
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chitosan gold nanoparticles molecularly imprinted polymer based ciprofloxacin sensor†
Sandeep G. Surya‡
 a,
Shahjadi Khatoon‡b,
Abdellatif Ait Lahcena,
An T. H. Nguyena,
Boris B. Dzantievc,
Nazia Tarannum*b and
Khaled N. Salama*a
a,
Shahjadi Khatoon‡b,
Abdellatif Ait Lahcena,
An T. H. Nguyena,
Boris B. Dzantievc,
Nazia Tarannum*b and
Khaled N. Salama*a
aSensors Lab, Advanced Membranes and Porous Materials Center, Computer, Electrical and Mathematical Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Saudi Arabia
bDepartment of Chemistry, Chaudhary Charan Singh University, Meerut, 250004, India
cA. N. Bach Institute of Biochemistry, Research Centre of Biotechnology of the Russian Academy of Sciences, Moscow, Russia. E-mail: khaled.salama@kaust.edu,sa; naz1012@gmail.com
First published on 31st March 2020
Abstract
In this work, we present a novel study on the development of an electrochemical biomimetic sensor to detect the ciprofloxacin (CIP) antibiotic. A chitosan gold nanoparticles decorated molecularly imprinted polymer (Ch-AuMIP) was used to modify the glassy carbon electrode (GCE) for preparation of the sensor. The Ch-AuMIP was characterized to understand various properties like chemical composition, morphology, roughness, and conduction using Fourier transform infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), atomic force microscopy (AFM) and cyclic voltammetry (CV) respectively. Several experimental conditions affecting the Ch-AuMIP/GCE sensor such as the CIP removal agent, the extraction time, the volume of Ch-AuMIP drop-cast onto GCE and the rebinding time were studied and optimized. The Ch-AuMIP sensor sensitivity was studied in the concentration range of 1–100 μmol L−1 exhibiting a limit of detection of 210 nmol L−1. The synergistic combination of Au nanoparticles and Ch-MIP helps detect the CIP antibiotic with good sensitivity and selectivity, respectively. We investigated the selectivity aspect by using some possible interfering species and the developed sensing system showed good selectivity for CIP with a 66% response compared to the other compounds (≤45% response). The proposed sensing strategy showed its applicability for successful detection of CIP in real samples like tap water, mineral water, milk, and pharmaceutical formulation. The developed sensor showed good selectivity towards CIP even among the analogue molecules of Norfloxacin (NFX) and Ofloxacin (OFX). The developed sensor was successfully applied to determine the CIP in different samples with a satisfactory recovery in the range of 94 to 106%.
Introduction
Ciprofloxacin (CIP) is a type of fluoroquinolone antibiotic of second generation having activity against Gram (−) and Gram (+) bacteria and is frequently used for treating bacterial infection.1 Due to the presence of a fluorine atom in the ring, the antimicrobial activity of CIP increases by facilitating the approach of the drug into the bacterial cell. The mode of fluoroquinolone activity is across binding DNA-gyrase enzyme.2 Fluoroquinolones cause phototoxicity in humans,3 condrotoxic effects in young animals, and tendon rupture.4 Godman et al. also observed allergic reactions to CIP5 and the excessive use of this antibiotic may lead to numerous problems in human health such as nausea, diarrhea, vomiting, change in liver function, headache etc.6 In the interest of public health, the European Union has set the maximum residue limits (MRLs) for a sum of enrofloxacin and CIP as 0.1 mg g−1 in muscle tissue, 0.2 mg g−1 in the liver and 0.3 mg g−1 in the kidney of chickens.7,8 Thus, an accurate, fast, sensitive method for the detection of CIP in food samples is crucial. Different methods have been developed so far for the detection of CIP quantitatively and qualitatively such as UV spectrophotometry, capillary electrophoresis, titration, and HPLC.9 Due to the low level of antibiotic residue in a real sample, it is necessary to find a good method for routine analysis. Molecular imprinting is a globally accepted technique that involves a template to create specific and selective memory complementary to the template in the polymer. The cavity formed after removing the template molecule helps to sense the analytes, which is similar in their structure and functionality. Molecular imprinting technique is also the substitute of natural receptors and successfully used as an artificial analytical receptor for molecular identification with binding properties as natural antibodies in immunoassays.10,11 Therefore, MIPs are termed as plastic anti-bodies. Literature reveals that including biological structure more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules such as inorganic ions, drugs, cells, nucleic acid, protein, and viruses have been imprinted successfully.11
000 molecules such as inorganic ions, drugs, cells, nucleic acid, protein, and viruses have been imprinted successfully.11
