
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural effect of oxazolone derivatives on the initiating abilities of dye-borate photoredox systems in radical polymerization under visible light†
F. Ścigalski and
B. Jędrzejewska *
*
Faculty of Chemical Technology and Engineering, UTP University of Science and Technology, Seminaryjna 3, 85-326 Bydgoszcz, Poland. E-mail: beata@utp.edu.pl
First published on 5th June 2020
Abstract
Three photoinitiating systems based on new oxazolone derivatives have been developed and their performance in initiation of radical polymerization of acrylate monomers has been tested by differential scanning calorimetry. The absorption characteristics of the oxazol-5(4H)-ones is compatible with the emission characteristics of different light sources like diode pulse solid state lasers. Thus, the dyes were used as sensitizers which are photoreduced during a photochemical reaction in the presence of phenyltriethylborate salt. Results showed that the increase in the dimensionality of the molecule extends the range of light absorption and increases the efficiency of the photoinitiation process. The photoreduction of the oxazolone–borate complex was studied using steady-state and nanosecond laser flash photolysis. The dye singlet and triplet were found to be quenched by the electron donor via an electron transfer process. Rate constants for the quenching of the excited states were high and were found to depend on the dye structure.
Introduction
Application of photopolymerization in nanotechnology, optics and imaging techniques, electronics, material processing and other fields of science and technology,1–3 leads to development of novel photoinitiators (PI),4 i.e. substances that participate in the photoinitiation of polymerization when exposed to radiation (UV or visible).5 Type I photoinitiators, which undergo direct photofragmentation into radicals, mostly require irradiation with ultraviolet light in the 300–400 nm range. Photoinitiators operating in the visible region usually generate radicals through bimolecular processes. In such a case a dye plays a role of a photosensitizer (PS) in some specific reaction.5,6 Thus, radicals which start a chain reaction may be generated through primary or subsequent reactions involving one or more components, followed dye excitation.5,6The decisive factor of radical polymerization in industrial applications is that it should typically be carried out under relatively undemanding conditions.3 From this standpoint, a visible light which is safe, cheap and easily available, is more attractive energy source than an ultraviolet light. Therefore, the design of novel dyes with excellent light absorption properties in the 380–800 nm region and high molar extinction coefficients is still a challenge.7
Several photoinitiator systems8 that are active in the visible light region have been developed. They are based on sensitizers such as cyanine and hemicyanine,9,10 coumarin,11,12 acridinedione,13,14 perylene,15,16 hexaarylbiimidazole,17 curcumin,18 carbazole,19 benzophenothiazine, benzophenoxazine,20,21 dithienothiophene22 derivatives and other,23–32 and different co-initiators.33 These systems initiate polymerization reaction by either free radical or cationic mechanism.1,6
Recently, much attention is paid to polymers that may be used in medicine and biochemistry. In this respect, polymeric materials containing the electrophilic oxazolone ring are of particular interest due to their hydrolytic stability and formation of stable covalent bonds with amine and thiol groups present in enzymes, proteins, and other biomolecules.34,35 For example, copolymers containing 4,4-dimethyl-5-oxazolone rings have been used to immobilize enzymes such as RNase A, deoxyribonuclease I, glucose oxidase, glucoamylase, and trypsin36 and proteins such as sericin,37 protein A, IgG, and bovine serum albumin (BSA).38 Kilbey et al. recently showed that a biomolecule like dansylcadaverine can be attached onto poly(2-vinyl-4,4-dimethyl-5-oxazolone).39,40 Lynn et al. revealed that the poly(2-vinyl-4,4-dimethyl-5-oxazolone) grafted surfaces may be used to prevent or promote mammalian cell adhesion and bacterial biofilm growth.35,41 Fontaine et al.35 disclosed the ability of 2-vinyl-4,4-dimethyl-5-oxazolone to produce block copolymers by reversible addition fragmentation chain transfer sequential polymerization with methyl acrylate, styrene, and methyl methacrylate. Apart from the biomedical applications, 4,4-dimethyl-5-oxazolone functionalized polymeric materials has been widely studied as nucleophile scavengers.42–45 Another interesting application of copolymers with oxazolone chromophore is their used as nonlinear optical materials in photonics and electronics. Sahraoui et al.46 revealed that film forming of oxazolone containing polymers show higher nonlinear optical effect than the corresponding oxazolone derivatives and offer great promise for practical device applications.
In general, polymeric materials containing the oxazolone moiety have attracted considerable attention because of their interesting biological application. However, to the best of our knowledge there have been no reports regarding the application of oxazolone derivatives in photochemistry as light sensitive photoinitiator for radical polymerization. This prompted us to study the effect of the dye structure on the photochemical properties and photoinitiating abilities of the photoinitiating system containing oxazolone dyes.
