
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-initiated rupture of azobenzene micelles to enable the spectroscopic analysis of antimicrobial peptide dynamics†
Matthew G.
Roberson
,
Julia M.
Duncan
,
Keveen J.
Flieth
,
Laina M.
Geary
 * and
Matthew J.
Tucker
*
* and
Matthew J.
Tucker
*
Department of Chemistry, University of Nevada, Reno, Reno, Nevada 89501, USA. E-mail: lgeary@unr.edu; mtucker@unr.edu
First published on 4th June 2020
Abstract
Antimicrobial peptides (AMPs) show promise for the treatment of bacterial infections, but many have undesired hemolytic activities. The AMP MP1 not only has broad spectrum bactericidal activity, but has been shown to have antitumor activity. The interaction between AMPs and cellular membranes gives rise to a peptide's cell-specificity and activity. However, direct analysis of the biophysical interactions between peptides and membrane is complex, in part due to the nature of membrane environments as well as structural changes in the peptide that occurs upon binding to the membrane. In order to investigate the interplay between cell selectivity, activity, and secondary structural changes involved in antimicrobial peptide activity, we sought to implement photolizable membrane mimics to assess the types of information available from infrared spectroscopic measurements that follow from photoinitiated peptide dynamics. Azo-surfactants (APEG) form micelles containing a photolizable azobenzene core, which upon irradiation can induce membrane deformation resulting in breakdown of micelles. Spectroscopic analysis of membrane deformation may provide insights into the physical behavior associated with unfolding and dissociation of antimicrobial peptides within a membrane environment. Herein, we synthesized and characterized two new azo-surfactants, APEGTMG and APEGNEt2MeI. Furthermore, we demonstrate the viability of azosurfactants as membrane mimics by examining both the membrane binding and dissociation induced secondary structural changes of the antimicrobial peptide, MP1.
Introduction
Antimicrobial peptides (AMPs) are short 15–40 amino acid peptides that are capable of selectively binding and disrupting cell membranes.1,2 AMPs are typically cationic and tend to be intrinsically disordered in an aqueous environment.3 However, peptide secondary structure changes occur upon interaction with a membrane that prompts peptide aggregation and formation of stabilized pores within a cell membrane, ultimately resulting in cell lysis.4–8 Knowledge of the structural evolution of membrane peptides and the dynamic motions of their side chains is pivotal to combating several diseases, the development of therapeutic drugs, and discovering biomaterial mimics. Several spectroscopic methods have provided information about rates at which populations interchange leading to the development of kinetic schemes, but usually with little or no structural resolution.9–11 The most common approach to analyzing AMP binding and folding is to utilize membrane mimics, such as micelles and vesicles. Commonly used monolayer mimics include sodium dodecylsulfate (SDS) and dodecylphosphocholine (DPC), while many bilayers have been constructed using glycerophospholipids.12–25 Unfortunately, examining the dynamics associated with AMP unbinding or unfolding in the presence of these traditional mimics is not trivial due to their ability to form stable micelles over broad high temperature ranges26 and ability to stabilize peptide secondary structure.27In contrast, photo initiation of conformational changes in proteins can assist in addressing many intriguing questions regarding structural dynamics. Current methods utilize covalent constraints that narrow the structure distribution. Irradiation releases these constraints, thereby propagating the molecule on a path to different equilibria distributions. Structural evolution, even when ultrafast, can be followed by infrared probe spectroscopy or two-dimensional infrared spectroscopy. In this way, fast phototriggering can help uncover early kinetics events in protein dynamics by providing a means to explore the free energy landscape of folding and misfolding, as well as elucidate intermediates along the folding pathway.
Several phototriggers have been developed and incorporated into peptides for this purpose,28–33 including disulfides, S,S-tetrazines, and azobenzenes. Disulfide bonds in peptides can be used as phototriggers with the aid of deep UV light, photolyzing in a few ps.28,29,32,34 However, the UV excitation required to dissociate the S–S bond also excites the peptide backbone. Furthermore, disulfide linkers can also dissociate homolytically into reactive species capable of undergoing geminate recombination, as well as side reactions with protein sidechains. The well-studied phototrigger, S,S-tetrazine, dissociates into nonreactive thiocyanate derivatives and N2 gas upon irradiation. Unfortunately, use of the tetrazine trigger often requires the addition of cysteine to proteins or peptides.35,36 Azobenzene undergoes fast and reversible photoisomerization upon visible irradiation.37–39 However, when azobenzene is used within peptides as a trigger, it is covalently linked to two residues within the peptide throughout the whole folding/unfolding process, limiting the accessible conformational space.40,41 This limitation has been observed in azobenzene mediated phototriggered activation of neuron channels and DNA binding.42,43
Alternatively, incorporation of a photoswitch into a membrane mimic, rather than into the native AMPs, would remove this limitation. Irradiation of the membrane mimic induces changes in the surroundings (e.g., solvation, dielectric constant, size and shape of the micelle, etc.) that, in turn, perturb the AMP system. Azobenzene-containing polyethylene glycol (APEG) surfactants and lipids have been previously synthesized and structurally characterized.44–51 Irradiation of APEG surfactants with UV-Vis light induces trans–cis isomerization resulting in a geometrical change from linear to bent. Furthermore, there exists a notable compression of the azobenzene core as shown by the decrease in distance from approximately 9.9 Å to 5.5 Å of the para-substituted carbon atoms, as well as an increase in dipole moment from 0.5 D to 3.1 D upon isomerization to the cis isomer.52,53 Therefore, packing of the monomers is expected to be less efficient for the cis isomer, and the critical micelle concentration (CMC) for the cis isomer will exceed that of the trans isomer.47,50 Previously synthesized APEG surfactants were found to have low solubility (μM).46,48,54 These low solubilities create difficulty when working at relevant peptide to surfactant concentrations.
The dynamics associated with protein folding and unfolding are expected to range over several orders of magnitudes (ps to μs).55 In order to monitor the ultrafast dynamics associated with protein folding, a viable photoswitch must be capable of changing, either via dissociation or isomerization, more rapidly than the associated structural changes being analyzed. The azobenzene core of APEG surfactants readily undergo isomerization on a sub-picosecond timescale, rendering ultrafast dynamics analyses feasible.56,57 Furthermore, Hamm et al. were able to follow the energy transfer induced by azobenzene isomerization along the peptide backbone. Upon isomerization, an instantaneous jump in temperature was recorded for the nearest neighboring probe. Aside from this, the azobenzene isomerization and peptide secondary structural changes were observed to be completely decoupled.58 Since the thermal effects of isomerizing an azobenzene covalently bound within a peptide were minimal, isomerization of APEG is expected to be decoupled from any peptide structural changes.
