
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu-catalysed oxidative cyclisation of 1,5-dienes: an unprecedented role for the co-oxidant†
Aqeel A. Hussein ab
ab
aSchool of Chemistry, University of Southampton, Southampton, Hampshire SO17 1BJ, UK. E-mail: aahh1f19@soton.ac.uk
bCollege of Dentistry, University of Al-Ameed, PO Box 198, Karbala, Iraq
First published on 17th April 2020
Abstract
The Ru-mediated oxidative cyclisation of 1,5-dienes to furnish 2,5-dihydroxyalkyl-substituted tetrahydrofuran-diols (THF-diols) represents a practical approach for the synthesis of many bioactive natural products. In the current study, we reported profound findings obtained by density functional theory (DFT) simulations, and they were consistent with the experimental conditions. The results set out a catalytic cycle within intermediacy of NaIO4-complexed Ru(VI) species. Importantly, the co-oxidant played a critical role in the cyclisation step and subsequently the release of THF-diols. Following the formation of Ru(VI) glycolate, cyclisation and THF-diol release proceeded through NaIO4-coordinated Ru(VI) intermediates, outpacing the Ru(VIII) glycolate or THF-diolate intermediates and subsequently entering “second cycle” type pathways. The results indicated a cycle involving Ru(VIII)/Ru(VI)/Ru(IV)/Ru(VI) rather than Ru(VIII)/Ru(VI)/Ru(VIII)/Ru(VI)/Ru(VIII). Additionally, the existence of an electron-withdrawing group (EWG) on one of the double bonds of 1,5-dienes revealed that the regioselectivity of the Ru-catalysed oxidative cyclisation was predominantly initiated at the electron-rich alkene. Overall, this study offers new insights, which were ignored by earlier experimentalists and theoreticians, into the Ru-catalysed functionalizations of alkenes and 1,5-dienes.
Introduction
Many bioactive natural products such as polyether ionophore antibiotics and annonaceous acetogenins contain THF-diols.1,2 An important approach for the synthesis of THF-diols is the oxidative cyclisation of 1,5-dienes using transition-metal–oxo species (MnO4−, OsO4, and RuO4). This approach has been shown to give structurally complex products,3 whereby up to four stereogenic centres can be formed with the control of relative stereochemistry in a single reaction.3–8 The synthesis of cis-THF-diols by the oxidative cyclisation of 1,5-dienes was first reported using potassium permanganate,9 which was followed by related stereoselective approaches using osmium and ruthenium.10,11 The first mechanistic proposal was set out for the permanganate-promoted reaction by Walba et al. and was based on the (2 + 2) suprafacial additions of metal–oxo species across the olefin double bonds. Baldwin proposed an alternative mechanism for Mn oxidative cyclisations based on (3 + 2) cycloadditions,6c,9a,12,13 which has become a general framework for oxidative cyclisations by Os and Ru–oxo species,3,10,11 and the Ru-catalysed version is the concern of this paper.14Previous studies on the RuO4 oxidative cyclisation of 1,5-dienes have reported mixtures of cis and trans THF-diol products.11a However, developments by Stark et al. led to cis-THF diols with high yields and stereoselectivities (Scheme 1).5b,11b,11d They suggested a mechanism analogous to the one presented by Baldwin.9a It is basically believed that RuO4 interacts with a double bond to form Ru(VI) glycolate 3, followed by cyclisation to give THF-diolate Ru(IV) 4 and subsequent hydrolysis to afford cis-THF-diol 2 (Scheme 1).
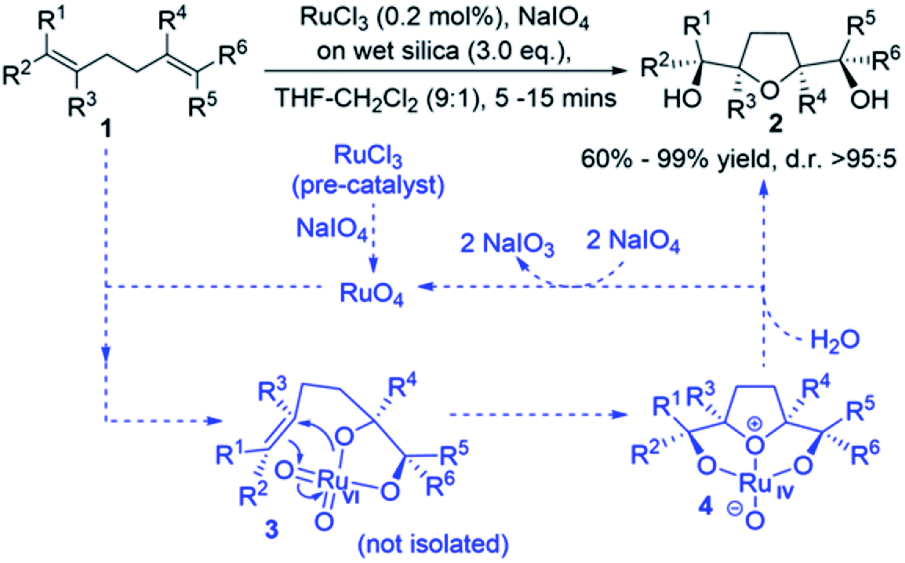 | ||
| Scheme 1 Ru-mediated oxidative cyclisation of 1,5-diene 1 to afford cis-THF-diol 2. | ||
Mechanistic computational studies on the oxidative cyclisation of 1,5-dienes by metal–oxo species have been reported, but studies concerning Ru-mediated reactions have not been fully investigated.13,15 Despite the reliable use of the reaction in the synthetic methods,11,14 only one step of the catalytic cycle, which is alkene oxidation to yield Ru(VI) dioxoglycolate, has been studied computationally.16 The mechanism of Ru-catalysed oxidative cyclisation involves several steps. These steps are as follows:
(a) alkene oxidation to generate Ru(VI) glycolate (the first reaction step, FRS);16
(b) cyclisation to form the Ru(IV) THF-diolate (the second reaction step, SRS);
(c) possible secondary reactions that may occur prior to cyclisation such as reoxidation, complexation with a co-oxidant, and hydration with water;
(d) hydrolysis to release the THF-diol; and
(e) other intermolecular (3 + 2) cycloaddition pathways that propagate the catalytic cycle (Fig. 1).
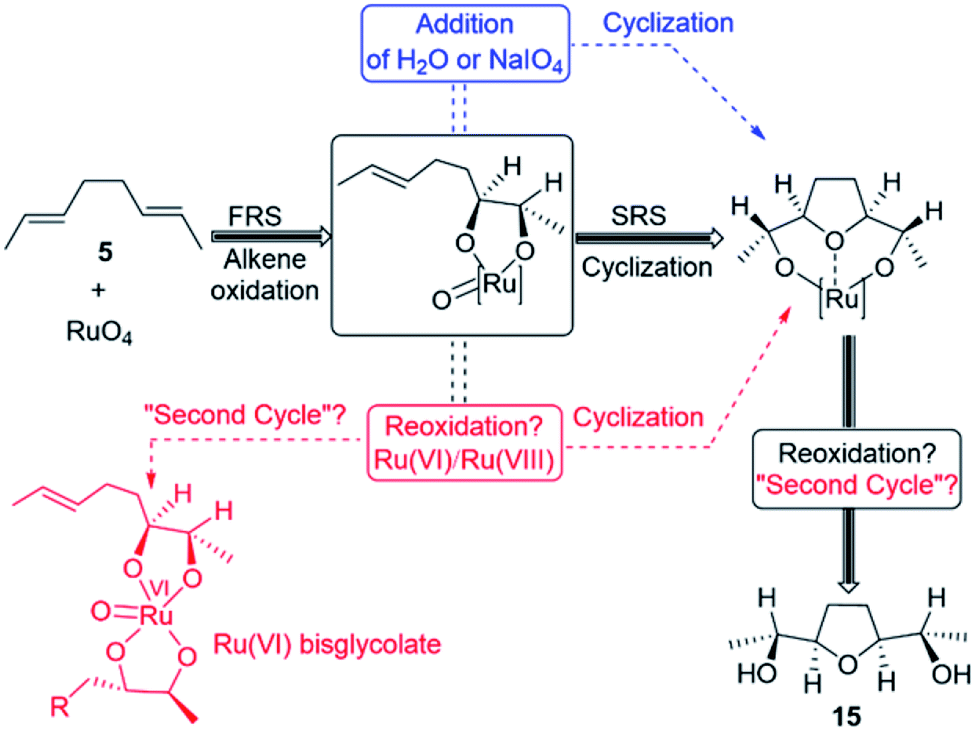 | ||
| Fig. 1 Pathways obtained for the Ru-catalysed oxidative cyclisation of 1,5-diene (5) in this study. | ||
In this context, almost no details of the mechanism's steps have appeared in the literature. This lack of mechanistic understanding frequently impedes further improvements in the catalytic studies. Therefore, the present study reports a detailed description of these steps, which were ignored by previous researchers, both experimentalists and theoreticians, using computational investigations. Based on the results, we proposed a catalytic cycle and determined the important role of the co-oxidant in the Ru-catalysed oxidative cyclisation of 1,5-dienes. Subsequently, the regioselectivity of the reaction was evaluated when an EWG was found on one of the double bonds of 1,5-dienes.
