
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolypyrrole decorated metal–organic frameworks for supercapacitor devices†
Nigel Pattersona,
Bo Xiaob and
Anna Ignaszak *a
*a
aDepartment of Chemistry, University of New Brunswick, 30 Dineen Drive (Toole Hall), Fredericton, NB, Canada. E-mail: anna.ignaszak@unb.ca
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road (David Kier Building), Belfast BT9 5AG, UK
First published on 27th May 2020
Abstract
Due to their large specific surface areas and porosity, metal–organic frameworks (MOFs) have found many applications in catalysis, gas separation, and gas storage. However, their use as electronic components such as supercapacitors is stunted due to their poor electrical conductivity. We report a remedy for this by combining the MOF structure with polypyrrole (PPy), a well-known conductive polymer. Three MOFs are studied for modification to this end: CPO-27-Ni and CPO-27-Co (M2DOBDC, M = Ni2+, Co2+, DOBDC = 2,5-dihydroxy-1,4-benzenedicarboxylate) and HKUST-1 (Cu3(BTC)2, BTC = 1,3,5 benzenetricarboxylate). The gravimetric capacitance of pure MOFs is boosted several orders of magnitude after reinforcement of PPy (e.g., from 0.679 to 185 F g−1 for HKUST-1 and PPy–HKUST-1, respectively), and is much higher than reported for pure PPy. In total, these PPy-d-MOFs exhibit specific capacitances up to 354 F g−1, retaining 70% of this value even after 2500 cycles. Among them, the highest capacitance is found for PPy–CPO-27-Ni (354 F g−1), followed by PPy–CPO-27-Co (263 F g−1) and PPy–HKUST-1 (185 F g−1). The maximum operating potential for these electrodes is 0.5 V, which is restricted by the contact of MOF with aqueous electrolyte and with extremely low PPy content. As a solution, higher PPy loading and rational adjustment of particle size and porosity of both MOF and PPy are recommended so that the MOF/electrolyte interface is limited, leading to more robust electrode. The work completed here describes a highly promising approach to tackling the electrically insulating nature of MOFs, paving the way for their use in electrochemical energy storage devices.
1 Introduction
Metal–organic frameworks (MOFs) are a class of highly ordered and structured compounds, leading to their classification as coordination polymers. These materials bridge the oft-separated fields of organic and inorganic chemistry as they combine metal ion clusters connected through organic ligands. This coordinated structure leads to an exceptionally porous material that has one of the highest specific surface areas of any known material.1 A variety of MOFs have been created providing many possibilities for different structures and porosities, which are themselves tunable. MOFs have been keenly studied for many different applications across fields including gas separation and storage, catalysis, and drug delivery.2–6 Their use in electrochemical energy applications however has come up against a significant roadblock: the MOF's inherently poor electronic conductivity. This stems from the hard metal ions and redox inactive ligands creating a high energy barrier for charge transport.7Several methods have so far been successful in addressing this problem including (1) the use of specific redox active ligands, (2) doping with redox active molecules or metallic nanoparticles creating MOF-based composites, and (3) reinforcement of conducting polymers into the MOF's framework.7,8 In approach (1) redox active ligands generate charge transport pathways upon coordination with metal cation, favouring charge delocalization resulting in improved electrical conductivity. An example of this strategy are 2D-MOFs as analogs of graphene such as Ni3(HITP)2 that allow for full charge delocalization across the 2D plane.9 A similar approach is to replace oxygen with sulfur atoms which works due to “redox matching” since the electronegativity of sulfur is closer than oxygen's to that of transition metals.10 Promising candidates are found among the tetrathiafulvalene family of ligands, for example Zn2(TTFTB).11 This strategy was utilized to synthesize analogues of MOF-74, Mn2(DSBDC)(DMF)2, where thiolated ligand H4DSBDC forms an infinite 1D metal–sulfur chain (–M–S– in contrast to typical –M–O– chains in MOF-74) that act as the charge transport pathways.12
Method (2) of addressing MOF's conductivity is by loading redox active compounds (ferrocene, iodine, and tetracyanoquinodimethane)13–15 introduced either during the synthesis or infused in various post-synthetic treatments. As a result, redox active sites are created inside the MOF's structure which help facilitate charge transfer. Likewise, metallic clusters of an ultra-small size including Ag, carbon nanotubes, graphene, and carbon quantum dots have also succeeded to this end.16–19 Finally, incorporation of conducting polymers20 such as polypyrrole, polyaniline, or poly(3,4-ethylenedioxythiophene) (PEDOT) as guest molecules in the pores of the MOF have also proven a viable tactic.21–23 One of the most spectacular results in this research was an increase in conductivity of the parent MOF by a billion-fold which was attributed to the formation of conductive pathways due to non-covalent interactions between the MOF and guest polymer. In addition, the morphology of polymer nanoparticles evenly scattered over large surface areas facilitate flow of electrons across the network. The resulting hybrid materials demonstrated excellent carrier mobility similar to common semiconducting inorganic materials.24
Hybridization of polypyrrole (PPy) with various MOFs has been extensively studied in last few years, providing working materials as electrodes for sensors (CuTRZMoO4@PPy-n), electro-catalysts in CO2 reduction (MgNi-MOF-74/PPy), water deionization membranes (ZIF-67/PPy), metal ion-selective membranes (HKUST-1/PPy), imaging and chemo-thermal cancer therapy (MIL-100(Fe)/PPy), optoelectronics ([Cd(NDC)0.5(PCA)]/PPy), various biomedical applications, and energy storage (ZIF-67-S/PPy).24–31 Particularly, in the past two years, MOF-reinforced PPy have been tested as electrode materials in supercapacitors. Taking advantage of both their large surface area and good electrical conductivity, these combined materials demonstrated very promising characteristics. When ZIF-67 was coated via an electro-polymerization of pyrrole it showed much higher specific capacitance (284 F g−1)32 than pure PPy (78–144 F g−1).33 The same was true for PPy-decorated bimetal-MOF Zn/Ni-MOF@PPy (80–160 F g−1).34 More recent discoveries which combine PPy–MOFs with carbon showed outstanding electrochemical performance, such as UiO-66/PPy on cotton fabric electrodes (565 F g−1), ZIF-PPy (598 F g−1), and NiCo-MOF nanosheets (1109 F g−1).35–37 Other recent work on PPy–MOF supercapacitor electrodes are reported with excellent areal capacitance such as UiO-66/PPy on carbon fibres (206 mF cm−2), NENU-5/PPy (5147 mF cm−2), and Cu-CAT MOF with PPy (251 mF cm−2).21,38,39
In this work, PPy is for the first time combined with CPO-27-M (M2DOBDC, M = metal, DOBDC = 2,5-dihydroxy-1,4-benzenedicarboxylate). CPO-27-M is a family of isostructural honeycomb MOFs capable of forming from a wide variety of different metal ions; the ones here being Ni2+ and Co2+. The structure of this material results in the metal ions belonging to a distorted octahedral environment. Of the ligands, five are oxygen atoms belonging to the organic ligand, and the sixth is a coordinated solvent molecule located in the MOF's pores. Upon desolvation, CPO-27-M is left with available sites along the 1-D pore channel which can then be used for incorporating guest molecules.40 The third MOF chosen is HKUST-1, a copper-based grid-shaped MOF. The central Cu2+ ion is bound on four sides by BTC ligands to make square shaped pores spanning 3-dimensions. The three MOFs were chosen for their stability, well-characterized nature, and relative ease of synthesis.3,41,42 They are also notedly insulating MOFs, which allows us to isolate improvements on conductivity exclusively due to incorporation of the conductive polymer. Both MOFs provide open metal sites for coordination with guest molecules, however the differences in shape and dimensionality of the pore network may control how PPy assembles within the MOF, thus influencing the final electronic properties.43,44
Although PPy/HKUST-1 has been already reported in literature as a conducting platform for ion selective membranes,28 it has not yet been utilized in energy storage applications. A major difference between our work and the aforementioned study is how the PPy/MOF system is synthesized. In their work, PPy was generated via electro-polymerization and further combined with Cu2+ precursor used in the formation of HKUST-1. For our approach, HKUST-1 was synthesized separately using established methods, and the PPy was decorated onto its surface using an oxidative polymerization with iodine. Nevertheless, our ultimate goal is to take advantage of the large surface areas of MOFs as a platform for common pseudocapacitive polymer (PPy) and to validate electrochemical characteristics of these combined materials as electrodes for energy storage applications. The need to find new types of efficient and reliable electrical energy storage devices is driven by the dwindling supply of fossil fuels and the harm they do to the environment. Supercapacitors are electrochemical capacitors, meaning capacitance is derived from electrochemical processes. Unlike in conventional batteries, these redox processes occur at the electrode–electrolyte interface,45 giving extremely fast charge–discharge time. Since MOFs have incredible amount of internal surface area, they are highly attractive to bring around for the purpose of supercapacitors.