Experimental section
Materials and methods
The synthesis of the tested compounds, (4Z)-4-[4-(diphenylamino)benzylidene]-2-phenyl-1,3-oxazol-5(4H)-one (1), (4Z)-N,N,N-tris[2-phenyl-4-benzylidene-1,3-oxazol-5(4H)-one]amine (2) and (4Z)-N,N,N-tris[2-phenyl-4-(4-phenylbenzylidene)-1,3-oxazol-5(4H)-one]amine (3) is described in our previous paper.47 The structures are presented in Chart 1. | ||
| Chart 1 The structure of dyes tested as light absorber and borate salt used as co-initiator in photoinitiated polymerization of TMPTA. | ||
All solvents (spectroscopic grade), monomer – trimethylolpropane triacrylate (TMPTA) were purchased from Aldrich Chemical co. and were used as received.
The steady-state absorption and emission spectra were recorded at room temperature using a Shimadzu UV-vis Multispec-1501 spectrophotometer and a Hitachi F-4500 fluorescence spectrophotometer, respectively. The solutions were prepared in solvents of different polarity with a concentration of the dye in the solution 1.0 × 10−5 M and 1.0 × 10−6 M, respectively.
The fluorescence quantum yields for the dyes in EtOAc were calculated using eqn (1). The absorbances (A) of both the dye and reference solution at an excitation wavelength (470 nm) was ca. 0.1. Fluorescein in 0.1 M NaOH (ϕref = 0.91 (ref. 48)) was used as reference.
 | (1) |
The fluorescence lifetimes were measured using a time-correlated single photon counting (TCSPC) system (FLS920P Spectrometers). The samples were excited at 466 nm using a picosecond diode laser (Edinburgh Instruments) generating pulses of about 81.5 ps. Its maximal average power is 5 mW. The instrument response function (IRF) was obtained by water in a quartz cuvette. The fluorescence decays were deconvoluted using FAST software. The fluorescence decays were fitted to two-exponentials.
Nanosecond laser flash photolysis (LFP) experiments were carried out using an LKS.60 Laser Flash Photolysis apparatus (Applied Photophysics). For the excitation, a Q-switched nanosecond Nd:YAG laser (λexc = 355 nm, pulse width about 4–5 ns, energy 65 mJ per pulse) from a Lambda Phisik/model LPY 150 was used. Transient absorbances at pre-selected wavelengths were monitored by a detection system consisting of a monochromator, a photomultiplier tube (Hamamatsu R955) and a pulsed xenon lamp (150 W) as a monitoring source. The signal from the photomultiplier was processed by a Helwett-Packard/Agilent an Agilent Infiniium 54810A digital storage oscilloscope and an Acorn compatible computer.
The oxidation or reduction potentials (Eox or Ered versus Ag/AgCl (3.0 M KCl) electrode) of the borate salt and the dyes, respectively, were measured in acetonitrile by cyclic voltammetry with tetrabutylammonium perchlorate (0.1 M) as the supporting electrolyte (Electroanalytical Cypress System Model CS-1090). The working electrode was a 1 mm platinum disk whereas a platinum wire was employed as counter electrode. The cyclic voltammograms were obtained from a one-compartment glass cell, where the scan rate was 100 or 400 mV s−1.
The free energy change ΔG for an electron transfer between oxazolone dyes and borate salt was calculated from the classical Rehm–Weller equation:49
| ΔGet = Eox − Ered − ES (or ET) + C | (2) |
Photopolymerization of TMPTA was studied by means of Differential Scanning Calorimeter, DSC 2010 (TA Instrument) with an argon ion laser Model Melles Griot 43 series. Approximately 0.035 ± 0.002 g sample mixture was placed in the aluminum DSC pans. Heat flow versus time (DSC thermogram) curves were recorded in an isothermal mode, cured at 25 °C by 488 nm light with an intensity of 100 mW cm−2. The reaction heat liberated in the polymerization was directly proportional to the number of vinyl groups reacting in the system.50 By integrating the area under the exothermic peak, the conversion of the vinyl groups (C) or the extent of reaction could be determined according to eqn (3).
 | (3) |
The polymerization rate (Rp) is directly related to the heat flow (dH/dt) according to eqn (4).
 | (4) |
The quantum yield of polymerization Φp was defined as the number of polymerized double bonds per absorbed photon.52
Polymerization solution was composed of 1-methyl-2-pyrrolidinone (MP; 1 mL), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA; 9 mL), oxazolone derivatives (A488 nm = 2) and B6 (0.0075 M). A reference formulation did not contain an electron donor (B6).