Results and discussion
The computational approach to the oxidative cyclisation of 1,5-diene was performed using DFT simulations with the (SMD/THF)-M06/aug-cc-pVDZ/LANL2DZ//M06/cc-pVDZ/LANL2DZ level of theory at 298.15 K.17 All calculations considered here account for singlet spin multiplicity.18 The following sections explain the mechanism of Ru-catalysed oxidative cyclisation and the regioselectivity of the reaction.Mechanism of Ru-catalysed oxidative cyclisation
The formation of THF-diolate from the oxidative cyclisation of 1,5-diene (5) has been realised through different reaction manifolds considered for SRS after the formation of the Ru(VI) dioxoglycolate intermediate 7 (Fig. 2). Generally, these manifolds either proceed without or with a co-oxidant (NaIO4).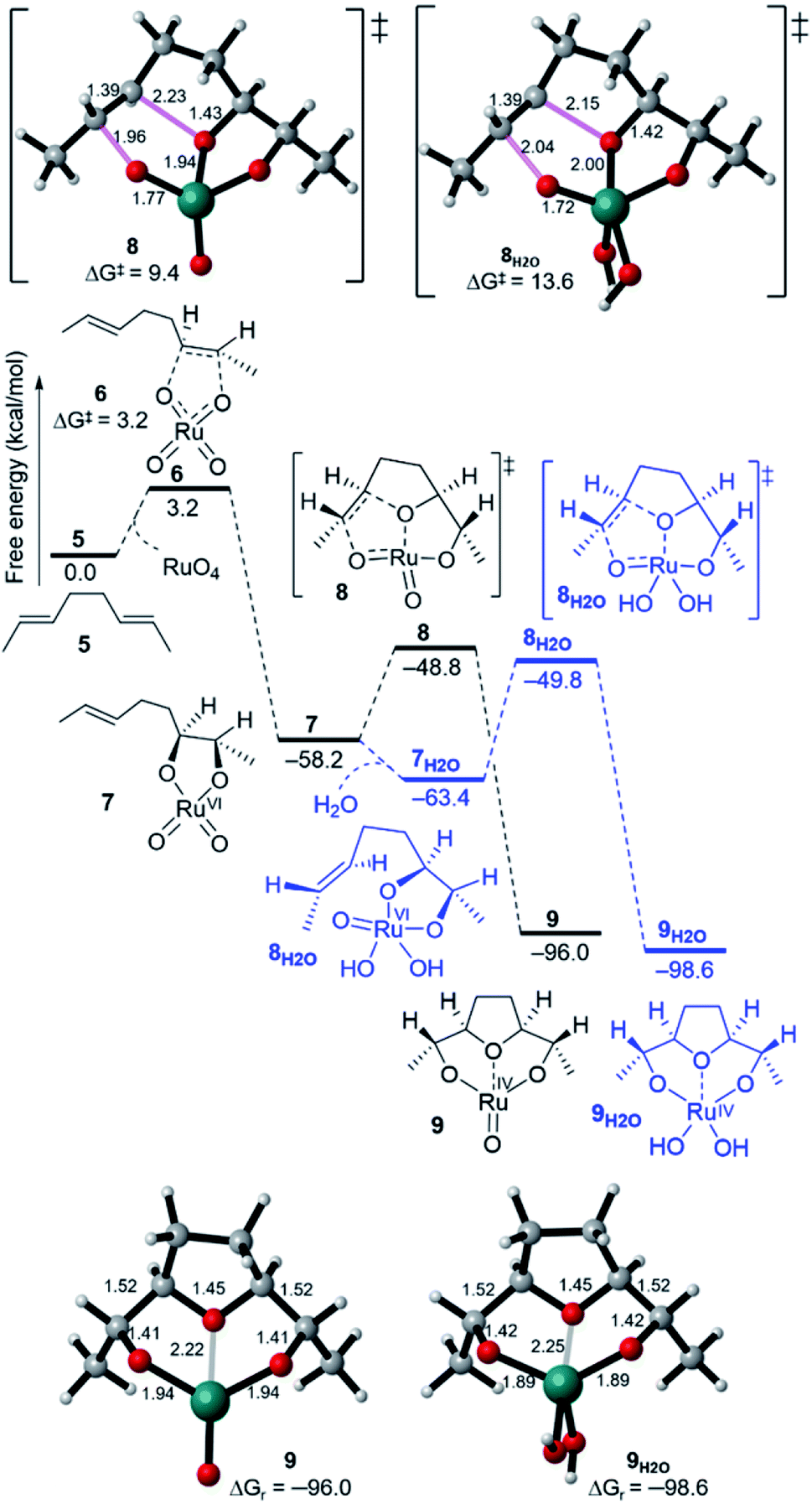 | ||
| Fig. 2 Free energy profiles for the oxidative cyclisation of 1,5-diene 5 by RuO4, showing cyclisation pathways without water (7 → 9) and with water (7H2O → 9H2O). | ||
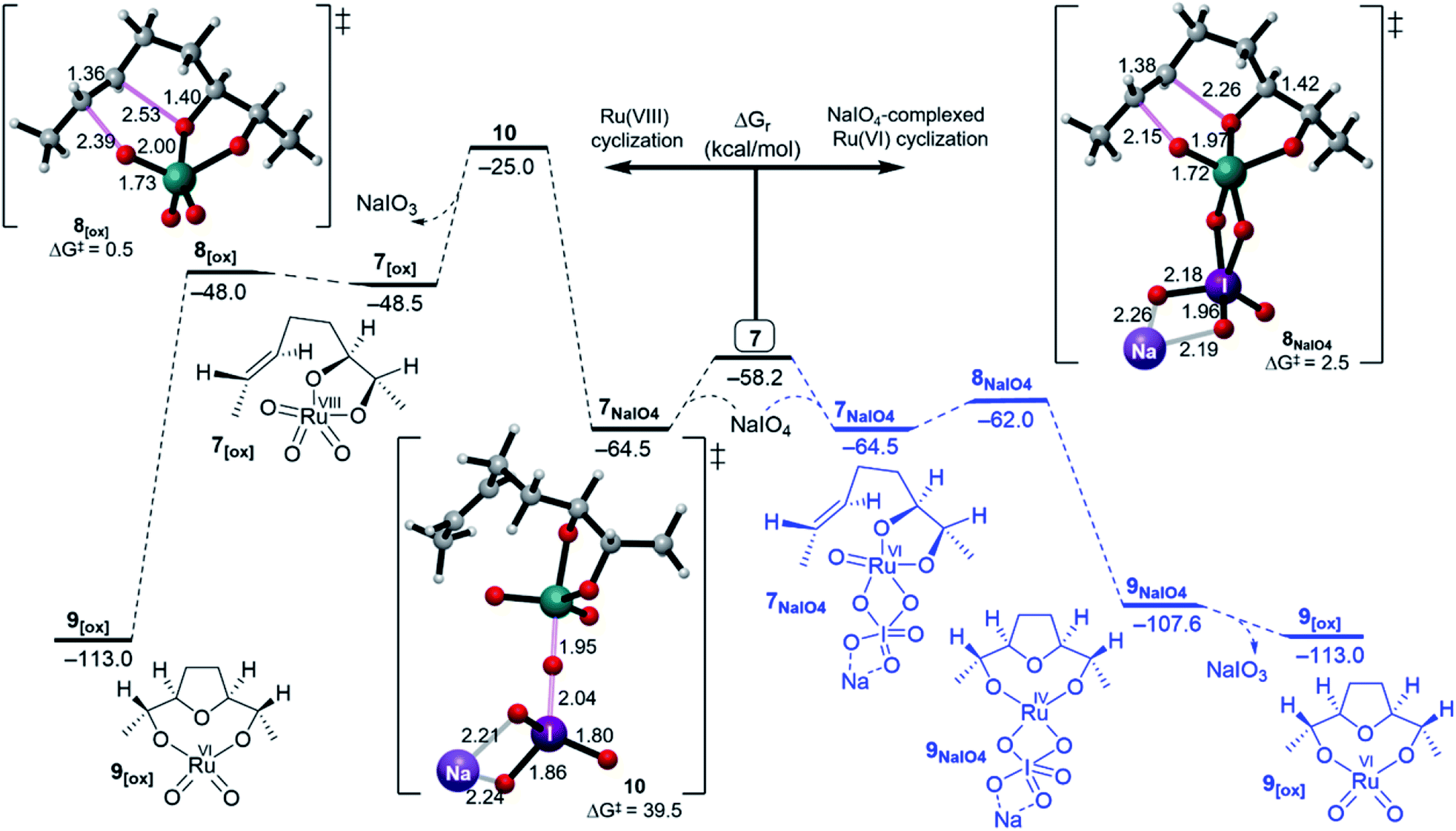 | ||
| Fig. 3 Free energy profiles for the Ru(VIII) (right) and NaIO4-complexed Ru(VI) (left) cyclisation pathways. | ||
The addition of a water molecule stabilized Ru(VI) 7 by 5.2 kcal mol−1 to give a distorted square pyramid Ru(VI) glycolate 7H2O (Fig. 2, blue pathway). However, this raised the barrier of cyclisation to 13.6 kcal mol−1 via a distorted trigonal bipyramidal (3 + 2) cycloaddition TS 8H2O to form the trigonal bipyramidal Ru(IV) diolate 9H2O (ΔGr = −35.2 kcal mol−1). The increased energy barrier can be understood by the effect of the decrease in the electrophilicity of the hydrated Ru(VI) glycolate 7H2O (see Fig. 4). The LUMO energy of 7H2O (LUMO = −3.7 eV, Egap = 3.1 eV) has been calculated to be higher than that of the non-hydrated Ru(VI) 7 (LUMO = −4.2 eV, Egap = 2.6 eV). The HOMO and LUMO orbitals are shown in Fig. 4.