Thus, our work here describes the synthesis and characterization of APEG scaffolds with improved solubility (mM), with more substantial differences in CMC values between cis and trans, tunability for specificity in peptides, and utility for 2D IR transient measurements. Finally, the overall applicability of utilizing these surfactant systems as potential membrane mimics to study protein folding dynamics is demonstrated.
Results and discussion
Synthesis of azo-surfactants
The known APEG scaffold54 was modified with two different polar head groups (Scheme 1). APEG was first activated towards nucleophilic functionalization by conversion to the tosylate, which was then reacted with tetramethylguanidine (TMG) to give APEGTMG in 75% yield over two steps. APEG was also converted to the cationic ammonium derivative in three steps. Reaction of the tosylated APEG with dimethylamine gave a neutral trisubstituted amine that was quaternized by reaction with iodomethane to give cationic APEGNEt2MeI in 64% overall yield. | ||
| Scheme 1 Synthesis of APEG derivatives APEGTMG and APEGNEt2MeI. (a) TsCl, NEt3, DCM, 0 °C – rt, 18 h, 83% yield; (b) TMG, K2CO3, THF, reflux, 48 h, 91% yield; (c) HNEt2, K2CO3, THF, reflex, 48 h, 91% yield; (d) MeI, THF, reflux, 24 h, 85% yield. | ||
Characterization of APEGNEt2MeI and APEGTMG azobenzene photoswitches via UV-Vis spectroscopy
To validate the utility of the azobenzene core of both APEG surfactants for photoswitching, UV-Vis spectra were obtained for each surfactant prior to and post irradiation with 365 nm light (Fig. 1). Prior to irradiation (blue spectra of Fig. 1), both surfactants exhibited spectral characteristics associated with the trans isomer of azobenzene. Specifically, each spectrum shows a broad peak centered at 350 nm, attributed to a π → π* transition, and a second peak observed at 460 nm characteristic of a n → π* transition.59–61 Isomerization to the cis isomer of the APEG derivatives was induced by irradiation with 365 nm (red spectra of Fig. 1). Upon trans–cis isomerization, a decrease in the broad 350 nm peak occurs, accompanied by a blue shift of the maximum to approximately 330 nm for the π → π* transition. Furthermore, the emergence of the 440 nm peak associated with the n → π* transition was observed for the cis isomer.53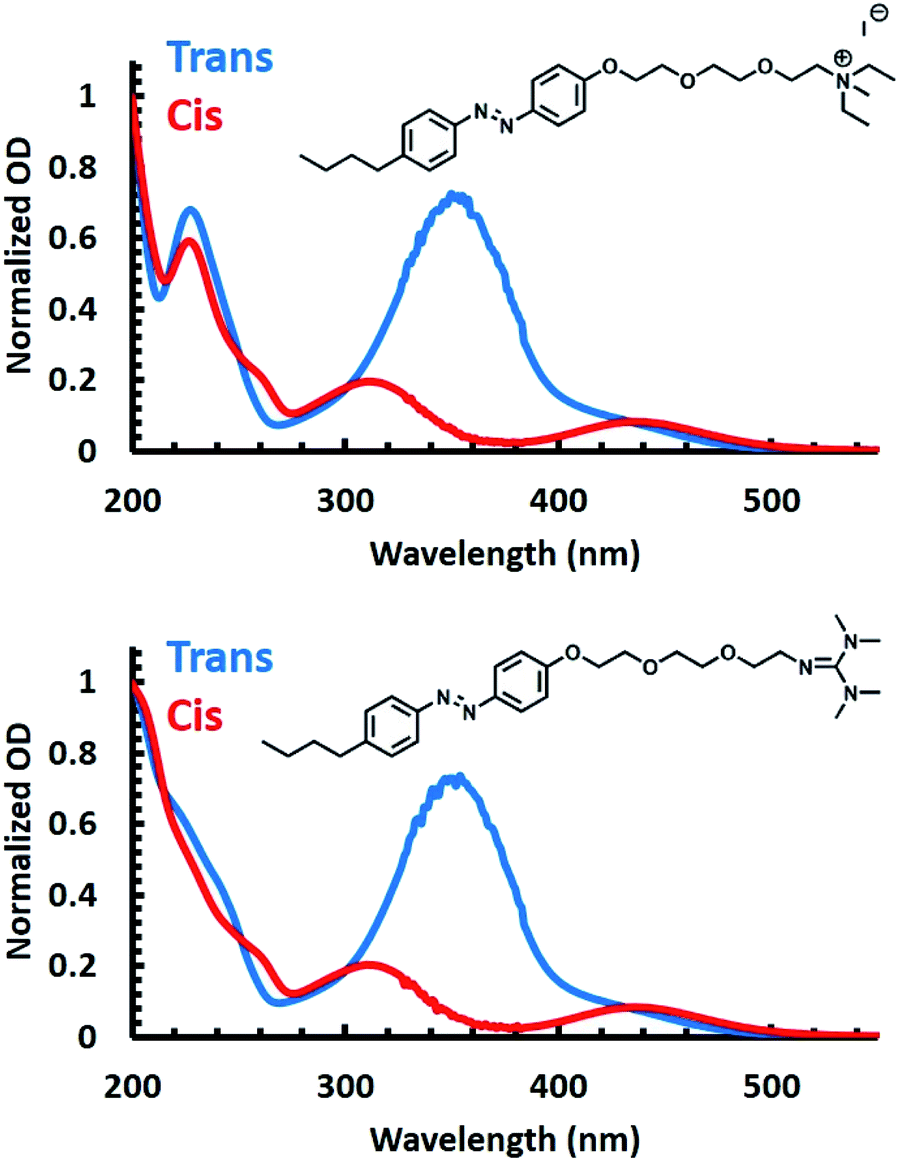 | ||
| Fig. 1 UV-Vis spectra of APEGNEt2MeI (top) and APEGTMG (bottom) demonstrating the electronic transitions associated with each isomer, trans (blue) and cis (red), obtained prior to irradiation with 365 nm and upon irradiating samples with 365 nm, respectively. | ||
To quantify the total surfactant concentration, [S]T, via UV-Vis, extinction coefficients were determined by utilizing an isobestic point at 226 nm (see ESI for details†). Using the isobestic point, concentrations of the surfactant can be measured despite any isomeric mixtures present. Thus, using Beer's law, the extinction coefficients of 16.7 ± 0.2 and 16.5 ± 0.4 mM−1 cm−1 were obtained for APEGNEt2MeI and APEGTMG, respectively, at 226 nm.