Results and discussion
Spectroscopic and physico-chemical properties
Panchromatic sensitization of polymerization usually requires the type II photoinitiators consisting of a suitable dye as a primary absorber and a co-initiator (usually an amine).1,2,5,6 In the present work three oxazolone derivatives were used as new sensitizers because they show absorption over a wide region of the visible spectrum which is the common for different light sources like diode pulse solid state lasers (DPSS) with the emission at 457 nm, 473 nm, 488 nm or 514 nm. Their electronic absorption spectra in ethyl acetate (EtOAc) are depicted in Fig. 1 and S3–S5† whereas Fig. S1 and S2† illustrate the spectra in toluene. | ||
| Fig. 1 UV-vis absorption spectra of 1, 2 and 3 in EtOAc solution (the concentration was 1.9 × 10−5 M for 1, 7.1 × 10−6 M for 2 and 7.8 × 10−6 M for 3). | ||
As indicated from Fig. 1, the main bands at 466.5 nm (ε = 4.24 × 104 M−1 cm−1), 496 (ε = 10.37 × 104 M−1 cm−1) and 454 nm (ε = 11.06 × 104 M−1 cm−1) for 1, 2 and 3, respectively, are attributed to π, π* transition. The absorption peaks in the 300–365 nm regions corresp5orption spectrum observed in going from 1 to 2 is due to the increase of electron affinity of the electron-accepting moiety of the light absorbing chromophore. However, the main absorption peak of 3 is slightly blue shifted. As described earlier,47,53 this trend is connected with an elongation of the π system which results in a decrease of the effectiveness of charge transfer between the electron donor (triphenylamine) and the electron acceptor (2-phenyl-oxazolone) moieties caused by the internal rotation.
The fluorescence spectra of compounds 1, 2 and 3 are centered at 571 nm, 573.2 nm and 627 nm in ethyl acetate, respectively. The fluorescence quantum yield ϕfl is higher for the branched dyes 2 and 3 (0.4 and 0.38) than for a linear push–pull compound (ca. 0.095) indicating that the nonradiative pathways are reduced in such structures.47,53
The photochemical reactivity of new photoinitiating systems was investigated by steady state photolysis experiments upon DPSS laser exposure in ethyl acetate as described previously.47,53 The maximum of their absorption is not sensitive to the irradiation, there are no significant changes in band position and its intensity upon 473 nm irradiation. On the other hand, for oxazolone derivatives/borate solution, the absorption bands intensity strongly decreases upon the irradiation, as resulting from the photosensitized decomposition of the dyes (Fig. S6†) which may indicate formation of a radical anion of the dye.
Besides bleaching of the absorption also a strong fluorescence quenching was observed in the dye/B6 couples (Fig. S7†). It indicated that there may be intermolecular fluorescence quenching and energy transfer between B6 and the tested dyes, and B6 can reduce the fluorescence emission intensity of oxazolones.
If the fluorescence of the tested dyes is quenched by electron transfer reaction, the rate constant interaction between dye/borate salt can be calculated from the Stern–Volmer coefficient KSV according to the eqn (5):
 | (5) |
 | ||
| Fig. 2 Stern–Volmer plots for 1/B6, 2/B6 and 3/B6 couples in EtOAc (the dye concentration was 5 × 10−6 M; Ex = 466 nm; Em = 560 nm). | ||
The fluorescence decay for given dyes was bi-exponential with shorter component of few hundred picoseconds and the longer one of the nanosecond time scale as described earlier.47,53 They were connected with emission from non-relaxed and relaxed transfer states. The calculated averaged fluorescence lifetimes in the absence of quencher were 88 ns, 2.5 ns and 3 ns for compounds 1, 2 and 3, respectively. Thus, the bimolecular quenching rate constants were 1.52 × 1010 M−1s−1, 1.6 × 1010 M−1s−1 and 1.82 × 1010 M−1s−1, respectively. These very high fluorescence quenching rate constants are in an agreement with the favorable ΔG found for the electron transfer reaction. The ΔGet was estimated based on the well-known Rehm–Weller eqn (2) (ref. 49) giving the negative values (Table 1). The Eox and Ered of both photoredox pair components were determined from cyclovoltametric measurements (Table 1). The calculations show that for the tested photoredox pairs the electron transfer process is thermodynamically allowed.54,55 This, in turn, allows to predict that the oxidation of the oxazolones by borate salt (B6) leads to the generation of a free radical that can start polymerization of the acrylic monomers.9,10