MIP is a significant and widely used synthetic polymer with selectivity and sensitivity of predetermined ligand.12,13 According to the template adding methodology, the synthesis of MIPs is divided into two categories covalent and non-covalent. The covalent approach is stoichiometric and interaction between the functional group of template and monomer recognition sites developed a reversible covalent interaction. These interactions do not favour fast binding and removal of the template. Whereas, the non-covalent approach offers fast binding and removing of the template molecule.14,15 An electrochemical sensor is a promising method for the detection of antibiotic residues in food samples because of its characteristics such as easy-to-operate, fast response, high sensitivity, inexpensive and good stability.16 In this method, MIP is used as a sensing material and is a kind of template accommodated synthesis by using functional monomer, crosslinker, and template molecule.17,18 The template will be extracted in a polymeric matrix by the chemical or physical methods, which are suitable for specific cases from the imprinted polymer matrix. Washing exposed recognition cavities which is complementary to the template molecules neither in shape and size but also in interaction sites.19,20 MIP that was synthesized by this technique has high sensitivity and selectivity for the target molecule. For the application in MIP, the polymer template must be easily synthesizable and possess good conductivity for the preparation of electrochemical sensors. The prepared MIP has a wide range of applications because of its low cost, high sensitivity and specificity for the analyte, easy-to-handle, recyclable, stable and ease of operation. Some factors determine the sensitivity of the sensors that include: (i) availability of cavities in the imprinted polymer (ii) capability of electron transfer from recognition sites to the surface of electrode21 and ability of electrocatalytic oxidation of electrode to the template molecule.22 Because of these interesting characteristics, MIPs have been used in catalysis, sensing, separation, isolation and drug delivery.23 Nanotechnology enhances efficiency in the performance of the MIPs sensor. The nanomaterial provides the conductivity of polymer, large surface area, high adsorptive ability, and biocompatibility, easy and inexpensive production. Nanomaterial deposited on GCE increased the sensitivity of the electrode and facilitating their merge into transducer.24 Nanoparticles have been extensively used in the fabrication of nanocomposites.25 They can homogenize with material containing many functional groups such as CN, NH2, SH and form covalent bonds.26,27
Numerous studies have been reported on the use of imprinted thin film and nanoparticles to get the enhanced sensitivity and quick response for antibiotics.23,24 Electrochemical MIP sensor hybrid with nanomaterial is the one current trend,25 most favourable and sensitive of electrochemical sensing methods. In this context, a novel molecularly imprinted electrochemical sensor was reported by Wei et al. (2014) and synthesized for the detection of ampicillin (AMP) antibiotic.28 The sensor was fabricated with multi-walled carbon nanotubes (MWCNTs), gold nanoparticles (AuNPs), modified electrode to increase the sensor's sensitivity and electronic conducting ability. Lian et al. (2012) proposed a molecularly imprinted electrochemical sensor on the gold electrode fabricated by chitosan-platinum nanoparticles (CS-PtNPs) and graphene-gold nanoparticles (GR-AuNPs) nanocomposites for sensitive and convenient detection of erythromycin in real spiked samples.29 The sensor was successfully applied for the detection of ERY and showed excellent stability, selectivity and good reproducibility. Lian et al. (2013) prepared an electrochemical sensor with a gold electrode recast by multilayer films of chitosan-multi-walled carbon nanotube composite (CS-MWCNTs) and gold nanoparticles (AuNPs) for detection of oxytetracycline (OTC).30 Xu et al. (2009) prepared a sensitive molecularly imprinted electrochemical sensor for a particular diagnosis of tricyclic antidepressant imipramine by adding Au nanoparticles (Au-NPs) with a thin film of MIPs.31 The sensor was coated onto the ITO electrode and displayed excellent selectivity towards the targeted imipramine molecule. This technique was efficiently applied to the detection of imipramine in drug tablets. The aim of this work is to develop a molecularly imprinted electrochemical sensor modified by chitosan gold nanocomposite for sensitive and selective detection of CIP antibiotics. The prepared sensor has high-specificity toward the template molecule. Chitosan-based gold nanocomposites (Ch-AuNPs) used for the construction of highly efficient MIP sensors. In the fabrication of an imprinted sensor, gold nanocomposite is used as the supporting material. This modification augments the electron transferability to the surface electrode from imprinted cavities.