2 Experimental
2.1 Materials and reagents
From Alfa Aesar, the following were acquired: nickel(II) acetate tetrahydrate (Ni(OAc)2·4H2O, 98%), cobalt(II) acetate tetrahydrate (Co(OAc)2·4H2O, 98%), 2,5-dihydroxy-1,4-benzenedicarboxylic acid (H4DOBDC, 97%), 1,3,5-benzenetricarboxylic acid (H3BTC, 98%), poly(vinylidene fluoride) (PVDF), pyrrole (98%), and iodine (99%). Sigma Aldrich supplied copper(II) nitrate hemi(pentahydrate) (Cu(NO3)2·2.5H2O, 98%), isopropanol (99.5%), and tetrahydrofuran (THF, HPLC grade 99.9%). Ethanol (99.8%) was purchased from Fisher, n-hexane from T.E. Laboratories, carbon black from Leco Corporation, and deionized water (DI water, 18.2 MΩ cm) was purified using an in-house distill. All reagents purchased from these commercial sources, except pyrrole, were used as received without further purification. Pyrrole was distilled prior to use.2.2 Materials characterization
Powder X-ray diffraction (PXRD) was collected on an X'Pert PRO PANalytical diffractometer using Cu Kα1 radiation (λ = 1.5406 Å). Analysis was performed in 2θ range from 5–50°. Experimental spectra were compared against reported literature found through the Cambridge Crystallographic Data Centre (CCDC). Attenuated total reflectance fourier-transform infrared spectroscopy (ATR-FTIR) was carried out on an Agilent Technologies Cary 630 FTIR with diamond ATR attachment. The spectral range was from 650–4000 cm−1. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) elemental mapping were conducted using both an FEI Quanta FEG 250 – Environmental SEM Oxford Ex-ACT scanning electron microscope and JEOL 2011 Scanning Transmission Electron Microscope. The instrument was operating under high vacuum (10−6 Torr) with 20 kV accelerating voltage. Optical microscope images were from a Zeiss West Germany model using 10× magnifier. Photographs through the eyepiece were taken on an iPhone 6S camera. Adsorption isotherms for N2 at 77 K were measured on a Micromeritics TriStar II 3020. Specific surface area (SBET) was calculated using the Brunauer–Emmett–Teller (BET) method using N2 adsorption. The as prepared samples were pretreated for 8–12 hours at 393 K prior to measurement. Thermogravimetric analysis (TGA) was run on a PerkinElmer TGA 8000. Non-activated samples were taken from 298–873 K at a rate of 10 K min−1. The carrier gas used was N2 flowing at a rate of 40 mL min−1. Elemental analysis (EA) was carried out on a Perkin Elmer PE2400CHNS instrument.2.3 Synthesis of CPO-27-M and HKUST-1
Synthesis of MOFs was carried out using the solvothermal technique adapted from the literature.3,40 For CPO-27-M MOFs, inorganic salts Ni(OAc)2·4H2O or Co(OAc)2·4H2O (2.5 mmol, 0.633 g) were dissolved into 6 mL of DI water. Organic linker H4DOBDC (1.25 mmol, 0.247 g) was dissolved into 6 mL of THF. Synthesis of CPO-27-M often follows two distinct recipes, one discovered by Yaghi et al. based on DMF/water as the solvent mix,46 and the other by Dietzel et al. using a THF/water solvent mix.47 While synthesis in DMF/water occurs readily and at room temperature41 (vs. 72 h at 110 °C in THF/water), the trade-off comes during removal of DMF from the pores, which necessitates a methanol exchange step prior to activation under harsher conditions.For HKUST-1, Cu(NO3)2·2.5H2O (7.5 mmol, 1.74 g) and H3BTC (5 mmol, 1.05 g) were dissolved in 10 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol/water mixture. The reactant solutions were combined in a 25 mL Teflon-lined stainless steel autoclave, sealed, and stirred for 60 min using a magnetic stir bar before being placed in a 110 °C oven for 72 hours. After this period the products were filtered and washed with their respective pure solvent mixtures, then left to dry on the filter paper overnight in the fumehood. The dried powder was collected without activation in glass vials. For activation to remove coordinated water molecules, the glass vials were stoppered with cotton wool and covered with a perforated plastic cap. These were placed in a Schlenk tube under vacuum and immersed in a 120 °C oil bath overnight (8–12 hours). The appearance of the CPO-27-M powders changed from light yellow to yellow ochre for CPO-27-Ni and from red to dark brown for CPO-27-Co; HKUST-1 went from sky to deep blue.
:1 ethanol/water mixture. The reactant solutions were combined in a 25 mL Teflon-lined stainless steel autoclave, sealed, and stirred for 60 min using a magnetic stir bar before being placed in a 110 °C oven for 72 hours. After this period the products were filtered and washed with their respective pure solvent mixtures, then left to dry on the filter paper overnight in the fumehood. The dried powder was collected without activation in glass vials. For activation to remove coordinated water molecules, the glass vials were stoppered with cotton wool and covered with a perforated plastic cap. These were placed in a Schlenk tube under vacuum and immersed in a 120 °C oil bath overnight (8–12 hours). The appearance of the CPO-27-M powders changed from light yellow to yellow ochre for CPO-27-Ni and from red to dark brown for CPO-27-Co; HKUST-1 went from sky to deep blue.
2.4 Loading and polymerization of pyrrole
In the typical operation,24 activated MOF samples (0.2 g) were soaked in 0.5 mL freshly distilled pyrrole monomers. These were sealed under N2 and kept in the fridge (5 °C) for 24 hours. At the end of this period, excess pyrrole was removed and a solution of 0.05 M iodine in hexane was introduced as the oxidant to initiate polymerization. The reactants were reintroduced to a N2 atmosphere, stirred for 10 min and then returned to the fridge to polymerize over 72 hours. The final products were filtered under vacuum and washed several times with pure hexane.2.5 Electrochemical measurements
Electrochemical measurements were performed on a CHI Instruments 760e. Inks were made using a suspension of 9:1 isopropanol:DI water containing 2 mg mL−1 of PVDF as binder, 2 mg mL−1 carbon black to aid conductivity, and 17 mg mL−1 of the PPy-d-MOF sample. They were always prepared fresh before testing to avoid potential for degradation of the MOF structures. Inks were cast onto a glassy carbon working electrode in 5 μL portions till fully coating the electrode (approximately four to five times), allowing the ink to fully dry in between each. A Ag/AgCl reference electrode was used along with a platinum wire counter electrode and 1.0 M KCl electrolyte. Before recording data, the samples were allowed to equilibrate with the electrolyte by running 100 cyclic voltammograms (CV) between +0.5 V and −0.5 V at 200 mV s−1. CV was performed in the range of −0.5 to +0.5 V at varying scan rates from 200 mV s−1 to 25 mV s−1 in 25 mV s−1 increments. The results collected here were used to calculate specific capacitance and diffusion coefficients. Long term capacitance retention was conducted over 2500 CV cycles at 500 mV s−1 scan rate across the same potential range. Differential pulse voltammetry (DPV) was run forwards then backwards between −0.5 V and +0.5 V at a rate of 4 mV s−1. Galvanostatic charge discharge (GCD) ran under constant current of 250 μA. For electrochemical impedance spectroscopy (EIS), the samples were charged by bulk electrolysis for 10 min. After charging, the sample was allowed to relax during which time the open circuit potential (OCP) was measured. This was followed by CV (by the same parameters previously mentioned) then impedance measured from 0.1 to 10000 Hz. The samples continued this cycle, increasing each time the charge by increments of 0.1 V starting from 0.1 V and ending at 1.0 V.