| Photoinitiator system | |||
|---|---|---|---|
| 1/B6 | 2/B6 | 3/B6 | |
| a ϕt – yield of triplet formation; Ered – reduction potential of the dyes; Eox – oxidation potential of B6, Eox (B6) = 0.764 V; Rp max av – average maximal polymerization rate; Cf av – average final conversion; Hmax av – average maximal heat flow; Tmax – average time to reach maximal heat flow. | |||
| ϕt | 0.086 | 0.24 | 0.04 |
| ES1 (eV) | 2.43 | 2.33 | 2.35 |
| Ered (A/A˙−) (V) | −1.010 | −1.036 | −0.908 |
| ΔGet (eV) | −0.657 | −0.533 | −0.677 |
| Rp max av (μmol s−1) | 2.1 | 2.4 | 3.3 |
| Cf av (%) | 22.3 | 26.0 | 30.6 |
| Hmax av (mW mg−1) | 4.54 | 5.10 | 6.96 |
| Tmax av (s) | 30.3 | 29.7 | 27.9 |
Polymerization kinetics studies
Fig. 3 and 4 illustrate the photo-DSC profiles of TMPTA initiated by oxazolone/B6 systems. The data for the maximal heat flow (Hmax av), maximal polymerization rate (Rp max) and the final conversion of TMPTA (Cf) are summarized in Table 1. | ||
| Fig. 3 Photo-DSC profiles for 1/B6, 2/B6 and 3/B6 in TMPTA, cured at r.t. by visible light (488 nm) with an intensity of 100 mW cm−2; the B6 concentration is 0.0075 M. | ||
 | ||
| Fig. 4 Rate vs. conversion for photopolymerization of TMPTA initiated by 1/B6, 2/B6 and 3/B6, cured r.t. by visible light (488 nm) with an intensity of 100 mW cm−2 (The photoinitiator concentration is 0.0075 M). | ||
Obviously, compounds 1, 2 and 3 does not work alone as photoinitiators. However, when coupled with an electron donor, they initiate polymerization of TMPTA. In all the photoinitiator pairs the tetramethylammonium phenyltriethylborate (B6) was chosen as co-initiator. Thus, the efficiency of photoinitiation of polymerization should be addressed to the structure of the dyes. The results verify that 3/B6 exhibits more preferable a photoinitiation of polymerization behavior than 2/B6 and 1/B6. Evidently, the final conversion of TMPTA is higher and it takes less time to reach the maximum polymerization rate (Rp max) for 3/B6 photoinitiator system. The enhanced efficiency may be attributed to the increase of electron affinity of the electron-accepting moiety of the light-absorbing chromophore for the three-branched oxazolones. In 2/B6 and 3/B6, the intermolecular fluorescence quenching and energy transfer between dye and borate salt are more efficient and accelerate the intersystem conversion of the dyes from the excited singlet state to the excited triplet state,56 which is in favor of the increase in photoinitiating efficiency of the systems.
Triplet state properties of dye/borate salt were investigated by flash photolysis (355 nm excitation) of an argon saturated toluene solution (Adye ≈ 0.5). The results show a readily detectable transient absorption spectra of 1, 2 and 3 (see Fig. 5 and S8†) peaking at 530 nm, 550 nm and 540 nm, respectively, with the decay displayed a first order kinetic. The lifetimes calculated based on the data were equaled 2.1 μs, 2.8 μs and 3.0 μs. Therefore, we concluded that the transient absorption corresponds to the triplet absorption of the oxazolones.
 | ||
| Fig. 5 Laser flash photolysis spectra of the dye 2 in toluene recorded after irradiation with laser pulses of 355 nm. Delay times shown in legends. Inset: kinetic curves for the growth (left) and the decay (right) of absorbance (550 nm) of 2 in toluene. | ||
The quantum yields of triplet state formation were determined using methodology given by Waluk et al.57 and by us.58 The method is based on quantitative analysis of the ground state recovery and its deconvolution that concerns an instrumental response function. Typical example of such approach is shown in Fig. 6 and obtained data are summarized in Table 1. According to the authors, the yield of triplet formation (ϕt) is proportional to the ratio described by eqn (6):
 | (6) |
 | ||
| Fig. 6 Analysis of ΔA(t) curve recorded for 2 in toluene at 510 nm. | ||
The transients were quenched by electron donors like N-phenylglycine (NPG) and trimethylammonium phenyltriethylborate (B6) (Fig. 7 and S9†).