Herein, we report the preparation of Ch-AuNPs based MIP nanocomposite to detect CIP for the first time. The electrochemical sensor prepared is selective, stable, sensitive for CIP, easy to use, economical, and can be altered for different antibiotics for facile applications. Further, we demonstrated an electrochemical sensor with Ch-AuNP based MIPs (as a sensitive element) for the detection of CIP. We characterized the morphology of these synthesized Ch-AuNPs-MIPs/NIPs using FT-IR, SEM, and AFM. First, we tried to understand the binding phenomenon of the extracted MIP adducts by subjecting them to various extraction times. Second, we optimized the volume of these MIPs (7.5 μL) that can be deposited on 3 mm GCEs to deliver uniform coverage while being sensitive. Third, we further studied the incubation time for the CIP solution (10 min) and used the optimized time value for selectivity studies. Randles–Sevick equation was used to calculate the surface area of the receptor and was compared to the bare electrode area, which helps understand sensitivity improvement. The developed sensor showed good selectivity towards CIP even among the analogue molecules of Norfloxacin (NFX) and Ofloxacin (OFX). Finally, we conducted multiple experiments with the above conditions to check the sensitivity of the sensors. The developed sensing system capability to detect CIP in real samples was checked using mineral and tap water samples, milk, and pharmaceutical tablets. ANOVA was applied to see the difference between the results obtained data from real samples.
Experimental section
Chemicals and reagent
Ciprofloxacin hydrochloride monohydrate (CIP) was purchased from HiMedia. Norfloxacin (NFX) 98%, Ofloxacin (OFX) were purchased from Sigma Aldrich. Dopamine (C8H11NO2), uric acid (C5H4N4O3), ascorbic acid (C6H8O6), glucose (C6H12O6), potassium ferricyanide K3[Fe(CN)6], phosphate buffer saline (PBS), methanol (CH3OH), ethanol (C2H5OH), acetic acid (AAc), sodium hydroxide (NaOH), ethylene glycol dimethacrylate (EGDMA), α,α′-azobisisobutyronitrile (AIBN) were all purchased from Sigma Aldrich. Methacrylic acid (MAA) was purchased from Fisher Scientific. Chitosan flakes (low molecular weight), and chloroauric acid (gold chloride) were procured from Molychem and other chemicals were of analytical grade. Triple distilled water was used in all experiments and deionized water from the Milli-Q system. Pharmaceutical tablets (Ciprogen 500 mg) containing CIP and HCl to carry out the real-time study were obtained from a local drug store.Apparatus and instruments
All the electrochemical experiments of CV and differential pulse voltammetry (DPV) measurements were carried out by employing an electrochemical instrument PalmSens (BV Houten-Netherlands) connected to a computer and controlled by software named PsTrace 5.5. An electrochemical cell containing a three-electrode system was used throughout all the studies. GCE with a diameter of 3 mm was used as a working electrode. The reference electrode was Ag/AgCl electrode (saturated with KCl) and a platinum wire as the counter electrode, were employed. The pH values of PBS were measured with an Accumet pH meter. In this work, we used the ultrapure water (18.2 MΩ cm−1) from a Milli-Q ultrapure system to prepare the aqueous solutions. FT-IR spectroscopy (Agilent Cary 630) was used to characterize and analyze (Agilent resolution pro) the structure of properties of MIP, NIP and adduct. The morphology studies of the thin films were carried out by SEM from the Zeiss Merlin instrument. VEECO Dimension icon tool was used to do the AFM studies to understand the surface topology and roughness of the Ch-AuNPs based MIPs and NIPs (Ch-AuMIPs and Ch-AuNIPs). Absorbance study was performed using Systronics T2201 UV-vis spectroscopy.Preparation of chitosan-based gold nanoparticles (Ch-AuNPs)
As reported previously,32 we followed the same approach and did the synthesis of gold nanoparticles for the target composite. Initially, chitosan flakes were dissolved in 0.1 M AAc under magnetic stirring using an oil bath at 60 °C until a transparent solution was obtained (∼12 h). Then gold chloride (HAuCl4) solution (5 mM in volume ratio CS:HAuCl4 = 5:2) was added drop by drop under constant stirring and heating at 60 °C (4 h). This resulted in a rapid change in the color from pale yellow to purple indicating the formation of chitosan-based gold nanoparticles. The gold nanocomposite solution was kept at 4 °C when not in use.