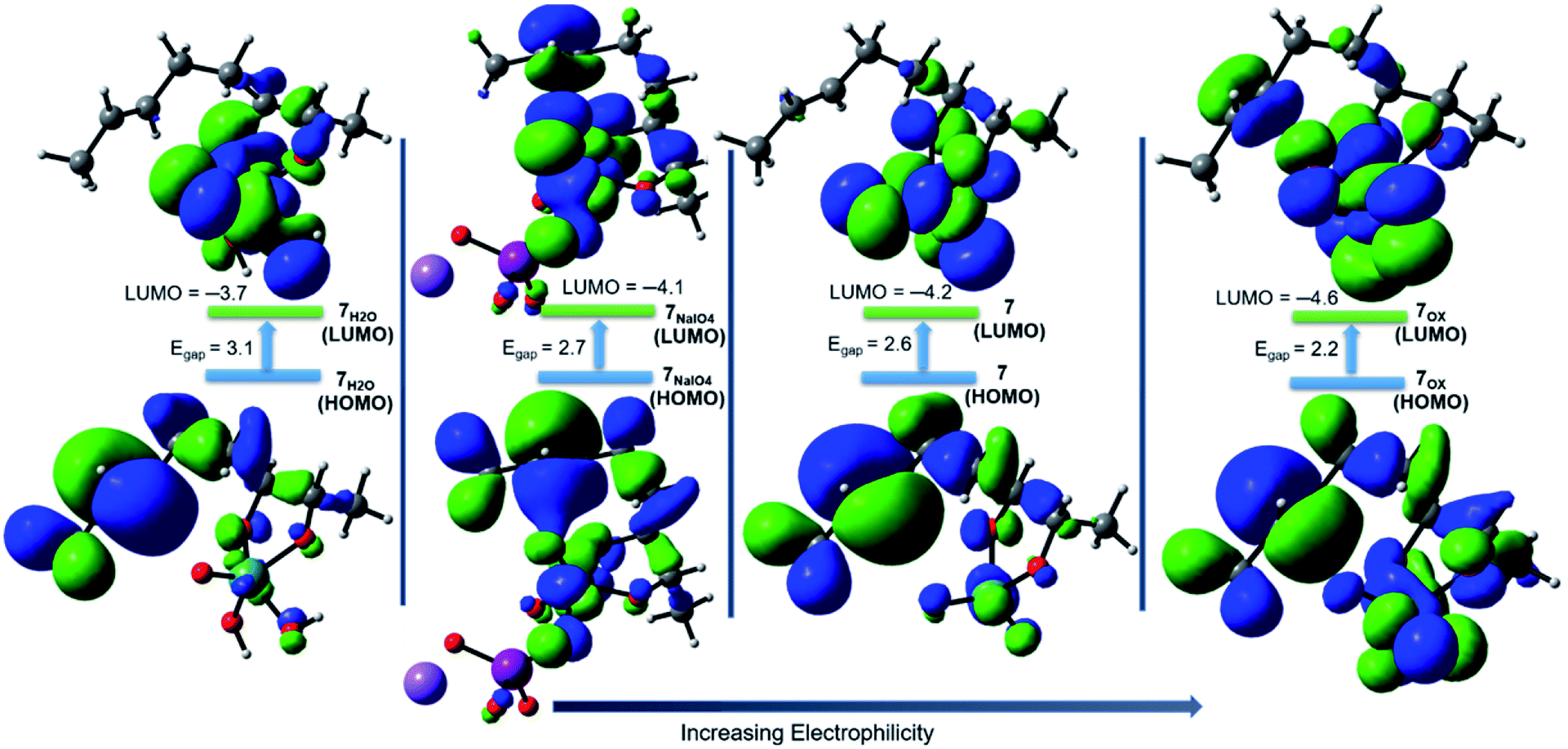 | ||
| Fig. 4 Visualization (0.02 isovalue surface) of the HOMO (located on the alkene site) and LUMO (located on the Ru site) orbitals of Ru(VI) 7, 7H2O, and 7NaIO4 and Ru(VIII) 7[ox] glycolates during the oxidative cyclisation of 1,5-diene (5) by RuO4. | ||
A comparison between Ru(VIII), hydrated Ru(VI), and naked Ru(VI) cyclisation is clearly evident. The Ru(VI) glycolates 7 and 7H2O have two d electrons, and these can lead to electronic repulsions with the two oxo ligands.26 Consequently, this will decrease the electrophilicity and increase the cyclisation barrier. In contrast, this does not occur in 7[ox] and thus, a remarkable increase in the electrophilicity (LUMO = −4.6 eV, Egap = 2.2 eV, Fig. 4) is seen to stimulate barrierless cycloaddition.
It is noteworthy to mention that reoxidation may be competitive when only alkenes are applied and not 1,5-dienes. However, whether the Ru(VIII) intermediate is viable during the Ru-catalysed dihydroxylation of alkenes is still disputed.27 In this work, DFT simulations showed that reoxidation was excluded due to the high barrier needed. Therefore, there is no possibility of entering a “second cycle” type pathway from the reaction of 7[ox] with another alkene to obtain the Ru(IV) bisglycolate (see Fig. 1).4c,11f,28–30
Cyclisation through NaIO4-complexed Ru(VI) glycolate displayed a low barrier of 2.5 kcal mol−1 via distorted trigonal bipyramidal TS 8NaIO4 and was a favourably exergonic step with ΔGr = −43.1 kcal mol−1 (Fig. 3, right).31 When the NaIO4-complexed pathway (7NaIO4 → 8NaIO4 → 9NaIO4) and the reoxidation pathway (7NaIO4 → 7[ox] → 8[ox] → 9[ox]) were compared, the former was exceedingly preferred. Molecular orbital analysis revealed that although the energy gap for 7NaIO4 was 2.7 eV (Fig. 4), which was slightly lower than that for 7, its TS 8NaIO4 had a significantly lower barrier. This may be attributed to the favourable interactions between the reacting moieties with less geometrical changes or reorganizations around the Ru centre due to the exchangeable oxo ligands between the I and Ru centres that lead to a more stabilized TS to give Ru(IV) THF-diolate 9NaIO4. The calculations show that Ru(IV) THF-diolate 9NaIO4 is simultaneously dissociated to Ru(VI) dioxodiolate 9[ox] in a favoured step (ΔGr = −6.6 kcal mol−1) (Fig. 3, right).
In summary, in the presence of NaIO4, cyclisation through NaIO4-coordinated Ru(VI) glycolate 7NaIO4 outpaces cyclisation via hydrated Ru(VI) 7H2O and reoxidized Ru(VIII) 7[ox]. In this regard, the simulations showed that reoxidation was energetically unfavourable and thus, the formation of a “second cycle” type pathway product, Ru(VI) bisglycolate, was totally outpaced.
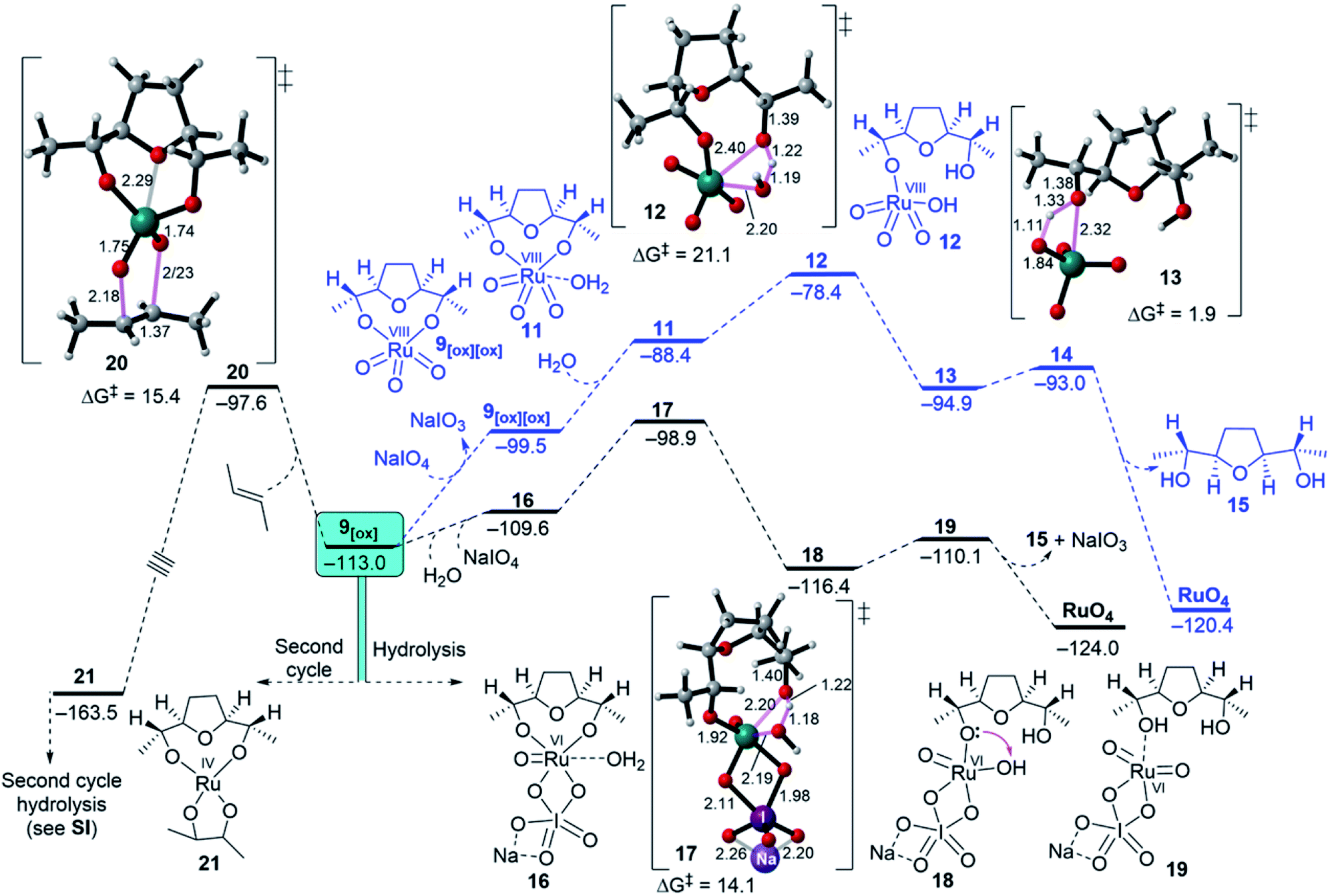 | ||
| Fig. 5 Free energy evaluations for divergent reactions of 9[ox], showing hydrolysis via Ru(VI) or Ru(VIII) or entering a second cycle through another ruthenylation. E-But-2-ene was used as a model for 5 to reduce the computational cost. | ||
First, the oxidation of Ru(IV) THF-dioxodiolate 9[ox] to Ru(VIII) trioxodiolate 9[ox][ox] is considered with an overall endergonicity of 13.5 kcal mol−1 (Fig. 5, blue pathway).32 In this regard, Ru(VIII) THF-trioxodiolate 9[ox][ox] can overcome a barrier of 21.1 kcal mol−1 via TS 12 to break the first O–[Ru] diolate in a modestly exergonic step (ΔGr = −6.5 kcal mol−1, see 13, Fig. 5), which is followed by the release of THF-diol 15 as an overall exergonic hydrolysis pathway (ΔGr = −20.9 kcal mol−1). Instead, DFT simulations indicate that Ru(VI) THF-dioxodiolate 9[ox] coordinates with NaIO4 and then H2O to yield 16 via a low endergonic step (ΔGr = 3.4 kcal mol−1, Fig. 5). Also, 16 needs a comparatively low barrier of 10.7 kcal mol−1 via a TS 17 to give intermediate 18 via an exergonic process (ΔGr = −3.4 kcal mol−1). In the opposite mode to the hydrolysis of Ru(VIII), the hydrolysis of the second O–[Ru] diolate in intermediate 18 was calculated to be unfavourable when compared to the experimental conditions. The located TS 22, shown in ESI Fig. 4,† requires a high energy barrier of 21.7 kcal mol−1 to transfer the proton from the oxo ligand (HO–[Ru]) to the second O–[Ru] diolate bond although it is an exergonic step (ΔGr = −7.6 kcal mol−1). Obviously, proton transfers involving electronegative atoms are known to be fast and diffusion controlled and therefore, getting intermediate 19 via a slightly endergonic step (ΔGr = 6.3 kcal mol−1) is suggested. At this point, hydrolysis through Ru(VI) followed by oxidation to regenerate RuO4 is more accessible than reoxidation, i.e., Ru(VI) → Ru(VIII) followed by hydrolysis.
Second, it is interesting to note that Ru(VI) dioxodiolate 9[ox] is a competent ruthenylating agent in the hydrolysis pathway and is capable of reacting with another alkene via a so-called “second cycle” type pathway (Fig. 5, left pathway).33 The energy barrier of the (3 + 2) cycloaddition of 9[ox] with E-but-2-ene was found to be 15.4 kcal mol−1 via TS 19 to afford a tetraester 20 via an exergonic reaction (ΔGr = −50.5 kcal mol−1). This would reveal a barrier difference of 1.3 kcal mol−1 between intermolecular ruthenylation and hydrolysis with the hydrolysis being predominant.