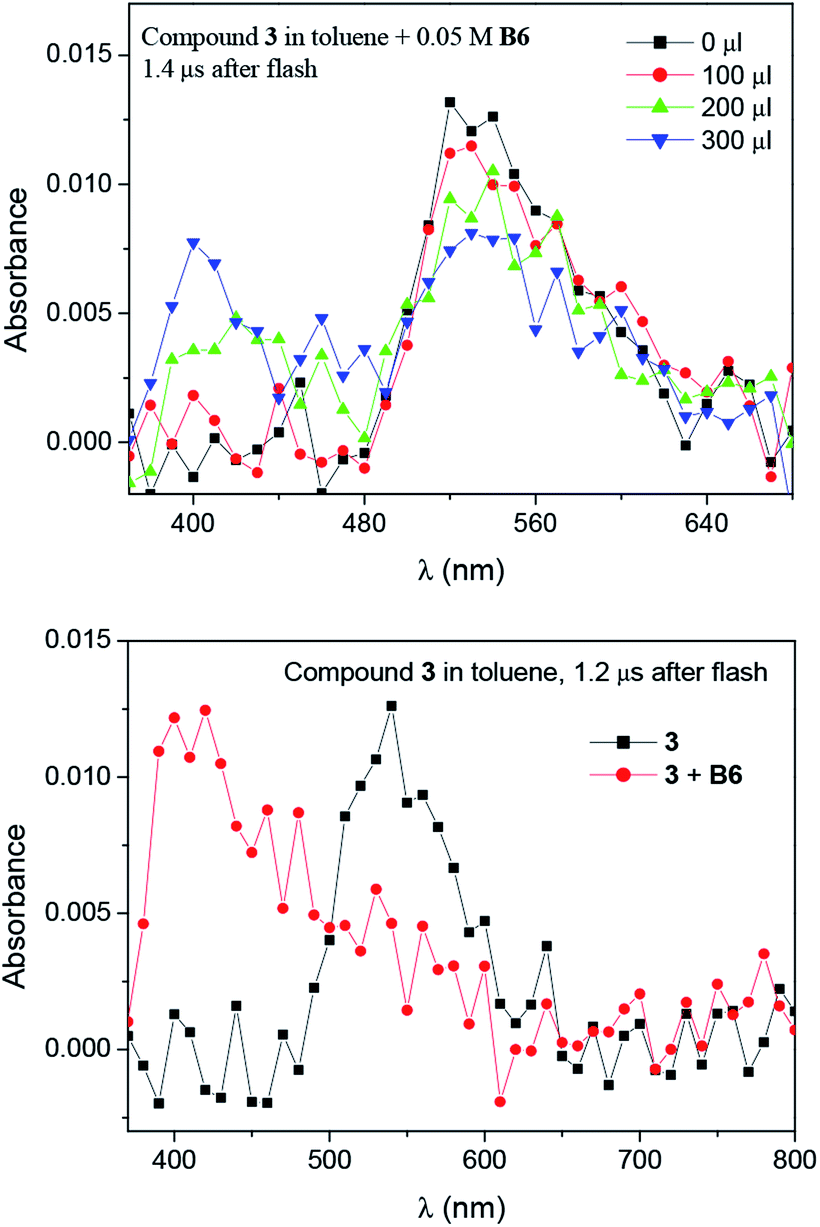 | ||
| Fig. 7 Laser flash photolysis spectra of the dye 3 (A = 0.5 at 355 nm; 3 mL) in toluene recorded after irradiation with laser pulses of 355 nm in the presence of B6 (0.05 M). Delay time marked in figure. | ||
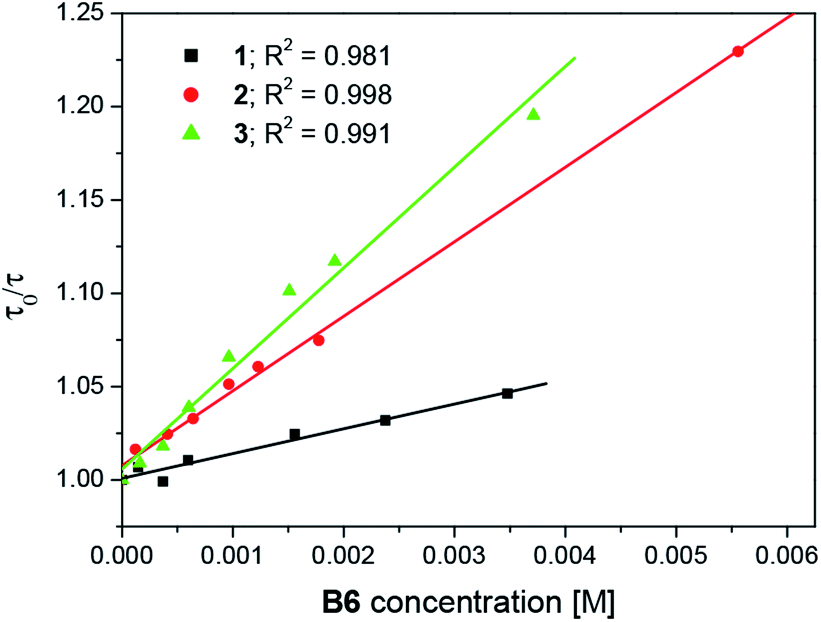
Moreover, the transient absorption spectra recorded at different times after laser excitation of the oxazolone solution containing tetramethylammonium phenyltriethylborate salt showed formation of a new peaks at ∼350–360 nm and ∼400–420 nm; this suggests that by quenching of the triplet, new transients are formed at these wavelengths (Fig. 7 and S9–S10†). The new bands may be assigned to the dye radical anion. However, to our knowledge, there is no literature data, which could confirm our suspicion. Whether, the rate constants for the quenching were determined by measuring the effects of the additive on the lifetimes of the dye triplet state. The linear Stern–Volmer plots were observed (Fig. 8). The quenching rate constants obtained by linear fitting of the data in Fig. 8 are 2.85 × 107 M−1s−1, 3.27 × 108 M−1s−1, and 3.61 × 108 M−1s−1, for the 1/B6, 2/B6, and 3/B6 photoredox pairs, respectively. The results may indicate that the limiting step in the polymerization photoinitiated by the oxazolone-borate pairs was the process related to the intermolecular electron transfer. The quenching rates are affected by the dye structure.