To accurately determine the extinction coefficients of the trans isomer, UV-Vis spectra were collected prior to any irradiation with the assumption that [S]T was equal to the concentration of the trans isomers. For the cis isomer, UV-Vis spectra were collected post irradiation with 365 nm light for 15 minutes, and we assumed that [S]T equaled the concentration of the cis isomers. Under these assumptions, extinction coefficients were found to be 17.9 ± 0.3 and 1.63 ± 0.04 mM−1 cm−1 at 351 nm for trans and cis isomer of APEGNEt2MeI, respectively. Extinction coefficients of APEGTMG were found to be 19.0 ± 1.8 and 2.31 ± 0.10 mM−1 cm−1 at 348 nm for trans and cis isomers, respectively (see Table 1). With those data, we determined the reversibility, or conversion efficiency, of isomerization of the APEG surfactants under multiple irradiation cycles (see the ESI†). An initial decrease in the conversion efficiency was observed where approximately 70% of the original trans isomer population was recovered for both surfactants. However, complete conversion was observed following consecutive irradiation cycles (shown in Fig. S1 in ESI†). The cis isomer, however, exhibited no change in concentration from the approximately quantitative population upon further irradiation with 365 nm light.
| APEGTMG | APEGNEt2MeI | |||
|---|---|---|---|---|
| trans | cis | trans | cis | |
| ε 226 | 16.5 ± 0.4 | 16.7 ± 0.2 | ||
| ε 348 | 19.0 ± 1.8 | 2.31 ± 0.10 | ||
| ε 351 | 17.9 ± 0.3 | 1.63 ± 0.04 | ||
| CMC | 1.45 ± 0.08 | 1.83 ± 0.02 | 0.52 ± 0.02 | 2.69 ± 0.19 |
CMC determination of APEG micelles via NMR
In order to utilize the azo-surfactants as membrane mimics, the CMC of each isomer of both surfactants must be measured (Fig. 2).62–64 Ideally, the difference between the CMCs of each isomer for a given surfactant should be substantial. In this way, by photoswitching the azobenzene from the trans to the cis isomer, the concentration of micelles would significantly reduce due to the shifting equilibrium.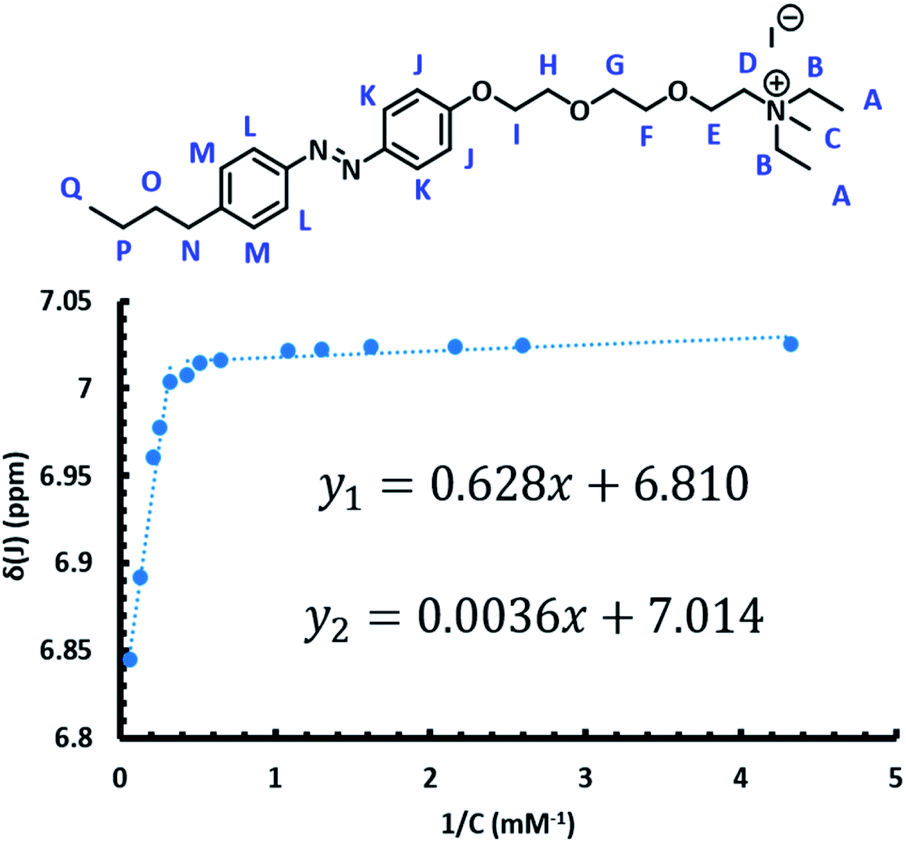 | ||
| Fig. 2 CMCs of the trans and cis isomers of the APEG surfactants were calculated by monitoring the changes in chemical shifts (depicted here as the chemical shifts of the proton labelled as “J”) as a function of surfactant concentration using 1H NMR. | ||
Various methods such as dynamic light scattering, fluorescence dye quenching, and differential scanning calorimetry have been utilized previously to determine CMCs of various surfactants.65–68 Herein, 1H NMR was utilized in order to determine the CMC of the trans and cis isomers for both APEG surfactants (Fig. 2).69
The CMC of each isomer of APEGNEt2MeI and APEGTMG was determined by monitoring changes in the chemical shifts at different concentrations in D2O, ranging from 0.18–18.6 mM (ESI†). To determine the CMCs of the trans isomers, samples were irradiated with visible light for one hour immediately prior to spectra collection. The horizontal intercept of two linear functions was then found for each proton. The CMCs of the trans isomers were determined to be 1.45 ± 0.08 mM and 0.52 ± 0.02 mM for APEGTMG and APEGNEt2MeI, respectively. Similarly, the cis isomers were obtained by irradiating samples with 365 nm light for 30–60 minutes immediately prior to spectra collection. The horizontal intercept of a linear function and a cubic function were estimated via Newton's method for each sample. The CMCs of the cis isomers were determined to be 1.83 ± 0.02 mM and 2.69 ± 0.19 mM for APEGTMG and APEGNEt2MeI, respectively (see Table 1).