Finally, based on the above-mentioned results, a catalytic cycle for the Ru-catalysed oxidative cyclisation of 1,5-diene 5 was proposed (Fig. 6). The complexation of Ru(VI) glycolate 7 with a co-oxidant (NaIO4) is an essential feature found for SRS. The reoxidation of NaIO4-coordinated Ru(IV) THF-diolate 9NaIO4 to Ru(VI) THF-dioxodiolate 9[ox] followed by another coordination with both water and NaIO4 gives 17, releasing its THF-diol 15 and regenerating RuO4 to continue the iterative cycle.
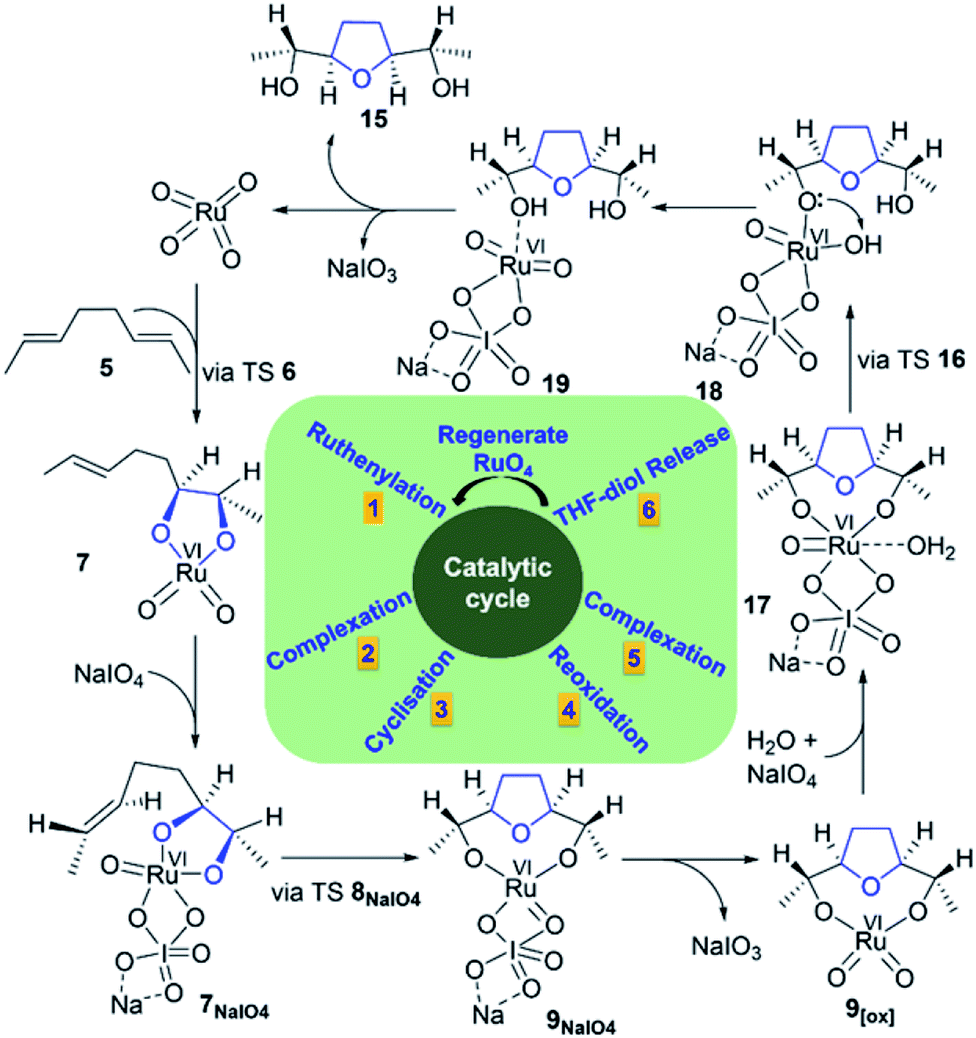 | ||
| Fig. 6 Proposed catalytic cycle for Ru-catalysed oxidative cyclisation of 1,5-diene 5. | ||
Regioselectivity of the Ru-catalysed oxidative cyclisation
The regioselectivity of RuO4 is considered when an EWG such as a methyl ester group is found on one of the double bonds of 1,5-diene, as indicated in methyl dienoate 5a shown in Fig. 7.11b Based on the aforementioned findings, DFT calculations have been performed for FRS and SRS (Fig. 7). For FRS, the favourability of RuO4 to initiate the electron-rich alkene (ERA) pathway over the electron-deficient alkene (EDA) pathway is highly evident. For instance, the ERA Ru(VI) glycolate 7a (ΔGr = −62.4 kcal mol−1) is formed via a barrierless (3 + 2) cycloaddition TS 6a of ΔG‡ = 1.7 kcal mol−1 (Fig. 7, right), whereas the formation of the EDA Ru(VI) glycolate d-7a requires a higher barrier of 4.9 kcal mol−1 via TS d-6a via a slightly less exergonic step (ΔGr = −55.1 kcal mol−1 (Fig. 7, left).34 Thus, the ERA product is kinetically (ΔΔG‡ = −3.2 kcal mol−1) and thermodynamically (ΔΔGr = −7.3 kcal mol−1) favoured with a greater development of the C–O bond formation for ERA TS 6a over EDA TS d-6a.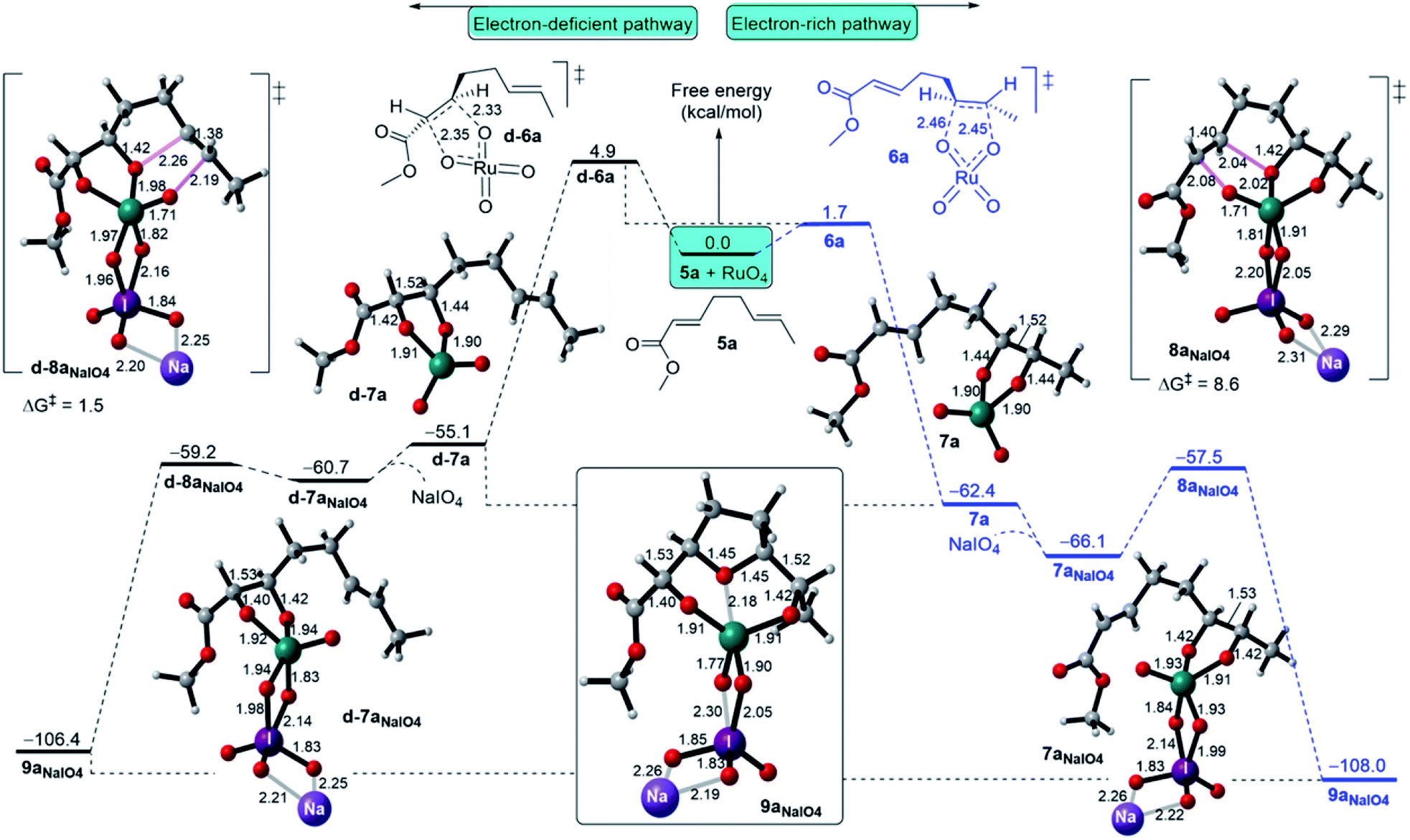 | ||
| Fig. 7 Free energy bifurcation for the regioselectivity of the Ru-catalyzed oxidative cyclisation of methyl 1,5-dienoate (5a) initiated at the electron-rich and electron-deficient alkenes. | ||
On the cyclisation step, the ERA Ru(VI) glycolate 7aNaIO4 cyclises, via TS 8aNaIO4 with a barrier of ΔG‡ = 8.6 kcal mol−1, and this is slower than cyclisation through EDA Ru(VI) glycolate pathway (TS d-8aNaIO4, ΔG‡ = 1.5 kcal mol−1). A reasonable explanation for this is that the oxo ligands in d-7aNaIO4 are more electrophilic than those in 7aNaIO4 mainly due to the electron-withdrawing nature of the carbonyl group with a possible minor contribution from complexation with NaIO4. This would lead to a lower barrier for cyclisation because it has lower LUMO energy on the Ru site and higher HOMO energy on the alkene site. However, in the ERA Ru(VI) glycolate 7aNaIO4, cyclisation occurs from the highly electron-deficient alkene site to the slightly electron-deficient Ru site, subsequently ensuring that the cyclisation has a higher barrier. In comparison with the symmetric 1,5-diene (5), the cyclisation via the ERA NaIO4-complexed Ru(VI) glycolate requires a three-fold higher energy barrier than the cyclisation with the symmetric 1,5-diene (5). Overall, the existence of an EWG on one of the alkenes in the 1,5-diene structure results in the Ru-catalysed oxidative cyclisation being predominantly initiated at the electron-rich alkene.
Conclusions
The first detailed mechanistic study on the Ru-catalysed oxidative cyclisation of 1,5-dienes has been performed with the (SMD/THF)-M06/aug-cc-pVDZ/LANL2DZ//M06/cc-pVDZ/LANL2DZ level of theory. The DFT simulations are in excellent agreement with the experimental conditions and consequently, complications accompanied throughout the Ru-catalysed oxidative cyclisation of 1,5-dienes are resolved.The results set out the catalytic cycle for THF-diol formation through the intermediacy of Ru(VI) with RuO4 as the active catalytic species. Importantly, the co-oxidant NaIO4 has been evidently shown to play a critical role in the cyclisation and hydrolysis steps through complexation with Ru rather than reoxidation to a higher oxidation state. Following the initially formed Ru(VI) dioxoglycolate, the cyclisation overcomes a favourable NaIO4-coordinated TS to give the THF-diolate ring, excluding the cyclisation or entering the second cycle through the intermediacy of Ru(VIII) trioxoglycolate 7[ox]. Thereafter, the dissociation of the NaIO4-coordinated Ru(IV) THF-diolate 9NaIO4 to Ru(VI) THF-dioxodiolate 9[ox] was calculated to undergo hydrolytic release to give THF-diol 15 and RuO4 to continue the iterative cycle. The release of THF-diol 15 from Ru(VI) THF-dioxodiolate 9[ox] was found to be favourable over reoxidation to Ru(VIII) THF-trioxodiolate 9[ox][ox] or entering the so-called “second cycle” type pathway. This protocol has evidently conducted a cycle of Ru(VIII)/Ru(VI)/Ru(IV)/Ru(VI) rather than Ru(VIII)/Ru(VI)/Ru(VIII)/Ru(VI)/Ru(VIII).