 | ||
| Fig. 8 Concentration dependence of the triplet decay rate of the dyes in toluene in the presence of borate salt. | ||
The photolysis of the oxazolone dyes in the presence of B6 leads to the formation of two kinds of radicals, radical anion produced from the dyes, and the other kind of radical derived from the borate (as shown below). The photopolymerization of acrylic monomers is usually initiated by the ethyl radical, generated from the phenyltriethylboranyl radical while the radical anion is usually not reactive toward double bonds of the monomer due to the steric hindrance and the delocalization of the unpaired electron. However, the dye affects the efficiency of the photoinduced electron transfer process and radical formation.9,10

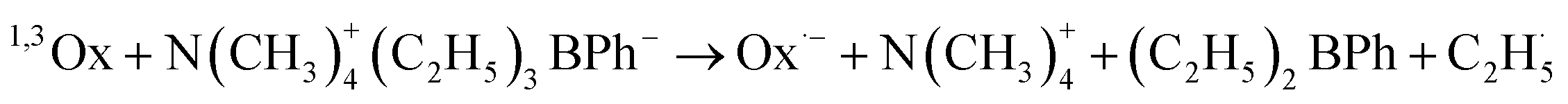

The 2-phenyl-oxazolone group is an electron-withdrawing group, which is helpful to photoinduced electron transfer between dyes and co-initiator B6, therefore, the photolysis of 3/B6 is more efficient than 2/B6 and 1/B6. Thus, the ethyl radical is more effectively generated from the electron donor molecule, thereby increasing the polymerization rate. Accordingly, for the 1/B6, 2/B6, and 3/B6 photoinitiating systems, the final conversion of the double bonds in TMPTA increases with increased dimensionality of the dye structure; the rates of polymerization become faster, the time to reach Rp max becomes shorter, and the values of Rp max increases.
The reaction sequence proposed above simplifies the possible mechanism of the initiation of the acrylates' polymerization by oxazolone dye – phenyltriethylborate salt. The preliminary results outlined in this paper indicate that the photoinitiated oxidation of tetramethylammonium phenyltriethylborate leads to formation of species active to start a chain reaction. Further studies are under way to deeply understand the photochemistry of the system, especially intermediate products formed after the electron transfer process and to define the proposed mechanism more clearly.
Conclusions
The oxazolone derivatives in combination with a borate salt can be an efficient photoinitiating system under visible light to initiate the radical polymerization of TMPTA. Considering that all samples have the same electron donor, variation in the polymerization behaviors was solely attributed to the differences in oxazolones structure and can be interpreted as being caused by the increase in the effectiveness of radical formation as a result of an acceleration of the intersystem conversion of the dyes from the excited singlet state to the excited triplet state. The photolysis of the oxazolone dyes in the presence of B6 confirmed an efficient quenching of the dye singlet and triplet as a result of an effective electron transfer from B6 to the excited singlet or triplet state of the oxazolones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Ministry of Science and Higher Education (MNiSW), through an internal grant provided by the UTP University of Science and Technology (grant no. 7/2019).References
- P. Xiao, F. Dumur, J. Zhang, B. Graff, F. Morlet-Savary, J.-P. Fouassier, D. Gigmes and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2860–2866 CrossRef CAS.
- J.-P. Fouassier and J. Lalevée, Photoinitiators for polymer synthesis-scope, reactivity, and efficiency, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, 2012 Search PubMed.
- K. Nakamura, Photopolymers: Photoresist Materials, Processes, and Applications, CRC Press, Boca Raton, London, New York, 2018 Search PubMed.
- J. Lalevée and J.-P. Fouassier, Photopolymerisation Initiating Systems, Royal Society of Chemistry, Croydon, UK, 2018 Search PubMed.
- J. P. Fouassier and X. Allonas, Dyes and Chromophores in Polymer Science, ISTE Ltd and John Wiley & Sons, Inc, 2015 Search PubMed.
