
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbiotic reduction of p-chloronitrobenzene by sulfate green rust: influence factors, products and mechanism†
Ying Han ,
Junkai Huang,
Hongyuan Liu*,
Yue Wu,
Zhao Wu,
Kemin Zhang and
Qingjie Lu
,
Junkai Huang,
Hongyuan Liu*,
Yue Wu,
Zhao Wu,
Kemin Zhang and
Qingjie Lu
College of Civil Engineering and Architecture, Zhejiang University of Technology, Hangzhou 310023, P. R. China. E-mail: lhyzyy@zjut.edu.cn
First published on 20th May 2020
Abstract
The reduction of p-chloronitrobenzene (p-CNB) by sulfate green rust (GRSO4) was systematically studied. The results revealed that GRSO4 has a good removal effect on p-CNB. The removal efficiencies of p-CNB by GRSO4 improved with the increase of the pH value. The removal efficiencies in the presence of ions were better than that of GRSO4 alone, while natural organic matter (NOM) could adsorb p-CNB, which competed with GRSO4. The reductions of p-CNB by GRSO4 under different conditions followed pseudo-first-order reaction kinetics except for the reactions in the presence of NOM. p-CNB was converted into p-chloroaniline (p-CAN), which produced p-nitrosochlorobenzene and p-chlorophenylhydroxylamine as the intermediate products. The results of the X-ray diffraction (XRD), scanning electron microscopy (SEM) and transmission electron microscopy (TEM) showed GRSO4 was gradually transformed into goethite. Fe(II) in the GRSO4 structure was the main electron donor involved in the reaction.
Introduction
As a widely used raw material, p-chloronitrobenzene (p-CNB) is an important intermediate in the production of fine chemicals, pesticides, drugs and rubber.1,2 Due to its high toxicity and strong bioaccumulation, p-CNB may cause cancer and affect the normal functioning of the central nervous system. If not disposed properly, it will eventually reach the aquatic environment and soil sediments, which poses a great threat to human beings and wildlife.3–6 Therefore, it has been listed as a priority pollutant by many countries and regions.7In recent decades, a lot of research has been carried out on the treatment of chlorinated nitrobenzene wastewater. The methods mainly include physical adsorption,8 biodegradation,9 advanced oxidation10,11 and catalytic reduction.12 The methods mentioned above have excellent removal efficiencies. However, the physical adsorption has difficulty in recovery. Biodegradation organisms are susceptible to interference with the concentration of substances. Advanced oxidation methods have the risks of secondary pollution and high cost, chemical stability and deactivation of the catalyst. In addition, due to the presence of strong electron withdrawing group chlorine substituent and nitro substituent on the aromatic ring, the oxidation processes require more energy.
In contrast, the chlorine substituent and the nitro substituent are thermodynamically favorable for the reduction reaction and enhance the p-CNB removal performance.13 The application of catalytic reduction with H2 may cause the concern of the safety of H2 and the high price of catalyst. While, the natural subsurface anaerobic and hypoxic environment not only provide a variety of potential reduction conditions for the abiotic transformation of pollutants, but also provide reduction agents. For example, iron, as the fourth most abundant element in the earth's crust, is highly reductive in its elemental state and is often used as a reductant.14,15 Although iron is a good reductant, in its natural state most of it exists as an oxide of iron.16 Some minerals containing structural Fe(II) have been shown to reduce chlorinated hydrocarbons,17 nitroaromatic hydrocarbons,18 and pesticides.19 Green rust (GR) as a kind of iron oxide with layered bimetallic hydroxide (LDH) structure,20 obeys the general formula [FeII(1−x)FeIIIx(OH)2]x+[(x/n)An−·mH2O]x− including An− is anion intercalated (An− = Cl−, SO42−, CO32−), m is the number of inserted water molecules and x is the molar fraction of Fe(III) {[Fe(III)]/[Fe(total)]}.21,22 GR not only has good adsorption performance, but also has strong reduction ability.23 The excellent reduction strength of GR is attributed to the high Fe(II) content and the rapid electron transfer between Fe(II) and Fe(III) in the hydroxide tablets by polaron jumps.24 Due to its reductive reactivity and ability to absorb contaminants, GR suspension is considered a promising reductant for soil and groundwater remediation by in situ injection of GR into pollutant sources and plume.25 The reductive transformation of o-chloronitrobenzene (o-CNB) by green rust was investigated, including reducing capacity of GR, effect of pH value, products and pathway. There were no kinetic studies, and the characterization of green rust was inadequate.26 The reduction of p-CNB with Fe(II) bound to GR was investigated, mainly on the degradation kinetics.16 There is a lack of the systematic study of p-CNB with GR solely. This study attempts to investigate the main influence factors on the reduction process of p-CNB removal by GR, the degradation products and explore the reaction mechanism.
