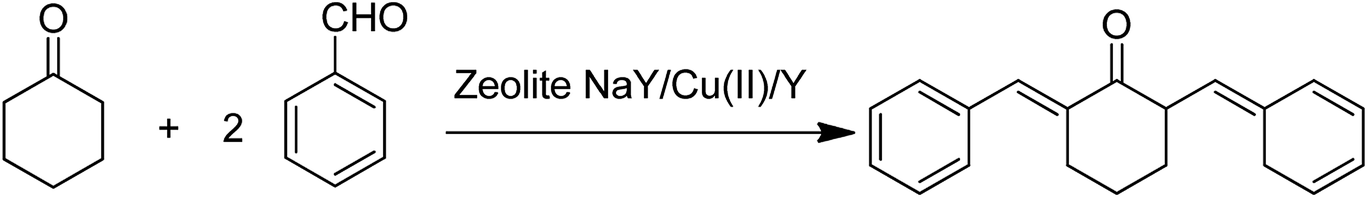 | ||
| Scheme 35 Aldol condensation of cyclohexanone and benzaldehyde using zeolite NaY, Cu(II)/Y. | ||
Metal-imidazole-salen complexes were encapsulated in situ in the cavities of Y zeolite by Song and co-workers 2018.83 The zeolite-Y encapsulated metal-salen was designated as M-salenY 34. The chemical structures and synthetic routes of the metal-salen-comprising samples are shown in Scheme 36. The catalytic activities of MY, M-salen and encapsulated M-salenIIY samples for the selective oxidation of benzyl alcohol were explored. The results demonstrated that the activity of the catalyst followed the order: Cu-salenII > Cu-salenI > Zn-salenII > Zn-salenI. For the similar ligand, copper(II)-salen complexes were more active than Zn salen. These results showed that the inherent properties of the M-salen complexes, including their electronic configuration and geometry, exhibited a key role in identifying their catalytic activities. In addition, the oxidation mechanism may be different for copper(II) and zinc(II) complexes, with Zn(II) salen(I/II) catalysts containing a redox inactive central ion. Thus, Cu-salen(II), comprising lower oxidation state and a planar tetragonal structure, illustrated the highest benzyl alcohol oxidation activity.
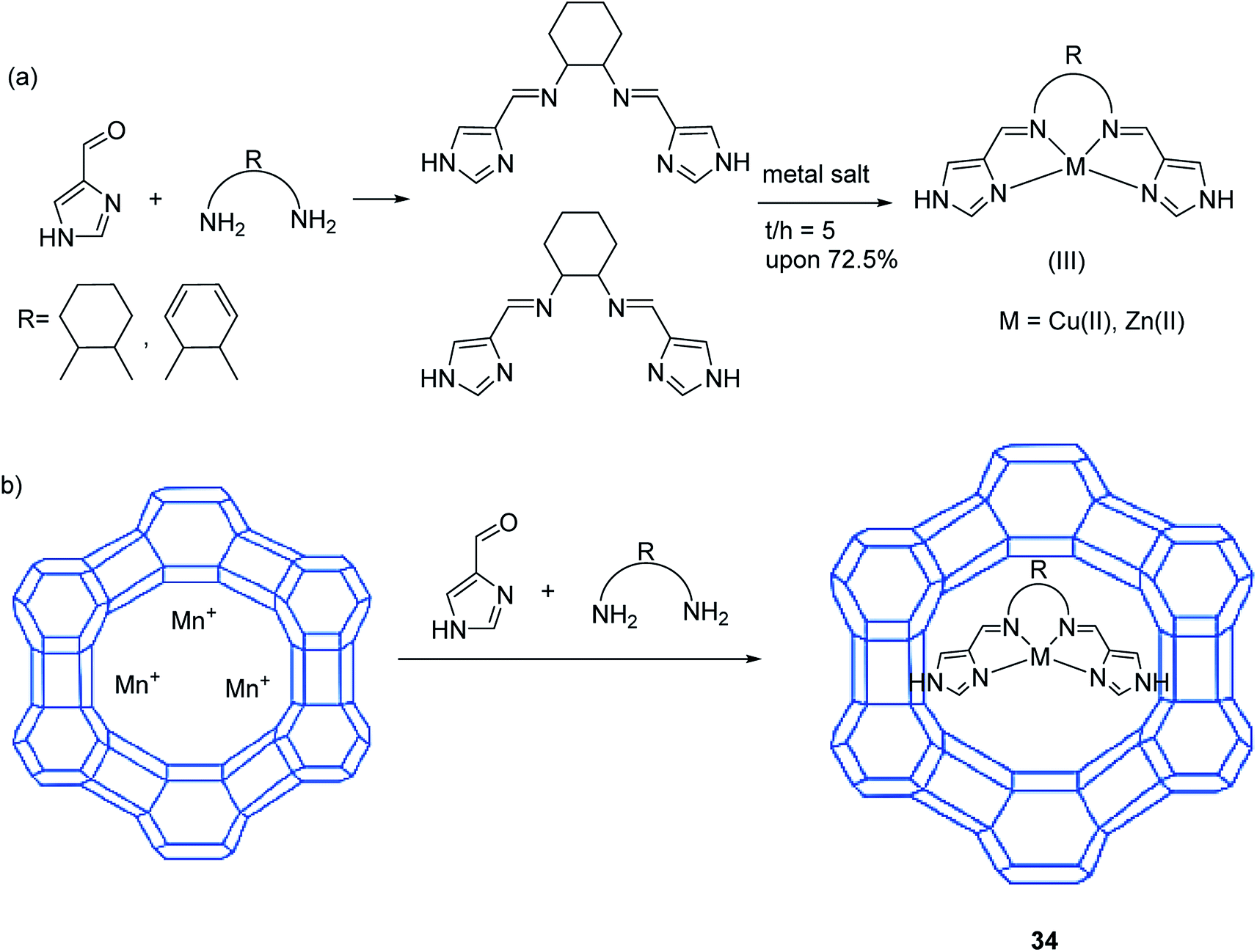 | ||
| Scheme 36 Construction of the zeolite-encapsulated metal-salen complexes. Reprinted from (ref. 83) with permission of the Royal Society of Chemistry. | ||
Copper(II) complexes of the 12-membered macrocyclic ligands bearing three different donating atoms (N2S2 (35), N2O2 (36) and N4 (37)) in the macrocyclic ring were encapsulated into the nanocavity of zeolite-Y via the flexible ligand method (Scheme 37).84 It is noteworthy that the copper(II) complexes having macrocyclic ligands were entrapped in the nanocavity of zeolite-Y. Remarkably, the neat and encapsulated complexes exhibited satisfactory catalytic activity in the oxidation of ethylbenzene at 333 K, using tert-butylhydroperoxide (TBHP) as the oxidant. Moreover, acetophenone was the major product synthesized when using small amounts of o- and p-hydroxyacetophenones, showing that the carbon–hydrogen bond activation occurs both at the benzylic and aromatic ring carbon atoms (Table 15). Noticeably, ring hydroxylation reaction was more over the neat complexes in comparison to over the encapsulated complexes.
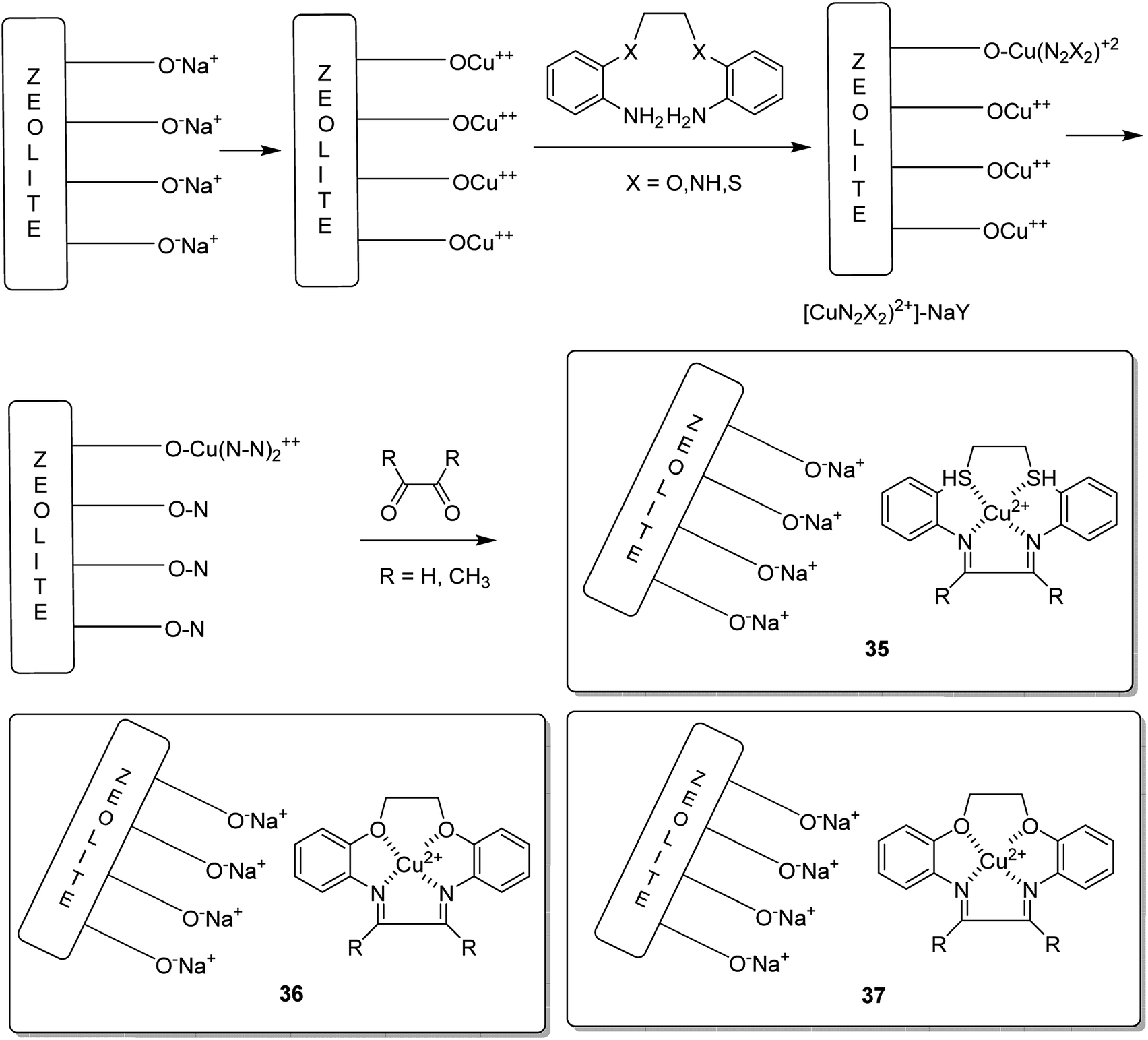 | ||
| Scheme 37 The formation of Cu(II) complexes of the 12-membered macrocyclic ligand. | ||
| Catalyst | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|
| Acetophenone | o- /p-Hydroxyacetophenone | Benzaldehyde, benzoic acid and 1-phenylethanol | ||
| a Reaction condition: catalyst, TBHP, ethylenedichloride, CH3CN, T = 333 K. | ||||
| Cu(II)-NaY, [Cu([H]2-N4)]2+-NaY, [Cu([H]2-N4)]2+-NaY, [Cu([H]2-N4)]2+-NaY, [Cu([CH3]2-N4)]2+-NaY, [Cu([H]2-N2O2)]2+-NaY, [Cu([CH3]2-N2O2)]2+-NaY, [Cu([H]2-N2S2)]2+-NaY, [Cu([CH3]2-N2S2)]2+-NaY | 30.7–58.2 | 62.7–97.3 | 2.7–9.8 | 40.5 |
The encapsulation of various copper(II) complexes including [12]ane N4: 1,4,7,10-tetraazacyclododecane-2,3,8,9-tetraone [Cu([12]aneN4)]2+ (38); [14]aneN4: 1,4,8,11-tetraazacyclotetradecane-2,3,9,10-tetraone[Cu([14]aneN4)]2+ (39); Bzo2[12]aneN4: dibenzo-1,4,7,10-tetraazacyclododecane-2,3,8,9-tetraone and Bzo2[14]aneN4 [Cu(Bzo2[12]oneN4)]2+ (40) and dibenzo-1,4,8,11-tetraazacyclotetradecane-2,3,9,10-tetraone[Cu(Bzo2[14]oneN4)]2+ (41) into the nanopores of zeolite-Y (42–45) was achieved using the in situ one-pot condensation reaction (Schemes 38 and 39).85
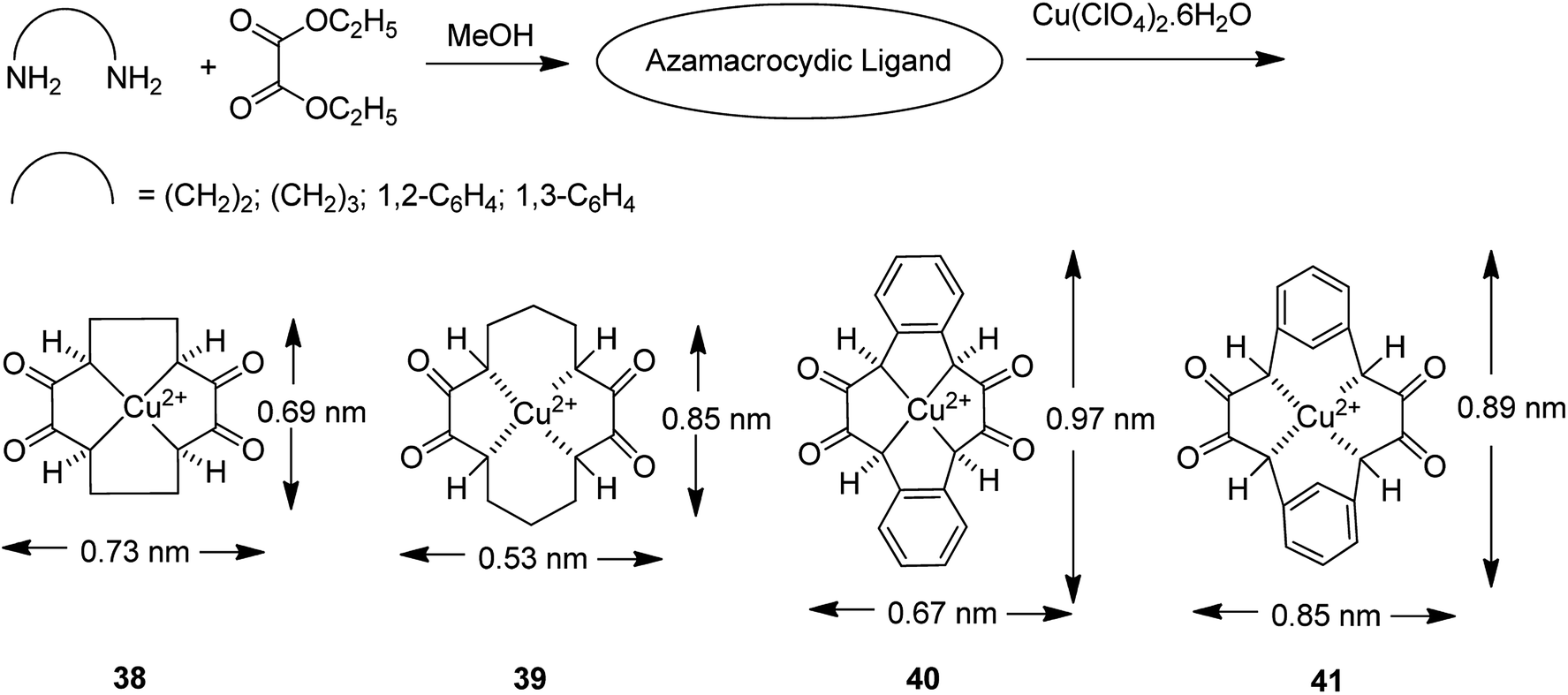 | ||
| Scheme 38 Synthesis of [Cu(azamac.)]2+. | ||
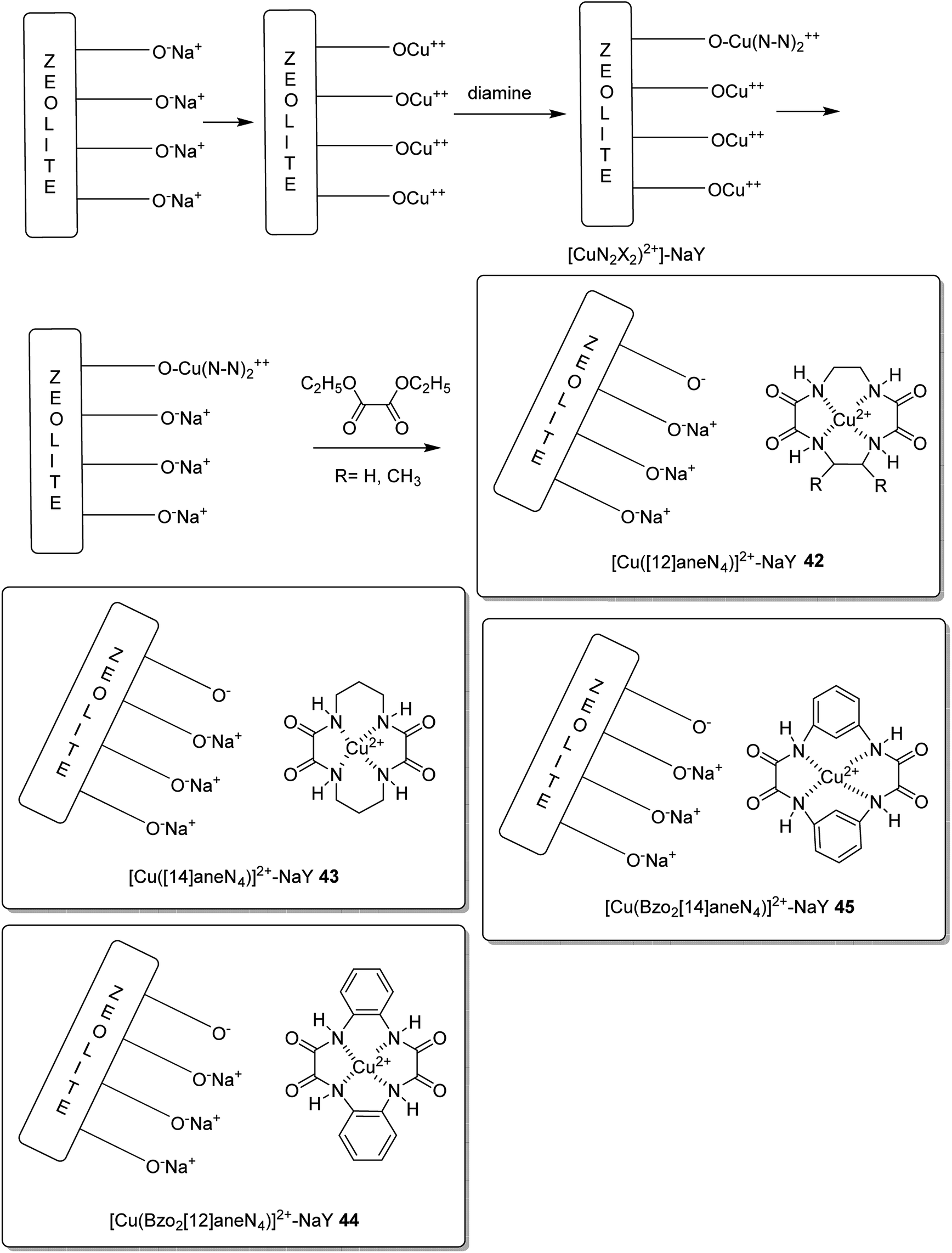 | ||
| Scheme 39 Encapsulation of [Cu(azamac.)]2+ into the nanocavity of zeolite-Y. | ||
The neat and encapsulated complexes exhibited satisfactory catalytic activity in the oxidation of ethylbenzene using tert-butyl hydroperoxide (TBHP) as the oxidant at 333 K. Remarkably, acetophenone was the main product synthesized using small amounts of o- and p-hydroxyacetophenones, exhibiting that C–H bond activation occurs both at the aromatic and benzylic ring carbon atoms. The ring hydroxylation performed much better than the neat complexes, in comparison to its performance over that of the encapsulated complexes. Transformation for copper(II) complexes with different azamacrocyclic ligands decreased in the order: Bzo2[14]aneN4 > [12]aneN4 > Bzo2[14]aneN4 > [14]aneN4 (for neat and encapsulated complexes). The encapsulated complexes demonstrated higher activity in comparison with the neat complexes (Tables 16 and 17).
| Catalyst | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|
| Acetophenone | o- /p-Hydroxyacetophenone | Benzaldehyde | ||
| a Reaction condition: ethylbenzene in ethylenedichloride, catalyst, TBHP, acetonitrile, T = 333 K. | ||||