3 Results and discussion
3.1 Spectroscopic characterization
Confirmation of successful MOF and final PPy-d-MOF products were attained by PXRD and ATR-FTIR. The X-ray patterns generated from PXRD were compared to simulated patterns taken from the literature.3,43,48,49 The characteristic peaks for CPO-27-M appear at 2θ = 6.8° and 11.8° corresponding to the (210) and (300) lattice planes (Fig. 1d and f). For HKUST-1, its identifying peaks are found at 6.7°, 9.5°, and 11.6° which correspond to the (200), (220), and (222) lattice planes (Fig. 1b). Upon inclusion of PPy, the overall spectra had more noise than the plain MOFs due to PPy being an amorphous compound. The supporting MOF structure is undisturbed by this treatment, as the sharp peaks in the spectra prove they are intact. Aside from the increased noise, the lower 2θ region of the spectra consistently had a much higher intensity which tapered off with increasing 2θ in all PPy-d-MOF samples.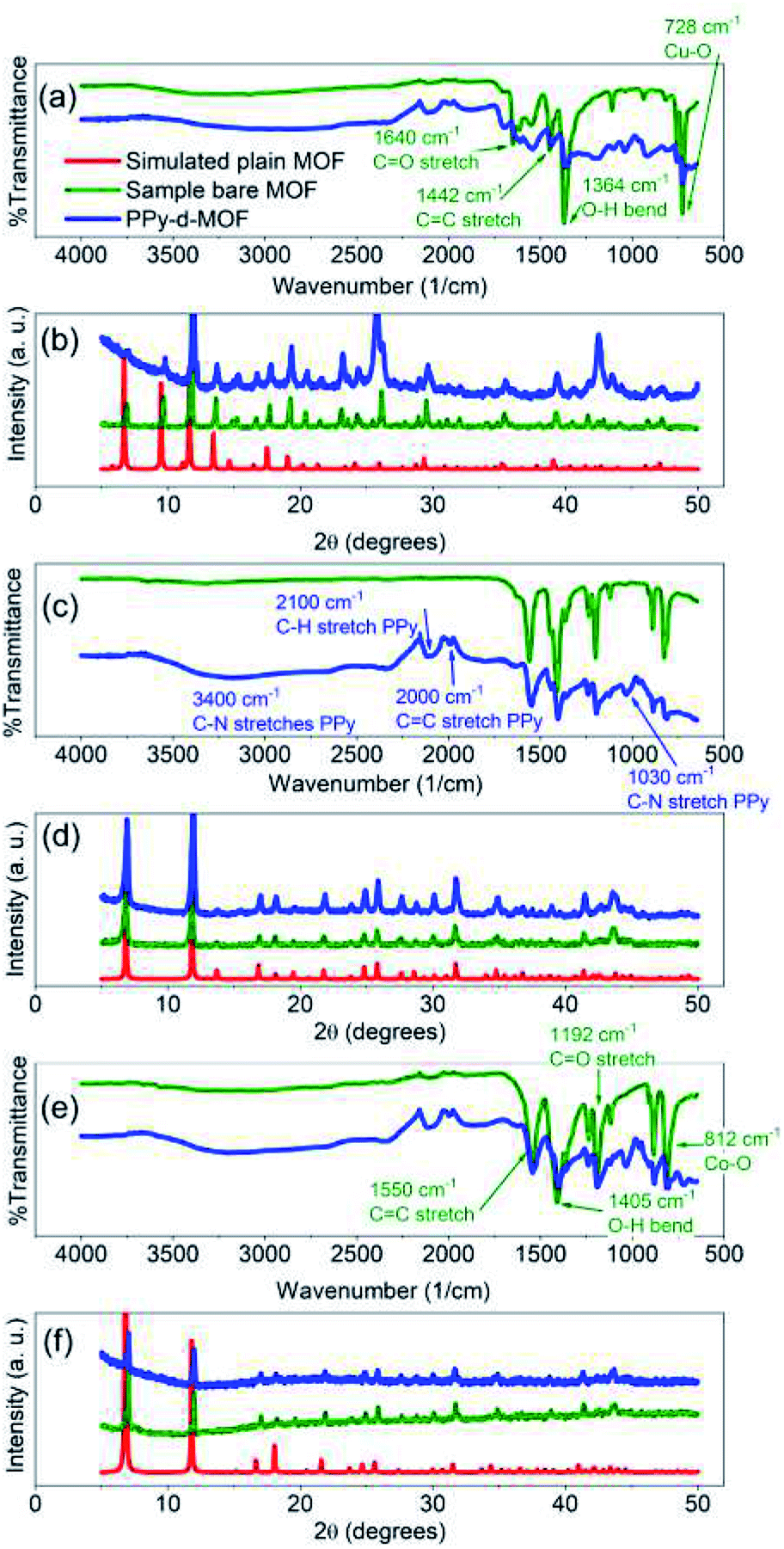 | ||
| Fig. 1 FTIR (a, c and e) and PXRD (b, d and f) spectra of synthesized MOFs and PPy-d-MOFs. (a and b) (PPy–)HKUST-1, (c and d) (PPy–)CPO-27-Ni, (e and f) (PPy–)CPO-27-Co. | ||
ATR-FTIR as well conclusively identified the incorporation of PPy into MOFs by comparing the resulting spectra of samples before and after PPy loading. For CPO-27-Co, identifying peaks of the MOF structure can be found at 1192 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch), 1405 cm−1 (O–H bend), and 1550 cm−1 (CC stretch), as part of the DOBDC ligand (Fig. 1e).43 These same peaks appear in CPO-27-Ni since it shares the same ligand as its cousin (Fig. 1c). In both, the peak at 812 cm−1 points to the metal–oxygen interaction (i.e. Ni–O or Co–O) in the structure.50 HKUST-1, having a different organic ligand has its own distinct peaks to be found at 1364 cm−1 (O–H bend), 1442 cm−1 (CC stretch), and 1640 cm−1 (CO stretch) (Fig. 1a).51 Meanwhile the Cu–O interaction appears at 728 cm−1. With the possible exception of CPO-27-Ni, the other two plain MOFs have a slight dip from level between 2300–3200 cm−1. While the MOFs were activated to remove solvent prior to measurement, it was impossible to fully remove them from contact with ambient humidity. Thus this slight yet broad peak is indicative of trace amount of water coordinated to the pores in the MOF.
O stretch), 1405 cm−1 (O–H bend), and 1550 cm−1 (CC stretch), as part of the DOBDC ligand (Fig. 1e).43 These same peaks appear in CPO-27-Ni since it shares the same ligand as its cousin (Fig. 1c). In both, the peak at 812 cm−1 points to the metal–oxygen interaction (i.e. Ni–O or Co–O) in the structure.50 HKUST-1, having a different organic ligand has its own distinct peaks to be found at 1364 cm−1 (O–H bend), 1442 cm−1 (CC stretch), and 1640 cm−1 (CO stretch) (Fig. 1a).51 Meanwhile the Cu–O interaction appears at 728 cm−1. With the possible exception of CPO-27-Ni, the other two plain MOFs have a slight dip from level between 2300–3200 cm−1. While the MOFs were activated to remove solvent prior to measurement, it was impossible to fully remove them from contact with ambient humidity. Thus this slight yet broad peak is indicative of trace amount of water coordinated to the pores in the MOF.