Furthermore, DFT simulations on asymmetric 1,5-dienes, such as methyl 1,5-dienoate (5a), have provided further mechanistic descriptions when an electron-withdrawing group is attached to one of the alkenes. The calculations revealed that the Ru-catalysed oxidative cyclisation is predominantly initiated at the electron-rich alkene. The cyclisation from the ERA NaIO4-complexed Ru(VI) glycolate was shown to be slower than the cyclisation from an electron-deficient Ru(VI) intermediate. In comparison with symmetric 1,5-diene (5), the barrier for cyclisation from an asymmetric 1,5-diene was calculated to be more than three times higher than the barrier for symmetric cyclisation.
Overall, this computational study gives significantly important insights into this key synthetic method, which may offer a viable pathway towards advancing an efficient protocol for alkene oxidation.
Computational details
All calculations were performed using Gaussian 09 (ref. 35) with the global hybrid functional approximations M06 (ref. 36) used with the augmented correlation-consistent polarized valence double-ζ (DZ) basis set (cc-pVDZ and aug-cc-pVDZ)37 for C, H, O, Si, and Na, whereas an effective core potential basis step LANL2DZ38 was used for Ru and I. All geometry optimisations, even for triplet and quintet spin multiplicities, shown in the ESI,† were performed with the basis set cc-pVDZ/LANL2DZ, in which all minima intermediates were verified by the absence of negative eigenvalues in the vibrational frequency analysis. Restricted spin Hartree–Fock calculations were used for singlet spin multiplicity, whereas unrestricted spin calculations were used for triplet and quintet spin multiplicity. All of the transition state structures were found using the Berny algorithm,39 verified by vibrational analysis, and visualised by animating the negative eigenvector coordinate. In order to account for the effect of the solvent, single-point energies of the optimised geometries were evaluated with the M06/aug-cc-pVDZ/LANL2DZ level of theory in tetrahydrofuran (THF) as a representative solvent medium via the solvation model based on density (IEFPCM-SMD).40 To obtain the free energies at 298.15 K and 1 atm, the thermal corrections were evaluated from the unscaled vibrational frequencies at the M06/cc-pVDZ/LANL2DZ level of theory and were then added to the electronic energies calculated from the M06/aug-cc-pVDZ/LANL2DZ level of theory. Intrinsic reaction coordinate (IRC) calculations were performed for the identified transition states to confirm the reaction path proceeding in both directions (reactant and product), in which the Hessian was recomputed every 3 predictor steps with a step size along the reaction path of 0.05 Bohr.41 All activation free energies are quoted relative to infinitely separated reagents and the optimised structures were illustrated using CYLview.42Conflicts of interest
There are no conflicts to declare.Acknowledgements
I acknowledge the computational resources from the iridis4 supercomputer supported by the University of Southampton. I highly acknowledge the University of Southampton/School of Chemistry for providing the visitor-status research position (2717441/EB00-VISIT). Many thanks to Prof. Richard C. D. Brown for his valuable proofreading and his precious support.Notes and references
- For reviews of THF-containing natural products see. Polyether ionophores: (a) J. Rutkowski and B. Brzezinski, Structures and Properties of Naturally Occurring Polyether Antibiotics, BioMed Res. Int., 2014, 21, 1559–1581 Search PubMed; (b) A. Huczynski, Polyether ionophores-promising bioactive molecules for cancer therapy, Bioorg. Med. Chem. Lett., 2012, 22, 7002–7010 CrossRef CAS PubMed; (c) D. A. Kevin, D. A. F. Meujo and M. T. Hamann, Polyether ionophores: broad-spectrum and promising biologically active molecules for the control of drug-resistant bacteria and parasites, Expert Opin. Drug Discovery, 2009, 4, 109–146 CrossRef CAS PubMed; (d) C. J. Dutton, B. J. Banks and C. B. Cooper, Polyether ionophores, Nat. Prod. Rep., 1995, 12, 165–181 RSC; (e) A. Acetogenins, R. Tundis, J. B. Xiao and M. R. Loizzo, Annona species (Annonaceae): a rich source of potential antitumor agents?, Ann. N. Y. Acad. Sci., 2017, 1398, 30–36 CrossRef PubMed; (f) C. C. Liaw, J. R. Liou, T. Y. Wu, F. R. Chang and Y. C. Wu, Acetogenins from Annonaceae, Prog. Chem. Org. Nat. Prod., 2016, 101, 113–230 CAS; (g) A. Bermejo, B. Figadere, M. C. Zafra-Polo, I. Barrachina, E. Estornell and D. Cortes, Acetogenins from Annonaceae: recent progress in isolation, synthesis and mechanisms of action, Nat. Prod. Rep., 2005, 22, 269–303 RSC; (h) M. Leboeuf, A. Cavé, P. K. Bhaumik, B. Mukherjee and R. Mukherjee, The phytochemistry of the annonaceae, Phytochemistry, 1980, 21, 2783–2813 CrossRefTHF-containing macrolides: (i) A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Alvarez, Tetrahydrofuran-containing macrolides: a fascinating gift from the deep sea, Chem. Rev., 2013, 113, 4567–4610 CrossRef CAS PubMed.