| [Cu([12]aneN4)](ClO4)2 [Cu([12]aneN4)](ClO4)2, [Cu(Bzo2[12]aneN4)](ClO4), [Cu(Bzo2[12]aneN4)](ClO4)2[Cu(Bzo2[12]aneN4)](ClO4)2, [Cu(Bzo2[12]aneN4)](ClO4)2[Cu(Bzo2[12]aneN4)](ClO4)2 | 31.9–56.5% | 86.3% | 22.7% | 19.7% |
| Catalyst | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|
| Acetophenone | o- /p-Hydroxyacetophenone | Benzaldehyde | ||
| a Reaction condition: ethylbenzene, catalyst, TBHP in ethylenedichloride, acetonitrile, T = 333 K. | ||||
| [Cu([12]aneN4)]2+-NaY, [Cu([12]aneN4)]2+-NaY, [Cu(Bzo2[12]aneN4)]-NaY, [Cu(Bzo2[12]aneN4)]-NaY, [Cu(Bzo2[12]aneN4)]-NaY, [Cu(Bzo2[12]aneN4)]NaY, [Cu(Bzo2[12]aneN4)]-NaY | 31.4–52.9% | 99.0% | 3.8% | 40.5% |
The [Cu(terpy)]2+ complexes in the Na-Y zeolite ([Cu(terpy)]2+@Y) (terpy = 2,2′:6′,2′′-terpyridine) were encapsulated and their catalytic activity was explored in the oxidation reaction of sulfides with hydrogen peroxide.86 The oxidation of thioanisole, benzene and 2-phenethylamine in the presence of [[Cu(terpy)]2+@Y]*, generated by the reaction of [Cu(terpy)]2+@Y and hydrogen peroxide, was accomplished as shown in Scheme 40.
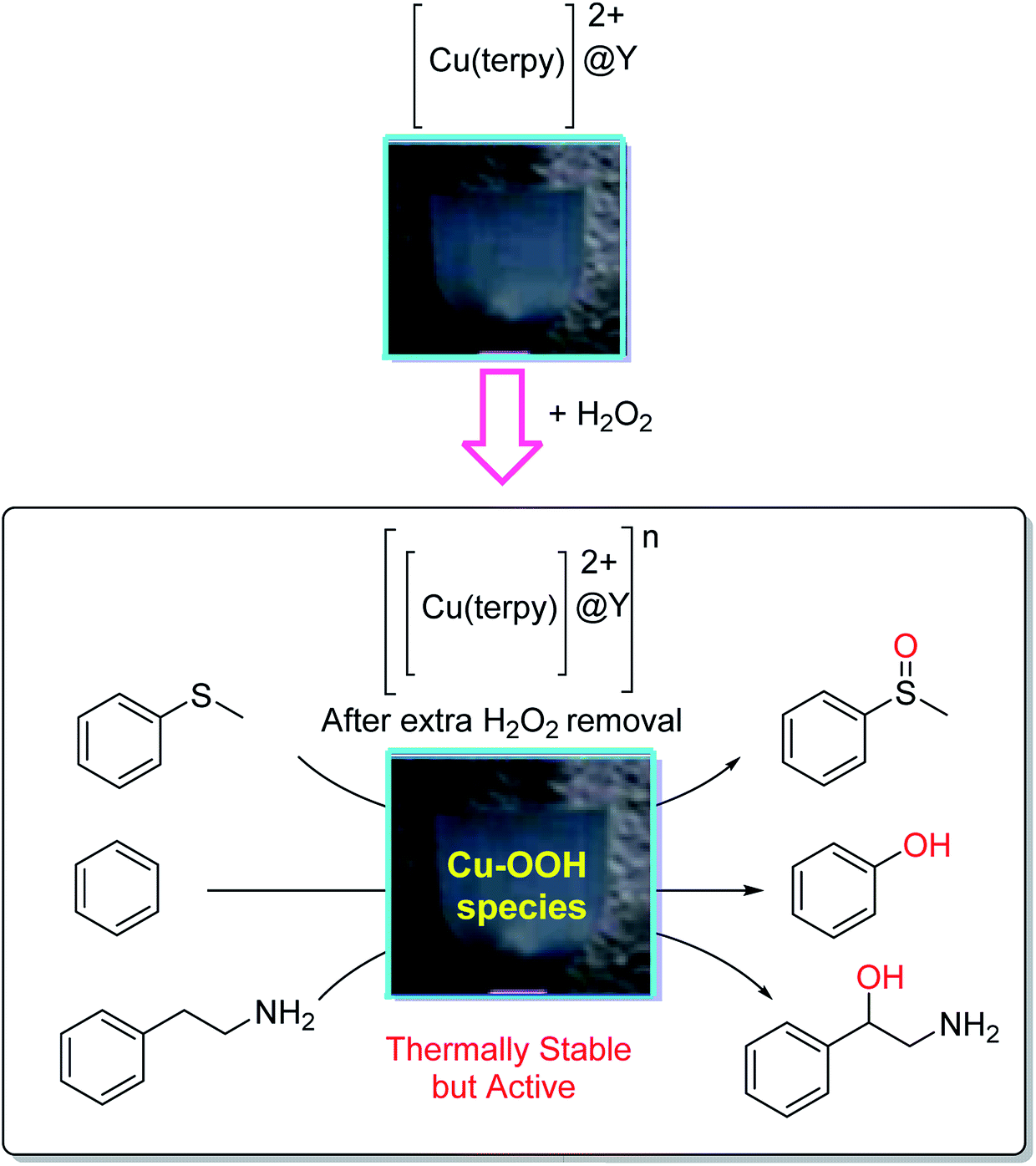 | ||
| Scheme 40 Reaction of [Cu(terpy)]2+@Y and hydrogen peroxide to form the thermally stable, but active, CuII-OOH species in [Cu(terpy)]2+@Y. Reprinted from (ref. 86) with permission of the Royal Society of Chemistry. | ||
CuCl2 complexes were easily prepared within the pores of zeolite X, with the modified zeolite found to serve as a heterogeneous catalyst for the oxygenation reactions of various enamines using molecular oxygen (Scheme 41).87 It is noteworthy that this catalyst can be recycled without leaching the Cu species, and the catalyst preserved its excellent catalytic activity and selectivity in this oxygenation reaction.
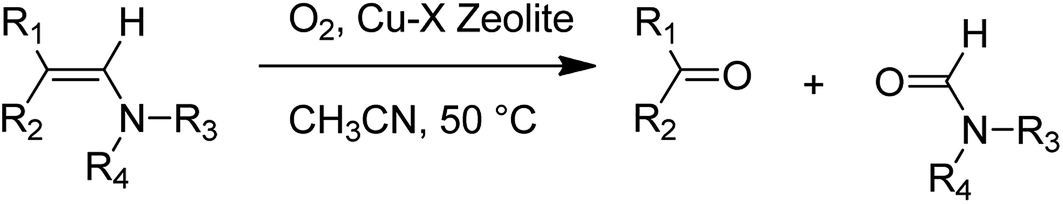 | ||
| Scheme 41 Oxygenation reaction of enamines catalyzed by the Cu(Cl)-X zeolite. | ||
Kaneda and co-workers in 1982 demonstrated that the oxygenation reaction catalyzed by Cu(Cl)-X might contain a one-electron transfer from the enamine to molecular O2 in a ternary Cu2+ complex attached to enamine and O2, followed by the formation of Cu2+ and a dioxetane intermediate.88 These important results for the oxygenation reaction of enamines with Cu-X in MeCN-DCE are demonstrated in Table 18. The carbon double bonds of different enamines were easily eliminated to afford the corresponding ketones and amides. For bulky enamines (for instance, 1-(4-morpholino)-2,2-diphenylethene), the oxygenation rate with Cu-X was slower in comparison with that of a homogeneous copper(II) chloride catalyst. This different activity between the heterogeneous Cu-X and the homogeneous copper(II) chloride catalyst might be attributed to a shape selective effect of the Cu2+ species in the three-dimensional zeolite pores. In addition, the Cu-X catalyst was active for the oxygenation reaction of 2,3-dimethylindole.
| Substrate | t/h | Conversion (%) | Yield (%) | Products (%) |
|---|---|---|---|---|
| Amide | Ketone | |||
| a Reaction condition: CuX, MeCN, 50 °C, O2 atmosphere. | ||||
| 1-(2-Methylprop-1-en-1-yl)piperidine, 4-(2-methylprop-1-en-1-yl)morpholine, (E)-4-(2-methylpent-1-en-1-yl)morpholine, 4-(2-ethylbut-1-en-1-yl)morpholine, (cyclohexylidenemethyl)morpholine, 4-(2,2-diphenylvinyl)morpholine, 2,3-dimethyl-1H-indole | 5 | 91–100% | 84–97% | 66–97% |
Salavati-Niasari and co-workers in 2003 demonstrated the formation of various copper(II) complexes of 14-membered hexaazamacrocyclic ligand “1,3,6,8,10,13-hexaazacyclotetradecane, 1,8-dimethyl-“[Cu((Me)2[14]aneN6)](ClO4)2” 47, 1,8-diethyl-“[Cu((Et)2[14]aneN6)](ClO4)2” 48, 1,8-dibuthyl-“[Cu((Bu)2[14]aneN6)](ClO4)2” 49 and 1,8-dibenzyl-1,3,6,8,10,13-hexaazacyclotetradecane “[Cu((benzyl)2[14]aneN6)](ClO4)2” 50”.89 Notably, these complexes were entrapped in the supercage of zeolite Y via a two-step route in the liquid phase, including the inclusion of a Cu(II) precursor complex, [Cu(en)2]2+-NaY 53, and the spontaneous one-pot reaction of the copper(II) precursor complex using an amine and formaldehyde (Scheme 42). These complexes 55–57 were applied in the oxidation reaction of tetrahydrofurane using H2O2 as the oxygen donor. Remarkably, the resultant encapsulated complexes gave mostly THF-2-ol and a slight quantity of THF-2-one. While using their homogenous counterparts, the selectivities to THF-2-one are better.
 | ||
| Scheme 42 The formation of Cu(II) complexes of the 14-membered hexaazamacrocyclic ligand. | ||
The 14-membered hexaazamacrocycle Cu(II) encapsulated complexes in zeolite (54–57) were synthesized using a stepwise approach via flexible ligand method.9 As a result, the copper(II) complexes of hexaaza ligand encapsulated into the zeolite pore were synthesized efficiently (Scheme 43).