Evidence for PPy can be seen in unique peaks found in the PPy-d-MOF spectras (Fig. 1b, d and f). Well visible in PPy–CPO-27-M samples are new peaks at 960 cm−1 (C–H wagging), 1030 cm−1 (C–N stretch), and 1630 cm−1 (CN stretch).52,53 The PPy bipolaron located at 917 cm−1 and 1185 cm−1, is most visible in PPy–HKUST-1.54 This sample also presents a peak at 1680 cm−1 related to CN stretching in PPy. All three samples exhibit peaks at 2000 cm−1 (CC stretch) and 2100 cm−1 (C–H stretch) of the PPy ring.52,55 Finally, a broad peak centered at 3400 cm−1 is attributed to N–H stretches.53
3.2 Images, optical microscopy, and SEM/EDS
The morphology and crystal structure of the MOFs were determined under both optical and electron microscopes. As seen by the eye and more clearly through the optical microscope, each MOF presents itself as a brightly coloured powder (Fig. 2). CPO-27-Co takes on a red-orange hue crystalline body, CPO-27-Ni appears as a bright yellow powder, and HKUST-1 as a bright blue powder (Fig. 2a–c). Of the three, CPO-27-Co forms the largest crystals, while the others take on a more powdery form with their smaller, sand-like crystals. The size of each individual grain in the latter two is harder to discern. Once combined with PPy, the major differences between the MOFs are lost and are all but indistinguishable from each other (Fig. 2d–f). Where once colourful, the materials have all taken on a shiny black appearance due to infiltration of PPy into the MOF structure. Conductive polymers such as PPy are known to take on a metallic sheen under certain conditions, such as when iodine is used to induce polymerization.56 To separate any effects of iodine on colour change, MOFs were immersed in purely iodine solution absent from PPy monomers. These produced darker shades of the MOF, but nowhere near as black as with PPy present (Fig. S1†). Indeed it is still possible to tell the I2-d-MOF samples apart based on their colour, something that is impossible to do with PPy-d-MOFs. The effects of PPy polymerization within the MOF changes its optical properties. | ||
| Fig. 2 Images taken of the MOFs (a) CPO-27-Co, (b) CPO-27-Ni, (c) HKUST-1 and PPy-d-MOF (d) PPy–CPO-27-Co, (e) PPy–CPO-27-Ni, (f) PPy–HKUST-1 powders under an optical microscope. Inset in each is a image of the bulk samples. | ||
To further discover the differences between the coated and uncoated MOFs, the samples were analyzed by an electron microscope (Fig. 3). The large crystals of CPO-27-Co vs. the tinier ones in CPO-27-Ni are reflected even at this small scale (Fig. 3a and b). Rod-like crystals of CPO-27-Co are near an order of magnitude larger than its nickel based cousin. Crystals of HKUST-1 observed were the largest and also formed very well-defined octahedrons (Fig. 3c). The sizes and shapes of these materials is consistent with reports in the literature.3,46,57
 | ||
| Fig. 3 Images taken of the MOFs (a) CPO-27-Co, (c) CPO-27-Ni, (e) HKUST-1 and PPy-d-MOF (b) PPy–CPO-27-Co, (d) PPy–CPO-27-Ni, (f) PPy–HKUST-1 powders under an electron microscope. | ||
PPy-d-MOFs under SEM show no differences to the structure of the MOFs themselves. However, the presence of PPy is clearly shown by small globular islands covering the once smooth surface of the MOFs (Fig. 3d–f). These islands are most easily seen in PPy–CPO-27-Co and PPy–HKUST-1, where the size of the MOF crystal relative to the PPy islands is significant. For the much smaller PPy–CPO-27-Ni, the scale of the crystal size, as well as its shape does not lend itself to easily discerning the two. The overall shape can be said to be rounded out once coated in PPy, as compared to the sharper edges in the pure CPO-27-Ni crystal. These results clearly show that PPy is loaded within the MOF, and that the MOF crystal morphology is left intact afterwards. This is consistent with the high crystallinity indicated by PXRD.
EDS results were taken concurrently with SEM on the plain MOFs (Fig. S2†). These show the atomic composition of the MOF, especially with regards to the metals they contained. Aside from the metals, significant amount of carbon and oxygen were found and are attributed to the organic ligands. However, when it comes to identifying PPy in the PPy-d-MOF EDS is a poor choice due to limitations of the instrument.58 The unique atom, nitrogen, produces a signal which is often obscured by carbon and oxygen. In the case of our scaffolding MOF, as well as PPy being an organic compound, the significant abundance of C and O masks any evidence of pyrrolic nitrogens.
3.3 Thermogravimetric analysis
TGA allowed us to validate the presence of PPy in the PPy-d-MOF samples, from changes in weight as a result of thermal decomposition. Common to every sample is an early weight loss appearing near 100 °C which is attributed to evaporation of water captured in the pores. For the pure MOFs, each has a distinct weight loss onset which corresponds to decomposition of the organic ligand; CPO-27-Ni decomposes at 400 °C, CPO-27-Co at around 480 °C, and HKUST-1 at 340 °C (Fig. 4). The first derivative overlaid curves are able to show where maximum weight loss occurs precisely by looking at the peaks in the curves.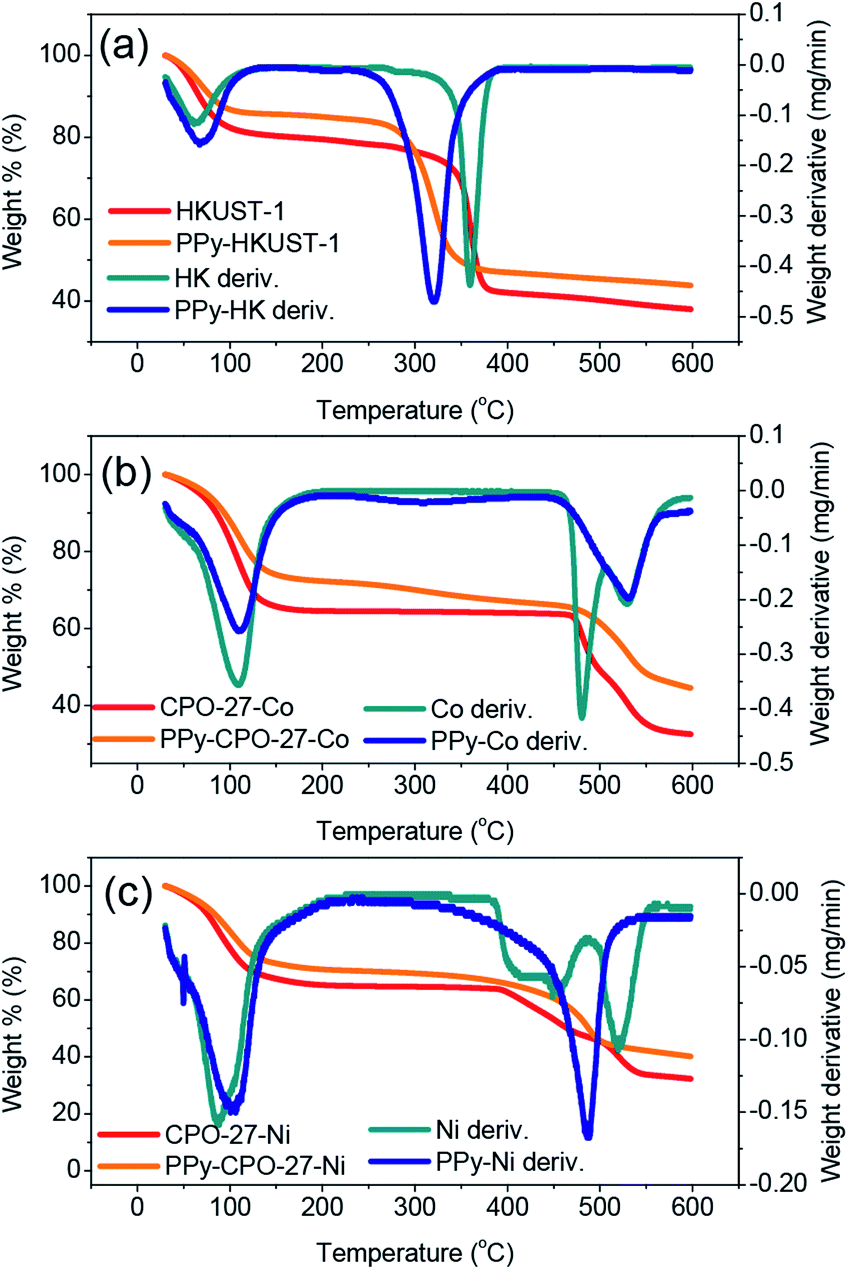 | ||
| Fig. 4 TGA and their first derivative curves for the (a) (PPy–)HKUST-1, (b) (PPy–)CPO-27-Co, (c) (PPy–)CPO-27-Ni. | ||
In the PPy-d-MOF samples, there is a new region of decomposition with much more gradual weight change. Occurring at around 320 °C the curves show very slight decreases in weight percent, different from their once steady weight in this temperature region. The first derivative curves show clearly that this peak is attributed to PPy as it is matched perfectly with the decomposition of pure PPy (Fig. S3†). Due to the fact that there is no real variation in the onset degradation temperature, it is safe to say that no chemical interaction exists between the PPy and MOF phases, that they are simple mixed in with one another. For HKUST-1, its decomposition temperatures somewhat masks the decomposition of PPy as they exist in an overlapping range. CPO-27-Ni and CPO-27-Co have much higher decomposition temperatures so there is no mistaking the presence of PPy. It should be noted that although CPO-27-Ni had the smallest weight change in the expected region for PPy, especially when comparing the lack of significant peak in the first derivative curve, weight loss does commence at this temperature. Compare that with the plain MOF where the temperature curve is completely level passing through the 300 °C range while in PPy–CPO-27-Ni it begins to drop off.