- For reviews of THF synthesis see: (a) A. de la Torre, C. Cuyamendous, V. Bultel-Ponce, T. Durand, J. M. Galano and C. Oger, Recent advances in the synthesis of tetrahydrofurans and applications in total synthesis, Tetrahedron, 2016, 72, 5003–5025 CrossRef CAS; (b) J. D. Rainier, Synthesis of Substituted Tetrahydrofurans. In Synthesis of Saturated Oxygenated Heterocycles I: 5- and 6-Membered Rings, Top. Heterocycl. Chem., 2014, 35, 1–41 CAS; (c) G. Jalce, X. Franck and B. Figadère, Diastereoselective synthesis of 2,5-disubstituted tetrahydrofurans, Tetrahedron: Asymmetry, 2009, 20, 2537–2581 CrossRef CAS; (d) J. P. Wolfe and M. B. Hay, Recent advances in the stereoselective synthesis of tetrahydrofurans, Tetrahedron, 2007, 63, 261–290 CrossRef CAS PubMed; (e) M. C. Elliott, Saturated oxygen heterocycles, J. Chem. Soc., Perkin Trans. 1, 2002, 2301–2323 RSC; (f) M. M. Faul and B. E. Huff, Strategy and methodology development for the total synthesis of polyether ionophore antibiotics, Chem. Rev., 2000, 100, 2407–2473 CrossRef CAS PubMed; (g) T. L. B. Boivin, Synthetic routes to tetrahydrofuran, tetrahydropyran, and spiroketal units of polyether antibiotics and a survey of spiroketals of other natural-products, Tetrahedron, 1987, 43, 3309–3362 CrossRef CAS.
- For further examples see: (a) A. Roushanbakhti, Y. F. Liu, P. C. M. Winship, M. J. Tucker, W. M. Akhtar, D. S. Walter, G. Wrigley and T. J. Donohoe, Cobalt versus Osmium: Control of Both trans and cis Selectivity in Construction of the EFG Rings of Pectenotoxin 4, Angew. Chem., Int. Ed., 2017, 56, 14883–14887 CrossRef CAS PubMed; (b) N. S. Sheikh, Synthetic endeavours towards oxasqualenoid natural products containing 2,5-disubstituted tetrahydrofurans-eurylene and teurilene, Nat. Prod. Rep., 2014, 31, 1088–1100 RSC; (c) T. J. Donohoe and R. M. Lipiński, Interplay of Cascade Oxidative Cyclisation and Hydride Shifts in the Synthesis of the ABC Spiroketal Ring System of Pectenotoxin-4, Angew. Chem., Int. Ed., 2013, 52, 2491–2494 CrossRef CAS PubMed; (d) H. Göksel and C. B. W. Stark, Total Synthesis of cis-Solamin: Exploiting the RuO4-Catalysed Oxidative Cyclisation of Dienes, Org. Lett., 2006, 8, 3433–3436 CrossRef PubMed; (e) B. Lygo, D. Slack and C. Wilson, Synthesis of neodysiherbaine, Tetrahedron Lett., 2005, 46, 6629–6632 CrossRef CAS.
- For reviews see: (a) J. Adrian, L. J. Gross and C. B. W. Stark, The direct oxidative diene cyclisation and related reactions in natural product synthesis, Beilstein J. Org. Chem., 2016, 12, 2104–2123 CrossRef CAS PubMed; (b) N. S. Sheikh, Comparative perspective and synthetic applications of transition metal mediated oxidative cyclisation of 1,5-dienes towards cis-2,5-disubstituted tetrahydrofurans, Org. Biomol. Chem., 2014, 12, 9492–9504 RSC; (c) V. Piccialli, Ruthenium tetroxide and perruthenate chemistry. Recent advances and related transformations mediated by other transition metal oxo-species, Molecules, 2014, 19, 6534–6582 CrossRef PubMed; (d) B. S. Pilgrim and T. J. Donohoe, Osmium-catalysed oxidative cyclisation of dienes and their derivatives, J. Org. Chem., 2013, 78, 2149–2167 CrossRef CAS PubMed; (e) V. Piccialli, Oxidative cyclisation of dienes and polyenes mediated by transition-metal-oxo species, Synthesis, 2007, 2585–2607 CrossRef CAS.
- For other oxidative cyclisations of 1,4-dienes, 1,6-dienes and 1,7-dienes see: (a) V. Piccialli, N. Borbone and G. Oliviero, Ruthenium-catalysed oxidative cyclisation of 1,7-dienes. A novel diastereoselective synthesis of 2,7-disubstituted trans-oxepane diols, Tetrahedron Lett., 2007, 48, 5131–5135 CrossRef CAS; (b) S. Roth and C. B. W. Stark, Efficient oxidative cyclisation of 1,6-dienes: a highly diastereoselective entry to substituted tetrahydropyrans, Angew. Chem., Int. Ed., 2006, 45, 6218–6221 CrossRef CAS PubMed; (c) A. R. L. Cecil and R. C. D. Brown, Stereoselective synthesis of cis-2,6-bis-hydroxyalkyl-tetrahydropyrans by the permanganate promoted oxidative cyclisation of 1,6-dienes, Tetrahedron Lett., 2004, 45, 7269–7271 CrossRef CAS; (d) B. Travis and B. Borhan, Oxidative cyclisation of 1,4-dienes to yield 2,3,5-trisubstituted tetrahydrofuran-diols, Tetrahedron Lett., 2001, 42, 7741–7745 CrossRef CAS; (e) V. Piccialli, RuO4-catalysed oxidative cyclisation of 1,6-dienes to trans-2,6-bis(hydroxymethyl)tetrahydropyranyldiols. A novel stereoselective process, Tetrahedron Lett., 2000, 41, 3731–3733 CrossRef CAS.
- (a) A. M. Al Hazmi, N. S. Sheikh, C. J. R. Bataille, A. A. M. Al-Hadedi, S. V. Watkin, T. J. Luker, N. P. Camp and R. C. D. Brown, trans-2-Tritylcyclohexanol as a chiral auxiliary in permanganate-mediated oxidative cyclisation of 2-methylenehept-5-enoates: application to the synthesis of trans-(+)-linalool oxide, Org. Lett., 2014, 16, 5104–5107 CrossRef CAS PubMed; (b) R. C. D. Brown and P. J. Kocienski, A synthesis of salinomycin 1. Synthesis of key fragments, Synlett, 1994, 415–417 CrossRef CAS; (c) D. M. Walba, M. D. Wand and M. C. Wilkes, Stereochemistry of the permanganate oxidation of 1,5-dienes, J. Am. Chem. Soc., 1979, 101, 4396–4397 CrossRef CAS.
- R. C. D. Brown and J. F. Keily, Asymmetric permanganate-promoted oxidative cyclisation of 1,5-dienes by using chiral phase-transfer catalysis, Angew. Chem., Int. Ed., 2001, 40, 4496–4498 CrossRef CAS PubMed.