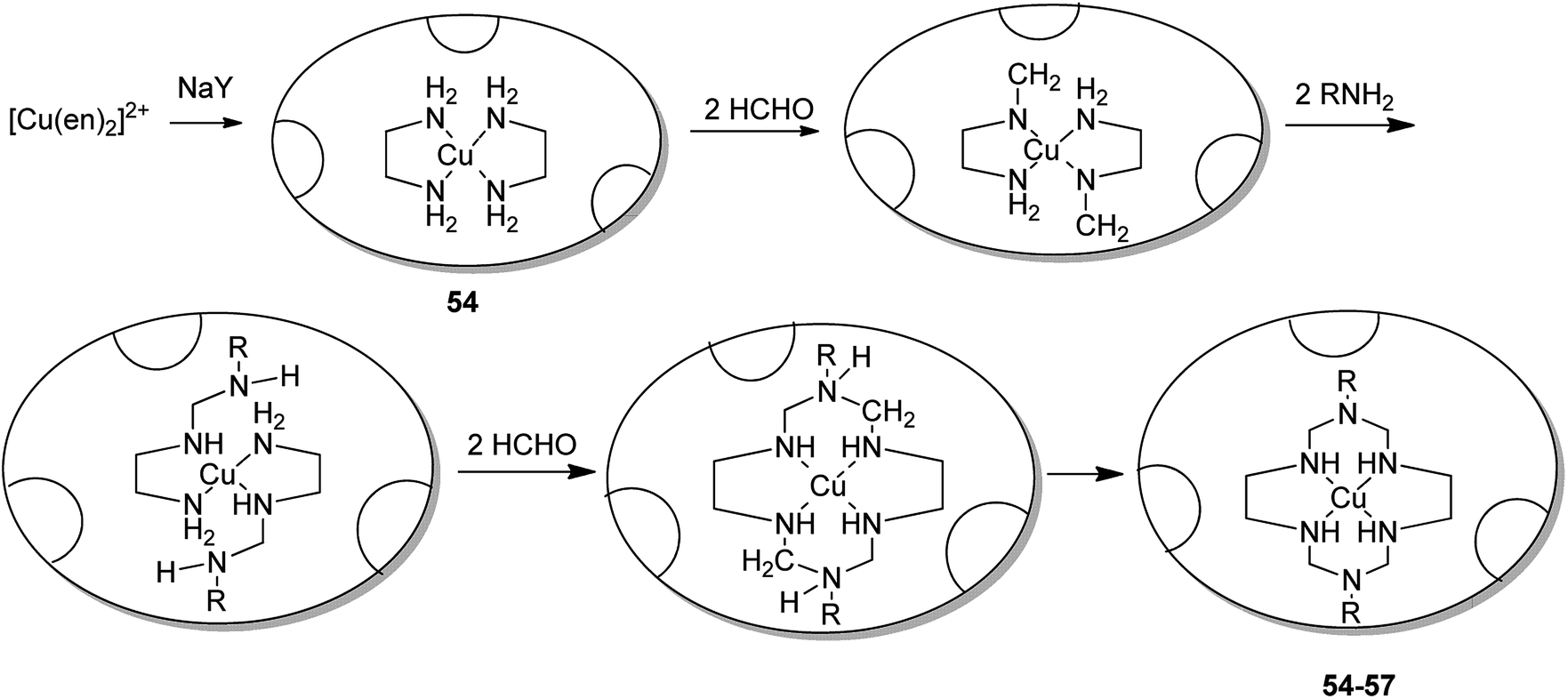 | ||
| Scheme 43 The encapsulation of Cu(II) complexes of the hexaaza ligand inside the zeolite pore. | ||
Schiff base ligand O,O′-trimethyl bis(salicylidene isonicotinoyl hydrazone) (H2L) (60), synthesized by treatment of 1,3-bis(2-carboxyaldehydephenoxy) propane (SAL) (58) and isonicotinyl hydrazine (INH) (59) with Ni(II) and Cu(II) perchlorates (Scheme 44),90 was encapsulated into the cavities of the zeolite via a fixed ligand method. Both free complexes and encapsulated complexes have been examined in the liquid phase oxidation reaction of benzhydrol/hydrogen peroxide, and the photodegradation of rhodamine-B (RhB) using UV/visible (H2O2) irradiation. The catalytic activities of the benzhydrol oxidation and rhodamine-B degradation were greater in comparison with those of the neat complexes and encapsulated complexes, respectively. It is noteworthy that in most cases, the copper(II) [Cu(II)L-Y, Cu(II)L2ClO4] complexes demonstrated greater activity in comparison with the nickel(II) complexes [Ni(II)L2ClO4, Ni(II)L-Y] in both benzhydrol and rhodamine-B degradation reactions.
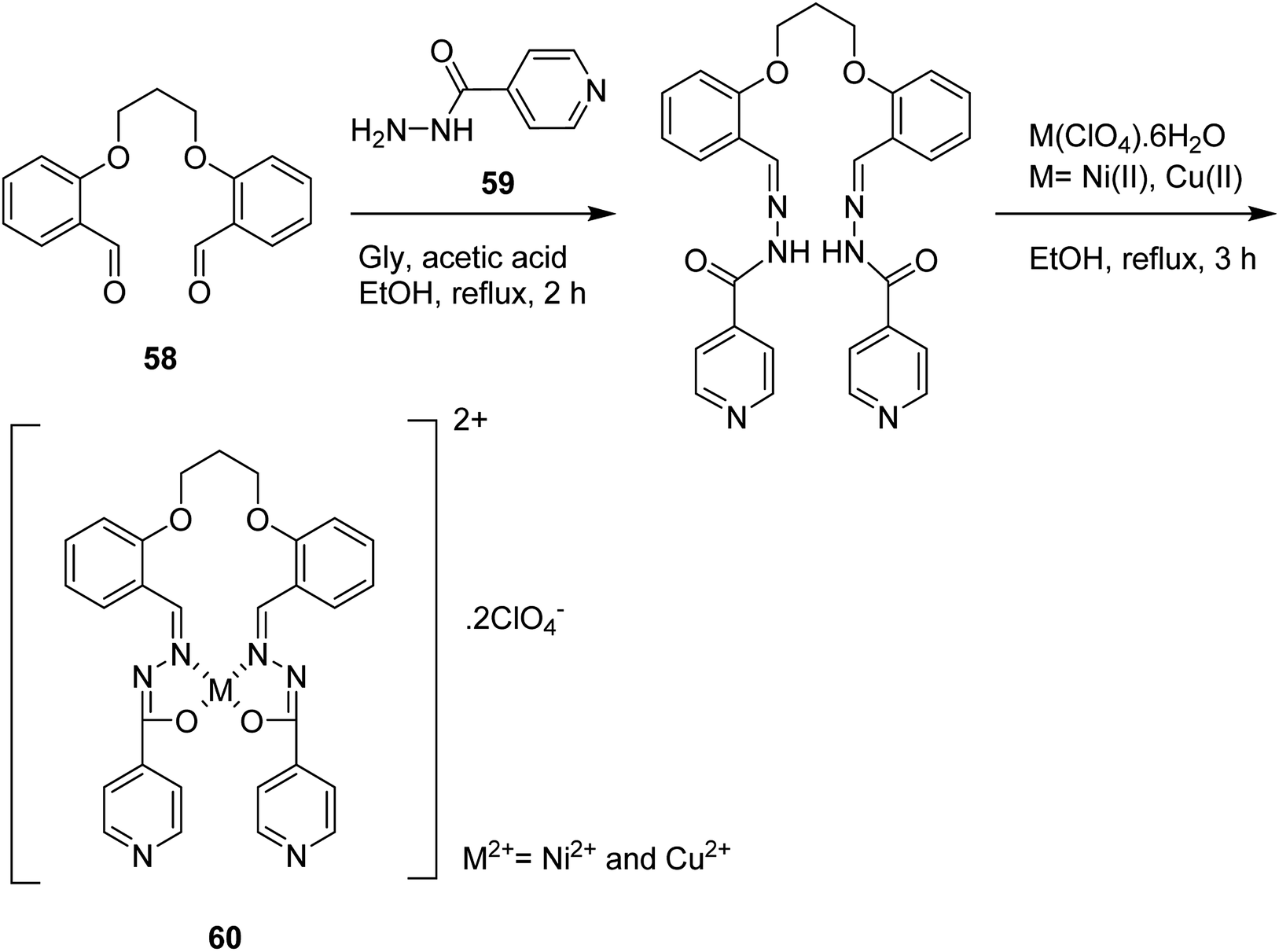 | ||
| Scheme 44 The suggested probable mechanism of the nickel(II) and copper(II) complexes having a tetradentate N2O4 Schiff base moiety. | ||
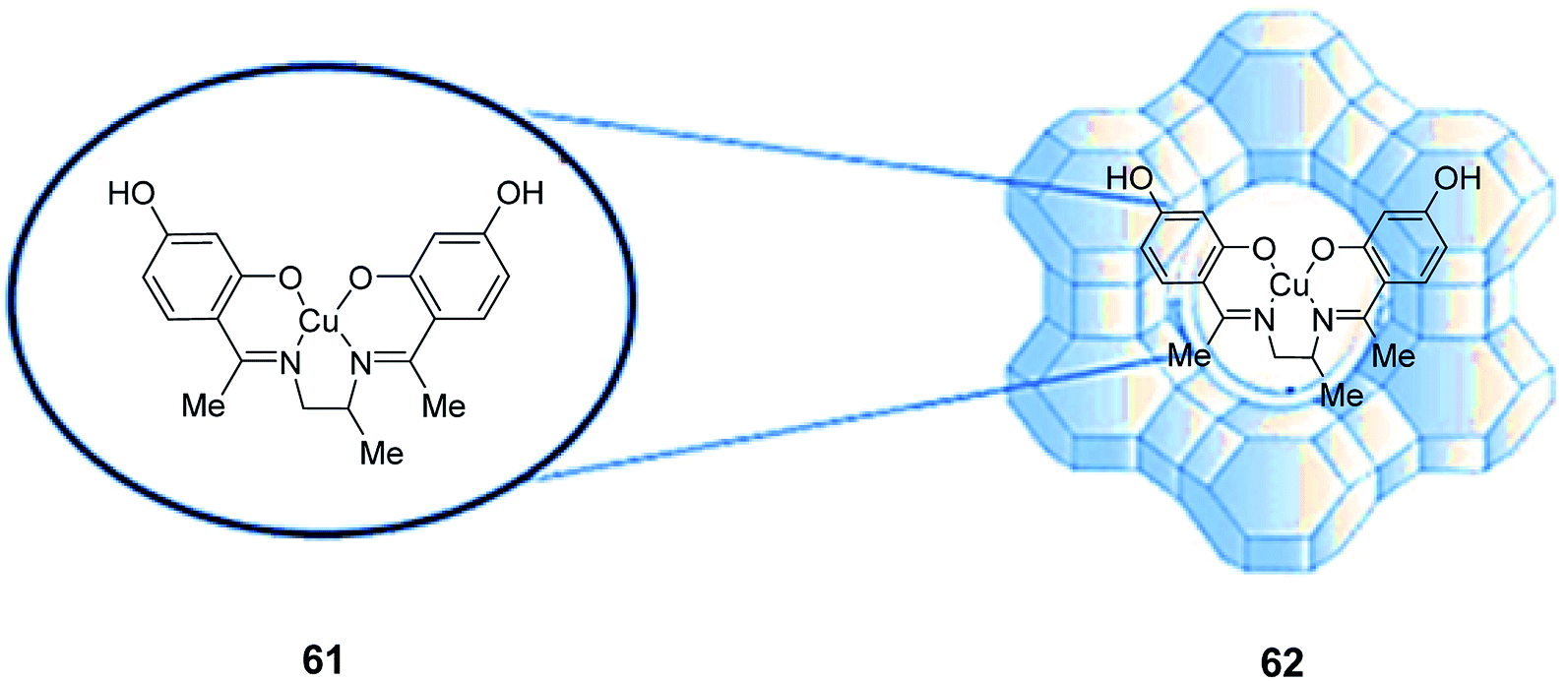
The a tetradentate (N2O2) Schiff base ligand (H2L) (61) and its related copper(II) complex (CuL) (62) were synthesized and encapsulated in the cavities of zeolite-Y via a fixed ligand method (Scheme 45).91
 | ||
| Scheme 45 Suggested scaffold structure of the zeolite-encapsulated metal Schiff base complexes. Reprinted from (ref. 91) with permission of the Royal Society of Chemistry. | ||
The catalytic and electrochemical activity of the synthesized encapsulated complex was examined in the oxidation of a wide range of olefins and sulfides using H2O2 in ethanol (Tables 19 and 20). The catalyst showed excellent activity in the oxidation of styrene in ethanol. Remarkably, various olefins afforded the epoxide products with high selectivity and conversion (Table 19).
| Substrates | Conversion (%) | Selectivity (%) |
|---|---|---|
| CuL-Y | (Sulfoxide) (CuL-Y) | |
| a Reaction conditions: catalyst:sulfide:H2O2:1:80:800 at 25 °C for a 20 min reaction time. |
||
 |
100 | 100 |
 |
97 | 90 |
 |
100 | 100 |
 |
93 | 95 |
 |
55 | 100 |
 |
100 | 100 |
 |
100 | 100 |
 |
95 | 100 |
 |
100 | 100 |
 |
100 | 100 |
| Substrate | Products | Conversion (%) | Epoxide selectivity (%) |
|---|---|---|---|
| (CuL1-Y) | (CuL1-Y) | ||
| a The molar ratio of the catalyst:alkene:H2O2 was 1:50:500 in EtOH at 55 °C for a one-hour reaction time. |
|||
 |
 |
100 | 100 |
 |
 |
100 | 100 |
 |
 |
100 | 95 |
 |
 |
98 | 94 |
 |
 |
97 | 95 |
 |
 |
100 | 90 |
 |
 |
100 | |
 |
 |
100 | 98 |
 |
 |
78 | 100 |
 |
 |
100 | 100 |
Raja and Ratnasamy in 1997 explored the low temperature and selective oxidation of toluene, benzene, naphthalene and ethyl benzene over Cl- and NO2-substituted phthalocyanine complexes of Cu encapsulated in zeolites X and Y, with molecular oxygen and aqueous H2O2 as the oxidants.10 It is noteworthy that the catalytic activity of the copper atoms is greater in the encapsulated state in comparison with the neat copper phthalocyanines. Once benzene was oxidized to phenol, both side chain oxidation and ring hydroxylation were identified in the case of the alkyl aromatics. Cl- and NO2-substituted complexes of copper(II), cobalt(II) and iron encapsulated in molecular sieves are selective and favorable oxidation catalysts. Both ring hydroxylation of alkyl aromatics and side chain oxidation were identified in the oxidation of various aromatic substrates using CuCI14Pc-Na-Y (0.17) with molecular oxygen or H2O2 as the oxidants (Table 21).
| Substrate | Oxidant | Conversion (%) | Products (%, wt) |
|---|---|---|---|
| a Reaction condition: substrate/H2O2 (3 mol), r.t., t = 8 h, acetonitrile, T = 384 K, catalyst = CuCl14Pc-Na-Y (0.17). | |||
| Benzene | H2O2 | 6.2 | Phenol (95) (Cat + HQ) |
| O2 | 5.4 | Phenol (100) | |
| Toluene | H2O2 | 23.6 | CH2OH (68.6); CHO (8.8); COOH (16.5); cresols (5.9) |
| O2 | 22.9 | CH2OH (4.3), CHO (15.7), cresols (80) | |
| Ethyl benzene | H2O2 | 31.6 | COCH3 (19.7), CHOHCH3 (20.9), ortho + para hydroxyl ethyl benzenes (58.3), para hydroxyl acetophenone (0.9) |
| O2 | 16.2 | COCH3 (80.2), CHOHCH3 (9.27), ortho + para hydroxyl ethyl benzenes (4.3), ortho + para hydroxyl acetophenone (6.2) | |
| Naphthalene | H2O2 | 17.9 | α-Naphthol (7.8), β-naphthol (53.6), naphthaquinone (1.1), phthalic anhydride (37.4) |
| β-Naphthol (100) | |||
| O2 | 5.4 | ||
In the oxidation of benzene, catechol and hydroquinone were synthesized using the oxidation reaction of phenol. For toluene, the ring hydroxylation was the main reaction when molecular oxygen was used as the oxidant (80 wt%, yield of cresols). The oxidation reaction of the methyl group to benzaldehyde, benzyl alcohol, and benzoic acid (total selectivity = 94.1% wt) predominated using H2O2 as the oxidant.