3.4 Elemental analysis and BET surface area
While the presence of expected metal, C, and O atoms was shown through EDS (Fig. S2†), the exact amounts of each could not accurately be determined, nor could strict evidence for N (indicating PPy) be seen due to the limitations of the technique. Additional EA was performed on the PPy-d-MOF samples, quantifying the relative amounts of C, H, and N present (Table 1). From the % N we were able to have an idea on the amount of PPy present in these MOFs. Overall, the low % N detected is in agreement with the small % weight change found by TGA, indicating a relatively sparse amount of PPy is present in the samples.| Sample | Elemental analysis (%) | BET surface area (m2 g−1) | |||
|---|---|---|---|---|---|
| C | H | N | MOF | PPy–MOF | |
| PPy–CPO-27-Ni | 25.25 | 3.76 | 2.51 | 364.9 | 19.93 |
| PPy–CPO-27-Co | 22.83 | 4.10 | 1.52 | 506.2 | 61.78 |
| PPy–HKUST-1 | 34.00 | 3.67 | 4.32 | 1105 | 36.51 |
As stated previously, the aim was to take advantage of the high surface area of MOFs to benefit supercapacitance in the overall material. Comparisons were made on the SBET between the pure MOFs and PPy-d-MOFs (Table 1), while the plots can be found in the ESI (Fig. S4†). While the MOFs themselves show the characteristic high surface area upwards of 360 m2 g−1, the PPy-d-MOFs do not retain this high value. SBET has diminished rather rapidly to some tens of m2 g−1. Judging by the amount of PPy determined through EA and TGA, it is unlikely therefore that the pores are fully permeated with the active polymer. What instead is more likely occurring, is that the pores have only been infiltrated at their entrance by PPy, blocking access to the inner recesses. In the future this may be combated by utilizing larger pore MOFs or by adjusting the viscosity/hydrophobicity of the pyrrole loading solution.
3.5 Electrochemical characteristics
To characterize the electrochemical performance of these new materials, and to judge their potential as supercapacitors, a multitude of electrochemical tests were performed. First it was necessary to characterize the differences between the plain and PPy treated MOFs. These differences are shown in Fig. 5 which uses HKUST-1 as an example. The comparative behaviours of CPO-27-Ni/Co before and after can be found in the ESI (Fig. S5 and S6†). Straight away it is clear that there is a marked difference between the two. Cyclic voltammetry (CV) (Fig. 5a) shows a much higher current for the PPy-d-MOF than the plain. Its shape can also be described as boxy, which is a common attribute of supercapacitors measured in this way.59 | ||
| Fig. 5 Comparison of electrochemical properties of plain MOF vs. PPy-d-MOFs (PPy–HKUST-1 shown here). (a) CV, (b) CV of PPy-d-MOF at varying scan rates, (c) GCD, (d) DPV. | ||
One other difference between the two voltammograms is the appearance of reversible peaks for the PPy-d-MOF CV located at around 0.25 V, vs. the singular oxidation peak of the MOF at −0.15 V. These peaks are even more clearly shown by the differential pulse voltammetry (DPV) traces (Fig. 5d), which also shows higher current for PPy-d-MOFs than plain MOFs. To reiterate, this MOF is HKUST-1, which comprises copper as its metallic building unit. Thus the peak located at −0.15 V is the oxidation of this metal, which without any reduction peak implying regeneration will lead to the eventual degradation of the MOF. On the other hand, metal oxidation peaks are not seen in either of the two CPO-27-M MOFs (Fig. S5d and S6d†). That is not to say these MOFs are immune to leaching and degradation, only that the metal centre oxidation could not be measured. The redox peaks for PPy are clearly observed hovering around 0.2 V for PPy–HKUST-1 and PPy–CPO-27-Ni (Fig. 5d and S5d†). PPy–CPO-27-Co has a strong reversible redox peak shifted to −0.2 V, which could be a result having to do with interaction with the MOF. Aside from this, galvanostatic charge–discharge (GCD) behaviour of the materials is also significantly varied. Without any real charge storage ability, the plain MOF does not charge and the high frequency of the curve is an indication of this. In contrast, the PPy-d-MOF produces a triangular GCD trace. This is indicative of the characteristic response of PPy-d-MOFs as supercapacitive materials.
To further clarify the effect of PPy in MOFs, samples were prepared in the iodine solution but without PPy monomers. Iodine doping is a fairly common strategy for inducing conductivity in MOFs, which could be a significant contributor to supercapacitance.60,61 We isolate the effect of either PPy or iodine on overall supercapacitance, plain MOFs were activated following the regular procedure, and then immersed in 0.05 M iodine in hexane solution for 48 hours (without having undergone pyrrole loading). The same set of electrochemical tests were run, and the data collected showed that these iodine-doped MOFs (I2-d-MOFs) behaved more similarly to the plain MOFs than to PPy-d-MOFs, successfully ruling out any influence of iodine doping on the MOF itself (Fig. S7†).
Comparing all three PPy-d-MOFs to each other followed many of the same characterizations used to differentiate the plain from PPy-d-MOFs. CV was again used, but this time to help quantify the specific capacitance (Cs) of each (Fig. 6). The area bound by the CV is related to Cs (F g−1) by the eqn (1), where i(E) is the instantaneous current of the CV (W), ν is the scan rate (V s−1), V is the potential window (V), and m is the mass of active material (g). In the case of PPy-d-MOFs, the active material is taken to be the amount of PPy present, as determined from % N by EA (Fig. 2).
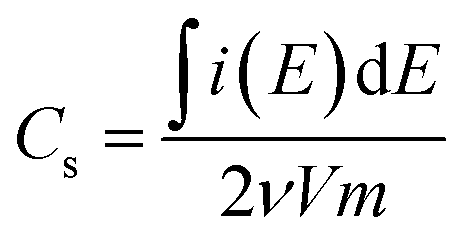 | (1) |
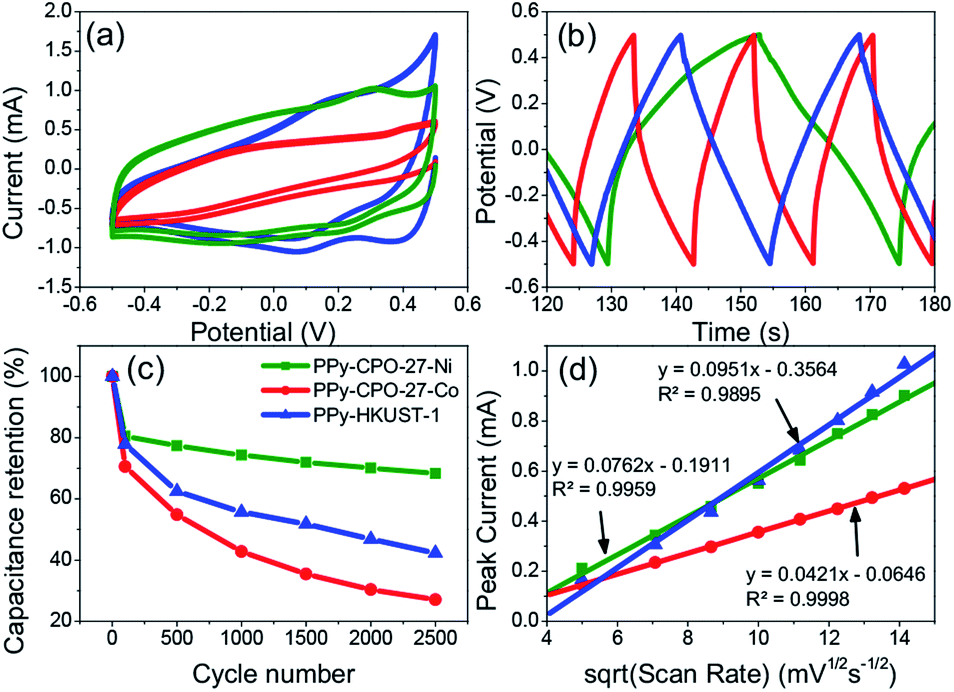 | ||
| Fig. 6 Comparison of electrochemical properties of all PPy-d-MOFs with each other. (a) CV, (b) GCD, (c) long term charge retention, (d) diffusion coefficient. | ||
The Cs values for not only the PPy-d-MOFs, but the previously mentioned plain MOFs and I2-d-MOFs are given in Table 2. The PPy-d-MOFs show a rather impressive increase in the supercapacitance compared to their base MOF and PPy (78–144 F g−1).33 Comparing PPy–CPO-27-Ni and PPy–CPO-27-Co, although of the same family one exhibits near 33% higher Cs than the other. One reason for this difference is attributed to the stability of PPy–CPO-27-Ni which is better at retaining its capacitance than PPy–CPO-27-Co. The pretreatment step which involved moderate CV cycling may have impacted the integrity of PPy–CPO-27-Co more than it did the Ni MOF. We may also be seeing an effect of the different metal atom in each of these MOFs. They already show completely different sizes and morphologies in the pure MOF, so it is very possible that this small change plays a large part on the overall electronics of the material. Although PPy–HKUST-1 had the largest crystal size as well as highest incorporation of PPy, it had the lowest measured Cs. Between the two families of MOFs, PPy–CPO-27-M outperformed HKUST. If not due to the different metal atoms in each of them, it is possible that the pore size or shape affects the way incorporated PPy forms and interacts with the open metal sites. The overall size of HKUST-1 is also much larger than CPO-27-M, which lowers the surface area to volume ratio for the crystal. This creates a harder time to allow the PPy monomers to fully penetrate the MOF during the loading step, or even of the electrolyte during measurements. Comparison of the Cs values in this work to others found in the literature are reported in the ESI (Table S1†).