Amide I and amide II IR spectral properties of APEG micelles
Due to the inherently high extinction coefficients of APEG, circular dichroism was not a highly capable method of peptide secondary structural analysis for these systems. Furthermore, the high absorbance of the APEG surfactants in the UV region does not permit fluorescence binding studies of peptides using natural chromophores such as tryptophan, tyrosine, or phenylalanine. Thus, infrared spectroscopy (IR) was utilized to follow peptide secondary structural changes associated with interactions of the APEG micelles.Numerous linear and nonlinear infrared (IR) spectroscopic methods have been utilized to follow peptide structural changes.35,70–88 Of specific interest, the amide I and II region (1600–1700 cm−1 and 1540–1600 cm−1, respectively) were analyzed for each isomer of both surfactants, since these regions are mainly attributed to hydration of the peptide backbone and secondary structure of AMPs.89–101 In order for the APEG surfactants to be viable candidates as membrane mimics utilized for FTIR, the vibrational modes of the surfactants were assessed.
IR peak analyses were performed for the monomeric units of each isomer of APEGTMG and APEGNEt2MeIvia standard geometrical optimization and frequency calculation in the gas phase via DFT calculations (see ESI†). The frequency calculations revealed that the experimentally observed FTIR spectra for both APEG surfactants share a common set of peaks in the amide II region (maxima at ∼1585 and 1603 cm−1), which arise primarily from the ring modes and C–O stretches associated with the glycol chains of the APEG scaffold.
Analysis of the amide I region revealed no peaks for APEGNEt2MeI. APEGTMG, however, exhibited a somewhat broad peak centered at 1675 cm−1 (Fig. S3†). This peak is attributed to the tetramethylguanidine head group of APEGTMG, and was found to be essentially invariant between isomers. Although most of the infrared transition are negligible at concentrations of interests, a minimal peak contribution in the amide I was noted for APEGTMG at the desired concentration of APEGTMG (∼1.74 mM) between the CMCs of the different isomers. Since all spectra of interest are difference spectra, the residual signals coming from the surfactants within the amide I region are subtracted out, with the exception of the green difference spectrum of Fig. 3.
 | ||
| Fig. 3 Linear IR spectra of MP1/D2O (blue solid), MP1 with trans APEGTMG micelles (red dashed). The green difference spectrum is associated with changes in MP1 structure upon incorporation of trans APEGTMG micelles. | ||
FTIR analyses of MP1 structural changes induced by APEGTMG micelles
To assess the applicability of the new photolizable APEG surfactants for peptide structural analysis, infrared studies of the amide I region were invoked to monitor peptide secondary structural changes upon membrane association and dissociation. It should be noted that no significant changes in pH were measured in the sample upon irradiation, suggesting the observed structural changes are not a result of any pH differences (see ESI for more details†). Specifically, we examined structural changes associated with the interactions of APEGTMG micelles and the antimicrobial peptide MP1 (IDWKKLLDAAKQIL-NH2).102 As shown below, APEGTMG was chosen due to its ability to readily bind the synthesized MP1 peptide. MP1 exhibited no observable binding affinity for APEGNEt2MeI micelles. This lack of binding affinity is likely attributed to repulsive interactions between the cationic AMP and the cationic headgroup of APEGNEt2MeI. Upon removal of these repulsive interactions, other factors, such as hydrophobicity, can play a more significant role in the peptide/micelles interplay for the APEGTMG micelles. Thus, the observed higher binding affinity of MP1 to APEGTMG is most likely a result of the limited/neutral charge of the surfactant headgroup allowing the forces associated with the hydrophobic contacts to become more pronounced. However, we plan to utilize APEGNEt2MeI to analyze other membrane peptides that have more anionic character in future studies.First, the FTIR spectra (Fig. 3 top) in the presence and absence (in D2O) of trans micelles were measured to observe the folding induced by MP1 (1.9 mM) associating with APEGTMG micelles (∼1.74 mM). A difference spectrum (Fig. 3 – green) was constructed by subtracting the MP1/D2O spectrum (blue) from the IR spectrum of MP1 with trans APEGTMG micelles (red).
In this difference spectrum, the presence of structural change is observable from the transition with maximum around 1651 cm−1. The two other maxima present (1601 and 1673 cm−1) correspond to spectral features due to the presence of trans APEGTMG. The maximum at 1651 cm−1 arises from the induced structural changes of MP1 by the trans micelles, which are indicative of a gain in α-helical content.82,89,92,103,104 By closer examining the FTIR spectra used to construct the green difference spectrum, the amide I bandwidth of MP1/D2O (Fig. 3, blue curve) is broader than the red spectrum obtained for MP1 in the presence of trans APEGTMG micelles. The phenomenon of band narrowing further supports that the micelles induce a population gain in helical content.92,103,104 This structural change has also been shown by circular dichroism for MP1 in the presence of various other micelles.105
It should be noted that the maximum of the green difference spectrum at 1651 cm−1 falls below the x-axis. This spectral feature is likely due to the hydrophobic environment resulting in the unsolvated helical content. A blue shift from 1645 to 1651 cm−1 in the transition maximum is observed in the FTIR spectra upon association with the micelles. This shift is accompanied by a slight decrease in OD. These two phenomena are most likely associated with the extent of hydration of the MP1 amide I backbone. Specifically, loss of hydration of the backbone is known to increase the force constant of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch.92 This increase of force constant results in a blue shift of the peak maximum and a decrease in the OD due to stronger excitonic coupling.92,106–108
O stretch.92 This increase of force constant results in a blue shift of the peak maximum and a decrease in the OD due to stronger excitonic coupling.92,106–108
As mentioned above, the maxima of the green difference spectrum of Fig. 3 suggest an increase in helical content due to the presence of APEGTMG micelles. Examination of the minima reveal structural content lost due to this change. Peaks with minima are observed in the lower frequency amide I region (1616–1640 cm−1), at 1661 cm−1, and from 1687–1700 cm−1 as a result of the loss of intrinsically disordered residual backbone structure of MP1 in solution. Using standard peak analysis,71,90,92 these minima correspond predominately to a loss in β-content and random coil in MP1 upon interaction with the APEGTMG micelles, which likely occur in an AMP of this length possessing a decent degree of hydrophobicity.109,110
The surfactant concentration was close to the CMC of the trans isomer (∼1.74 mM) so that upon photoswitching the micelles would likely break up since the cis CMC is much higher. After observing that APEGTMG invoked secondary structural changes within MP1, specifically a gain in α-helical content with a subsequent loss in β-content and random coil, the sample cell was irradiated with 365 nm light prompting micelle dissociation. This result revealed a convoluted spectrum (see the ESI†) but still suggested the potential loss of α-helical structure.