The liquid-phase direct catalytic oxidation reaction of benzene to phenol was performed using a copper(II) complex in a Faujasite-type zeolite with sucrose as a reducing agent and molecular oxygen as an oxidant.92 The construction of phenol using the copper(II) complex encapsulated in Y-zeolite was also examined using sucrose in the aqueous solution of acetic acid. Sucrose was an effective additive as a reducing agent since it was hydrolyzed into two reducing sugars, including fructose and glucose. The results of the reusing experiments for CuPA-Y, CuQA-Y, CuPCA-Y and Cu-Y, using Cu/Y (Imp) as a reference, are shown in Table 22.
| Catalyst | Copper content (wt%) | Yield (%) | Phenol (%) |
|---|---|---|---|
| Run 1 | Run 2 | ||
| Y-zeolite | 0.0 | n.d. | n.d. |
| Imp. Cu/Y | 15.0 | 5.3 | 1.9 |
| Cu-Y | 6.18 | 4.6 | 1.7 |
| CuPA-Y | 7.00 | 7.0 | 4.4 |
| CuQA-Y | 6.00 | 7.4 | 5.2 |
| CuPCA-Y | 6.09 | 5.1 | 3.3 |
Nair and co-workers in 2016 examined the degradation of 4-chloro-3-methylphenol (PCMC) using zeolite-encapsulated copper(II) complexes of N,N′-disalicylidene-1,2-phenylenediamine as significant heterogeneous catalysts via a Fenton-type improved oxidation reaction (Scheme 46).93 The results demonstrated that at low acidic pH, nearly complete removal of 4-chloro-3-methylphenol was performed using the iron(III), nickel(II), and copper(II) catalysts with hydrogen peroxide. The Fe(III), Ni(II) and Cu(II)-N,N′-disalicylidene-1,2-phenylenediamine complexes were efficiently encapsulated via the ship-in-a-bottle approach into the zeolite Y supercage.
 | ||
| Scheme 46 The degradation of 4-chloro-3-methylphenol using the zeolite-encapsulated copper(II) complexes of N,N′-disalicylidene-1,2-phenylenediamine as the catalyst. Reprinted from (ref. 93) with permission of the Royal Society of Chemistry. | ||
The reductions in the percentage degradation upon ten cycles for the Ni(II)-, and Cu(II)- and Fe(III)-based catalysts were 25%, 21%, and 14%, respectively. However, decreases in the catalytic activities were detected. The heterogeneous catalytic performances of the catalysts were not lost since the zeolite building block avoided leaching the metal-salophen complexes. Notably, these results demonstrated that these catalysts could be recycled at least three to five times without significant loss of activity.
Researchers worldwide have focused their interest on providing approaches for the removal of pollutants. These pollutants include dyes arising from various industries, such as leather, textile and others. It is expected that about 1–15% of the dye is lost during the dyeing methods, and is released in wastewaters.94 Removal of organic pollutants (such as dyes) with a Fenton reagent containing homogenous iron ions and H2O2, was found to be very active.95 Copper was found to undergo a Fenton-type reaction, and was used successfully to react with various organic pollutants.96
The Fenton kind of process is a very favorable oxidation method in which wastewater is reacted with hydrogen peroxide in a non-pressurized reactor at low temperature with a catalyst, affording CO2 and H2O or other oxidation products.97
The catalytic wet H2O2 oxidation reaction of an anionic dye was explored in this research. The copper(II) complex of N,N′-ethylene bis(salicylidene-aminato) (salenH2) was encapsulated into supercages of zeolite-Y via flexible ligand method. The effect of various factors, including the catalyst, pH and hydrogen peroxide concentration, on the oxidation of dye were explored. Complete color removal is accomplished at 60 °C in less than 1 hour, using catalyst and hydrogen peroxide. Investigations on the marketable tannery wastewaters show that the catalyst acts well, even when using other chemicals in the processing.
The oxidation/decomposition of various organic materials using H2O2 is enhanced with the addition of a catalyst, as the latter activates the construction of ˙OH from hydrogen peroxide. The reaction occurring in a Fenton method can be produced as follows: the restrictive stage in these types of oxidations is the construction of the hydroxyl radicals.98 The ˙OH species is generated, as shown in Scheme 47, and then attacks the organic substrates in the wastewater. This investigation was related to the construction and usage of a Cu salen-incorporated zeolite as a remarkable catalyst for the decolorization of a synthetic dye solution.
 | ||
| Scheme 47 The construction of hydroxyl radicals. | ||
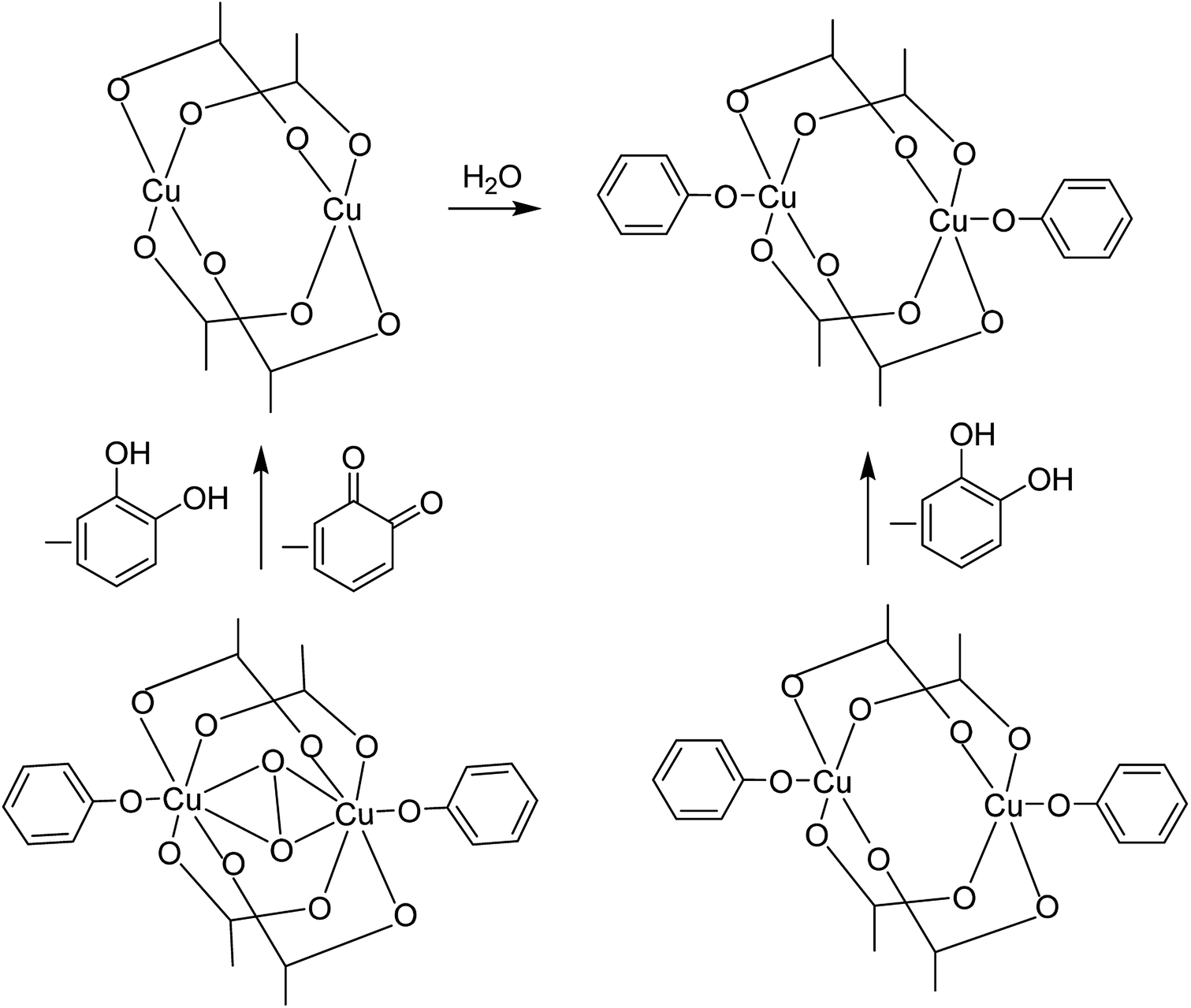 | ||
| Scheme 48 The suggested probable mechanism of oxidation that phenols exist in the phenolate form. | ||
Copper(II), nickel(II) and zinc(II) complexes of the amidate ligand 1,2-bis(2-hydroxybenzamido) ethane(H2hybe) (63) were encapsulated into the supercages of zeolite-Y ([Cu(hybe)]-Y) (Scheme 49).101 These complexes catalyzed the liquid-phase hydroxylation reaction of phenol to catechol as a major product, and hydroquinone as a major product using hydrogen peroxide. Based on the optimized conditions, ([Cu(hybe)]-Y) exhibited the maximum conversion of 40% after 6 hours.
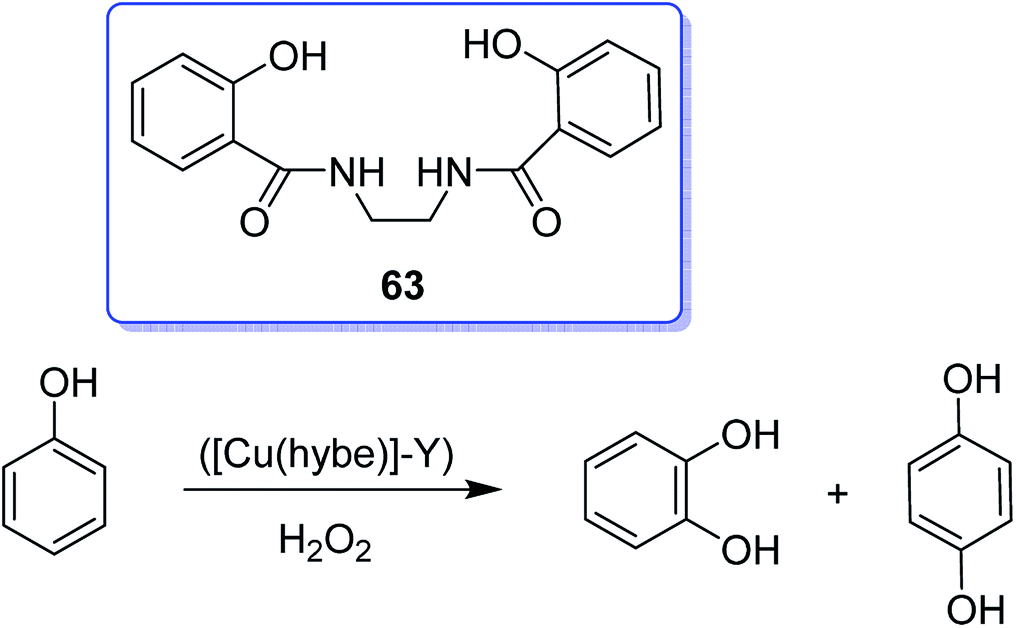 | ||
| Scheme 49 Hydroxylation of phenol in the presence of [Cu(hybe)]-Y and hydrogen peroxide. | ||
Table 23 shows the results along with the Turnover Frequency (TOF) for the phenol conversion upon 6 hours of reaction time. The selectivity of the catechol construction is comparable (89–91%) in all cases. However, the conversion of phenol changed from catalyst to catalyst. Fascinatingly, this great selectivity of the catechol construction is preserved even with 24 hours of reaction time.
| Catalyst | Conversion% | TOF h−1 | Selectivity% | |
|---|---|---|---|---|
| Catechol | Hydroquinone | |||
| [Cu(hybe)]-Y | 40.14 | 201.5 | 90.9 | 9.1 |
The copper(II) complex of 2,2-(((butane-1,4-diylbis(oxy))bis(2,1-phenylene)bis(azanylylidene) bis(methanylylidene)diphenol (H2L) was encapsulated into the supercages of zeolite NaY (64) via the flexible ligand method (Fig. 6).102 The selective hydroxylation reaction of phenol was accomplished using the free and encapsulated complexes. The catalytic efficacy of Cu(II)-Y was lower in comparison with that of the encapsulated complex. The results showed that the catalytic activity and stability of the complex was increased via encapsulation.
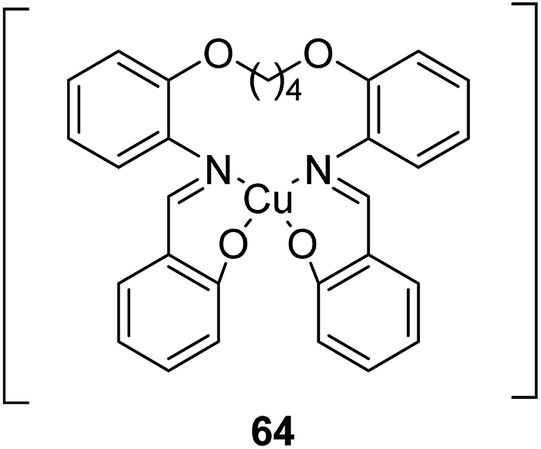 | ||
| Fig. 6 Proposed structure for the free and encapsulated complexes. | ||
To examine the influence of the solvent, various solvents such as CHCl3, CH2Cl2, MeOH, and MeCN were applied. The results demonstrated that for the hydroxylation of phenol, the optimal solvent is MeCN because the great polarity of MeCN provided a completely homogeneous reaction medium.