- For cis-selective THF-diol formation from oxidative cyclisation of dihydroxyalkenes using metal oxo agents (Ru, Os, and Cr). For Ru oxo-species: (a) H. Cheng and C. B. W. Stark, A double donor-activated ruthenium(VII) catalyst: synthesis of enantiomerically pure THF-diols, Angew. Chem., Int. Ed., 2010, 49, 1587–1590 CrossRef CAS PubMed; (b) S. Gohler and C. B. W. Stark, Catalytic diastereo- and position selective oxidative mono-cyclisation of 1,5,9-trienes and polyenes, Org. Biomol. Chem., 2007, 5, 1605–1614 RSCOs oxo-species: (c) T. J. Donohoe, K. M. P. Wheelhouse, P. J. Lindsay-Scott, P. A. Glossop, I. A. Nash and J. S. Parker, Pyridine-N-oxide as a mild reoxidant which transforms osmium-catalysed oxidative cyclisation, Angew. Chem., Int. Ed., 2008, 47, 2872–2875 CrossRef CAS PubMed; (d) T. J. Donohoe and S. Butterworth, Oxidative cyclisation of diols derived from 1,5-dienes: formation of enantiopure cis-tetrahydrofurans by using catalytic osmium tetroxide; Formal synthesis of (+)-cis-solamin, Angew. Chem., Int. Ed., 2005, 44, 4766–4768 CrossRef CAS PubMedCr oxo-species: (e) E. J. Corey and D. C. Ha, Total synthesis of venustatriol, Tetrahedron Lett., 1988, 29, 3171–3174 CrossRef CAS; (f) D. M. Walba and G. S. Stoudt, Oxidative cyclisation of 5,6-dihydroxyalkenes promoted by Cr(VI) oxo species: A novel cis-2,5-disubstituted tetrahydrofuran synthesis, Tetrahedron Lett., 1982, 23, 727–730 CrossRef CAS; (g) B. D. Hammock, S. S. Gill and J. E. Casida, Synthesis and morphogenetic activity of derivatives and analogs of aryl geranyl ether juvenoids, J. Agric. Food Chem., 1974, 22, 379–385 CrossRef CAS PubMed.
- (a) J. E. Baldwin, M. J. Crossley and E.-M. M. Lehtonen, Stereospecificity of oxidative cycloaddition reactions of 1,5-dienes, J. Chem. Soc., Chem. Commun., 1979, 918–920 RSC; (b) E. Klein and W. Rojahn, Permanganate oxidation of 1,5-diene compounds, Tetrahedron, 1965, 21, 2353–2358 CrossRef CAS; (c) A. Kötz and T. Steche, About the gradual oxidation of citronellol and geraniol, J. Prakt. Chem., 1924, 107, 193–210 CrossRef.
- (a) T. J. Donohoe and S. Butterworth, A general oxidative cyclisation of 1,5-dienes using catalytic osmium tetroxide, Angew. Chem., Int. Ed., 2003, 42, 948–951 CrossRef CAS PubMed; (b) T. J. Donohoe, J. J. G. Winter, M. Helliwell and G. Stemp, Hydrogen bonding control in the oxidative cyclisation of 1,5-dienes, Tetrahedron Lett., 2001, 42, 971–974 CrossRef CAS; (c) M. de Champdore, M. Lasalvia and V. Piccialli, OsO4-Catalysed oxidative cyclisation of geranyl and neryl acetate to cis-2,5-bis(hydroxymethyl)tetrahydrofurans, Tetrahedron Lett., 1998, 39, 9781–9784 CrossRef CAS.
- For cis and trans THF-diol mixture products from Ru-catalysed oxidative cyclisations of 1,5-dienes see: (a) V. Piccialli, T. Caserta, L. Caruso, L. Gomez-Paloma and G. Bifulco, RuO4-mediated oxidative polycyclisation of linear polyenes, a new approach to the synthesis of the bis-THF diol core of antitumour cis-cis adjacent bis-THF annonaceous acetogenins, Tetrahedron, 2006, 62, 10989–11007 CrossRef CAS; (b) S. Roth, S. Göhler, H. Cheng and C. B. W. Stark, A highly efficient procedure for ruthenium tetroxide catalysed oxidative cyclisations of 1,5-dienes, Eur. J. Org. Chem., 2005, 4109–4118 CrossRef CAS; (c) S. Roth, S. Göhler, H. Cheng and C. B. W. Stark, A Highly Efficient Procedure for Ruthenium Tetroxide Catalysed Oxidative Cyclisations of 1,5-Dienes, Eur. J. Org. Chem., 2005, 4109–4118 CrossRef CAS; (d) V. Piccialli and T. Caserta, Perruthenate ion. Another metal oxo species able to promote the oxidative cyclisation of 1,5-dienes to 2,5-disubstituted cis-tetrahydrofurans, Tetrahedron Lett., 2004, 45, 303–308 CrossRef CAS; (e) V. Piccialli and N. Cavallo, Improved RuO4-catalysed oxidative cyclisation of geraniol-type 1,5-dienes to cis-2,5-bis(hydroxymethyl)tetrahydrofuranyldiols, Tetrahedron Lett., 2001, 42, 4695–4699 CrossRef CAS; (f) L. Albarella, D. Musumeci and D. Sica, Reactions of 1,5-dienes with ruthenium tetraoxide: stereoselective synthesis of tetrahydrofurandiols, Eur. J. Org. Chem., 2001, 997–1003 CrossRef CAS; (g) P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, A greatly improved procedure for ruthenium tetraoxide catalysed oxidations of organic compounds, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS.
- Significantly, it was proposed that Mn(VI) or Mn(V) glycolate intermediate, and based on DFT calculations shown in ref. 13, enforces the 2,5-cis-relationship across the incipient THF ring during cyclisation step as indicated in Baldwin's proposed mechanism for the oxidative cyclisation of 1,5-dienes.
- A. Poethig and T. Strassner, The mechanism of the permanganate-promoted oxidative cyclisation of 1,5-dienes - A DFT study, Collect. Czech. Chem. Commun., 2007, 72, 715–727 CrossRef CAS.
- For RuO4 catalysed oxidative polycyclisation of isoprenoid polyenes for the synthesis of adjacently linked poly-THF rings see: (a) G. Bifulco, T. Caserta, L. Gomez-Paloma and V. Piccialli, RuO4-promoted syn-oxidative polycyclisation of isoprenoid polyenes: a new stereoselective cascade process, Tetrahedron Lett., 2002, 43, 9265–9269 CrossRef CAS; (b) G. Bifulco, T. Caserta, L. Gomez-Paloma and V. Piccialli, Corrigendum to “RuO4-promoted syn-oxidative polycyclisation of isoprenoid polyenes: a new stereoselective cascade process”, Tetrahedron Lett., 2002, 43, 9265–9269 (Tetrahedron Lett., 2003, 44, 3429) CrossRef CAS; (c) G. Bifulco, T. Caserta, L. Gomez-Paloma and V. Piccialli, RuO4-promoted oxidative polycyclisation of isoprenoid polyenes. A further insight into the stereochemistry of the process, Tetrahedron Lett., 2003, 44, 5499–5503 CrossRef CAS; (d) T. Caserta, V. Piccialli, L. Gomez-Paloma and G. Bifulco, RuO4-catalysed oxidative polycyclisation of squalene. Determination of the configuration of the penta-tetrahydrofuranyl diol product, Tetrahedron, 2005, 61, 927–939 CrossRef CAS; (e) V. Piccialli, N. Borbone and G. Oliviero, RuO4-catalysed oxidative polycyclisation of the Cs-symmetric isoprenoid polyene digeranyl. An unexpected stereochemical outcome, Tetrahedron, 2008, 64, 11185–11192 CrossRef CAS.
- (a) A. A. Hussein, M. J. S. Phipps, C.-K. Skylaris and R. C. D. Brown, Mechanism of Os-Catalysed Oxidative Cyclisation of 1,5-Dienes, J. Org. Chem., 2019, 84, 15173–15183 CrossRef CAS PubMed; (b) A. Poethig and T. Strassner, Stereoselective OsO4-catalysed oxidative cyclisation of 1,5-dienes, J. Org. Chem., 2010, 75, 1967–1973 CrossRef CAS PubMed; (c) P. J. di Dio, S. Zahn, C. B. W. Stark and B. Kirchner, Understanding Selectivities in Ligand-free Oxidative Cyclisations of 1,5-and 1,6-Dienes with RuO4 from Density Functional Theory, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 367–375 CAS.
- (a) J. Frunzke, C. Loschen and G. Frenking, Why Are Olefins Oxidized by RuO4 under Cleavage of the Carbon-Carbon Bond whereas Oxidation by OsO4 Yields cis-Diols?, J. Am. Chem. Soc., 2004, 126, 3642–3652 CrossRef CAS PubMed; (b) T. Strassner and M. Drees, Rutheniumtetraoxide oxidation of alkenes - a density functional theory study, J. Mol. Struct.: THEOCHEM, 2004, 671, 197–204 CrossRef CAS; (c) P. O. Norrby, H. C. Kolb and K. B. Sharpless, Calculations on the reaction of ruthenium tetroxide with olefins using density functional theory (DFT). Implications for the possibility of intermediates in osmium-catalysed asymmetric dihydroxylation, Organometallics, 1994, 13, 344–347 CrossRef CAS.
- For suitability of the DFT function M06 with Ru-containing system see: (a) S. Chen, Y. Zheng, T. Cui, E. Meggers and K. N. Houk, Arylketone π-Conjugation Controls Enantioselectivity in Asymmetric Alkynylations Catalysed by Centrochiral Ruthenium Complexes, J. Am. Chem. Soc., 2018, 140, 5146–5152 CrossRef CAS PubMed; (b) J. S. Cannon, L. Zou, P. Liu, Y. Lan, D. J. O'Leary, K. N. Houk and R. H. Grubbs, Carboxylate-Assisted C(sp3)-H Activation in Olefin Metathesis-Relevant Ruthenium Complexes, J. Am. Chem. Soc., 2014, 136, 6733–6743 CrossRef CAS PubMed; (c) Y.-F. Yang, L. W. Chung, X. Zhang, K. N. Houk and Y.-D. Wu, Ligand-Controlled Reactivity, Selectivity, and Mechanism of Cationic Ruthenium-Catalysed Hydrosilylations of Alkynes, Ketones, and Nitriles: A Theoretical Study, J. Org. Chem., 2014, 79, 8856–8864 CrossRef CAS PubMed; (d) H. Miyazaki, M. B. Herbert, P. Liu, X. Dong, X. Xu, B. K. Keitz, T. Ung, G. Mkrtumyan, K. N. Houk and R. H. Grubbs, Z-Selective Ethenolysis with a Ruthenium Metathesis Catalyst: Experiment and Theory, J. Am. Chem. Soc., 2013, 135, 5848–5858 CrossRef CAS PubMed.