- N. S. Allen, Photochemistry and photophysics of polymeric materials, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- J. P. Fouassier, X. Allonas and D. Burget, Prog. Org. Coat., 2003, 47, 16–36 CrossRef CAS.
- F. Dumur, Eur. Polym. J., 2020, 126, 109564 CrossRef CAS.
- B. Jędrzejewska, Colloid Polym. Sci., 2013, 291, 2225–2236 CrossRef PubMed.
- B. Jędrzejewska, M. Pietrzak and Z. Rafiński, Polymer, 2011, 52, 2110–2119 CrossRef.
- X. Allonas, J. P. Fouassier, M. Kaji and M. Miyasaka, J. Photopolym. Sci. Technol., 2000, 13, 237–241 CrossRef CAS.
- M. Abdallah, A. Hijazi, B. Graff, J.-P. Fouassier, G. Rodeghiero, A. Gualandi, F. Dumur, P. G. Cozzi and J. Lalevée, Polym. Chem., 2019, 10, 872–884 RSC.
- J.-P. Fouassier, F. Morlet-Savary, J. Lalevée, X. Allonas and C. Ley, Materials, 2010, 3, 5130–5142 CrossRef CAS PubMed.
- P. Xiao, F. Dumur, M.-A. Tehfe, B. Graff, D. Gigmes, J.-P. Fouassier and J. Lalevée, Polymer, 2013, 54, 3458–3466 CrossRef CAS.
- P. Xiao, F. Dumur, M. Frigoli, B. Graff, F. Morlet-Savary, G. Wantz, H. Bock, J.-P. Fouassier, D. Gigmes and J. Lalevée, Eur. Polym. J., 2014, 53, 215–222 CrossRef CAS.
- P. Xiao, F. Dumur, B. Graff, D. Gigmes, J.-P. Fouassier and J. Lalevée, Macromol. Rapid Commun., 2013, 34, 1452–1458 CrossRef CAS PubMed.
- Y.-H. Li and Y.-C. Chen, Polym. Chem., 2020, 11, 1504–1513 RSC.
- J. Zhao, J. Lalevée, H. Lu, R. MacQueen, S. H. Kable, T. W. Schmidt, M. H. Stenzel and P. Xiao, Polym. Chem., 2015, 6, 5053–5061 RSC.
- M. Sangermano, G. Malucelli, A. Priola, S. Lengvinaite, J. Simokaitiene and J. V. Grazulevicius, Eur. Polym. J., 2005, 41, 475–480 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3820–3829 CrossRef CAS.
- K. Podemska, R. Podsiadły, A. Orzeł, A. Kowalska, A. Maruszewska, A. Marcinek and J. Sokolowska, Color. Technol., 2014, 130, 250–259 CAS.
- B. Aydogan, A. S. Gundogan, T. Ozturk and Y. Yagci, Macromolecules, 2008, 41, 3468–3471 CrossRef CAS.
- J. Yu, Y. Gao, S. Jiang and F. Sun, Macromolecules, 2019, 52, 1707–1717 CrossRef CAS.
- J. Zhang, N. S. Hill, J. Lalevée, J.-P. Fouassier, J. Zhao, B. Graff, T. W. Schmidt, S. H. Kable, M. H. Stenzel, M. L. Coote and P. Xiao, Macromol. Rapid Commun., 2018, 39, 1800172 CrossRef PubMed.
- P. Xiao, J. Zhang, B. Graff, J.-P. Fouassier and J. Lalevée, Macromol. Chem. Phys., 2017, 218, 1700314 CrossRef.
- J. Kabatc, K. Kostrzewska, K. Jurek, M. Kozak, A. Balcerak and Ł. Orzeł, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 471–484 CrossRef CAS.
- G. Ding, C. Jing, X. Qin, Y. Gong, X. Zhang, S. Zhang, Z. Luo, H. Li and F. Gao, Dyes Pigm., 2017, 137, 456–467 CrossRef CAS.
- A. Al Mousawi, P. Garra, X. Sallenave, F. Dumur, J. Toufaily, T. Hamieh, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2018, 51, 1811–1821 CrossRef CAS.
- A. Al Mousawi, P. Garra, M. Schmitt, J. Toufaily, T. Hamieh, B. Graff, J. P. Fouassier, F. Dumur and J. Lalevée, Macromolecules, 2018, 51, 4633–4641 CrossRef CAS.
- M. Bouzrati-Zerelli, J. Kirschner, C. P. Fik, M. Maier, C. Dietlin, F. Morlet-Savary, J. P. Fouassier, J.-M. Becht, J. E. Klee and J. Lalevée, Macromolecules, 2017, 50, 6911–6923 CrossRef CAS.
- B. Jedrzejewska and B. Osmialowski, Polym. Bull., 2018, 75, 3267–3281 CrossRef CAS.