On the other hand, the concentration of H2O2 may affect the conversion of phenol. For this purpose, the hydroxylation of phenol was investigated using various molar ratios of oxidant to phenol. It was found that a 1:1 molar ratio was optimal to provide the maximum phenol conversion.
Moreover, atomic absorption spectroscopy demonstrated that no leaching of the encapsulated complex was detected. Remarkably, this catalyst can be recovered and reused, while the leaching of the Cu ions from Cu(II)-Y is constantly probable during the reaction.
Scheme 50 shows the suggested probable mechanism for the hydroxylation reaction of phenol. It was proposed that an electron transfer between Cu2+ and hydrogen peroxide gives M3+ and the hydroxyl radical. This high valence for the transition ions is definitely unusual. In addition, the cycle is closed when M3+ is reduced to M2+, with the cyclohexadienyl radical obtained from phenol.103
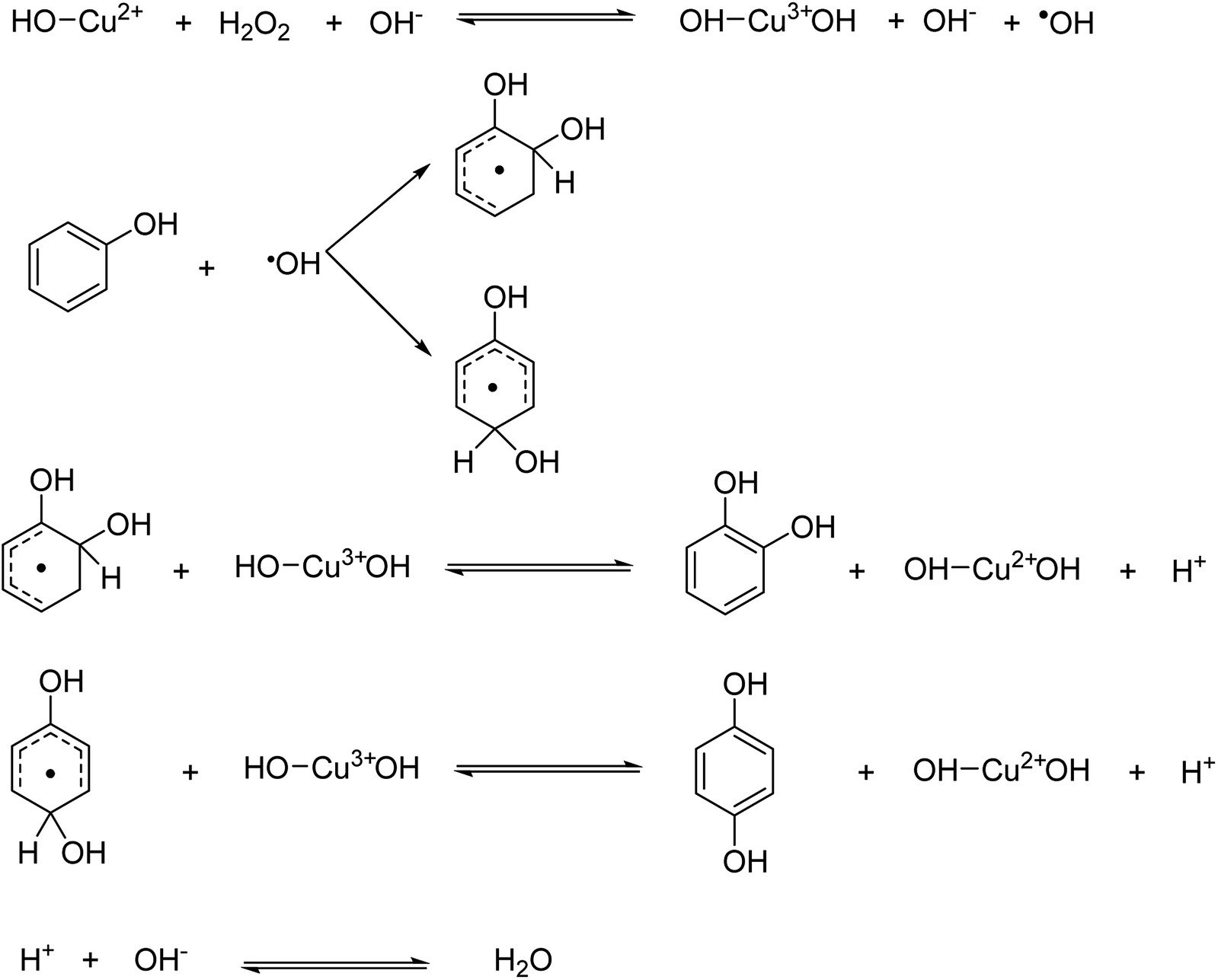 | ||
| Scheme 50 Suggested probable mechanism for the hydroxylation reaction of phenol. | ||
Cu(II) and Zn(II) complexes of 2-methylbenzimidazole (Mebzlh) were encapsulated into the supercages of zeolite-Y and ZSM-5 through the flexible ligand method.104 The catalytic activity of the encapsulated complexes was examined for the decomposition of hydrogen peroxide and the hydroxylation of phenol using hydrogen peroxide as an oxidant. Remarkably, the hydroxylation of phenol afforded hydroquinone and catechol as the major products (Scheme 51). A maximum conversion of about 52% was identified using the catalyst, [Cu(Mebzlh)]-Y.
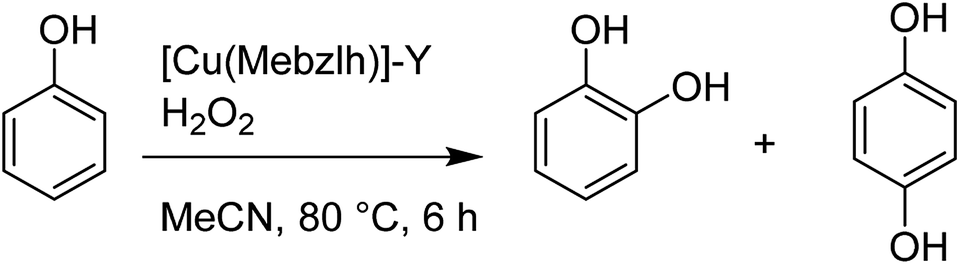 | ||
| Scheme 51 Hydroxylation reaction of phenol in the presence of [Cu(Mebzlh)]-Y. | ||
The results demonstrated that the decomposition of hydrogen peroxide is slow at first. However, it increases with time (Table 24). This is because of the fact that this encapsulated complex needs a comparatively prolonged time to show high catalytic activity as the number of metal centres is less than that of the neat complex.105
| Catalyst | Percentage of H2O2 reacted after 1 hour | TOF (h−1) | Percentage of H2O2 reacted | TOF (h−1) after 2 hours |
|---|---|---|---|---|
| [Cu(Mebzlh)]-Y | 0.673 | 159.42 | 1.360 | 159.42 |
| [Cu(Mebzlh)]-ZSM-5 | 0.600 | 538.89 | 0.600 | 269.44 |
Mahendra and co-workers in 2011 reported the encapsulation of the copper(II) complex of 2-(o-aminophenyl)benzimidazole (AmPhBzlH) (65) into the supercages of zeolite-Y and ZSM-5.106 The catalytic activity of the encapsulated complexes was investigated for the hydroxylation of phenol using hydrogen peroxide as an oxidant. The hydroxylation of phenol gave hydroquinone and catechol as the main products. [Cu(AmPhBzlH)]-Y exhibited the maximum phenol conversion with 82% selectivity for the catechol construction. It should be mentioned that the percentage selectivity for the formation of catechol is greater for all types of catalysts provided in comparison with that for hydroquinone. The catalytic hydroxylation reaction of phenol in the presence of various encapsulated complexes was investigated as a function of time using the aqueous hydrogen peroxide solution as the oxidant and MeCN as the solvent. Catechol and hydroquinone were provided as the major products (Scheme 52).
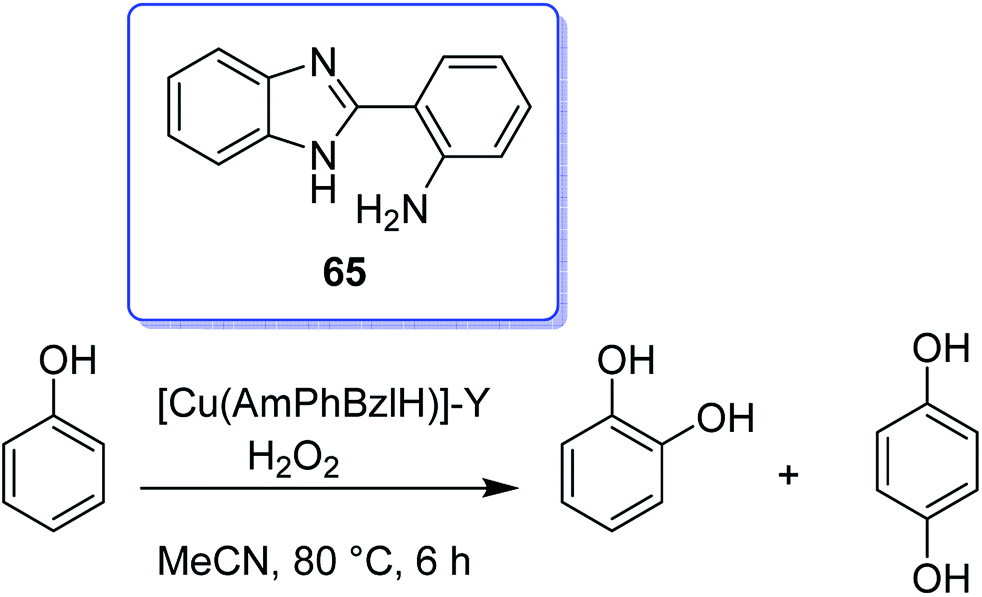 | ||
| Scheme 52 Hydroxylation reaction of phenol in the presence of AmPhBzlH as the catalyst. | ||
CuBA was linked covalently with APTES, and provided a multifunctional compound that involved both silicon alkoxides and Cu ions. Mostly, the protonated amine substituents contained the ability to catalyze silica construction. Therefore, the construction of Si–O–Si networks can be induced in situ without any additional basic or acidic catalysts. This occurrence assists the protection of the pH-sensitive organic Cu(II) salt. Notably, CuBA was covalently connected to APTES via a nucleophilic substitution reaction. This significant chemical bond can guarantee the molecular-level dispersion of Cu ions, which is vital for the generation of ultrasmall sized CuO NPs with a more dispersed spatial distribution. Lastly, Cu ions were transformed into CuO NPs spontaneously under thermal treatment. The encapsulation of the copper(II) complex of 3-phenylimino-1,3-dihydro-indol-2-one 66 into the supercages of zeolite-Y (Cu-Y-zeolite) was accomplished via flexible ligand method (Scheme 53).107
 | ||
| Scheme 53 The formation of 66. | ||
The acetylation reaction of p-cresol with several encapsulated complexes was investigated using Ac2O as the acetylating agent. The p-cresyl acetate 67 was provided as the main corresponding products (Scheme 54).
 | ||
| Scheme 54 Acetylation of p-cresol. | ||
The catalytic activity of the cationic exchanged zeolite, Cu(II) complex of ligand and complex encapsulated inside the zeolite was explored for the decomposition of hydrogen peroxide and for the acetylation of p-cresol. The maximum conversion of p-cresol was provided using the zeolite-encapsulated complex in a shorter reaction time in comparison with another catalyst. The order of the catalytic activity for the acetylation reaction of p-cresol was found to be as follows: heterogeneous catalyst (75%), [[Cu-Y] zeolite (55%), [[Na-Y] zeolite (50%), homogeneous catalyst (65%) and without catalyst (50%).
In this method, no leaching of metal ions was identified in the solution. The effect of various factors, including temperature, the quantity of catalyst and time, was investigated and these parameters exhibited various catalytic activities in the decomposition of hydrogen peroxide and the acetylation of p-cresol. This reaction was performed for all catalysts under the optimal reaction conditions (5 mmol p-cresol, acetic anhydride 7.5 mmol and 0.05 g catalyst at ambient temperature). Moreover, the influence of various catalysts on the percentage conversion of p-cresol was examined, and the results demonstrated the maximum p-cresol conversion using the heterogeneous catalyst.
 | ||
| Fig. 7 Copper(II) complexes of two biologically significant ligands, namely embelin (2,5-dihydroxy-3-undecyl-2,5-cyclohexadien 1,4-dione) (68) and 2-aminobenzimidazole (69). | ||
 | ||
| Scheme 55 Synthetic route for the construction of the Schiff base. | ||
 | ||
| Scheme 56 Synthesis of benzimidazoles. | ||
In an investigation to increase the yields, various solvents such as EtOH, DMF, MeCN, and CHCl3 were used for the reaction between benzaldehyde and o-phenylenediamine using [CuL]-Y under reflux. Significantly, polar solvents like MeCN and DMF were appropriate for this reaction, with moderately high yields. However, EtOH demonstrated the lowest yield, maybe due to its coordination to the Lewis acid sites, leading to a decrease in the catalytic efficacy.
The atomic absorption spectroscopy demonstrated that no leaching was detected using the encapsulated complex as a catalyst, while the moderate leaching of Cu ions happened once the reaction was accomplished using Cu(II)/Y.