- Calculations on high spin multiplicity triplet and quintet were considered and shown in ESI.†.
- (a) The ruthenate ester Ru(VI) was not isolated yet and there is no clear evidence of its formation although Sica and et al. had proposed the existence of Ru(VI) bisglycolate due to reoxidation Ru(VI) → Ru(VIII) glycolate after formation of Ru(VI) glycolate (see ref. 20); (b) Like osmylation reaction that gives osmate(VI) ester, the reaction of RuO4 with double bond is called ruthenylation because it gives ruthenate(VI) ester.
- (a) L. Albarella, F. Giordano, M. Lasalvia, V. Piccialli and D. Sica, Evidence for the existence of a cyclic ruthenium (VI) diester as an intermediate in the oxidative scission of (−)-α-pinene with RuO4, Tetrahedron Lett., 1995, 36, 5267–5270 CAS; (b) V. Piccialli, D. Sica and D. Smaldone, Reaction of 7-dehydrocholesteryl acetate with RuO4. First isolation of a cyclic ruthenium (VI) diester, Tetrahedron Lett., 1994, 35, 7093–7096 CrossRef CAS.
- (a) The low barrier and high exergonicity of reaction reflect the experimental reaction rate aspects which is comparingly more reactive than the other metal–oxo reagents MnO4− and OsO4 (see ref. 13 and 15a, b), where the typical reaction time for Ru-catalysed oxidative cyclisation is 5–15 mins; (b) The barriers calculated for FRS and SRS by Dio et al., using a model of two alkenes rather than 1,5-diene, were high to be compared to the reaction time in addition to the fact their study were in accord with the general mechanistic proposal for the oxidative cyclisation of 1,5-dienes by RuO4 (see ref. 15c).
- It is very desirable to mention that Ru(VI) complex ([(Me3tacn)-(CF3CO2)RuO2]ClO4) was isolated by Che et al. and used in cis-dihydroxylation of alkenes in which they also isolated the (3 + 2) cycloadduct intermediate Ru(IV) glycolate. (See ref. 23).
- W.-P. Yip, W.-Y. Yu, N. Zhu and C.-M. Che, Alkene cis-Dihydroxylation by [(Me3tacn)(CF3CO2)RuVIO2]ClO4 (Me3tacn = 1,4,7-Trimethyl-1,4,7-triazacyclononane): Structural Characterization of (3 + 2) Cycloadducts and Kinetic Studies, J. Am. Chem. Soc., 2005, 127, 14239–14249 CrossRef CAS PubMed.
- The energy barrier for the inner sphere oxidation of R(VIII) glycolate 7[ox] formation from 7NaIO4 via TS 10 is 39.5 kcal mol−1. If it is considered as an outer sphere pathway the barrier will be 33.2 kcal mol−1. From both manners, formation of Ru(VIII) glycolate is disfavoured.
- A. L. Tenderholt, K. O. Hodgson, B. Hedman, R. H. Holm and E. I. Solomon, Substrate and Metal Control of Barrier Heights for Oxo Transfer to Mo and W Bis-dithiolene Sites, Inorg. Chem., 2012, 51, 3436–3442 CrossRef CAS PubMed.
- (a) T. Ishizuka, H. Kotani and T. Kojima, Characteristics and reactivity of ruthenium-oxo complexes, Dalton Trans., 2016, 45, 16727–16750 RSC; (b) W. H. Leung and C. M. Che, High-valent ruthenium(IV) and -(VI) oxo complexes of octaethylporphyrin. Synthesis, spectroscopy, and reactivities, J. Am. Chem. Soc., 1989, 111, 8812–8818 CrossRef CAS.
- (a) B. Plietker, M. Niggemann and A. Pollrich, The acid accelerated ruthenium-catalysed dihydroxylation. Scope and limitations, Org. Biomol. Chem., 2004, 2, 1116–1124 RSC; (b) B. Plietker and M. Niggemann, The RuO4-catalysed dihydroxylation, ketohydroxylation and mono oxidation-novel oxidation reactions for the synthesis of diols and [small alpha]-hydroxy ketones, Org. Biomol. Chem., 2004, 2, 2403–2407 RSC; (c) B. Plietker and M. Niggemann, An Improved Protocol for the RuO4-Catalysed Dihydroxylation of Olefins, Org. Lett., 2003, 5, 3353–3356 CrossRef CAS PubMed.
- Any intermolecular (3 + 2) cycloaddition between Ru(VIII) 7[ox] and another alkene (E-but-2-ene was used as a model to reduce the computational cost) to get Ru(VI) bisglycolate as called “second cycle” type was not found. However, in the Os-catalysed dihydroxylation of alkenes or oxidative cyclisation of 1,5-dienes, the second cycle pathways were noticed (see ref. 15a and 29).
- (a) P. Dupau, R. Epple, A. A. Thomas, V. V. Fokin and K. B. Sharpless, Osmium-Catalysed Dihydroxylation of Olefins in Acidic Media: Old Process, New Tricks, Adv. Synth. Catal., 2002, 344, 421–433 CrossRef CAS; (b) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Catalytic Asymmetric Dihydroxylation, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS; (c) J. S. M. Wai, I. Marko, J. S. Svendsen, M. G. Finn, E. N. Jacobsen and K. B. Sharpless, A mechanistic insight leads to a greatly improved osmium-catalysed asymmetric dihydroxylation process, J. Am. Chem. Soc., 1989, 111, 1123–1125 CrossRef CAS.
- The Ru(VI) or Ru(VIII) glycolate have yet to be characterized or detected by any means due to the high instability. However, there is a week evidence on the existence of Ru(VI) bisglycolate. Sica et al. suggested the existence of Ru(VI) bisglycolate as an intermediate in the oxidative scission of (−)-α-pinene with RuO4 performed in acetone–water (2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) which gives α-ketol as the sole oxidation product (see ref. 20). Also, they proposed the same intermediate for reaction of 7-dehydrocholesteryl acetate with RuO4 to give consequently 1,2-diol and α-ketol as final oxidation products. Their evidence was based on fast experiments monitored by NMR in which they compared their 1H-NMR peaks with isolated Os(VI) bisglycolate.
:1) which gives α-ketol as the sole oxidation product (see ref. 20). Also, they proposed the same intermediate for reaction of 7-dehydrocholesteryl acetate with RuO4 to give consequently 1,2-diol and α-ketol as final oxidation products. Their evidence was based on fast experiments monitored by NMR in which they compared their 1H-NMR peaks with isolated Os(VI) bisglycolate. - For geometry of Ru(VI) complexes see: W. P. Griffith, The Chemistry of Ruthenium Oxidation Complexes, in Ruthenium Oxidation Complexes. Catalysis by Metal Complexes, Springer Netherlands, Dordrecht, 2011, vol. 34, pp. 1–134 Search PubMed.
- It was not possible to locate a TS for an intermolecular ruthenylation between 9[ox][ox], like 7[ox] glycolate, with E-but-2-ene whereas this is unlike to Os-catalysed oxidative cyclisation of 1,5-dienes.
- In comparison to Ru(VI) glycolate 7, the Ru(VI) glycolate 7 did not undergo intermolecular ruthenylation and this is believed
due to the geometrical difference, especially angle in O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) RuO, between 7 and 9[ox]. The angle of ORuO in Ru(VI) diolate 9[ox] is 119.4° and Ru(VI) glycolate 7 is 125.7°.
RuO, between 7 and 9[ox]. The angle of ORuO in Ru(VI) diolate 9[ox] is 119.4° and Ru(VI) glycolate 7 is 125.7°. - Due to the difficulty in locating the TS 6a using the DFT functional M06 (M06/cc-pVDZ/LANL2DZ), both TSs 6a and d-6a have been optimised in gas phase with M06-2X (M06-2X/cc-pVDZ/LANL2DZ) followed by single point energy calculations in THF with the M06 functional ((SMD)-M06//aug-cc-pVDZ/LANL2DZ).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2013 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Applications and validations of the Minnesota density functionals, Chem. Phys. Lett., 2011, 502, 1–13 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- (a) T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS; (b) R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS; (c) D. E. Woon and T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) W. R. Wadt and P. J. Hay, Ab initio effective core potentials for molecular calculations, Potentials for main group elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS; (c) P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- (a) H. B. Schlegel, Optimisation of equilibrium geometries and transition structures, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS; (b) X. Li and M. J. Frisch, Energy-Represented Direct Inversion in the Iterative Subspace within a Hybrid Geometry Optimisation Method, J. Chem. Theory Comput., 2006, 2, 835–839 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- (a) H. P. Hratchian and H. B. Schlegel, Accurate reaction paths using a Hessian based predictor-corrector integrator, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed; (b) M. A. Collins, Molecular potential-energy surfaces for chemical reaction dynamics, Theor. Chem. Acc., 2002, 108, 313–324 Search PubMed; (c) K. Fukui, The path of chemical reactions - the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Y. Legault, CYLview 1.0b, Universitéde Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional calculated reaction pathways, cartesian coordinates, absolute energies for reported structures. See DOI: 10.1039/d0ra02303e |
| This journal is © The Royal Society of Chemistry 2020 |