To further analyze peptide secondary structural changes induced by both association and dissociation with APEGTMG micelles, a slightly different experimental approach was adopted where the APEGTMG (1.74 mM) solution was prepared in D2O. This solution, which we used as our blank, was then used to dissolve the MP1 (4.8 mM). Upon initial sample preparation, the FTIR spectrum of MP1 in APEGTMG was obtained (Fig. 4 black dashed inset). Following this initial data collection, the linear IR spectrum was collected after 24 hours, without any sample irradiation (Fig. 4 red solid inset). This red spectrum accounts for any kinetic barriers related to binding, i.e. full equilibrium of the bound population to micelles. The pink difference spectrum of Fig. 4 (top) was constructed by subtracting the initial linear spectrum (Fig. 4 black dashed inset) from the final spectrum obtained prior to sample irradiation (Fig. 4 red solid inset). This difference spectrum is quite similar to the green curve of Fig. 3, which depicts the secondary structural changes of MP1 induced by the trans APEGTMG micelles.
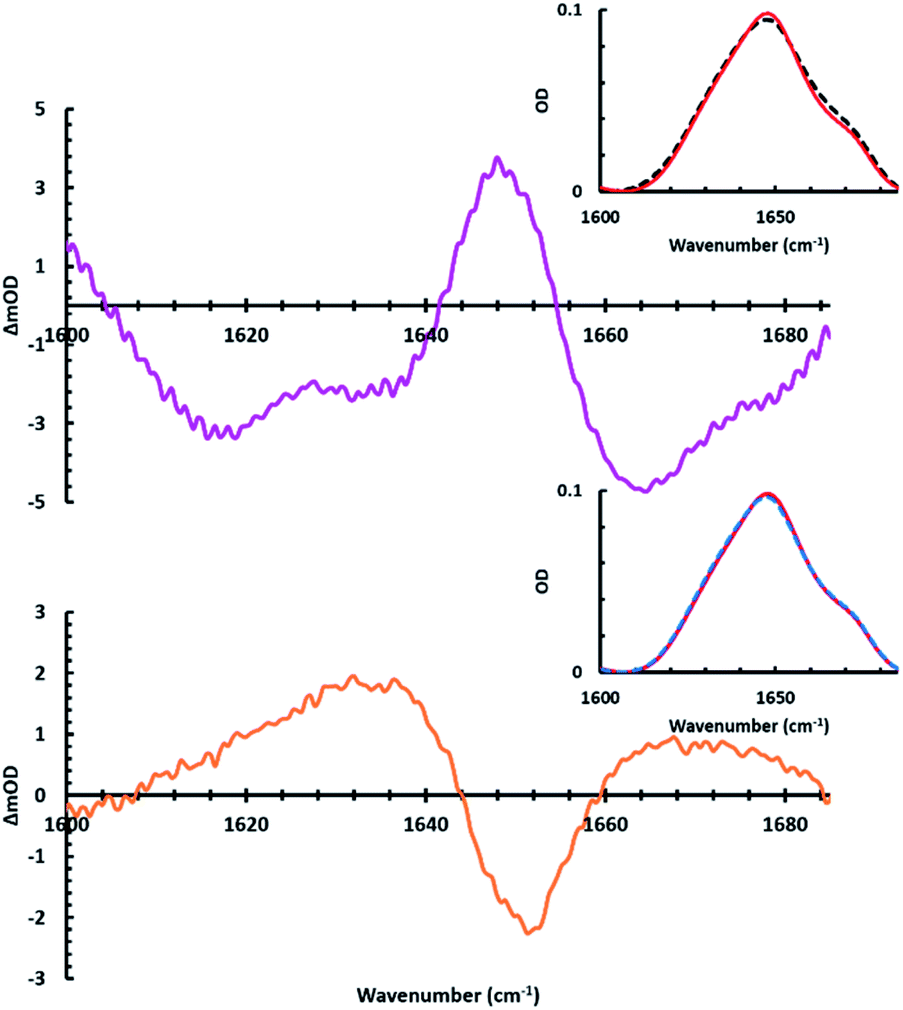 | ||
| Fig. 4 Linear IR spectra (insets) of MP1 (4.8 mM) in APEGTMG (1.74 mM) with trans APEGTMG micelles after initial preparation (t = 0 h) (black dashed inset), trans micelles after 24 h with no irradiation (red solid inset), and cis micelles after photoconversion (blue dashed inset) were collected. The pink difference spectrum (top) shows an increase of the bound peptide population (1651 cm−1) after 24 h equilibration period. The orange difference spectrum (bottom) shows the loss of the bound peptide population and return to the original state upon photoconversion (365 nm irradiation), which causes the micelles to rupture. | ||
As expected, the spectral changes observed for the pink spectrum agree with secondary structural changes of MP1 transitioning from an aqueous environment (D2O) to the presence of APEGTMG micelles. As described above, a loss of disordered structure and β-content upon micelle association (1618, 1636, and 1667 cm−1) are observed, followed by an increase in helical content (1651 cm−1). Furthermore, by using this approach, the pink difference spectrum was observed without any contributions of the peaks associated with the vibrational modes of the APEGTMG, unlike Fig. 3. This reduced influence from APEGTMG reveals a single larger peak maxima due to the increase in α-helical structure upon binding to the micelles, suggesting a larger population of MP1 associates with the APEGTMG micelles, given enough time to equilibrate.
After 24 hours, no further changes were observed in IR spectra of MP1 containing the trans isomer of APEGTMG. Thus, the sample was irradiated with 365 nm light inducing trans–cis isomerization, rupturing the micelles (blue dashed inset of Fig. 4). The orange difference spectrum of Fig. 4 was then constructed by subtracting the linear spectra (red and blue dashed insets of Fig. 4) of the sample before and after irradiation. Analysis of the orange spectrum suggests that the structural changes of MP1 associated with micelle rupture are a disappearance of the helical structure with a recovery of the disordered structure.
Peptide binding studies utilizing APEGTMG micelles
In order to verify APEG's ability to bind peptides, a fluorescently labelled variant of MP1 was synthesized, IDWKKLLDAAKQIL-NH2, where a cysteine was added to the N-terminus in order to label the peptide (TR-CMP1) with sulforhodamine 101 C2 maleimide, Texas Red.Prior to evaluating TR-CMP1's ability to bind APEGTMG micelles, fluorescence and UV-Vis experiments were performed in order to assess the viability of using sulforhodamine as an environmentally sensitive dye by monitoring bonding from an aqueous, unbound species, to a hydrophobic environment, bound species. In order for sulforhodamine to be an environmentally sensitive probe, observable spectral differences in either or both the absorbance and emission spectra should have been present depending on exposure to different solvents. These experiments were performed by varying the solvent, as well as the dielectric, in which sulforhodamine was dissolved. It was found that as the dielectric constant decreased, a blue shift and quenching occurred in the emission spectrum (see ESI†).
After confirming that sulforhodamine was environmentally sensitive, emission spectra of TR-CMP1 were obtained by excitation with 585 nm light in the absence and presence of APEGTMG micelles (Fig. 5). Incorporation of APEGTMG, resulted in a drastic decrease in the quantum yield of sulforhodamine accompanied with a blue shift in the emission maximum. The observation of the bound state of the peptide through the fluorescence measurements indicates that the micelle remains stable upon peptide binding. Otherwise, the observed fluorescence spectrum would resemble what is expected for bulk water (see ESI for more details on dye sensitivity†). These spectral changes are consistent with being exposed to a more hydrophobic environment as seen in the solvent analysis previously mentioned (see ESI†) and therefore support that MP1 was indeed bound to the APEGTMG micelles.
 | ||
| Fig. 5 Fluorescence spectra (top) and normalized fluorescence spectra (bottom) of TR-CMP1 (47 nM) were collected in the presence (red) and absence (blue) of TMG micelles (10 mM), to assess the ability of peptide binding towards APEGTMG micelles. Normalized fluorescence spectra were normalized to the maximum emission of each individual spectrum. | ||
Conclusions
The interplay between cell selectivity, activity, and peptide secondary structural changes associated with the lytic activity of many antimicrobial peptides has been a topic of intense debate. Herein, attempts to understand these phenomena have been carried out using various membrane mimics possessing photolizable constraints. Azobenzene has been used previously to “lock” peptides into a secondary structure that released via irradiation. Rather than perturb the peptide itself, azo-surfactants or APEG were utilized, which contain a photolizable azobenzene core which upon irradiation cause micellular breakdown. This breakdown was accompanied with spectroscopic analysis, suggesting great promise in the ability to monitor unfolding and disassociation of antimicrobial peptides from membranes. Herein, we have introduced and characterized two new water soluble azo-surfactants, APEGTMG and APEGNEt2MeI. Furthermore, we demonstrated the ability of antimicrobial peptides to selectively bind to these surfactants. Using these azo-surfactant, we were able to the follow MP1's structural changes induced upon membrane association and membrane dissociation. Initially, the MP1 was intrinsically disordered and had the ability to form aggregates. Upon micelle association, a gain in helical content with an accompanying loss in disorder and aggregate formation was observed. Finally, photo-initiated rupture of the micelles led to a major loss in helical population while the majority of the original disordered population as well as aggregates were recovered. These observations indicate that MP1's is involved in a broad range of conformational changes during the AMP pathway. Moreover, the interplay between sidechains and lipid components are both necessary and involved in the structural evolution of MP1. Future work will utilize nonlinear IR techniques to follow the dynamics of these systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the National Institutes of Health: R15GM1224597 to MJT and R15GM120738 to LMG.References
- K. L. Brown and R. E. W. Hancock, Curr. Opin. Immunol., 2006, 18, 24–30 CrossRef CAS
.
- R. E. W. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402–410 CrossRef CAS
- D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta, 2008, 1778, 357–375 CrossRef CAS
- H. W. Huang, Biochemistry, 2000, 39, 8347–8352 CAS
- M. J. Tucker, R. Oyola and F. Gai, J. Phys. Chem. B, 2005, 109, 4788–4795 CrossRef CAS
- M. Zasloff, Nature, 2002, 415, 389 CrossRef CAS
- R. Rathinakumar, W. F. Walkenhorst and W. C. Wimley, J. Am. Chem. Soc., 2009, 131, 7609–7617 CrossRef CAS
- K. J. Hallock, D.-K. Lee and A. Ramamoorthy, Biophys. J., 2003, 84, 3052–3060 CrossRef CAS
- H. Leontiadou, A. E. Mark and S. J. Marrink, J. Am. Chem. Soc., 2006, 128, 12156–12161 CrossRef CAS
- M. L. Gee, M. Burton, A. Grevis-James, M. A. Hossain, S. McArthur, E. A. Palombo, J. D. Wade and A. H. A. Clayton, Sci. Rep., 2013, 3, 1557 CrossRef
- M. Tang, A. J. Waring and M. Hong, J. Am. Chem. Soc., 2007, 129, 11438–11446 CrossRef CAS
- K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 1–10 CrossRef CAS
- K. Matsuzaki, O. Murase, N. Fujii and K. Miyajima, Biochemistry, 1996, 35, 11361–11368 CrossRef CAS
- K. Matsuzaki, O. Murase and K. Miyajima, Biochemistry, 1995, 34, 12553–12559 CrossRef CAS
- K. S. Moore, C. L. Bevins, M. M. Brasseur, N. Tomassini, K. Turner, H. Eck and M. Zasloff, J. Biol. Chem., 1991, 266, 19851–19857 CAS
- L. Monticelli, D. Pedini, E. Schievano, S. Mammi and E. Peggion, Biophys. Chem., 2002, 101–102, 577–591 CrossRef CAS
- N. Javkhlantugs, A. Naito and K. Ueda, Biophys. J., 2011, 101, 1212–1220 CrossRef CAS
- S. Toraya, N. Javkhlantugs, D. Mishima, K. Nishimura, K. Ueda and A. Naito, Biophys. J., 2010, 99, 3282–3289 CrossRef CAS
- S. Y. Shin, J. H. Kang, M. K. Lee, S. Y. Kim, Y. Kim and K.-S. Hahm, IUBMB Life, 2008, 44, 1119–1126 CrossRef
- D. Avrahami and Y. Shai, Biochemistry, 2002, 41, 2254–2263 CrossRef CAS
- G. Klocek, T. Schulthess, Y. Shai and J. Seelig, Biochemistry, 2009, 48, 2586–2596 CrossRef CAS
- T. Takahashi, F. Nomura, Y. Yokoyama, Y. Tanaka-Takiguchi, M. Homma and K. Takiguchi, Toxins, 2013, 5, 637–664 CrossRef CAS
- M. J. Tucker, J. Tang and F. Gai, J. Phys. Chem. B, 2006, 110, 8105–8109 CrossRef CAS
- G. Beschiaschvili and J. Seelig, Biochemistry, 1990, 29, 52–58 CrossRef CAS
- M. G. Roberson, D. K. Smith, S. M. White, I. S. Wallace and M. J. Tucker, Biophys. J., 2019, 116, 1064–1074 CrossRef CAS
- H.-U. Kim and K.-H. Lim, Colloids Surf., A, 2004, 235, 121–128 CrossRef CAS
- M. Stangl, A. Veerappan, A. Kroeger, P. Vogel and D. Schneider, Biophys. J., 2012, 103, 2455–2464 CrossRef CAS
- H. S. M. Lu, M. Volk, Y. Kholodenko, E. Gooding, R. M. Hochstrasser and W. F. DeGrado, J. Am. Chem. Soc., 1997, 119, 7173–7180 CrossRef CAS
- M. Volk, Y. Kholodenko, H. S. M. Lu, E. A. Gooding, W. F. DeGrado and R. M. Hochstrasser, J. Phys. Chem. B, 1997, 101, 8607–8616 CrossRef CAS
- K. C. Hansen, R. S. Rock, R. W. Larsen and S. I. Chan, J. Am. Chem. Soc., 2000, 122, 11567–11568 CrossRef CAS
- J. Bredenbeck, J. Helbing, R. Behrendt, C. Renner, L. Moroder, J. Wachtveitl and P. Hamm, J. Phys. Chem. B, 2003, 107, 8654–8660 CrossRef CAS
- C. Kolano, J. Helbing, M. Kozinski, W. Sander and P. Hamm, Nature, 2006, 444, 469–472 CrossRef CAS
- J. A. Ihalainen, J. Bredenbeck, R. Pfister, J. Helbing, L. Chi, I. H. M. van Stokkum, G. A. Woolley and P. Hamm, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5383–5388 CrossRef CAS
- C. Kolano, J. Helbing, G. Bucher, W. Sander and P. Hamm, J. Phys. Chem. B, 2007, 111, 11297–11302 CrossRef CAS
- M. J. Tucker, M. Abdo, J. R. Courter, J. Chen, S. P. Brown, A. B. Smith and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 17314 CrossRef CAS
- M. J. Tucker, M. Abdo, J. R. Courter, J. Chen, A. B. Smith and R. M. Hochstrasser, J. Photochem. Photobiol., A, 2012, 234, 156–163 CrossRef CAS
- R. Behrendt, C. Renner, M. Schenk, F. Q. Wang, J. Wachtveitl, D. Oesterhelt and L. Moroder, Angew. Chem. Int. Ed., 1999, 38, 2771–2774 CrossRef CAS
- J. Bredenbeck, J. Helbing, A. Sieg, T. Schrader, W. Zinth, C. Renner, R. Behrendt, L. Moroder, J. Wachtveitl and P. Hamm, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6452–6457 CrossRef CAS
- S. Sporlein, H. Carstens, H. Satzger, C. Renner, R. Behrendt, L. Moroder, P. Tavan, W. Zinth and J. Wachtveitl, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7998–8002 CrossRef
- C. Kolano, J. Helbing, G. t. Bucher, W. Sander and P. Hamm, J. Phys. Chem. B, 2007, 111, 11297–11302 CrossRef CAS
- D. G. Hogle, A. R. Cunningham and M. J. Tucker, J. Phys. Chem. B, 2018, 122, 8783–8795 CrossRef CAS
- M. Banghart, K. Borges, E. Isacoff, D. Trauner and R. H. Kramer, Nat. Neurosci., 2004, 7, 1381 CrossRef CAS
- L. Guerrero, O. S. Smart, G. A. Woolley and R. K. Allemann, J. Am. Chem. Soc., 2005, 127, 15624–15629 CrossRef CAS
- R. T. Buwalda, M. C. A. Stuart and J. B. F. N. Engberts, Langmuir, 2002, 18, 6507–6512 CrossRef CAS
- S. H. Donaldson, C. T. Lee, B. F. Chmelka and J. N. Israelachvili, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 15699 CrossRef CAS
- T. Shang, K. A. Smith and T. A. Hatton, Langmuir, 2003, 19, 10764–10773 CrossRef CAS
- T. Shang, K. A. Smith and T. A. Hatton, Langmuir, 2006, 22, 1436–1442 CrossRef CAS
- R. F. Tabor, M. J. Pottage, C. J. Garvey and B. L. Wilkinson, Chem. Commun., 2015, 51, 5509–5512 RSC
- S.-C. Wang and C. T. Lee, J. Phys. Chem. B, 2006, 110, 16117–16123 CrossRef CAS
- J. Zhang, S.-C. Wang and C. T. Lee, J. Phys. Chem. B, 2009, 113, 8569–8580 CrossRef CAS
- T. Koźlecki, A. Sokołowski and K. A. Wilk, Langmuir, 1997, 13, 6889–6895 CrossRef
- F. P. Nicoletta, D. Cupelli, P. Formoso, G. De Filpo, V. Colella and A. Gugliuzza, Membranes, 2012, 2, 134–197 CrossRef CAS
- E. R. Merino and M. Beilstein, J. Org. Chem., 2012, 8, 1071–1090 CAS
- M. Billamboz, F. Mangin, N. Drillaud, C. Chevrin-Villette, E. Banaszak-Léonard and C. Len, J. Org. Chem., 2014, 79, 493–500 CrossRef CAS
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC
- S. Spörlein, H. Carstens, H. Satzger, C. Renner, R. Behrendt, L. Moroder, P. Tavan, W. Zinth and J. Wachtveitl, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 7998 CrossRef
- J. A. Ihalainen, J. Bredenbeck, R. Pfister, J. Helbing, L. Chi, I. H. M. van Stokkum, G. A. Woolley and P. Hamm, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 5383 CrossRef CAS
- V. Botan, E. H. G. Backus, R. Pfister, A. Moretto, M. Crisma, C. Toniolo, P. H. Nguyen, G. Stock and P. Hamm, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 12749 CrossRef CAS
- H. Satzger, C. Root and M. Braun, J. Phys. Chem. A, 2004, 108, 6265–6271 CrossRef CAS
- C. M. Stuart, R. R. Frontiera and R. A. Mathies, J. Phys. Chem. A, 2007, 111, 12072–12080 CAS
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CAS
- R. Zana, M. Benrraou and R. Rueff, Langmuir, 1991, 7, 1072–1075 CAS
- J. N. Phillips, Trans. Faraday Soc., 1955, 51, 561–569 RSC
- P. R. Majhi and S. P. Moulik, Langmuir, 1998, 14, 3986–3990 CrossRef CAS
- G. C. Kresheck, Langmuir, 2000, 16, 3067–3069 CrossRef CAS
- Ö. Topel, B. A. Çakır, L. Budama and N. Hoda, J. Mol. Liq., 2013, 177, 40–43 CrossRef
- L. Piñeiro, M. Novo and W. Al-Soufi, Adv. Colloid Interface Sci., 2015, 215, 1–12 CrossRef
- T. Chakraborty, I. Chakraborty and S. Ghosh, Arabian J. Chem., 2011, 4, 265–270 CrossRef CAS
- J. Zhao and B. M. Fung, Langmuir, 1993, 9, 1228–1231 CrossRef CAS
- Y. S. Kim, L. Liu, P. H. Axelsen and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 17751 CrossRef CAS
- E. S. Manas, Z. Getahun, W. W. Wright, W. F. DeGrado and J. M. Vanderkooi, J. Am. Chem. Soc., 2000, 122, 9883–9890 CrossRef CAS
- A. M. Woys, A. M. Almeida, L. Wang, C.-C. Chiu, M. McGovern, J. J. de Pablo, J. L. Skinner, S. H. Gellman and M. T. Zanni, J. Am. Chem. Soc., 2012, 134, 19118–19128 CrossRef CAS
- D. B. Strasfeld, Y. L. Ling, S.-H. Shim and M. T. Zanni, J. Am. Chem. Soc., 2008, 130, 6698–6699 CrossRef CAS
- A. Remorino, I. V. Korendovych, Y. Wu, W. F. DeGrado and R. M. Hochstrasser, Science, 2011, 332, 1206 CrossRef CAS
- G. Montalvo, M. M. Waegele, S. Shandler, F. Gai and W. F. DeGrado, J. Am. Chem. Soc., 2010, 132, 5616–5618 CrossRef CAS
- Y. S. Kim, J. Wang and R. M. Hochstrasser, J. Phys. Chem. B, 2005, 109, 7511–7521 CrossRef CAS
- C.-Y. Huang, J. W. Klemke, Z. Getahun, W. F. DeGrado and F. Gai, J. Am. Chem. Soc., 2001, 123, 9235–9238 CrossRef CAS
- C.-Y. Huang, Z. Getahun, T. Wang, W. F. DeGrado and F. Gai, J. Am. Chem. Soc., 2001, 123, 12111–12112 CrossRef CAS
- D. Du, M. J. Tucker and F. Gai, Biochemistry, 2006, 45, 2668–2678 CrossRef CAS
- B. Ding, J. E. Laaser, Y. Liu, P. Wang, M. T. Zanni and Z. Chen, J. Phys. Chem. B, 2013, 117, 14625–14634 CrossRef CAS
- J. K. Chung, M. C. Thielges and M. D. Fayer, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 3578 CrossRef CAS
- A. Ghosh, J. S. Ostrander and M. T. Zanni, Chem. Rev., 2017, 117, 10726–10759 CrossRef CAS
- L. K. Tamm and S. A. Tatulian, Q. Rev. Biophys., 1997, 30, 365–429 CrossRef CAS
- T. Wang, Y. Zhu, Z. Getahun, D. Du, C.-Y. Huang, W. F. DeGrado and F. Gai, J. Phys. Chem. B, 2004, 108, 15301–15310 CrossRef CAS
- J. Ma, I. M. Pazos, W. Zhang, R. M. Culik and F. Gai, Annu. Rev. Phys. Chem., 2015, 66, 357–377 CrossRef CAS
- P. Hamm, M. Lim and R. M. Hochstrasser, J. Phys. Chem. B, 1998, 102, 6123–6138 CrossRef CAS
- Y. S. Lin, J. M. Shorb, P. Mukherjee, M. T. Zanni and J. L. Skinner, J. Phys. Chem. B, 2009, 113, 592–602 CrossRef CAS
- M. Khajehpour, J. L. Dashnau and J. M. Vanderkooi, Anal. Biochem., 2006, 348, 40–48 CrossRef CAS
-
L. K. Tamm and S. Tatulian, Infrared Spectroscopy of Proteins and Peptides in Lipid Bilayers, 1997 Search PubMed
- Z. Ganim, H. S. Chung, A. W. Smith, L. P. DeFlores, K. C. Jones and A. Tokmakoff, Acc. Chem. Res., 2008, 41, 432–441 CrossRef CAS
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS
- L. P. DeFlores, Z. Ganim, R. A. Nicodemus and A. Tokmakoff, J. Am. Chem. Soc., 2009, 131, 3385–3391 CrossRef CAS
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS
- C. Lee and M. Cho, J. Phys. Chem. B, 2004, 108, 20397–20407 CrossRef CAS
- E. Goormaghtigh, V. Raussens and J.-M. Ruysschaert, Biochim. Biophys. Acta Rev. Biomembr., 1999, 1422, 105–185 CAS
- A. Ghosh, M. J. Tucker and F. Gai, J. Phys. Chem. B, 2014, 118, 7799–7805 CrossRef CAS
-
C. Baiz, M. Reppert and r. Tokmakoff, An Introduction to Protein 2D IR Spectroscopy, 2012 Search PubMed
- C. R. Baiz, C. S. Peng, M. E. Reppert, K. C. Jones and A. Tokmakoff, Analyst, 2012, 137, 1793–1799 RSC
- J.-H. Choi and M. Cho, J. Chem. Phys., 2011, 134, 154513 CrossRef
- J.-H. Choi, S. Ham and M. Cho, J. Phys. Chem. B, 2003, 107, 9132–9138 CrossRef CAS
- D. S. Alvares, N. Wilke, J. Ruggiero Neto and M. L. Fanani, Chem. Phys. Lipids, 2017, 207, 38–48 CrossRef CAS
- J. Wang and R. M. Hochstrasser, Chem. Phys., 2004, 297, 195–219 CrossRef CAS
- M. C. Thielges and M. D. Fayer, Acc. Chem. Res., 2012, 45, 1866–1874 CrossRef CAS
- M. P. dos Santos Cabrera, S. T. B. Costa, B. M. de Souza, M. S. Palma, J. R. Ruggiero and J. Ruggiero Neto, Eur. Biophys. J., 2008, 37, 879 CrossRef CAS
- S. Ham and M. Cho, J. Chem. Phys., 2003, 118, 6915–6922 CrossRef CAS
- T. Hayashi, W. Zhuang and S. Mukamel, J. Phys. Chem. A, 2005, 109, 9747–9759 CrossRef CAS
- P. Bouř and T. A. Keiderling, J. Chem. Phys., 2003, 119, 11253–11262 CrossRef
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS
- W. F. DeGrado and J. D. Lear, J. Am. Chem. Soc., 1985, 107, 7684–7689 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic details, NMR spectra. See DOI: 10.1039/d0ra01920h |
| This journal is © The Royal Society of Chemistry 2020 |