2.2. Silica-encapsulated Cu NPs
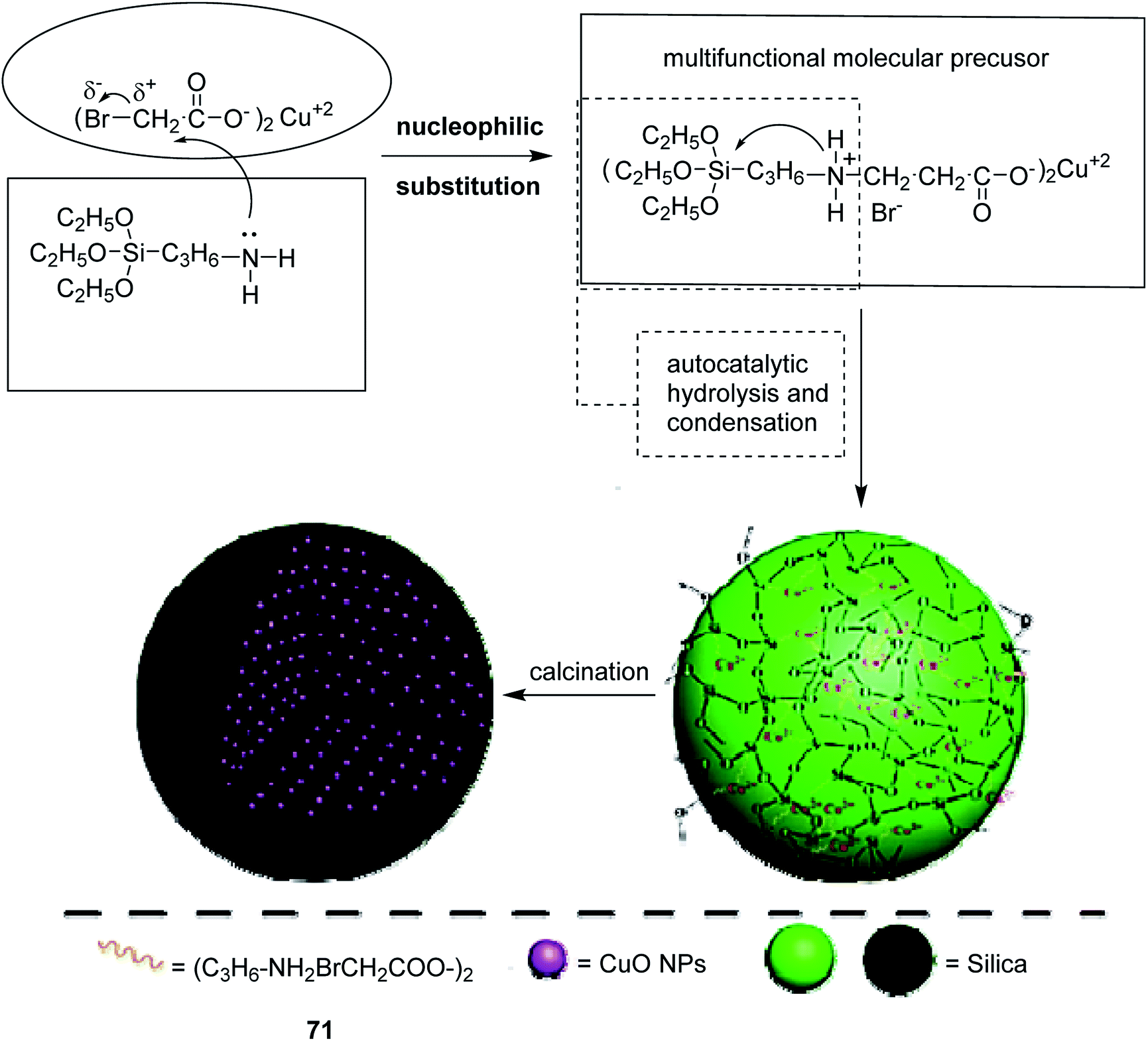 | ||
| Scheme 57 The formation of catalyst 71. Reprinted from (ref. 111) with permission of the Royal Society of Chemistry. | ||
Kooti and coworkers in 2019 examined the catalytic activity of the synthesized catalyst through immobilization of the Cu(proline)2 complex onto the surface of magnetic graphene in the aldol reaction.112
This group demonstrated the formation of a heterogeneous nanocatalyst bearing a Cu(proline)2 complex immobilized on a silica-coated graphene/MnFe2O4 (G/MF) composite. The unique nanocomposite [G/MF@SiO2@Cu(proline)2] was used as a significant catalyst for the enantioselective aldol reaction under solvent-free conditions.
The resultant nanocomposite bearing copper(II) center as a Lewis acid was used as a significant catalyst in the enantioselective aldol reaction to afford the aldol products in excellent yield and high ees (>90%).
By the way, proline was immobilized on different supports, involving metals, mesoporous materials, and layered compounds, to increase its activity for catalyzing asymmetric aldolization.113 Consequently, this group reported an eco-friendly approach for the one-pot aldol reaction of different aldehydes and ketones using G/MF@SiO2@Cu(proline)2 72 as an efficient catalyst (Scheme 58 and Table 25).
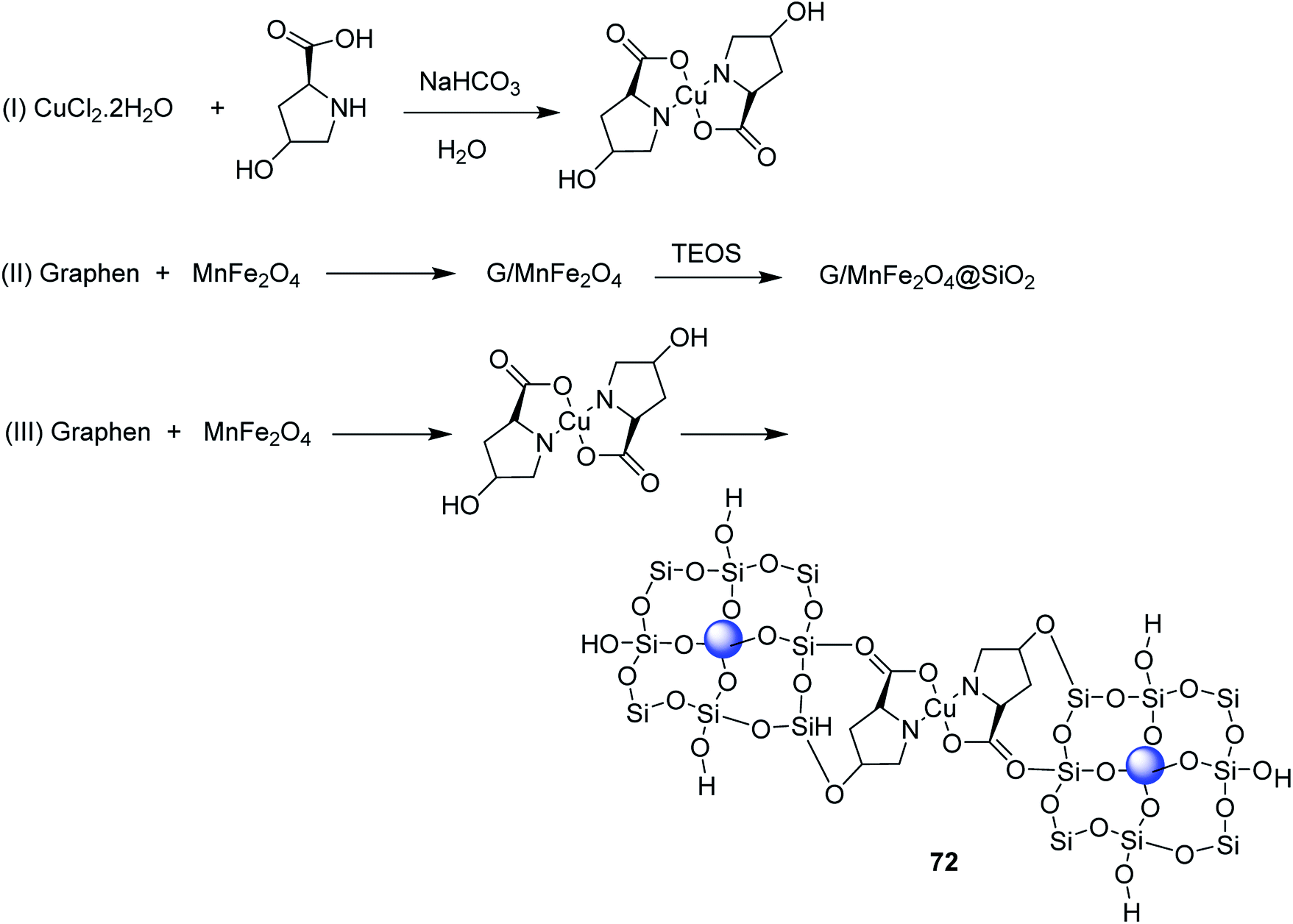 | ||
| Scheme 58 Stepwise construction of 72. | ||
| Aldehyde | Ketone | Yield | ee% (anti/syn) |
|---|---|---|---|
| 4-Nitrobenzaldehyde | Cyclohexanone propan-2-one acetophenone pentane-2,4-dione | 30–98% | 69/31–95/5 |
| 2-Hydroxybenzaldehyde | |||
| Benzaldehyde | |||
| Acetaldehyde | |||
| Formaldehyde | |||
| Cinnamaldehyde | |||
| Hexanal | |||
| Heptanal | |||
| 4-Methylbenzaldehyde | |||
| 3-Chlorobenzaldehyde | |||
| 3-Methoxybenzaldehyde | |||
| 2-Methoxybenzaldehyde | |||
| 3-Fluorobenzaldehyde | |||
| 4-(Dimethylamino)benzaldehyde | |||
| 2-Chlorobenzaldehyde | |||
| 2-Hydroxybenzaldehyde |
As aforementioned, proline and its derivatives can significantly catalyze asymmetric aldol reactions. The stepwise construction of catalyst 72 is depicted in Scheme 58. This catalyst exhibited high activity in the direct asymmetric aldol reaction. The enantioselectivity values of the anti-isomer, as the major enantiomer, for the reaction of cyclohexanone and aromatic aldehydes under a solvent-free condition in 5 hours were over 90%.
A suggested possible mechanism for the aldol reaction is shown in Scheme 59. At first, the carbonyl group of cyclohexanone is activated with the metal center of the Cu complex in the nanocatalyst. Hence, this interaction gives the α-hydrogen of the ketone more acidicity that results in the construction of the metal-enolate complex 75. Then, a nucleophilic attack of the C–C double bond of the enolate scaffold on the carbonyl substituent of benzaldehyde will successively occur to provide intermediate 76. Using the reorganization of bonding pairs, intermediate 77 is provided, and the protonation of that compound affords the aldol product.
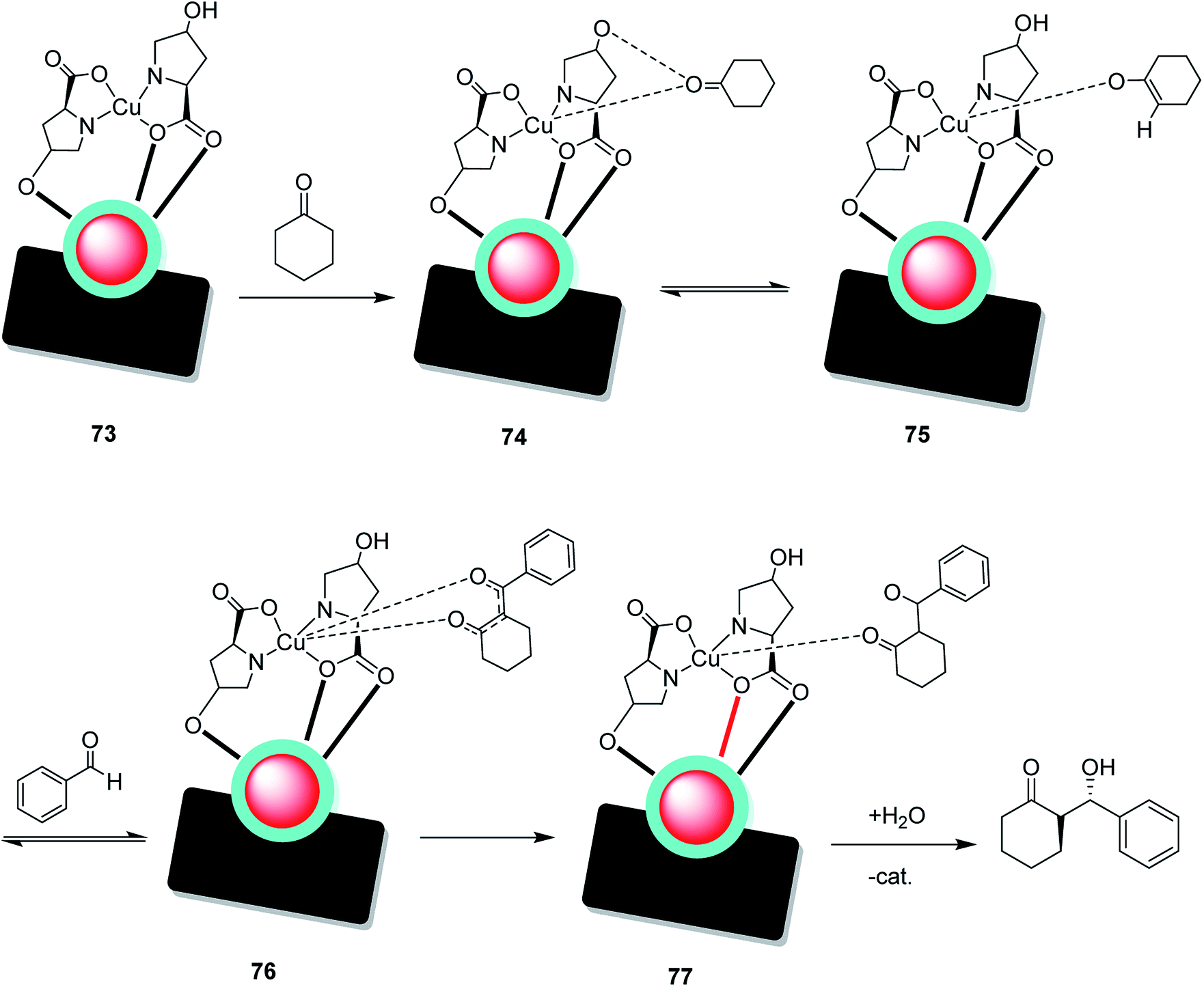 | ||
| Scheme 59 Suggested possible mechanism for the aldol reaction using 73. Reprinted from (ref. 112) with permission of the Royal Society of Chemistry. | ||
Moghadam and coworkers in 2018 demonstrated the formation of poly (N-heterocyclic carbene copper(II) complex) immobilized on nano-silica, (Cu(II)-NHCs) n@SiO2 (78). The catalytic activity of 78 was examined for the synthesis of 1,2,3-triazoles, benzimidazoles, benzothiazoles, and the Sonogashiro–Hagihara cross-coupling reaction. The suggested probable mechanisms for these reactions is shown in Scheme 60.114
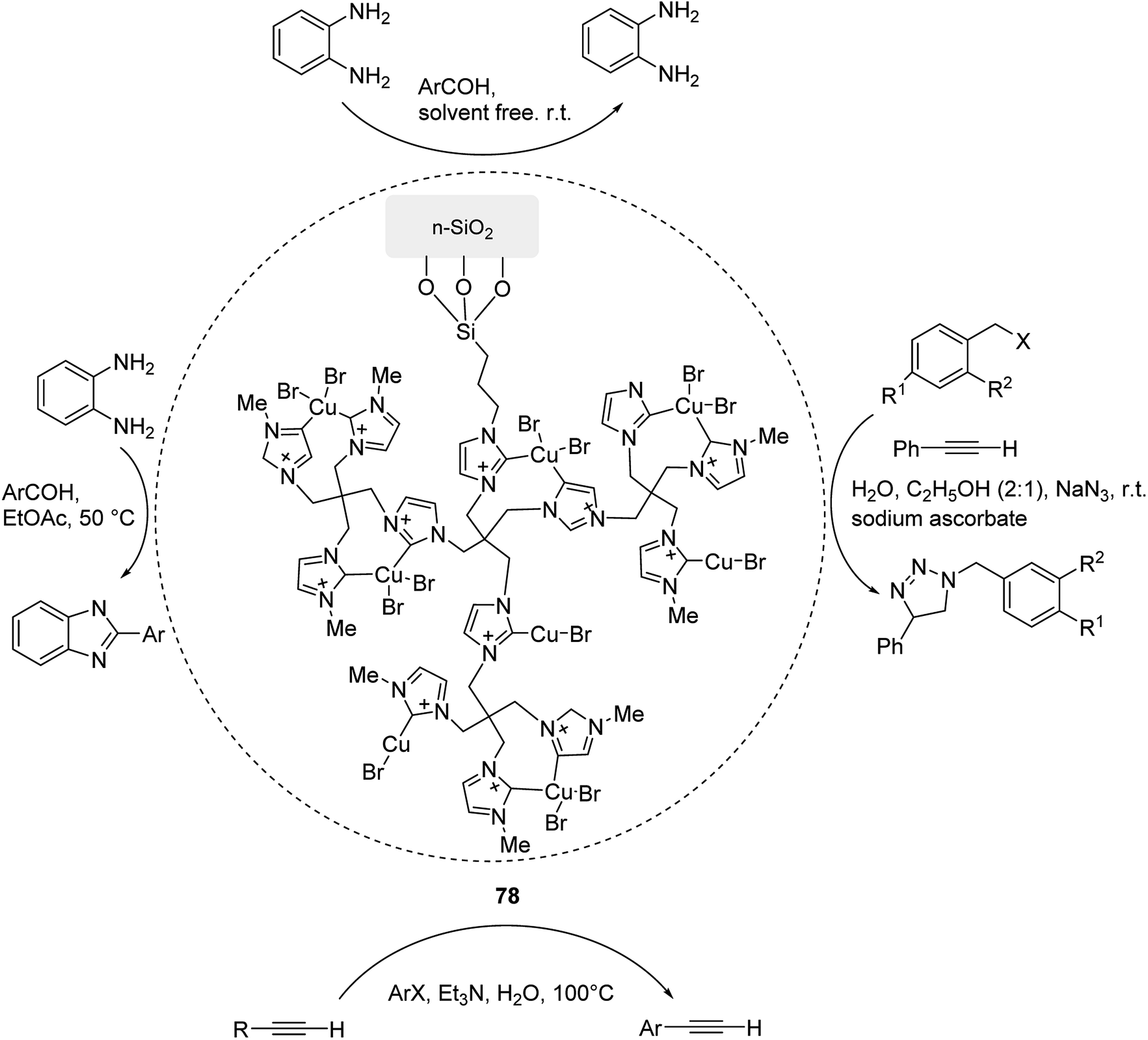 | ||
| Scheme 60 The formation of 1,2,3-triazoles, benzimidazoles, benzothiazoles, and the Sonogashiro–Hagihara cross-coupling reaction in the presence of 78 as an efficient catalyst. | ||
To examine the catalytic activity of 78 in the formation of benzimidazoles, the reactions of the substituted benzaldehydes and 1,2-diaminopropane were examined (Scheme 61). Remarkably, the catalyst showed excellent activity in this reaction and the relevant yields were 86–94%.