| Sample | Specific capacitance (F g−1) | Charge–discharge time (s) | Diffusion coefficient (cm2 s−1) | Ohmic drop (A) | Capacitance retention (%) | ||
|---|---|---|---|---|---|---|---|
| MOF | I2-d-MOF | PPy-d-MOF | |||||
| PPy–CPO-27-Ni | 0.281 | 0.289 | 354 | 23.5 | 1.45 × 10−6 | 0.0238 | 68.3 |
| PPy–CPO-27-Co | 263 | 9.30 | 8.00 × 10−7 | 0.125 | 27.1 | ||
| PPy–HKUST-1 | 0.679 | 0.493 | 185 | 13.8 | 1.81 × 10−6 | 0.0257 | 42.3 |
Looking at the GCD curves (Fig. 6b), charging time is rather similar for each sample. The PPy-d-MOF taking the longest to charge and discharge was PPy–CPO-27-Ni, which correspondingly had the highest Cs. Indeed there seems to be a correlation where the more capacitive the material, the longer it takes to charge fully. The GCD curves also allow for determining the internal resistance of the material by measuring the ohmic drop. The ohmic drop is a feature appearing at the moment of discharge as a sudden drop in the current. PPy–CPO-27-Ni most clearly demonstrates this in Fig. 6b. By applying Ohm's law (V = IR) we can see the inverse correlation of current to the resistivity of our materials. Table 2 gives the ohmic drop values for each PPy-d-MOF. From these results, PPy–CPO-27-Co has a much lower internal resistance than for PPy–CPO-27-Ni or PPy–HKUST-1, which are both similarly related to each other. Further analysis on internal resistance will be covered in the following section on EIS.
Diffusion coefficients (D) are useful to study the rate of doping and dedoping in PPy. Through the Randles–Sevcik equation (eqn (2)), a linear function of peak current (ip) vs. the square root of the scan rate (ν) is obtained (Fig. 6d). The plot of variable scan rates or the samples can be found in (Fig. 5b, S5b and S6b†).
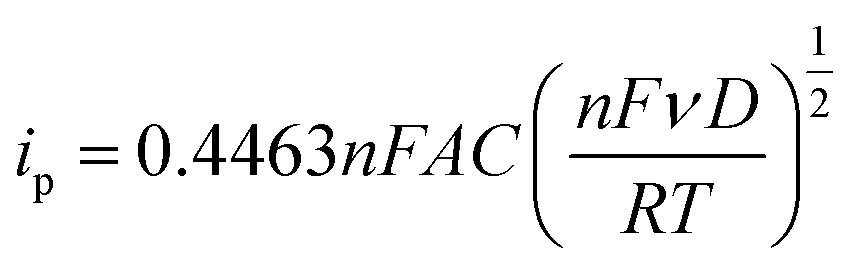 | (2) |
3.6 Impedance spectroscopy
AC impedance spectroscopy and the corresponding circuit modelling were carried out for all PPy-d-MOF electrodes in order to verify their electrochemical stability upon polarization (Fig. 7). An example of impedance spectra for PPy–HKUST-1 when a non-destructive potential (0.1 V) is applied is demonstrated in Fig. 7a. It composes a weakly resolved semicircle at high frequencies corresponding to resistance (R) and capacitance (C) of the carbon black additive, followed by resistance of PPy (poorly resolved in the impedance spectra prior to degradation since resistivities of doped PPy and the carbon additive are similar) and a diffusion resistor represented by straight line with a slope of 45° (mid frequencies). The low-frequency section of the impedance spectrum shows a classical capacitor represented by a nearly vertical line (non-ideal capacitor) which corresponds to the charge accumulation at the PPy–electrolyte interface.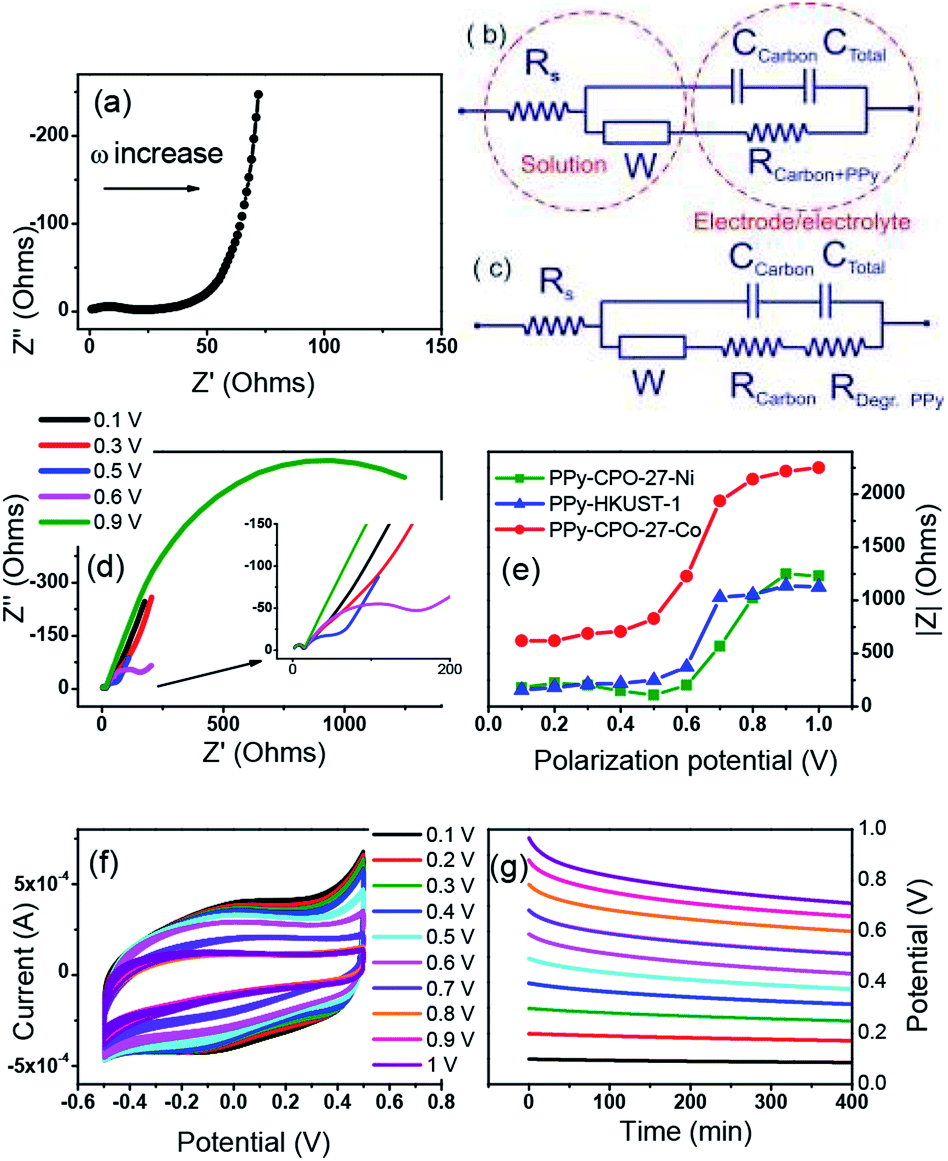 | ||
| Fig. 7 (a) The impedance spectrum of PPy–HKUST-1 polarized at 0.1 V, (b) an electrical equivalent circuit used for data fitting before degradation potential is applied, (c) and for degraded electrode, (d) evolution of impedance spectra of PPy–CPO-27-Ni electrode polarized at various potentials, (e) variation of absolute impedance for the PPy–MOFs electrodes polarized at various potentials (f) CVs, (g) and open circuit potential (OCP) of PPy–HKUST-1 electrode at different applied potentials. | ||
Two electrical equivalent circuits were used for data fitting, one for samples before degradation (Fig. 7b) and another for degraded electrodes (Fig. 7c). In both, the conductivity of electrolyte (Rs) is defined by a resistor whose magnitude estimated by the left-hand-side intercept of the semicircle. This is connected in series not only with the charge transfer resistance (RCT) (and capacitance) of carbon black but also the resistance of doped PPy (RCarbon+PPy). It is RCT and RCarbon+PPy that relate to the total conductivity of the bulk material. Furthermore, these resistivities are in parallel with an EDL capacitor that describes charge accumulation (of the ions from the electrolyte) at the surface of polarized PPy. The diffusion element called the Warburg resistor (W) was identified in the EIS spectra and is associated with both the diffusion of ions in electrolyte and inside the polymer. The total resistance (RCarbon+PPy) corresponding to charge transfer through conducting fractions is our parameter of interest as it provides information about the stability of the electrode upon polarization at increasing voltage. Fig. 7c represents a modified electrical circuit used for the fitting of Nyquist plots of degraded PPy–CPO-27-Ni electrode (Fig. 7d).