Experimental
Chemicals
FeCl3·6H2O was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd. FeSO4·7H2O was purchased from Sinopharm Chemical Reagent Co., Ltd. NaOH and HCl were purchased from Xiqiao Science and Technology Co., Ltd. p-CNB, NaCl, MgCl2, CaCl2, KCl, Na2SO4, NaHCO3, Na2CO3 and fulvic acid (FA) were purchased from Shanghai Aladdin Biotechnology Co., Ltd. Nitrobenzene, p-chloroaniline (p-CAN) and aniline were purchased from Shanghai Maclean Biotechnology Co., Ltd. All chemicals were used without further purification.Mineral synthesis
The sulfate green rust (GRSO4) in this study was synthesized according to the previous method27,28 of the co-precipitation of FeCl3·6H2O and FeSO4·7H2O in a glove box. 0.132 mol L−1 FeSO4·7H2O and 0.066 mol L−1 FeCl3·6H2O were added to 120 mL deoxygenated deionized water, stirring was continued until complete dissolution, 1 mol L−1 NaOH was dropwise titrated to the solution until the pH value reached 8.0. Then, solid and liquid separation was realized through suction filtration, and the filtered slurry was placed in a freeze-dried bottle, which was then freeze-dried. The dried powders were transferred to the glove box for grinding, and stored after 100 mesh screening. The obtained solid powders were confirmed to be GRSO4 by X-ray diffraction (XRD) (Fig. 4a).Reduction experiment
Reduction experiments were carried out in a glove box to test the ability of GRSO4 to reduce p-CNB in an anaerobic environment. First, 0.1 g L−1 of GRSO4, 150 mL of deoxygenated deionized water, and 500 μg L−1 of p-CNB were sequentially added to a 150 mL conical flask. The conical flask was shaked on a horizontal shaker at 120 rpm for 40 min. 1 mL sample was got from the flask and filtrated through a 0.22 μm membrane to terminate the reaction at each time point. Then, the water sample was got for analysis. The solid powders were freeze-dried under N2 protection and stored in a glove box for subsequent detection and characterization. The pH values of the deoxidized deionized water were adjusted first when the effects of initial pH value were investigated. For the experiments under different ions and NOM, ions or FA were added to water before p-CNB.The reduction procedure of p-CNB by Fe(II) was as follows. Fe2+ stock solution was added to the 150 mL conical flask first, then the deoxidized deionized water and p-CNB were added into the flask. The following steps were the same as the reduction of p-CNB by GRSO4.
Analysis and characterization
p-CNB, p-CAN, nitrobenzene and aniline were analyzed by high performance liquid chromatography (HPLC, Agilent 1200). For p-CNB, the v/v ratios of water and methanol were 35 to 65, and the ultraviolet wavelength was 295 nm. For p-CAN, the v/v ratios of water and methanol were 35 to 65, and the ultraviolet wavelength was 240 nm. For nitrobenzene, the v/v/v ratios of methanol, water and acetic acid were 70, 29 and 1, and the ultraviolet wavelength was 262 nm. For aniline, the v/v ratios of water and methanol were 35 to 65, and the ultraviolet wavelength was 231 nm. The p-chlorophenylhydroxylamine and p-nitrosochlorobenzene were analyzed by gas chromatography mass spectrometry (GC-MS, Agilent 7980), the details were described in Text S1.† The concentrations of Fe(II) in the solution were determined by the 1,10-phenanthroline method.29–31The solid powders before and after the reaction were characterized with X-ray diffraction (XRD, PANalytical B.V., X'Pert Pro) using Cu Kα radiation (λ = 0.1541 nm) at 40 kV and 40 mA, X-ray photoelectron spectroscopy (XPS, Shimadzu, KRATOS AXIS ULTRA DLD), scanning electron microscopy (SEM, ZEISS, GeminiSEM 300), and transmission electron microscopy (TEM, FEI, Tecnai G2 F30).
Kinetics
The kinetics of the p-CNB were described by a pseudo-first-order reaction (eqn (1)). C0 is the initial concentration, Ct is the concentration at time t, t is the reaction time, and kobs is the pseudo-first-order reaction rate constant.
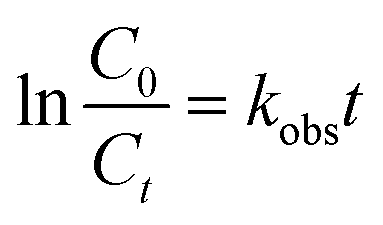 | (1) |
Results and discussion
Effect of initial pH value
The initial pH value is an essential factor which affects the reduction of p-CNB. The green rusts would be in varied states under different pH values and their abilities of reducing p-CNB are different. In this experiment, the pH values were set to 5.0, 6.5, 8.0 and 10.0, respectively, to investigate the effect of initial pH value on p-CNB reduction (Fig. 1). Interestingly, the final removals were higher with the increase of pH values. The pseudo-first-order reaction rates followed the same rule. The rates became higher with the increase of pH values (Table 1). This was significantly different from our common phenomena that lower pH values leaded to better reduction of pollutants with zero-valent iron (ZVI) and iron minerals.32 However, the similar phenomena happened when GR was used to reduce o-CNB under the alkaline condition.26 According to the Hayashi, this is caused by the different reactions of GRSO4 at different pH values.33 Besides, the rise of pH value could reduce the REDOX potential of green rust, thereby improving its reduction ability.34 | ||
| Fig. 1 Removal of p-CNB by GRSO4 at different initial pH values. [p-CNB]0 = 500 μg L−1, [GRSO4]0 = 0.1 g L−1, T = 20 °C. Error bars represent one standard deviation (n = 3). | ||
Effect of ions and NOM
There are some inorganic or organic substances in the natural groundwater. Effect of several common ions (Na+, K+, Ca2+, Mg2+, Cl−, SO42−, HCO3−, CO32−) and natural organic matter (NOM, fulvic acid) on the removal of p-CNB by GRSO4 were investigated (Fig. 2). The final removals in the presence of cationic ions and anionic ions were similar to the system with GRSO4 (Fig. 2a and b). All reaction rates were higher than GRSO4 system (Table 2). All the cations showed positive effects on the reaction. The reaction rates were similar with small differences (Table 2). All cations were added to the reaction system in the form of chlorine salts. These electrolytes could facilitate electron transfer.35 The higher rate in the presence of K+ than that of Na+ was caused by their conductivities.35 This also worked for Ca2+ and Mg2+. The systems containing Ca2+ and Mg2+ were better than that containing K+ and Na+. Because they have more moles of chlorine ions with the same mass concentration. The Cl− could enhance the electron transfer.35 The results of the system containing Cl− also illustrated this (Table 2). The addition of SO42− should also facilitate the electron transfer. However, the enhancement was just a little bit. This could be caused by the surface complexation of sulfate with green rust.36 The improvement with HCO3− could be due to its enhancement on electron transfer and the effect of pH buffer.37 In particular, the reaction rate of CO32− system was significantly higher than that of GRSO4 system (Table 2). The increase of reaction rate in the system with CO32− could be due to the substitution of SO42− in GRSO4 by CO32−, resulting in the conversion of GRSO4 to GRCO3.38 GRCO3 has lower reduction potential, and the pH value in the experiment increased due to the addition of CO32−.39 The reaction rate would also increase with increasing pH value as we discussed in the previous section (Fig. 1).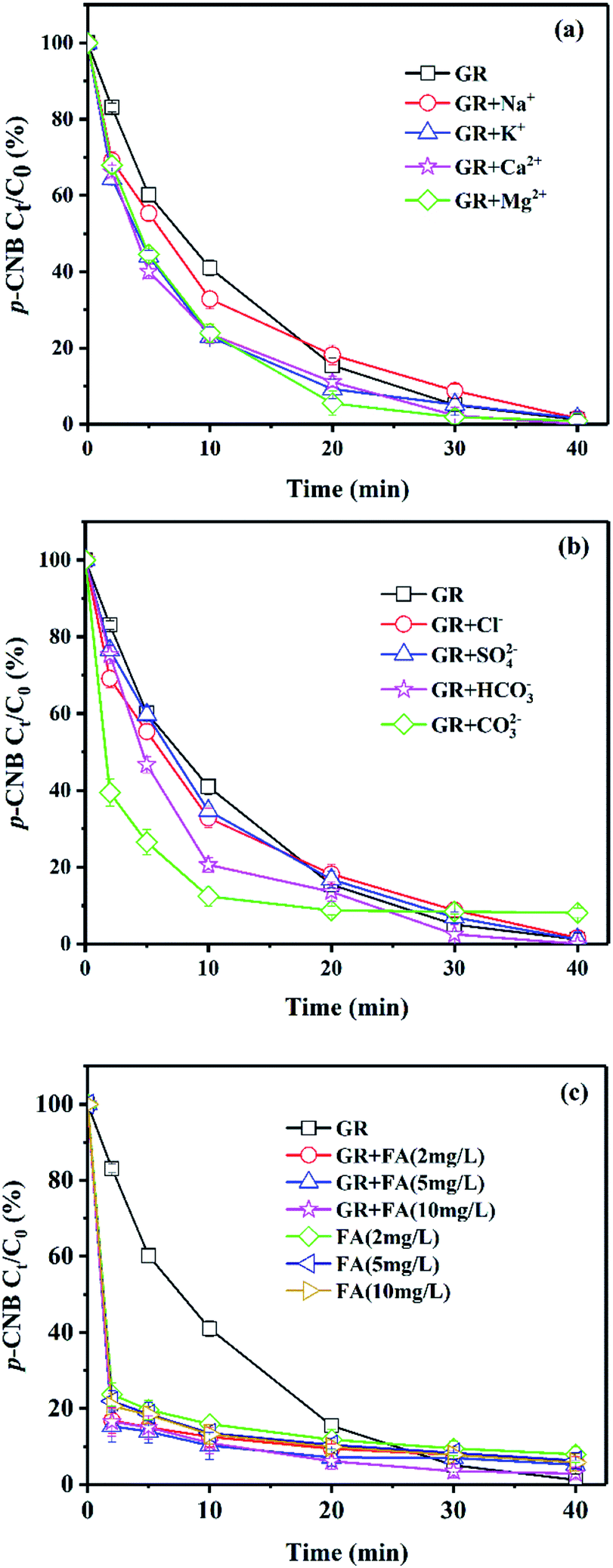 | ||
| Fig. 2 Effect of (a) cationic ions, (b) anionic ions and (c) FA on the removal of p-CNB by GRSO4. [p-CNB]0 = 500 μg L−1, [GRSO4]0 = 0.1 g L−1, T = 20 °C, pH = 6.5, [Na+]0 = [K+]0 = [Ca2+]0 = [Mg2+]0 = [Cl−]0 = [SO42−]0 = [HCO3−]0 = [CO32−]0 = 25 mg L−1. Error bars represent one standard deviation (n = 3). | ||
| Reaction conditions | kobs (×10−2)a (min−1) | R2 |
|---|---|---|
| a Uncertainties represent the standard deviation (n = 3). | ||
| GR only | 9.03 ± 0.46 | 0.99 |
| Na+ | 11.75 ± 0.52 | 0.99 |
| K+ | 12.02 ± 0.48 | 0.98 |
| Mg2+ | 12.72 ± 0.56 | 0.99 |
| Ca2+ | 13.14 ± 0.72 | 0.99 |
| Cl− | 11.75 ± 0.52 | 0.99 |
| SO42− | 9.26 ± 0.68 | 0.99 |
| HCO3− | 12.74 ± 0.65 | 0.98 |
| CO32− | 22.38 ± 2.80 | 0.89 |
Fulvic acid (FA) was used for the investigation of the effect of NOM. p-CNB was removed rapidly in a short time in the system of GRSO4 with FA (Fig. 2c). The reactions did not follow the pseudo-first-order reaction kinetics. The removals of p-CNB with FA only were a little bit lower than that with FA and GRSO4 (Fig. 2c). This indicated that the reaction mainly took place between p-CNB and FA, FA blocked the reduction reaction between GRSO4 and p-CNB. FA had shown the adsorption capacity on TNT.40 It is one component of humic acid (HA). HA had been used as the adsorbents for several organic contaminants.41,42 The adsorption of p-CNB by FA could account for the rapid removal of p-CNB by FA in the absence and presence of GRSO4. HA could complex or oxidize on the GR surface and hinder the access of p-CNB.37,43 The similar process may happen with FA. Therefore, the reduction of p-CNB by GR had been hindered. The small improvements for the p-CNB removals in the presence of GRSO4 and FA could be due to their joint effects.
Products
Based on the structure of p-CNB, it is expected to undergo nitro reduction and dechlorination. Possible products include nitrobenzene, aniline, p-nitrosochlorobenzene, p-chlorophenylhydroxylamine, p-CAN and chloride ions. Nitrobenzene and aniline were not detected. Therefore, GRSO4 could not dechlorinate p-CNB. p-CAN was the final product, which was similar as the former study on o-CNB.26 The concentration of p-CAN gradually increased with the decrease of the concentration of p-CNB (Fig. 3). But the total amount of nitrogen were unbalanced in the intermediate time periods. The removal of p-CNB basically completed in the reaction period of 40 minute, but p-CAN did not generate as much, indicating that the reaction was still going on. All p-CNB was converted to p-CAN after 120 min. The nitrogen loss should be due to the existence of the intermediate products. p-chlorophenylhydroxylamine was found (Fig. S1†). No p-chloronitrosobenzene was detected. Its existence time should be extremely short, which could be quickly reduced to p-chlorophenylhydroxylamine.16,44 This was similar to the reduction of nitrobenzene with Fe(II).44 | ||
| Fig. 3 Products of the removal of p-CNB by GRSO4. [p-CNB]0 = 3.17 μmol L−1 = 500 μg L−1, [GRSO4]0 = 0.1 g L−1, T = 20 °C, pH = 6.5. Error bars represent one standard deviation (n = 3). | ||
Characterization of GRSO4 powders
In order to observe the changes of phase compositions, surface morphologies and internal structures of GRSO4 powders before and after the reaction, the GRSO4 powders were analyzed by XRD, SEM and TEM. The XRD patterns were collected with a 2θ range from 10° to 80° at a step of 0.05°. The average crystallite sizes of the samples were calculated with the Scherrer equation based on the strongest hkl (002) diffraction peak of green rust. The results (Fig. 4) showed that the mineral powders converted from GRSO4 (Fig. 4a, PDF#13-0092, JCPDS) to goethite (Fig. 4c, PDF#29-0713, JCPDS). There was still unreacted GRSO4 in the presence of goethite after 15 min reaction (Fig. 4b). Goethite was also detected when o-CNB was reduced by GR, many other kinds of iron minerals were detected.26 However, only goethite was detected in this study. Through SEM images we directly saw that GRSO4 had a smooth layered structure, and the surface grew into strip crystals at 15 min, and finally became strip crystals at 40 min (Fig. S2†). In the low resolution TEM (Fig. 5), the similar phenomena occurred as the SEM (Fig. S2†), and the black part seen in Fig. 5a was due to the fact that the sample concentration was so high that the mineral particles were stacked and the GR particles were 90° away from the TEM probe. In the high resolution TEM after fourier transformation, the powders at 0 min were amorphous and the crystal structure was disordered (Fig. 5d). The powders at 40 min was polycrystalline (Fig. 5e). After the fourier transformation into concentric rings, the crystal structure was in a staggered and orderly state. After measuring the interplanar spacing in Fig. 5d, we found that it coincided with the 100 crystal plane of GRSO4 (PDF#13-0092, JCPDS), and Fig. 5e coincided with the 130 crystal plane of goethite (PDF#29-0713, JCPDS). The results were consistent with those of XRD. | ||
| Fig. 4 XRD pattern of GRSO4 powders at (a) 0 min, (b) 15 min and (c) 40 min. | ||
 | ||
| Fig. 5 TEM of powders at (a) 0 min, (b) 15 min and (c) 40 min and high-resolution TEM of powders at (d) 0 min and (e) 40 min. | ||
The surface chemical properties of the unreacted GRSO4 powders, the GRSO4 powders at 15 min and 40 min were analyzed by XPS. According to the spectra of the unreacted GRSO4 powders, Fe and O were found on the surface of the samples, as well as a small amount of residual S (about 3%) (Table S1†). The iron to oxygen ratio determined from utilising the peak area and sensitivity factor was close to 0.4 (Table S1 and eqn (S1)†), which was consistent with the Mullet.21
In order to further understand the changes of iron composition in GRSO4 before and after the reaction, spectral fitting is required. The fitting process of Fe (2p) domain spectra of iron (oxyhydro) oxides is usually complex, including multiple contributions and satellite characteristics.45 In mixed valence compounds, the complexity is enhanced by overlapping multiple peaks of Fe(II) and Fe(III) species.46 It was shown that the multiple peaks calculated by Gupta and Sen could well fit the Fe (2p3/2) peaks in Fe(II) and Fe(III) compounds.47 Therefore, they were used to study the changes of GRSO4 before and after reaction (Fig. 6, Tables S2 and S3†). When GRSO4 was not reacted, the photoelectron peaks of Fe (2p) and O (1s) were respectively at 710.9 ev and 531.4 ev. The Fe(II)/Fe(III) ratio of 2.0, and it was consistent with the results reported by Perez et al.48 The shape and position of Fe (2p3/2) photoelectron peaks were also similar to the previously reported XPS spectra of GRSO4.48 In addition, the relative values of Fe–O, O–H and adsorbed water of the O (1s) peak were 529.9, 531.3 and 532.1 eV, respectively (Fig. 6d and Table S3†), which were consistent with the values obtained by Mullet et al.21
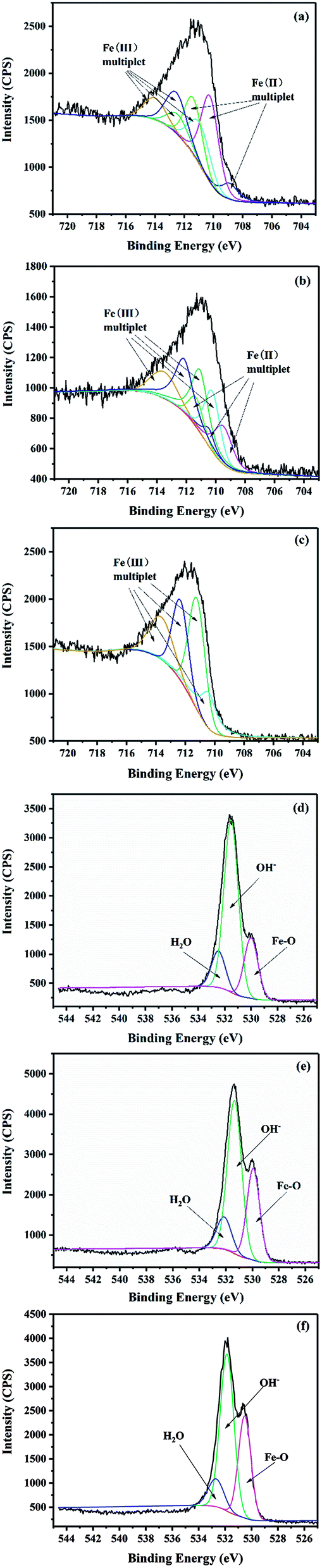 | ||
| Fig. 6 Fe 2p3/2 spectra of powders (a) unreacted, (b) 15 min, (c) 40 min and O 1s spectra of powders (d) unreacted, (e) 15 min, (f) 40 min. | ||
After the reaction, the photoelectron peaks of Fe (2p) and O (1s) all changed, in particular, the maximum value of the photoelectron peak of Fe (2p) migrated to 711.5 eV. The Fe(III) in solid minerals gradually increased, while Fe(II) gradually decreased and disappeared finally (Fig. 6a–c and Table S2†). The content of the Fe–O increased gradually (Fig. 6d–f and Table S3†). These all indicate that GRSO4 was oxidized, which was caused by electron transfer. The results were consistent with those of XRD, SEM and TEM.
Mechanism and pathway of the reaction
p-CNB, p-chlorophenylhydroxylamine and p-CAN were detected in this study (Fig. 3 and S1†). Dechlorination did not occur, the nitrogroup was reduced. It is generally thought that the reduction of aromatic nitrogroup to aniline occurs through a series of steps of electron addition and protonation, in which the corresponding aniline is the final product, nitrosobenzene and phenylhydroxylamine are intermediates respectively.49,50 Therefore, as was shown in Fig. 7, the intermediate products of this experiment should be p-nitrosochlorobenzene and p-chlorophenylhydroxylamine, and the final product should be p-CAN. The stepwise electron addition and protonation happened in this reaction system. In addition to the structural Fe(II) in GRSO4, dissolved Fe(II) were detected during the reaction (Fig. S3†). The sources of electron donors in the reaction can be divided into the following situations: Fe(II) in the solution, Fe(II) in the structure of GRSO4. Fe(II) ions with the same concentration of GRSO4 were investigated to reduce p-CNB (Fig. S4†). p-CNB hardly reacted with Fe(II) alone, the removal efficiencies were less than 4%. Based on the above data, the change of the mineral particles from GRSO4 to goethite and the transformation of Fe(II) to Fe(III) in solid minerals (Fig. 4–6), the electron donor should be structural Fe(II) in GRSO4, which was consistent with the former study on the reduction of o-CNB by GR.26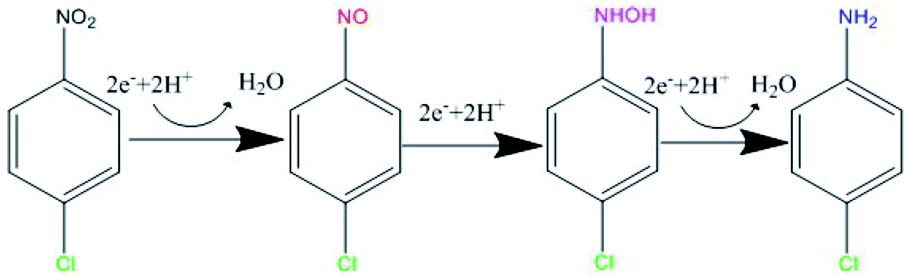 | ||
| Fig. 7 Reaction process of the reduction of p-CNB by GRSO4. | ||
Conclusions
GRSO4 could efficently reduce p-CNB. The removal efficiencies and reduction rates of p-CNB became higher with the increase of pH value. Ions improved the removals of p-CNB. The removal rates of p-CNB improved substantially with the presence of CO32−. The added NOM could have an adsorption with p-CNB. The reduction of p-CNB by GRSO4 can only reduce nitro group without dechlorination. The green rust was gradually transformed into goethite. The reduction depended mainly upon the structural Fe(II) in GRSO4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 51508510) and Zhejiang Provincial Natural Science Foundation of China (No. LQ15E080009).References
- C. R. Jones, Y. Y. Liu, O. Sepai, H. F. Yan and G. Sabbioni, Environ. Sci. Technol., 2006, 40, 387–394 CrossRef CAS PubMed.
- W. Q. Kong, J. Y. Lin, X. He, Y. Y. Cheng, X. S. Zhang, G. Z. Deng, R. S. Han and C. Wu, Chemosphere, 2017, 187, 62–69 CrossRef CAS PubMed.
- Z. Guo, S. Zheng, Z. Zheng, F. Jiang, W. Hu and L. Ni, Water Res., 2005, 39, 1174–1182 CrossRef CAS PubMed.
- B. Huang, W. Qian, C. Yu, T. Wang, G. Zeng and C. Lei, Chem. Eng. J., 2016, 306, 607–618 CrossRef CAS.
- X. Jiang, J. Shen, Y. Han, S. Lou, W. Han, X. Sun, J. Li, Y. Mu and L. Wang, Water Res., 2016, 88, 257–265 CrossRef CAS PubMed.
- X. Xu, J. Shao, M. Li, K. Gao, J. Jin and L. Zhu, Bioresour. Technol., 2016, 218, 1037–1045 CrossRef CAS PubMed.
- D. Colón, E. J. Weber and J. L. Anderson, Environ. Sci. Technol., 2006, 40, 4976–4982 CrossRef PubMed.
- Z. Guo, J. Liu and F. Liu, Microporous Mesoporous Mater., 2015, 213, 8–13 CrossRef CAS.
- L. Zhu, H.-z. Lin, J.-q. Qi, X.-y. Xu and H.-y. Qi, Water Res., 2012, 46, 6291–6299 CrossRef CAS PubMed.
- J. M. Shen, Z. L. Chen, Z. Z. Xu, X. Y. Li, B. B. Xu and F. Qi, J. Hazard. Mater., 2008, 152, 1325–1331 CrossRef CAS PubMed.
- M. Ye, Z. Chen, T. Zhang and W. Shao, Water Sci. Technol., 2012, 66, 479–486 CrossRef CAS PubMed.
- A. H. Pizarro, C. B. Molina, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2014, 158–159, 175–181 CrossRef CAS.
- B. Huang, J. Li, X. Cao, Y. Zhu, W. Chen and C. Lei, Electrochim. Acta, 2019, 296, 980–988 CrossRef CAS.
- M. Kobayashi, S. Kurosu, R. Yamaguchi and Y. Kawase, J. Environ. Manage., 2017, 200, 88–96 CrossRef CAS PubMed.
- Y. Zhang, G. B. Douglas, L. Pu, Q. Zhao, Y. Tang, W. Xu, B. Luo, W. Hong, L. Cui and Z. Ye, Sci. Total Environ., 2017, 598, 1140–1150 CrossRef CAS PubMed.
- M. Elsner, R. P. Schwarzenbach and S. B. Haderlein, Environ. Sci. Technol., 2004, 38, 799–807 CrossRef CAS PubMed.
- L. Yan and G. W. Bailey, J. Colloid Interface Sci., 2001, 241, 142–153 CrossRef CAS PubMed.
- J. Cervini-Silva, R. A. Larson, J. Wu and J. W. Stucki, Environ. Sci. Technol., 2001, 35, 805–809 CrossRef CAS PubMed.
- J. Cervini-Silva, J. Wu, R. A. Larson and J. W. Stucki, Environ. Sci. Technol., 2000, 34, 915–917 CrossRef CAS.
- B. C. Christiansen, T. Balic-Zunic, P. O. Petit, C. Frandsen, S. Mørup, H. Geckeis, A. Katerinopoulou and S. L. S. Stipp, Geochim. Cosmochim. Acta, 2009, 73, 3579–3592 CrossRef CAS.
- M. Mullet, Y. Guillemin and C. Ruby, J. Solid State Chem., 2008, 181, 81–89 CrossRef CAS.
- L. Fang, L. Xu, C. Liu, J. Li and L. Z. Huang, Environ. Int., 2019, 129, 299–307 CrossRef CAS PubMed.