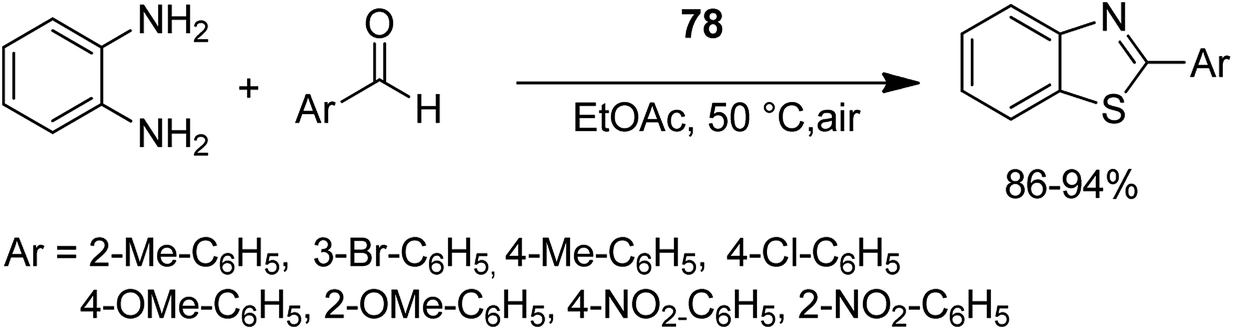 | ||
| Scheme 61 Synthesis of benzimidazoles in the presence of 78 as the catalyst. | ||
To examine the generality of this catalyst, various substituted benzaldehydes were reacted with 2-aminothiophenol to afford the desired 2-substituted benzothiazoles in high to excellent yields (85–95% yield) (Scheme 62).
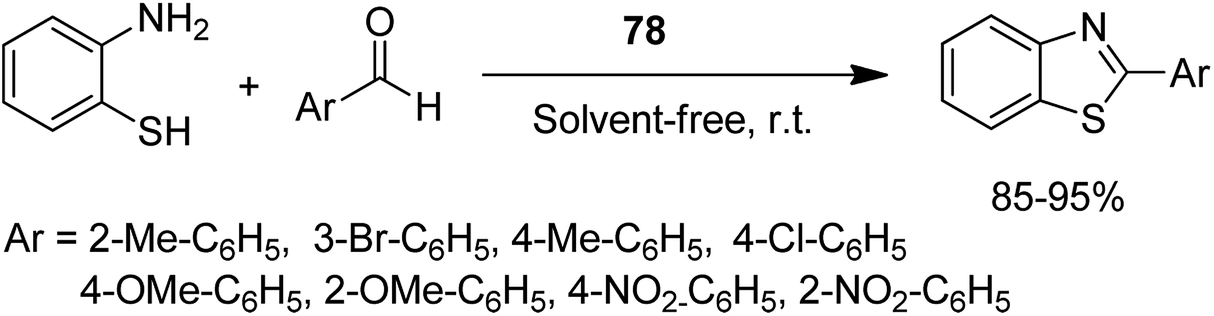 | ||
| Scheme 62 The formation of benzothiazole derivatives in the presence of 78 as an efficient catalyst. | ||
1,2,3-Triazoles were synthesized in the presence of 78 as the catalyst (Scheme 63).
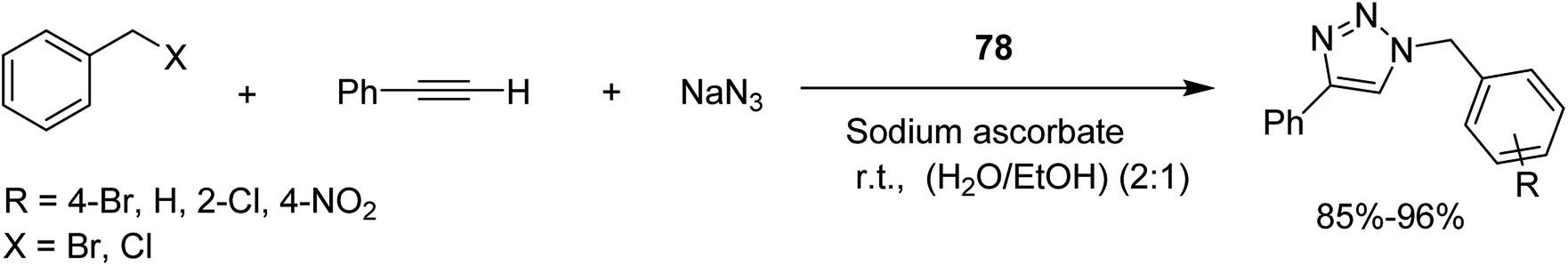 | ||
| Scheme 63 Synthesis of 1,2,3-triazoles in the presence of 78 as the catalyst. | ||
Next, the activity of catalyst 78 was examined in the Sonogashira–Hagihara cross-coupling reaction. Different aryl halides were treated with phenyl acetylene to afford the relevant diphenyl acetylenes in moderate to high yields (85–94%) (Scheme 64). Notably, aryl iodide is more active than aryl chlorides and bromides.
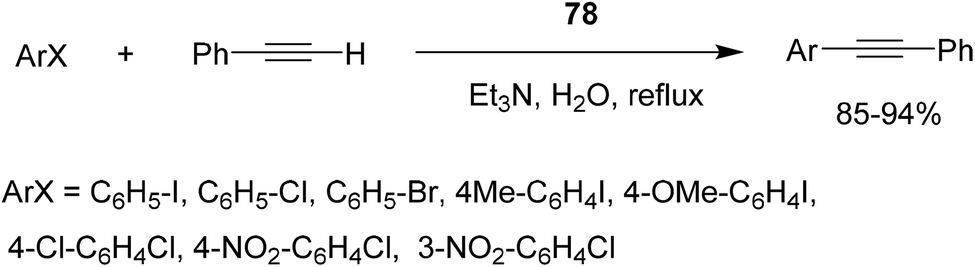 | ||
| Scheme 64 Sonogashire–Hagihara cross-coupling reaction of aryl halides and phenylacetylene in the presence of 78 as the catalyst. | ||
3. Conclusion
In summary, confinement of zeolite, silica and miscellaneous NPs within the cave of a nanoreactor not only disallowed the NPs from agglomeration, but also increased their consistency. It also facilitated the catalytic accomplishment of the Cu-encapsulated catalyst in terms of its selectivity, reactivity, and leaching of the metal oxide. As a result, this encapsulation significantly improved the catalyst dependability and controlled the Cu species leaching, maintaining its high catalytic originality and selectivity for the oxidation reaction. Different organic, inorganic and hybrid systems can be employed for the encapsulation of zeolites, silica and miscellaneous NPs. Frequently, mesoporous silica, zeolites, MOFs, dendrimers, and carbon materials are used for this purpose. We hope this review describes the merits of encapsulated zeolite, silica and miscellaneous NPs over their bare counterparts in terms of catalyzing different organic transformations.115Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this research from Alzahra University, Iran is gratefully acknowledged. M. M. H. is also thankful for the granted individual research chair given by the Iran National Science Foundation (INSF).References
- (a) L. R. Cox, Appl. Organomet. Chem., 2003, 17, 79 CrossRef CAS; (b) X. Lu and S. Ma, Transition Metal Catalyzed Reactions, Blackwell Science, Oxford, UK, 1999 Search PubMed.
- (a) I. Ojima, M. Tzamarioudaki, Z. Li and R. J. Donovan, Chem. Rev., 1996, 96, 635–662 CrossRef CAS PubMed; (b) M. L. Crawley and B. M. Trost, Applications of transition metal catalysis in drug discovery and development: an industrial perspective, John Wiley & Sons, 2012 CrossRef.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- (a) W. R. Moser and D. W. Slocum, Homogeneous Transition Metal Catalyzed Reactions, American Chemical Society, Washington, DC, United States, 1992 CrossRef; (b) Y. G. Shelke, A. Yashmeen, A. V. Gholap, S. J. Gharpure and A. R. Kapdi, Asian J. Chem., 2018, 13, 2991–3013 CrossRef CAS PubMed; (c) B. Cornils and W. A. Herrmann, J. Catal., 2003, 216, 23–31 CrossRef CAS; (d) S. Bhaduri and D. Mukesh, Homogeneous catalysis: mechanisms and industrial applications, John Wiley & Sons, 2014 Search PubMed.
- G. Parshall, J. Mol. Catal., 1978, 4, 243–270 CrossRef CAS.
- (a) D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed; (b) C. Copéret and J. M. Basset, Adv. Synth. Catal., 2007, 349, 78–92 CrossRef; (c) M. G. Petrucci and A. K. Kakkar, Adv. Mater., 1996, 8, 251–253 CrossRef CAS.
- A. Behr, A. J. Vorholt and T. Seidensticker, ChemBioEng Rev., 2015, 2, 6–21 CrossRef CAS.
- (a) A. Corma, H. García and F. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (b) C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef CAS PubMed; (c) D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- C. R. Jacob, S. P. Varkey and P. Ratnasamy, Microporous Mesoporous Mater., 1998, 22, 465–474 CrossRef CAS.
- R. Raja and P. Ratnasamy, Stud. Surf. Sci. Catal., Elsevier, 1997, vol. 105, pp. 1037–1044 Search PubMed.
- K. D. Karlin and Y. Gultneh, Prog. Inorg. Chem., 1987, 219–327 CAS.
- Y. Ukisu, A. Kazusaka and M. Nomura, J. Mol. Catal. B: Enzym., 1991, 70, 165–174 CrossRef CAS.
- A. Hay, H. Blanchard, G. Endres and J. Eustance, J. Am. Chem. Soc., 1959, 81, 6335–6336 CrossRef CAS.
- K. Kinoshita, Bull. Chem. Soc. Jpn., 1959, 32, 777–780 CrossRef CAS.
- (a) J. Pritchard, G. A. Filonenko, R. van Putten, E. J. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC; (b) A. Sivaramakrishna, P. Suman, E. Veerashekhar Goud, S. Janardan, C. Sravani, T. Sandeep, K. Vijayakrishna and H. S. Clayton, J. Coord. Chem., 2013, 66, 2091–2109 CrossRef CAS.
- S. Dang, H. Yang, P. Gao, H. Wang, X. Li, W. Wei and Y. Sun, Catal. Today, 2019, 330, 61–75 CrossRef CAS.
- (a) X. Zhao, X. Y. Bao, W. Guo and F. Y. Lee, Mater. Today, 2006, 9, 32–39 CrossRef CAS; (b) P. Barbaro and F. Liguori, Heterogenized Homogeneous Catalysts for Fine Chemicals Production, SSBM, 2010, vol. 33 CrossRef.
- L. Cao and R. Schmid, Application and Design, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, vol. 10 Search PubMed.
- S. Bhattacharyya and B. Basu, Green Techniques for Organic Synthesis and Medicinal Chemistry 2018, pp. 269–289 Search PubMed.
- M. Meldal, in Methods in enzymology, Elsevier, 1997, vol. 289, pp. 83–104 Search PubMed.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- Z. Xie, Z. Liu, Y. Wang, Q. Yang, L. Xu and W. Ding, Int. J. Mol. Sci., 2010, 11, 2152–2187 CrossRef CAS PubMed.
- J. A. Cecilia, R. Moreno Tost and M. Retuerto Millán, Mesoporous Materials: From Synthesis to Applications, Multidisciplinary Digital Publishing Institute, 2019 Search PubMed.
- Z. Wu and D. Zhao, Chem. Commun., 2011, 47, 3332–3338 RSC.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- M. Sohmiya, K. Saito and M. Ogawa, Sci. Technol. Adv. Mater., 2015, 16, 054201 CrossRef PubMed.
- O. Pagar, H. Nagare, Y. Chine, R. Autade, P. Narode and V. Sanklecha, Sci. Technol. Adv. Mater., 2018, 6, 1–12 Search PubMed.
- (a) B. M. Weckhuysen and J. Yu, Chem. Soc. Rev., 2015, 44, 7022–7024 RSC; (b) M. Mishra and S. K. Jain, Proc. Natl. Acad. Sci., India, Sect. B, 2011, 81, 250–259 Search PubMed.
- F. Fajula and D. Brunel, Microporous Mesoporous Mater., 2001, 48, 119–125 CrossRef CAS.
- C. Colella and A. F. Gualtieri, Microporous Mesoporous Mater., 2007, 105, 213–221 CrossRef CAS.
- S. Bhatia, Zeolite catalysts: principles and applications, CRC Press, 1989 Search PubMed.
- Z. Le Hua, J. Zhou and J. L. Shi, Chem. Commun., 2011, 47, 10536–10547 RSC.
- R. Mirsafaei, M. M. Heravi, T. Hosseinnejad and S. Ahmadi, Appl. Organomet. Chem., 2016, 30, 823–830 CrossRef CAS.
- F. Ebrahimpour-Malamir, T. Hosseinnejad, R. Mirsafaei and M. M. Heraviappl, Organomet. Chem., 2018, 32, e3913 CrossRef.
- A. M. Ealias and M. Saravanakumar, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 263, 032019 Search PubMed.
- Y. Suchorski and G. Rupprechter, Catal. Lett., 2018, 148, 2947–2956 CrossRef CAS PubMed.
- Y. Ukisu and A. Kazusaka, J. Mol. Catal., 1987, 43, 31–33 CrossRef CAS.
- L. D. Fernández, E. Lara and E. A. Mitchell, Eur. J. Protistol., 2015, 51, 409–424 CrossRef PubMed.