- M. Topa, F. Petko, M. Galek, K. Machowski, M. Pilch, P. Szymaszek and J. Ortyl, Polymers, 2019, 11, 1756 CrossRef CAS PubMed.
- W. G. Santos, F. Mattiucci and S. J. L. Ribeiro, Macromolecules, 2018, 51, 7905–7913 CrossRef CAS.
- S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3655–3677 CrossRef CAS.
- S. Pascual, T. Blin, P. J. Saikia, M. Thomas, P. Gosselin and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5053–5062 CrossRef CAS.
- S. P. Cullen, I. C. Mandel and P. Gopalan, Langmuir, 2008, 24, 13701–13709 CrossRef CAS PubMed.
- L. Fontaine, T. Lemele, J.-C. Brosse, G. Sennyey, J.-P. Senet and D. Wattiez, Macromol. Chem. Phys., 2002, 203, 1377–1384 CrossRef CAS.
- G. J. Drtina, S. M. Heilmann, D. M. Moren, J. K. Rasmussen, L. R. Krepski, H. K. Smith, R. A. Pranis and T. C. Turek, Macromolecules, 1996, 29, 4486–4489 CrossRef CAS.
- J. E. Barringer, J. M. Messman, A. L. Banaszek, H. M. Meyer and S. M. Kilbey, Langmuir, 2009, 25, 262–268 CrossRef CAS PubMed.
- J. M. Messman, B. S. Lokitz, J. M. Pickel and S. M. Kilbey, Macromolecules, 2009, 42, 3933–3941 CrossRef CAS.
- M. E. Buck, A. S. Breitbach, S. K. Belgrade, H. E. Blackwell and D. M. Lynn, Biomacromolecules, 2009, 10, 1564–1574 CrossRef CAS PubMed.
- D. Fournier, S. Pascual, V. Montembault, D. M. Haddleton and L. Fontaine, J. Comb. Chem., 2006, 8, 522–530 CrossRef CAS PubMed.
- A. Guyomard, D. Fournier, S. Pascual, L. Fontaine and J.-F. Bardeau, Eur. Polym. J., 2004, 40, 2343–2348 CrossRef CAS.
- C. Lucchesi, S. Pascual, G. Dujardin and L. Fontaine, React. Funct. Polym., 2008, 68, 97–102 CrossRef CAS.
- J. A. Tripp, J. A. Stein, F. Svec and J. M. J. Fréchet, Org. Lett., 2000, 2, 195–198 CrossRef CAS PubMed.
- V. Smokal, R. Czaplicki, B. Derkowska, O. Krupka, A. Kolendo and B. Sahraoui, Synth. Met., 2007, 157, 708–712 CrossRef CAS.
- B. Jędrzejewska, M. Gordel, J. Szeremeta, P. Krawczyk and M. Samoć, J. Org. Chem., 2015, 80, 9641–9651 CrossRef PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213 CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- S. Benedikt, J. Wang, M. Markovic, N. Moszner, K. Dietliker, A. Ovsianikov, H. Grützmacher and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 473–479 CrossRef CAS.
- J. Brandrup and E. H. Immergut, Polymer handbook, John Wiley & Sons, Inc., New York, Chichester, Brisbane, Toronto, Singapore, 3rd edn, 1989 Search PubMed.
- S. Zhang, B. Li, L. Tang, X.-D. Wang, D. Liu and Q. Zhou, Polymer, 2001, 42, 7575–7582 CrossRef CAS.
- B. Jędrzejewska, P. Krawczyk and M. Józefowicz, Spectrochim. Acta, Part A, 2017, 171, 258–267 CrossRef PubMed.
- J. Pączkowski, in Photochemistry and UV curing: New trends, ed. F. J. P, Research Signpost, Kerala, India 2006, p. 101 Search PubMed.
- J. P
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czkowski, M. Pietrzak and Z. Kucybała, Macromolecules, 1996, 29, 5057–5064 CrossRef.
czkowski, M. Pietrzak and Z. Kucybała, Macromolecules, 1996, 29, 5057–5064 CrossRef. - J. Qiu and J. Wei, J. Polym. Res., 2014, 21, 559 CrossRef.
- B. Lament, J. Karpiuk and J. Waluk, Photochem. Photobiol. Sci., 2003, 2, 267–272 RSC.
- F. Ścigalski and J. Pączkowski, Macromol. Chem. Phys., 2008, 209, 1872–1880 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Figures giving UV-vis and fluorescence spectra of 1–3 dyes, changes in the UV-vis and fluorescence spectra of compounds 1–3 upon irradiation and laser flash photolysis spectra. See DOI: 10.1039/d0ra02230f |
| This journal is © The Royal Society of Chemistry 2020 |