Clearly, the impedance of PPy-d-MOFs has changed upon increasing polarization and is manifested by an additional semicircle developing at mid frequencies. This refers to degradation of the conductive polymer through over-oxidation of PPy (0.59 V vs. Ag/AgCl) and results in both chemical modification of the polymer chain and structural changes such as pore collapse.62,63 This degradation is demonstrated by changes from purely capacitive behavior (vertical line at the lowest frequencies) to resistive systems formed upon applied potential higher than 0.6 V (Fig. 7d). This resistance (RDeg.PPy, Fig. 7c) increases at higher polarization of electrode and is represented by the increasing magnitude of the arc (RDeg.PPy). During this period the resistivity and capacitance of the carbon additive is not affected by the applied potential. Its capacitance has negligible contribution to the total capacitance of electrodes, as shown by a low current recorded for the MOF-based electrode with the same amount of carbon black yet without PPy (Fig. 5a).
In summary, for all electrodes the significant degradation represented by an increase in absolute resistivity takes place between 0.5 V and 0.6 V and remains at the constant level above 0.7 V (Fig. 7e). The trend for PPy–HKUST-1 and PPy–CPO-27-Ni is very similar and the overall impedance of these electrodes is almost identical. In contrast, PPy–CPO-27-Co shows significant increase in initial and final resistivity as compared to other electrodes, which is manifested by its higher internal resistivity (ohmic drop, shown in Table 2). Meanwhile, CVs recorded after polarization shows decreases in current with increasing applied potential (Fig. 7f), which is consistent with the increase in a total resistance of electrode due to PPy degradation (Fig. 7e). The specific gravimetric capacitance initially calculated for freshly prepared electrodes decreases at about 63% after the electrode is treated at 1 V and this decrease is more or less the same for all electrodes as shown in CVs after polarization (see Fig. 7f, S8b and S9b†). Fig. 7g represents open circuit potential (OCP) after polarization and indicates that the electrode is stable up to 0.5 V and undergoes slow self-discharge due to both instability of MOF and PPy. This trend is similar to PPy–CPO-27-Ni and PPy–HKUST-1 (compare Fig. 7g and S8a†). PPy–CPO-27-Co on the other hand appears to be less stable with rapid OCP lost at higher polarization potential (Fig. S9a†), which is also consistent with Fig. 6c showing the weakest capacity retention for PPy–CPO-27-Co. To rationalize this, we can assume that as CPO-27-Co has the lowest PPy content as per elemental analysis (% N related to total PPy content, Table 1), a larger proportion of CPO-27-Co is exposed to electrochemical (redox activity of metal center) processes. Ideally, continuous coating of MOF's surface with PPy will be recommended in order to improve its stability, which should be a subject of further development of these electrodes.
4 Conclusions
MOFs, with their high specific surface area, rigid pore structure, and diverse functionalities are an attractive material for employment as supercapacitors. The inherent electrical insulation brought upon by their typical construction is negated by the inclusion of PPy within these pores. Characterizations by PXRD and ATR-FTIR of the MOFs and PPy-d-MOFs show not only the successful synthesis of MOFs, but also clear inclusion of PPy. While the amount of PPy is low, as seen from quantification by TGA and CHNS analysis, this amount is sufficient to attain supercapacitive behaviour. SEM shows that PPy can be found on the surface of the MOF crystals, while BET supports that the polymer can also be found within the pores of the MOF. Our method of synthesis therefore allows us to take advantage of the high surface area. Electrochemical testing shows a marked difference in the plain vs. PPy-d-MOFs. Specific capacitance was boosted from 0.2 F g−1 in plain MOFs to over 185 F g−1. These PPy-d-MOF samples showed limited cycling stability, at best retaining 70% of the initial capacitance after 2500 cycles. However, all electrodes demonstrated through EIS that the maximum operating potential for these systems is 0.5 V and that the PPy is not stabilized by underlying MOF. The electrochemical stability of MOFs is restricted by their contact with liquid electrolyte. As a remedy to this, higher PPy content and rational adjustment of particle surface hydrophobicity/hydrophilicity are recommended so that the MOF/electrolyte interface is limited, resulting in more stable electrode materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank for their financial support the Fredrik and Catherine Eaton Graduate Fellowship, Mitacs Globalink Research Award (IT15169), The New Brunswick Innovation Foundation NBIF-RAI (RAI2019-009) and Queen's University Belfast internal funding. Special thanks to the following people for their help on this work: Angela Brownlie for running and training on PXRD, SEM, EDS, and TGA, Steven Cogswell for additional SEM, and Masoud Haeri for BET.References
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS.
- Z. Zhao, X. Ma, A. Kasik, Z. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2013, 52, 1102–1108 CrossRef CAS.
- K. S. Lin, A. K. Adhikari, C. N. Ku, C. L. Chiang and H. Kuo, Int. J. Hydrogen Energy, 2012, 37, 13865–13871 CrossRef CAS.
- D. Zhou, J. Ni and L. Li, Nano Energy, 2019, 57, 711–717 CrossRef CAS.
- I. Agirrezabal-Telleria, I. Luz, M. A. Ortuño, M. Oregui-Bengoechea, I. Gandarias, N. López, M. A. Lail and M. Soukri, Nat. Commun., 2019, 10, 1–8 CrossRef CAS.
- M. Al Haydar, H. R. Abid, B. Sunderland and S. Wang, Drug Des., Dev. Ther., 2017, 11, 2685–2695 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dincǎ, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS.
- S. K. Bhardwaj, N. Bhardwaj, R. Kaur, J. Mehta, A. L. Sharma, K.-H. Kim and A. Deep, J. Mater. Chem. A, 2018, 6, 14992–15009 RSC.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincǎ, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS.
- B. J. Holliday and T. M. Swager, Chem. Commun., 2005, 23–36 RSC.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dincǎ, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS PubMed.
- L. Sun, C. H. Hendon, M. A. Minier, A. Walsh and M. Dincǎ, J. Am. Chem. Soc., 2015, 137, 6164–6167 CrossRef CAS PubMed.
- R. Heck, O. Shekhah, O. Zybaylo, P. Weidler, F. Friedrich, R. Maul, W. Wenzel and C. Wöll, Polymers, 2011, 3, 565–1574 CrossRef.
- Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Chem. Mater., 2010, 22, 4120–4122 CrossRef CAS.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- S. Han, S. C. Warren, S. M. Yoon, C. D. Malliakas, X. Hou, Y. Wei, M. G. Kanatzidis and B. A. Grzybowski, J. Am. Chem. Soc., 2015, 137, 8169–8175 CrossRef CAS PubMed.
- H. A. Schulze, B. Hoppe, M. Schäfer, D. P. Warwas and P. Behrens, ChemNanoMat, 2019, 5, 1159–1169 CrossRef CAS.
- D. Kim, D. W. Kim, W. G. Hong and A. Coskun, J. Mater. Chem. A, 2016, 4, 7710–7717 RSC.
- J.-M. Yang, X.-W. Hu, Y.-X. Liu and W. Zhang, Microporous Mesoporous Mater., 2019, 274, 149–154 CrossRef CAS.
- T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef PubMed.
- K. Qi, R. Hou, S. Zaman, Y. Qiu, B. Y. Xia and H. Duan, ACS Appl. Mater. Interfaces, 2018, 10, 18021–18028 CrossRef CAS PubMed.
- C.-C. Lin, Y.-C. Huang, M. Usman, W.-H. Chao, W.-K. Lin, T.-T. Luo, W.-T. Whang, C.-H. Chen and K.-L. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 3400–3406 CrossRef CAS PubMed.
- B. Le Ouay, M. Boudot, T. Kitao, T. Yanagida, S. Kitagawa and T. Uemura, J. Am. Chem. Soc., 2016, 138, 10088–10091 CrossRef CAS PubMed.
- B. Dhara, S. S. Nagarkar, J. Kumar, V. Kumar, P. K. Jha, S. K. Ghosh, S. Nair and N. Ballav, J. Phys. Chem. Lett., 2016, 7, 2945–2950 CrossRef CAS PubMed.
- K. Zhou, D. Shen, X. Li, Y. Chen, L. Hou, Y. Zhang and J. Sha, Talanta, 2020, 209, 120507 CrossRef CAS PubMed.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- Z. Wang, X. Xu, J. Kim, V. Malgras, R. Mo, C. Li, Y. Lin, H. Tan, J. Tang, L. Pan, Y. Bando, T. Yang and Y. Yamauchi, Mater. Horiz., 2019, 6, 1433–1437 RSC.
- P. Wang, X. Du, T. Chen, X. Hao, A. Abudula, K. Tang and G. Guan, Electrochim. Acta, 2019, 306, 35–44 CrossRef CAS.
- X. Chen, M. Zhang, S. Li, L. Li, L. Zhang, T. Wang, M. Yu, Z. Mou and C. Wang, J. Mater. Chem. B, 2017, 5, 1772–1778 RSC.
- Z. Neisi, Z. Ansari-Asl, S. Jafarinejad-Farsangi, M. E. Tarzi, T. Sedaghat and V. Nobakht, Colloids Surf., B, 2019, 178, 365–376 CrossRef CAS PubMed.
- P. Geng, S. Cao, X. Guo, J. Ding, S. Zhang, M. Zheng and H. Pang, J. Mater. Chem. A, 2019, 7, 19465–19470 RSC.
- Y. Liu, N. Xu, W. Chen, X. Wang, C. Sun and Z. Su, Dalton Trans., 2018, 47, 13472–13478 RSC.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- Y. Jiao, G. Chen, D. Chen, J. Pei and Y. Hu, J. Mater. Chem. A, 2017, 5, 23744–23752 RSC.
- C. Zhang, J. Tian, W. Rao, B. Guo and L. Fan, Cellulose, 2019, 26, 3387–3399 CrossRef CAS.
- X. Xu, J. Tang, H. Qian, S. Hou, Y. Bando, M. S. A. Hossain, L. Pan and Y. Yamauchi, ACS Appl. Mater. Interfaces, 2017, 9, 38737–38744 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, Y. Chen, C. Wang and L. Guo, Appl. Surf. Sci., 2020, 507, 145089 CrossRef CAS.
- H.-N. Wang, M. Zhang, A.-M. Zhang, F.-C. Shen, X.-K. Wang, S.-N. Sun, Y.-J. Chen and Y.-Q. Lan, ACS Appl. Mater. Interfaces, 2018, 10, 32265–32270 CrossRef CAS.
- R. Hou, M. Miao, Q. Wang, T. Yue, H. Liu, H. S. Park, K. Qi and B. Y. Xia, Adv. Energy Mater., 2020, 10, 1901892 CrossRef CAS.
- A. Bétard, D. Zacher and R. A. Fischer, CrystEngComm, 2010, 12, 3768–3772 RSC.
- D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS.
- M. H. Yap, K. L. Fow and G. Z. Chen, Green Energy and Environment, 2017, 2, 218–245 CrossRef.
- C. Chen, X. Feng, Q. Zhu, R. Dong, R. Yang, Y. Cheng and C. He, Inorg. Chem., 2019, 58, 2717–2728 CrossRef CAS PubMed.
- G. Xu, P. Nie, H. Dou, B. Ding, L. Li and X. Zhang, Mater. Today, 2017, 20, 191–209 CrossRef CAS.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- P. D. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354–6358 CrossRef CAS PubMed.
- B. Pato-Doldán, M. H. Rosnes and P. D. C. Dietzel, ChemSusChem, 2017, 10, 1710–1719 CrossRef PubMed.
- A. A. Yakovenko, J. H. Reibenspies, N. Bhuvanesh and H.-C. Zhou, J. Appl. Crystallogr., 2013, 46, 346–353 CrossRef CAS.
- H. Qiao, Z. Wei, H. Yang, L. Zhu and X. Yan, J. Nanomater., 2009, 2009, 5 Search PubMed.
- F. A. Sofi, K. Majid and O. Mehraj, J. Alloys Compd., 2018, 737, 798–808 CrossRef CAS.
- W. Reusch, Infrared Spectroscopy, 2013, https://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/Spectrpy/InfraRed/infrared.htm Search PubMed.
- M. A. Chougule, S. G. Pawar, P. R. Godse, R. N. Mulik, S. Sen and V. B. Patil, Soft Nanosci. Lett., 2011, 01, 6–10 CrossRef CAS.
- M. Radtke, D. G. G. McMillan, B. Schröter, S. Höppener, B. Dietzek, U. S. Schubert and A. Ignaszak, Polymer, 2015, 77, 289–296 CrossRef CAS.
- A. Yussuf, M. Al-Saleh, S. Al-Enezi and G. Abraham, Int. J. Polym. Sci., 2018, 2018, 8 Search PubMed.
- K. K. Kanazawa, A. F. Diaz, R. H. Geiss, W. D. Gill, J. F. Kwak, J. A. Logan, J. F. Rabolt and G. B. Street, J. Chem. Soc., Chem. Commun., 1979, 854–855 RSC.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, R. Castledine, N. Parkinson, I. Haley and R. E. Morris, Dalton Trans., 2016, 45, 618–629 RSC.
- M. Gazulla, M. Rodrigo, E. Blasco and M. Orduña, X-Ray Spectrom., 2013, 42, 394–401 CrossRef CAS.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1–19 Search PubMed.
- Y. Jiang, I. Oh, S. H. Joo, O. Buyukcakir, X. Chen, S. H. Lee, M. Huang, W. K. Seong, S. K. Kwak, J. W. Yoo and R. S. Ruoff, J. Am. Chem. Soc., 2019, 141, 16884–16893 CrossRef CAS PubMed.
- D. Y. Lee, E. K. Kim, N. K. Shrestha, D. W. Boukhvalov, J. K. Lee and S. H. Han, ACS Appl. Mater. Interfaces, 2015, 7, 18501–18507 CrossRef CAS PubMed.
- H. L. Mosch, S. Höppener, R. M. Paulus, B. Schroter, U. S. Schubert and A. Ignaszak, Phys. Chem. Chem. Phys., 2015, 17, 13323 RSC.
- C. Johne, R. Fritzsch and A. Ignaszak, Electroanalysis, 2014, 26, 1560–1572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, EDS, BET, PPy–CPO-27-Ni/Co electrochemical performance, EIS, I2-doping effect. See DOI: 10.1039/d0ra02154g |
| This journal is © The Royal Society of Chemistry 2020 |