- (a) L. Guo, X. Zhao, R. Zhang, C. Chen, J. Chen, A. Chen, X. Liu and Z. Hou, J. Supercrit. Fluids, 2016, 107, 715–722 CrossRef CAS; (b) C. Sener, T. Dogu and G. Dogu, Microporous Mesoporous Mater., 2006, 94, 89–98 CrossRef CAS; (c) Y. Hou, X. Ji, G. Liu, J. Tang, J. Zheng, Y. Liu, W. Zhang and M. Jia, Catal. Commun., 2009, 10, 1459–1462 CrossRef CAS; (d) V. S. Garcia-Cuello, L. Giraldo and J. C. Moreno-Piraján, Catal. Lett., 2011, 141, 1659 CrossRef CAS.
- (a) J. Garcia-Martinez, N. Linares, S. Sinibaldi, E. Coronado and A. Ribera, Microporous Mesoporous Mater., 2009, 117, 170–177 CrossRef CAS; (b) P. Wang, Z. Wang, J. Li and Y. Bai, Microporous Mesoporous Mater., 2008, 116, 400–405 CrossRef CAS; (c) Á. Mastalir, B. Rác, Z. Király and Á. Molnár, J. Mol. Catal. A: Chem., 2007, 264, 170–178 CrossRef; (d) S. Ghosh, R. Dey, S. Ahammed and B. C. Ranu, Clean Technol. Environ. Policy, 2014, 16, 1767–1771 CrossRef CAS; (e) J. N. Park, A. J. Forman, W. Tang, J. Cheng, Y. S. Hu, H. Lin and E. W. McFarland, Small, 2008, 4, 1694–1697 CrossRef CAS PubMed; (f) G. Budroni, A. Corma, H. García and A. Primo, J. Catal., 2007, 251, 345–353 CrossRef CAS; (g) Y. Wan, H. Wang, Q. Zhao, M. Klingstedt, O. Terasaki and D. Zhao, J. Am. Chem. Soc., 2009, 131, 4541–4550 CrossRef CAS PubMed; (h) K. Bendahou, L. Cherif, S. Siffert, H. Tidahy, H. Benaissa and A. Aboukais, Appl. Catal., A, 2008, 351, 82–87 CrossRef CAS; (i) S. Banerjee, V. Balasanthiran, R. T. Koodali and G. A. Sereda, Org. Biomol. Chem., 2010, 8, 4316–4321 RSC.
- (a) E. Hashemi, Y. S. Beheshtiha, S. Ahmadi and M. M. Heravi, Transition Met. Chem., 2014, 39, 593–601 CrossRef CAS; (b) M. M. Heravi, S. Sadjadi, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, Catal. Commun., 2008, 9, 504–507 CrossRef CAS.
- S. Sadjadi and M. Heravi, RSC Adv., 2016, 6, 88588–88624 RSC.
- (a) S. Sabaqian, F. Nemati, M. M. Heravi and H. T. Nahzomi, Appl. Organomet. Chem., 2017, 31, e3660 CrossRef; (b) T. Baie Lashaki, H. A. Oskooie, T. Hosseinnejad and M. M. Heravi, J. Coord. Chem., 2017, 70, 1815–1834 CrossRef CAS; (c) M. M. Heravi, H. Hamidi and V. Zadsirjan, Curr. Org. Synth., 2014, 11, 647–675 CrossRef CAS.
- K. J. Balkus and A. G. Gabrielov, in Inclusion chemistry with zeolites: nanoscale materials by design, Springer, 1995, pp. 159–184 Search PubMed.
- R. Raja and P. Ratnasamy, J. Catal., 1997, 170, 244–253 CrossRef CAS.
- A. Corma, Zeolite microporous solids: synthesis, structure and reactivity, ed. E. Derouane, F. Lemos, C. Naccache and F. Ribeiro, Kluwer, 1992, vol. 352, p. 373 Search PubMed.
- C. R. Jacob, S. P. Varkey and P. Ratnasamy, Appl. Catal., A, 1998, 168, 353–364 CrossRef CAS.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 270, 225–235 CrossRef CAS.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 278, 12–21 CrossRef CAS.
- Z. Li, S. Wu, Y. Ma, H. Liu, J. Hu, L. Liu, Q. Huo, J. Guan and Q. Kan, Transition Met. Chem., 2013, 38, 243–251 CrossRef CAS.
- Y. Yang, H. Ding, S. Hao, Y. Zhang and Q. Kan, Appl. Organomet. Chem., 2011, 25, 262–269 CrossRef CAS.
- Y. Yang, Y. Zhang, S. Hao, J. Guan, H. Ding, F. Shang, P. Qiu and Q. Kan, Appl. Catal., A, 2010, 381, 274–281 CrossRef CAS.
- I. Kuźniarska-Biernacka, M. Carvalho, I. C. Neves, A. M. Fonseca, A. Lisińska-Czekaj and D. Czekaj, Arch. Metall. Mater., 2013, 58, 1291–1294 Search PubMed.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 263, 227–237 CrossRef CAS.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 274, 192–201 CrossRef CAS.
- E. Shilpa and V. Gayathri, J. Environ. Chem. Eng., 2016, 4, 4194–4206 CrossRef CAS.
- M. R. Maurya, S. J. Titinchi and S. Chand, J. Mol. Catal. A: Chem., 2003, 201, 119–130 CrossRef CAS.
- M. Maurya, S. Titinchi and S. Chand, Appl. Catal., A, 2002, 228, 177–187 CrossRef CAS.
- I. Kuźniarska-Biernacka, M. M. M. Raposo, R. Batista, P. Parpot, K. Biernacki, A. L. Magalhães, A. M. Fonseca and I. C. Neves, Microporous Mesoporous Mater., 2016, 227, 272–280 CrossRef.
- B. K. Kundu, V. Chhabra, N. Malviya, R. Ganguly, G. S. Mishra and S. Mukhopadhyay, Microporous Mesoporous Mater., 2018, 271, 100–117 CrossRef.
- B. Xiao, H. Hou and Y. Fan, J. Organomet. Chem., 2007, 692, 2014–2020 CrossRef CAS.
- A. Mobinikhaledi, M. Zendehdel and P. Safari, Transition Met. Chem., 2014, 39, 431–442 CrossRef CAS.
- R. Raja and P. Ratnasamy, Stud. Surf. Sci. Catal., Elsevier, 1996, Vol. 101, pp. 181–190 Search PubMed.
- G. Endres, A. Hay and J. Eustance, J. Org. Chem., 1963, 28, 1300–1305 CrossRef CAS.
- N. Oyama, T. Ohsaka, Y. Ohnuki and T. Suzuki, J. Electrochem. Soc., 1987, 134, 3068–3073 CrossRef CAS.
- W. Waters, J. Am. Oil Chem. Soc., 1971, 48, 427–433 CrossRef CAS.
- M. Salavati-Niasari, J. Mol. Catal. A: Chem., 2008, 283, 120–128 CrossRef CAS.
- B. Fan, H. Li, W. Fan, C. Jin and R. Li, Appl. Catal., A, 2008, 340, 67–75 CrossRef CAS.
- M. Salavati-Niasari and M. Shaterian, J. Porous Mater., 2008, 15, 581–588 CrossRef CAS.
- M. Salavati-Niasari, M. Shakouri-Arani and F. Davar, Microporous Mesoporous Mater., 2008, 116, 77–85 CrossRef CAS.
- M. Salavati-Niasari, Z. Salimi, M. Bazarganipour and F. Davar, Inorg. Chim. Acta, 2009, 362, 3715–3724 CrossRef CAS.
- I. Kuźniarska-Biernacka, K. Biernacki, A. Magalhães, A. Fonseca and I. Neves, J. Catal., 2011, 278, 102–110 CrossRef.
- H. Figueiredo, B. Silva, C. Quintelas, M. M. M. Raposo, P. Parpot, A. Fonseca, A. Lewandowska, M. Bañares, I. C. Neves and T. Tavares, Appl. Catal., B, 2010, 94, 1–7 CrossRef CAS.
- M. R. Maurya, C. Haldar, S. Behl, N. Kamatham and F. Avecilla, J. Coord. Chem., 2011, 64, 2995–3011 CrossRef CAS.
- M. Lashanizadegan, S. Rayati and Z. Dejparvar Derakhshan, Chin. J. Chem., 2011, 29, 2439–2444 CrossRef CAS.
- M. R. Maurya, P. Saini, C. Haldar, A. K. Chandrakar and S. Chand, J. Coord. Chem., 2012, 65, 2903–2918 CrossRef CAS.
- M. Lashanizadegan and E. Parvizi, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 45, 1154–1158 CrossRef CAS.
- A. Banaei and B. Rezazadeh, J. Coord. Chem., 2013, 66, 2129–2140 CrossRef CAS.
- A. R. Silva, H. Albuquerque, A. Fontes, S. Borges, Â. Martins, A. P. Carvalho and J. Pires, Ind. Eng. Chem. Res., 2011, 50, 11495–11501 CrossRef CAS.
- K. Xavier, J. Chacko and K. M. Yusuff, Appl. Catal., A, 2004, 258, 251–259 CrossRef CAS.
- K. N. Bhagya and V. Gayathri, J. Porous Mater., 2013, 20, 257–266 CrossRef CAS.
- A. Mobinikhaledi, M. Zendehdel and P. Safari, J. Porous Mater., 2014, 21, 565–577 CrossRef CAS.
- F. Li, D. Hu, Y. Yuan, B. Luo, Y. Song, S. Xiao, G. Chen, Y. Fang and F. Lu, Mol. Catal., 2018, 452, 75–82 CrossRef CAS.
- M. Salavati-Niasari, J. Mol. Catal. A: Chem., 2008, 284, 97–107 CrossRef CAS.
- M. Salavati-Niasari and S. Abdolmohammadi, J. Porous Mater., 2009, 16, 19–26 CrossRef CAS.
- S. Yamaguchi, A. Suzuki, M. Togawa, M. Nishibori and H. Yahiro, ACS Catal., 2018, 8, 2645–2650 CrossRef CAS.
- K. Ebitani, K. Nagashima, T. Mizugaki and K. Kaneda, Chem. Commun., 2000, 869–870 RSC.
- K. Kaneda, T. Itoh, N. Kii, K. Jitsukawa and S. Teranishi, J. Mol. Catal., 1982, 15, 349–365 CrossRef CAS.
- M. Salavati-Niasari, J. Mol. Catal. A: Chem., 2004, 217, 87–92 CrossRef CAS.
- G. R. Reddy, S. Balasubramanian and K. Chennakesavulu, J. Mater. Chem. A, 2014, 2, 19102 RSC.
- S. Rayati, E. Khodaei and M. Jafarian, J. Coord. Chem., 2017, 70, 2736–2750 CrossRef CAS.
- A. Okemoto, K. Ueyama, K. Taniya, Y. Ichihashi and S. Nishiyama, Catal. Commun., 2017, 100, 29–32 CrossRef CAS.
- S. L. Hailu, B. U. Nair, M. Redi-Abshiro, I. Diaz, R. Aravindhan and M. Tessema, Chin. J. Catal., 2016, 37, 135–145 CrossRef CAS.
- C. Galindo, P. Jacques and A. Kalt, Chemosphere, 2001, 45, 997–1005 CrossRef CAS PubMed.
- J. Prousek, Chem. Listy, 1996, 90, 229–237 CAS.
- (a) H. C. Sutton and C. C. Winterbourn, Free Radical Biol. Med., 1989, 6, 53–60 CrossRef CAS; (b) M. H. Robbins and R. S. Drago, J. Catal., 1997, 170, 295–303 CrossRef CAS; (c) A. H. Gemeay, I. A. Mansour, R. G. El-Sharkawy and A. B. Zaki, J. Mol. Catal. A: Chem., 2003, 193, 109–120 CrossRef CAS.
- M. Neamtu, C. Zaharia, C. Catrinescu, A. Yediler, M. Macoveanu and A. Kettrup, Appl. Catal., B, 2004, 48, 287–294 CrossRef CAS.
- O. Legrini, E. Oliveros and A. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS.
- S. Chavan, D. Srinivas and P. Ratnasamy, J. Catal., 2000, 192, 286–295 CrossRef CAS.
- J. M. Thomas, Nature, 1994, 368, 289 CrossRef.
- M. R. Maurya, S. J. Titinchi and S. Chand, Catal. Lett., 2003, 89, 219–227 CrossRef CAS.
- S. M. B. Hosseini-Ghazvini, P. Safari, A. Mobinikhaledi and M. Zendehdel, React. Kinet., Mech. Catal., 2015, 115, 703–718 CrossRef CAS.
- M. Zendehdel, H. Khanmohamadi and M. Mokhtari, J. Chin. Chem. Soc., 2010, 57, 205–212 CrossRef CAS.
- B. P. Nethravathi and K. N. Mahendra, J. Porous Mater., 2010, 17, 107 CrossRef CAS.
- M. R. Maurya, S. J. Titinchi and S. Chand, J. Mol. Catal. A: Chem., 2003, 193, 165–176 CrossRef CAS.
- B. Nethravathi, K. Mahendra and K. R. K. Reddy, J. Porous Mater., 2011, 18, 389–397 CrossRef CAS.
- B. Nethravathi, P. Manjunathan and K. Mahendra, J. Porous Mater., 2016, 23, 1305–1310 CrossRef CAS.
- R. Abraham and K. Yusuff, J. Mol. Catal. A: Chem., 2003, 198, 175–183 CrossRef CAS.
- A. Mobinikhaledi, M. Zendehdel, S. M.-B. Hosseini-Ghazvini and P. Safari, Transition Met. Chem., 2015, 40, 313–320 CrossRef CAS.
- A. Mobinikhaledi and M. Jabbarpour, Res. Chem. Intermed., 2015, 41, 511–523 CrossRef CAS.
- L. Xu, J. Zhang, Z. Li, Q. Ma, Y. Wang, F. Cui and T. Cui, New J. Chem., 2019, 43, 520–526 RSC.
- M. Kooti, F. Kooshki and E. Nasiri, Res. Chem. Intermed., 2019, 45, 2641–2656 CrossRef CAS.
- (a) Z. An, Y. Guo, L. Zhao, Z. Li and J. He, ACS Catal., 2014, 4, 2566–2576 CrossRef CAS; (b) S. M. Islam, A. S. Roy, R. C. Dey and S. Paul, J. Mol. Catal. A: Chem., 2014, 394, 66–73 CrossRef CAS; (c) A. Lu, T. P. Smart, T. H. Epps III, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2011, 44, 7233–7241 CrossRef CAS PubMed.
- M. Khajehzadeh, M. Moghadam and S. Jamehbozorgi, Inorg. Chim. Acta, 2019, 485, 173–189 CrossRef CAS.
- (a) A. Zhu, P. Tan, L. Qiao, Y. Liu, Y. Ma, X. Xiong and J. Pan, Inorg. Chem. Front., 2017, 4, 1748–1756 RSC; (b) L. Hu, F. Peng, D. Xia, H. He, C. He, Z. Fang, J. Yang, S. Tian, V. K. Sharma and D. Shu, ACS Sustainable Chem. Eng., 2018, 6, 17391–17401 CrossRef CAS; (c) B. Sahoo, A. E. Surkus, M. M. Pohl, J. Radnik, M. Schneider, S. Bachmann, M. Scalone, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11242–11247 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |