
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual role for acetohydrazide in Pd-catalyzed controlled C(sp3)–H acetoxylation of aldehydes†
Juan Chen,
Chaolumen Bai,
XingWen Tong,
Dan Liu and
Yong-Sheng Bao *
*
College of Chemistry and Environmental Science, Inner Mongolia Key Laboratory of Green Catalysis, Inner Mongolia Normal University, Hohhot, 010022, China. E-mail: sbbys197812@163.com; Tel: +86-471-4392442
First published on 25th March 2020
Abstract
The palladium catalyzed aldehyde directed acetoxylation of C(sp3)–H bonds was realized by a transient directing group approach for the first time. Crucial to the successful outcome of this reaction is the dual role of acetohydrazide as a directing group for the catalytic C(sp3)–H activation process and as a protecting group for the CHO functional group. The applicable methodology exhibits good functional group tolerance and occurs readily under mild conditions.
Introduction
Due to the ubiquitous nature of C–H bonds in organic molecules, selective functionalization of C(sp3)–H bonds emerged as a powerful tool in step-economical organic synthesis, featuring applications in pharmaceutical agent construction, bioactive molecules, and materials.1 Recently, the direct formation of a C–O bond via C(sp3)–H activation, particularly acetoxylation, has attracted much attention in organic synthesis2 because organic molecules bearing acetoxy group(s) are important structural features in drugs3 and agricultural chemicals.4 Various directing groups, such as oxazoline,2a,5 o-methyl oxime,6 pyridine,6,7 Boc-protected amines,8 Bts-protected amines,9 primary amines,10 quinazolinones11 and bidentate auxiliary groups,12 have been successfully employed for stoichiometric chelate-directed acetoxylation of C(sp3)–H bonds (see Scheme 1). Despite this significant progress, the development of oxidizable groups, such as aldehydes, for the directed acetoxylation of C(sp3)–H bonds is arguably highly desirable. There are a number of remaining challenges in the selective C(sp3)–H acetoxylation of aldehydes: (1) the tendency of CHO functional groups to undergo undesired oxidation; (2) competitive metal insertions into formyl C–H bonds; and (3) the weak coordinating ability of this group. To date only one example of aldehyde-directed ortho-hydroxylations of benzaldehydes via C(sp2)–H activation has been reported.13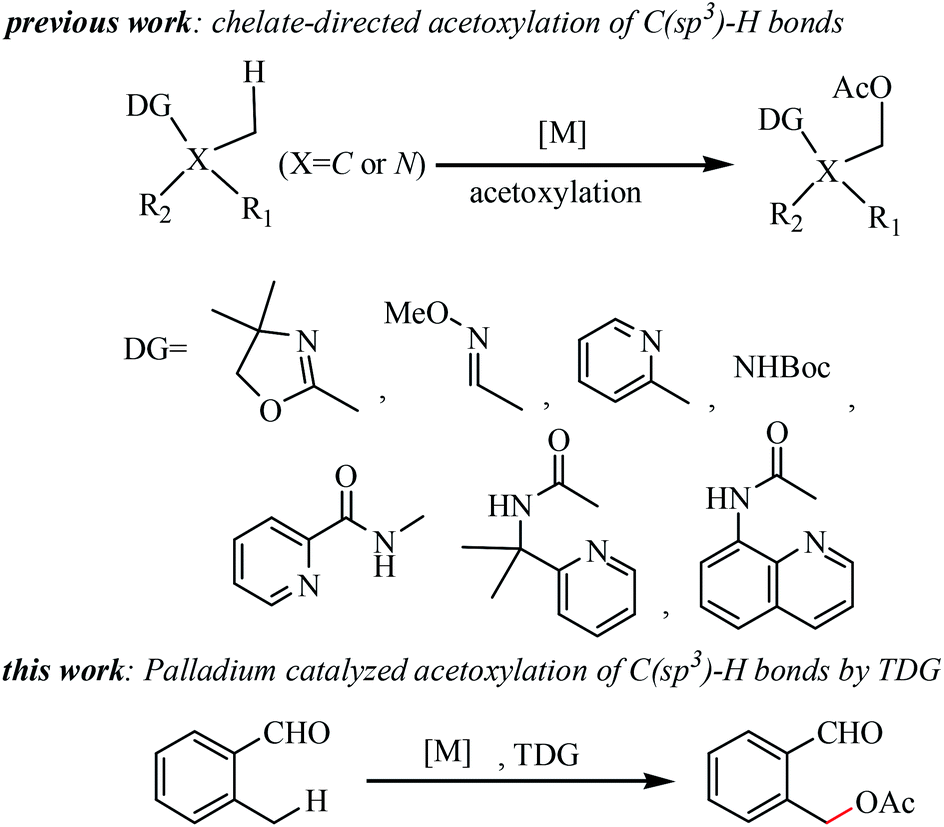 | ||
| Scheme 1 The development of C(sp3)–H acetoxylation. | ||
Very recently, the transient directing group (TDG) approach has been extended to the palladium-catalyzed ortho-C(sp3)–H bond arylations of aldehydes by the groups of Yu,14 Hu15, Chen,16 Bull,17 Ge,18 Wang19 and Wei.20 Notably, all these processes called for stoichiometric quantities of silver salts as oxidants and the CHO functional group is not oxidized in the reaction process. In these processes, the TDG plays a dual role, one role is to act as a directing group of the catalytic cycle, another is to act as a protecting group of the CHO functional group. Inspired by this concept, we questioned whether aldehyde-directed C(sp3)–H oxidation could be achieved using the TDG approach.
Herein, we describe a novel palladium catalyzed TDG approach for the direct and selective C(sp3)–H acetoxylation of aldehydes. This developed methodology provides a catalysis route for C–O bond formation in a straightforward fashion, which successfully suppresses the undesired oxidation of the –CHO group and the acetoxylation products could be transformed into various biologically active compounds such as indeno[1,2-b]quinolines,21 five-membered azacyclic compounds,22 mycophenolic acid which is an important antiparasitic, antineoplastic and antiviral agent,23 an intermediate of coleophomone which has antifungal activity and shows inhibition of human heart chymase,24 varitriol which is associated with high levels of biological activity toward renal, CNS, and breast cancer cell lines25 and an s-trans-heterodiene framework26 (see Scheme 2). Therefore, the methodology of aldehyde-directed selective acetoxylation of C(sp3)–H bonds can greatly reduce the steps of the total synthesis of these biologically active compounds.
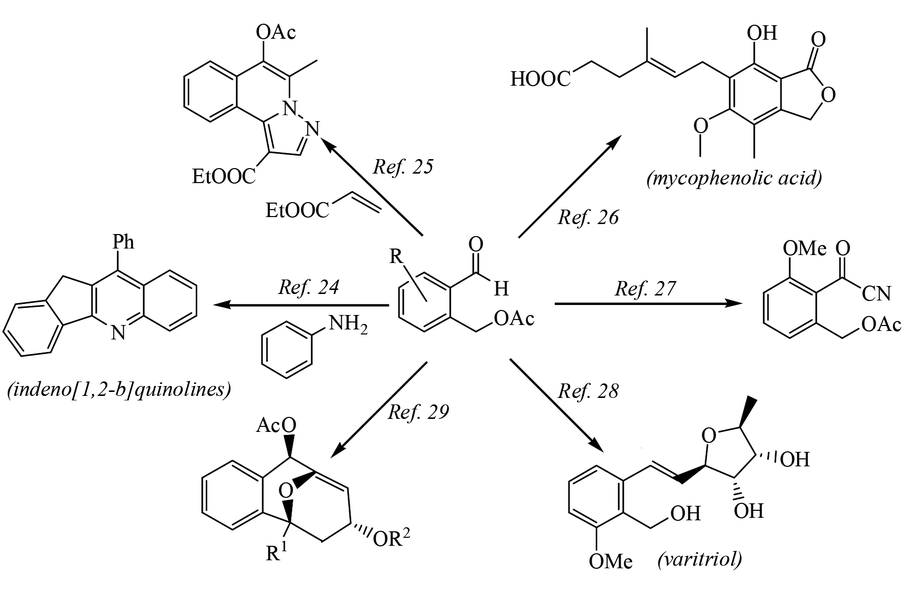 | ||
| Scheme 2 Application of the acetoxylation product. | ||
Experimental
General procedure for Pd(OAc)2 catalyzed C(sp3)–H acetoxylation reaction
A mixture of o-methylbenzaldehyde 1a (0.20 mmol), Pd(OAc)2 (4.5 mg, 10 mol%), acetohydrazide (40 mol%), MCM-48 (24 mg, 0.40 mmol), H2O (9 μL, 0.50 mmol) and K2S2O8 (108.2 mg, 0.40 mmol) in AcOH (2.0 mL) was added to a 25 mL oven dried reaction tube. The reaction mixture was heated and refluxed for 48 h at 110 °C. After cooling to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 1:10 as an eluent) to afford the desired product 2a. All the products were also confirmed by comparing the 1H NMR and 13C NMR data with authentic samples.
:5 to 1:10 as an eluent) to afford the desired product 2a. All the products were also confirmed by comparing the 1H NMR and 13C NMR data with authentic samples.
Gram-scale synthesis
In a gram-scale reaction, o-methylbenzaldehyde 1a (1 g, 8.33 mmol), Pd(OAc)2 (186.67 mg, 10 mol%), acetohydrazide (246.67 mg, 40 mol%), MCM-48 (1 g, 16.67 mmol), H2O (375.0 μL, 0.50 mmol) and K2S2O8 (4.50 g, 16.67 mmol) in AcOH (60.0 mL) were added to a 100 mL oven dried round flask. The reaction was carried out at 110 °C for 48 h in an oil bath under air conditions. After being cooled to room temperature, the reaction solution was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:10 as an eluent) to afford the desired product 2a (904.8 mg, 61% yield).
Results and discussion
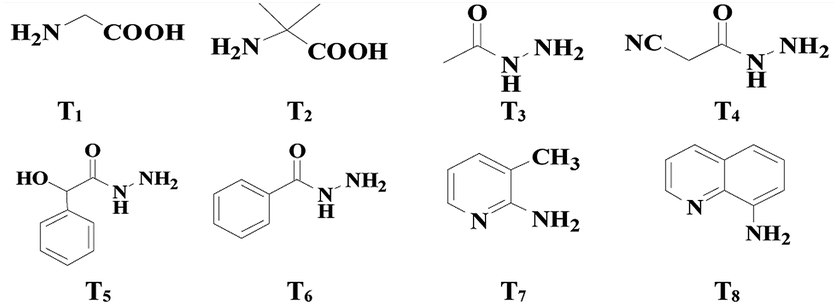
The study commenced with 2-methylbenzaldehyde (1a) as a model substrate. We initially employed the PhI(OAc)2/Pd(OAc)2 catalyzed system which has been shown to be a favourable system for C–H bond acetoxylation27 using glycine T1 as the TDG (see ESI, Table S1†). Unfortunately, we found that PhI(OAc)2 as an oxidant and acetate source was totally unreactive (see Table 1, entry 1). Other oxidants and acetate sources were screened and finally AcOH was chosen to be the external acetate source and solvent (see ESI, Table S1†). Gratifyingly, when K2S2O8 was used as the inorganic peroxide-based oxidant,28 the desired C(sp3)–H acetoxylation product, o-formylbenzyl acetate (2a) was isolated in 35% yield after refluxing 1a in AcOH (entry 2). Using AcOH as the acetate source, organic peroxides and H2O2 all can facilitate the acetoxylation reaction albeit with low yields of 2a (entries 3–5). The results indicated that the novel C(sp3)–H acetoxylation of aldehydes probably involved a radical process. Then various TDGs, including amino acids, hydrazides, aminopyridine and aminoquinoline, were tested and acetohydrazide T3 gave the best performance (entries 6–12). A control experiment indicated that the transient directing group had a vital role in this reaction (entry 13). With the aim of overcoming the lipophobicity of the inorganic oxidant, a molecular sieve (MCM-48) was added to the reaction solution and the yield of 2a increased to 61% (entry 14). Adding small amounts of water also effectively promotes the reaction because the addition of water reduces the concentration of the imine intermediate and prevents decomposition during the reaction14 (entry 15).
|
|||
|---|---|---|---|
|
|||
| Entry | TDG | Oxidant | Yieldb % |
| a Reaction conditions: 1a (0.2 mmol), Pd(OAc)2 (10 mol%), TDG (40 mol%), and AcOH (2.0 mL), 110 °C, 48 h.b Isolated yields.c MCM-48 (0.4 mmol, 24 mg) was added.d MCM-48 (0.4 mmol, 24 mg) and H2O (9 μL, 0.5 mmol) were added. | |||
| 1 | T1 | PhI(OAc)2 | NP |
| 2 | T1 | K2S2O8 | 35 |
| 3 | T1 | H2O2 | 11 |
| 4 | T1 | Cumene hydroperoxide | 21 |
| 5 | T1 | CH3CO3H | 15 |
| 6 | T2 | K2S2O8 | 22 |
| 7 | T3 | K2S2O8 | 46 |
| 8 | T4 | K2S2O8 | NP |
| 9 | T5 | K2S2O8 | 9 |
| 10 | T6 | K2S2O8 | 23 |
| 11 | T7 | K2S2O8 | NP |
| 12 | T8 | K2S2O8 | NP |
| 13 | K2S2O8 | NP | |
| 14c | T3 | K2S2O8 | 61 |
| 15d | T3 | K2S2O8 | 73 |
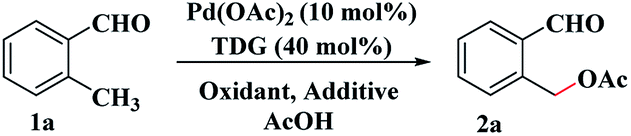
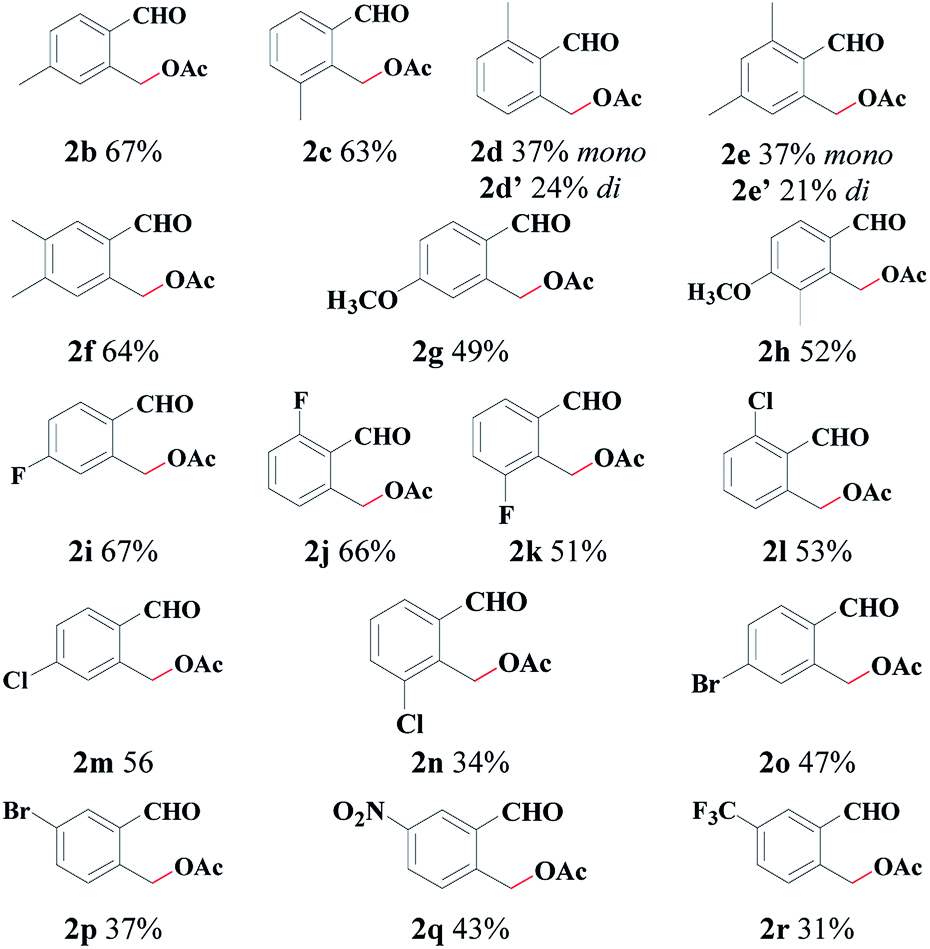
With the optimal reaction conditions in hand, we explored the scope with respect to the o-methylbenzaldehyde derivatives (see Table 2). The reaction can tolerate various functional groups, including alkyl, alkoxy, halogen (F, Br and Cl), trifluoromethyl, and nitro groups. Both electron-withdrawing (2b–2h) and electron-donating substituents (2i–2r) were tolerated in the C(sp3)–H acetoxylation reaction, and their reactivities did not exhibit a significant difference. Substrates with a 3-substituted group (2c, 2h, 2k, 2n) were well-tolerated, thus indicating a high steric tolerance of this system. When there is a methyl group at both the ortho-positions of the formyl group, mono-acetoxylation (2d, 2e) and bis-acetoxylation products (2d′, 2e′) were isolated.
|
|---|
| a Reaction conditions: 1a (0.2 mmol), Pd(OAc)2 (0.02 mmol), acetohydrazone (0.08 mmol), MCM-48 (0.4 mmol), H2O (0.5 mmol), AcOH (2.0 mL), 110 °C, 48 h. |
 |
To further establish the general utility of this transformation and shed some light on the reaction mechanism, some control experiments were conducted. To our delight, the gram-scale synthesis of the acetoxylation product 2a was carried out without significant lost in efficiency under the same reaction conditions (see Scheme 3A). As shown in Scheme 3B, propionic acid and butyric acid were all moderate reaction partners with a minor modification of the standard reaction conditions giving the corresponding product propionate 2s and butyrate 2t, respectively, and no acetoxylation product was observed, which confirmed that the acetate group of Pd(OAc)2 is not involved in the reaction process. A radical trapping experiment was carried out to judge the possibility of a radical process (Scheme 3C). When the model reaction was carried out in the presence of TEMPO [(2,2,6,6-tetramethyl-piperidin-1-yl)oxy, 0.4 mmol], a radical scavenger, only a trace (<5% yield) of the acetoxylation product was observed, but the captured benzyl radical was not detected. This result further confirmed that the reaction probably involves a radical process but it may be irrelevant to the C–H activation step. When Ac2O was employed as the solvent and acetate source instead of AcOH, the reaction could not proceed (Scheme 3D). In view of the fact that Ac2O is a more effective acetate source than AcOH under the palladium catalyst,6b,8,12b,12d we have reason to believe that AcOH may participate in the radical process while it is harder for Ac2O to generate radicals.
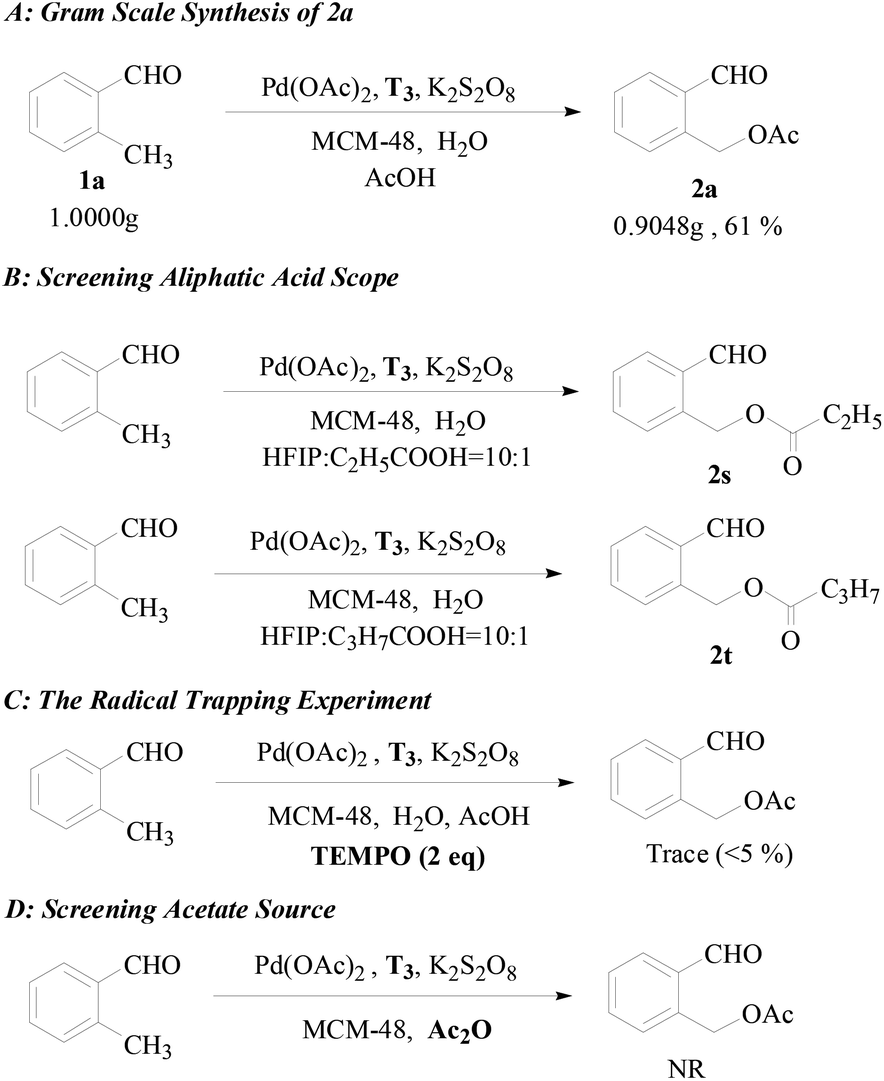 | ||
| Scheme 3 (A) Gram scale synthesis of 2a, (B) screening the aliphatic acid scope, (C) the radical trapping experiment, and (D) screening the acetate source. | ||
According to the present experimental results and the commonly accepted mechanism from the literature, a plausible reaction mechanism is proposed (see Scheme 4). First, o-methylbenzaldehyde reacts with acetohydrazide to form acetohydrazone I, which serves as a directing group in the next step. Second, bidentate coordination of the acetohydrazone moiety in I to Pd(OAc)2 occurs to form the five-membered palladium-ring II and then generate intermediate III via C–H activation. Then, intermediate III reacts with an acetate radical which resulted from the reaction of AcOH with K2S2O8, leading to an PdIV intermediate IV.29 The final step consists of the reductive elimination of intermediate IV to release compound V and PdII to complete the catalytic cycle. Compound V is then hydrolyzed to the desired product 2 and acetohydrazide.
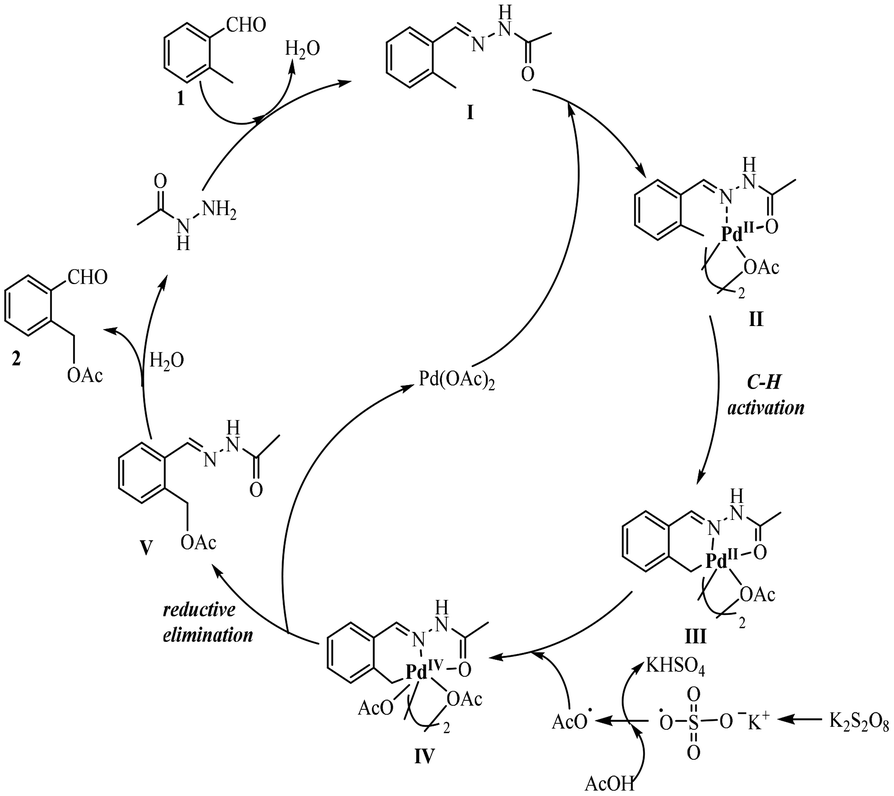 | ||
| Scheme 4 A plausible catalytic cycle. | ||
Conclusions
In summary, we solved the puzzle of oxidizable group-aldehyde directed selective acetoxylation of C(sp3)–H bonds for the first time. Using Pd(OAc)2 as a catalyst and acetohydrazide as both the TDG and protecting group, a broad scope of o-formylbenzyl acetates were synthesized under neutral and mild conditions. The methodology has good generality and tolerates various functional groups, including alkyl, alkoxy, halogen (F, Br and Cl), trifluoromethyl, and nitro groups. A mechanism study indicated that the reaction may involve a radical process. Further synthetic applications and additional mechanistic studies are currently under investigation in our laboratory and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Foundation of China (21861030), the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-17-A22) and Research Innovation Fund of Inner Mongolia Normal University Graduate (CXJJS18075).Notes and references
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (b) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed; (c) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (d) F. Bellina and R. Rossi, Chem. Rev., 2009, 110, 1082 CrossRef PubMed; (e) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (f) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- For examples, see: (a) R. Giri, J. Liang, J. G. Lei, J. J. Li, D. H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J. Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS PubMed; (b) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (c) D. H. Wang, X. S. Hao, D. F. Wu and J. Q. Yu, Org. Lett., 2006, 8, 3387 CrossRef CAS PubMed.
- J. A. May, H. Ratan, J. R. Glenn, W. Losche, P. Spangenberg and S. Heptinstall, Platelets, 1998, 9, 227 CrossRef CAS PubMed.
- E. P. Fuerst, C. J. Arntzen, K. Pfister and D. Penner, Weed Sci., 1986, 34, 344 CrossRef CAS.
- R. Giri, X. Chen and J. Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS PubMed.
- (a) H. Jiang, H. Chen, A. Wang and X. Liu, Chem. Commun., 2010, 46, 7259–7261 RSC; (b) J. Zhang, E. Khaskin, N. P. Anderson, P. Y. Zavalij and A. N. Vedernikov, Chem. Commun., 2008, 3625–3627 RSC.
- D. H. Wang, X. S. Hao, D. F. Wu and J. Q. Yu, Org. Lett., 2006, 8, 3387–3390 CrossRef CAS PubMed.
- Y. Zheng, W. Song, Y. Zhu, B. Wei and L. Xuan, J. Org. Chem., 2018, 83, 2448–2454 CrossRef CAS PubMed.
- K. Chen, D. Wang, Z. W. Li, Z. Liu, F. Pan, Y. F. Zhang and Z. J. Shi, Org. Chem. Front., 2017, 4, 2097–2101 RSC.
- D. N. Garad and S. B. Mhaske, J. Org. Chem., 2017, 82, 10470–10478 CrossRef CAS PubMed.
- (a) Z. Wang, Y. Kuninobu and M. Kanai, Org. Lett., 2014, 16, 4790–4793 CrossRef CAS PubMed; (b) L. Ju, J. Yao, Z. Wu, Z. Liu and Y. Zhang, J. Org. Chem., 2013, 78, 10821–10831 CrossRef CAS PubMed; (c) M. Wang, Y. Yang, Z. Fan, Z. Cheng, W. Zhu and A. Zhang, Chem. Commun., 2015, 51, 3219–3222 RSC; (d) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724 CrossRef CAS PubMed.
- F. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285–11288 CrossRef CAS PubMed.
- F. L. Zhang, K. Hong, T. J. Li, H. Park and J. Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed.
- F. Ma, M. Lei and L. Hu, Org. Lett., 2016, 18, 2708–2711 CrossRef CAS PubMed.
- B. B. Gou, H. F. Liu, J. Chen and L. Zhou, Org. Lett., 2019, 21, 7084–7088 CrossRef CAS PubMed.
- S. St John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC.
- K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS PubMed.
- X. L. Zhang, G. F. Pan, X. Q. Zhu, R. L. Guo, Y. R. Gao and Y. Q. Wang, Org. Lett., 2019, 21, 2731–2735 CrossRef CAS PubMed.
- C. Dong, L. Wu, J. Yao and K. Wei, Org. Lett., 2019, 21, 2085–2089 CrossRef CAS PubMed.
- M. Chen, N. Sun and Y. Liu, Org. Lett., 2013, 15, 5574–5577 CrossRef CAS PubMed.
- D. B. Huple, C. H. Chen, A. Das and R. S. Liu, Adv. Synth. Catal., 2011, 353, 1877–1882 CrossRef CAS.
- (a) A. Covarrubias-Zúnniga, Tetrahedron, 2003, 59, 1989–1994 CrossRef; (b) A. Covarrubias-Zúnniga and A. Gonzalez-Lucas, Tetrahedron Lett., 1998, 39, 2881–2882 CrossRef; (c) M. V. Paradkar, S. A. Kulkarni, A. R. Joseph and A. A. Ranade, J. Chem. Res., 2000, 364–366 CrossRef CAS; (d) A. Covarrubias-Zúnniga, J. Diaz-Dominguez and J. Olguín-Uribe, Synth. Commun., 2001, 31, 1373–1381 CrossRef.
- (a) K. C. Nicolaou, T. Montagnon, G. Vassilikogiannakis and C. J. N. Mathison, J. Am. Chem. Soc., 2005, 127, 8872–8888 CrossRef CAS PubMed; (b) K. C. Nicolaou, T. Montagnon and G. Vassilikogiannakis, Chem. Commun., 2002, 2478–2479 RSC.
- G. Sudhakar and J. Raghavaiah, J. Org. Chem., 2013, 78, 8840–8846 CrossRef CAS PubMed.
- T. M. Teng, A. Das, D. B. Huple and R. S. Liu, J. Am. Chem. Soc., 2010, 132, 12565–12567 CrossRef CAS PubMed.
- (a) Z. Ren, J. E. Schulz and G. Dong, Org. Lett., 2015, 17, 2696–2699 CrossRef CAS PubMed; (b) H. Zhang, R. B. Hu, X. Y. Zhang, S. X. Lia and S. D. Yang, Chem. Commun., 2014, 50, 4686–4689 RSC; (c) C. S. Buettner, D. Willcox, B. G. N. Chappell and M. J. Gaunt, Chem. Sci., 2019, 10, 83–89 RSC; (d) A. K. Cook, M. H. Emmert and M. S. Sanford, Org. Lett., 2013, 15, 5428–5431 CrossRef CAS PubMed; (e) F. R. Gou, X. C. Wang, P. F. Huo, H. P. Bi, Z. H. Guan and Y. M. Liang, Org. Lett., 2009, 11, 5726–5729 CrossRef CAS PubMed.
- L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141–1144 CrossRef CAS PubMed.
- G. W. Wang, T. T. Yuan and X. L. Wu, J. Org. Chem., 2008, 73, 4717–4720 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for the products, 1H NMR and 13C NMR spectra of the products. See DOI: 10.1039/d0ra01927e |
| This journal is © The Royal Society of Chemistry 2020 |