- C. Bhave and S. Shejwalkar, Int. J. Environ. Sci. Technol., 2017, 15, 1243–1248 CrossRef.
- M. C. F. Wander, K. M. Rosso and M. A. A. Schoonen, J. Phys. Chem. C, 2007, 111, 11414–11423 CrossRef CAS.
- W. Yin, J. Ai, L. Z. Huang, D. J. Tobler and H. C. B. Hansen, Environ. Sci. Technol., 2018, 52, 7876–7883 CrossRef CAS PubMed.
- D. Wu, W. Wang and L. Ma, J. Tongji Univ. Nat. Sci., 2010, 38, 1473–1477 CAS.
- J. M. R. Génin, A. A. Olowe, P. Refait and L. Simon, Corros. Sci., 1996, 38, 1751–1762 CrossRef.
- A. Géhin, C. Ruby, M. Abdelmoula, O. Benali, J. Ghanbaja, P. Refait and J. M. R. Génin, Solid State Sci., 2002, 4, 61–66 CrossRef.
- S. Koch and G. Ackermann, Talanta, 1992, 39, 687–691 CrossRef CAS.
- A. S. Anastácio, B. Harris, H.-I. Yoo, J. D. Fabris and J. W. Stucki, Geochim. Cosmochim. Acta, 2008, 72, 5001–5008 CrossRef.
- H. Fadrus and J. Maly, Analyst, 1975, 100, 549–554 RSC.
- Y. Liu and J. Wang, Sci. Total Environ., 2019, 671, 388–403 CrossRef CAS PubMed.
- H. Hayashi, K. Kanie, K. Shinoda, A. Muramatsu, S. Suzuki and H. Sasaki, Chemosphere, 2009, 76, 638–643 CrossRef CAS PubMed.
- C. Ruby, C. Upadhyay, A. Gehin, G. Ona-Nguema and J. M. R. Genin, Environ. Sci. Technol., 2006, 40, 4696–4702 CrossRef CAS PubMed.
- J. H. Fan, H. W. Wang, D. L. Wu and L. M. Ma, J. Chem. Technol. Biotechnol., 2010, 85, 1117–1121 CrossRef CAS.
- P. Larese-Casanova and M. M. Scherer, Environ. Sci. Technol., 2008, 42, 3975–3981 CrossRef CAS PubMed.
- C. Le, J. H. Wu, S. B. Deng, P. Li, X. D. Wang, N. W. Zhu and P. X. Wu, Water Sci. Technol., 2011, 63, 1485–1490 CrossRef CAS PubMed.
- C. Ruby, A. Gehin, R. Aissa, J. Ghanbaja, M. Abdelmoula and J. M. R. Génin, Hyperfine Interact., 2006, 167, 803–807 CrossRef CAS.
- P. Refait, S. H. Drissi, J. Pytkiewicz and J. M. R. Génin, Corros. Sci., 1997, 39, 1699–1710 CrossRef CAS.
- Q. Hao, H. Qiao, C. Zhou, K. Zhang, W. Peng, S. Lu and G. Fu, Chin. J. Environ. Eng., 2016, 10, 3267–3274 CAS.
- I. D. Kovaios, C. A. Paraskeva and P. G. Koutsoukos, J. Colloid Interface Sci., 2011, 356, 277–285 CrossRef CAS PubMed.
- Y. Zhang, M. Yin, X. Sun and J. Zhao, Bioresour. Technol., 2020, 307, 123183 CrossRef CAS PubMed.
- D. Colon, E. J. Weber and J. L. Anderson, Environ. Sci. Technol., 2008, 42, 6538–6543 CrossRef CAS PubMed.
- J. Klausen, S. P. Troeber, S. B. Haderlein and R. P. Schwarzenbach, Environ. Sci. Technol., 1995, 29, 2396–2404 CrossRef CAS PubMed.
- P. C. J. Graat, M. A. J. Somers and E. J. Mittemeijer, Appl. Surf. Sci., 1998, 136, 238–259 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- R. P. Gupta and S. K. Sen, Phys. Rev. B: Solid State, 1975, 12, 15–19 CrossRef CAS.
- J. P. H. Perez, H. M. Freeman, J. A. Schuessler and L. G. Benning, Sci. Total Environ., 2019, 648, 1161–1170 CrossRef CAS PubMed.
- L. Han, L. Yang, H. Wang, X. Hu, Z. Chen and C. Hu, J. Hazard. Mater., 2016, 308, 208–215 CrossRef CAS PubMed.
- D. Naka, D. Kim and T. J. Strathmann, Environ. Sci. Technol., 2006, 40, 3006–3012 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02113j |
| This journal is © The Royal Society of Chemistry 2020 |