Preparation/synthesis of MIPs and non-molecularly imprinted polymer (NIPs)
To prepare CIP-imprinted polymer, 0.4 g MAA and 0.1 g CIP were mixed in 11 mL of dimethylformamide/methanol in (2:3) ratio at room temperature. The mixture was sonicated for 10 min and then stirred for 60 min until a homogenous solution was obtained.33 The resultant solution was then mixed with 40 mL of Ch-AuNPs and stirred for 60 min at room temperature to obtain a homogenous solution. After adjusting the pH 9.0 by adding 2 M NaOH, 189 μL (1 mmol) EGDMA (crosslinking agent) and 20 mg AIBN (radical initiator) was added into the aforementioned solution and then stirred for 60 min.34 Finally, the CIP-imprinted polymer solution was obtained by bubbling nitrogen for 10 min to remove dissolved oxygen (Fig. 1).
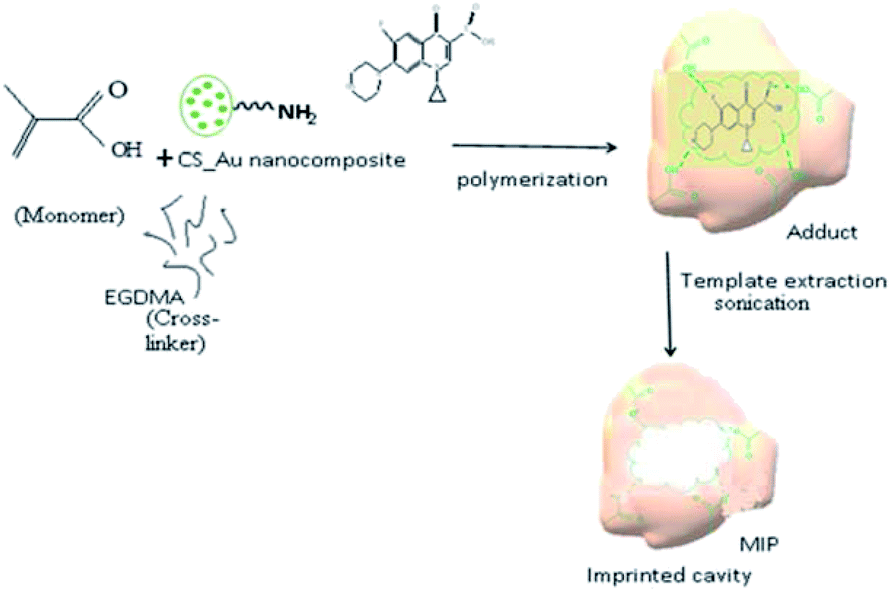 | ||
| Fig. 1 Schematic representation of the MIP synthesis using different precursors. | ||
Subsequently, to prepare Ch-AuNIPs same procedure was followed except adding the template molecule. Finally, both the materials were subjected to absorbance studies (UV-vis spectroscopy) and the corresponding results are presented in Fig. S1.†
Solutions
PBS of pH 7.4; 0.01 M was prepared by dissolving one tablet in 200 mL of deionized water. Regarding the preparation of the stock solution of CIP, the adequate amount for 0.01 M was dissolved in 50 mL of deionized water. A stock solution of (10–2 M) NFX was prepared by dissolving the adequate amount in AAc/deionized water (3:7). OFX stock solution was prepared by dissolving in isopropanol/deionized water (1:1). Glucose, dopamine, ascorbic acid solutions were prepared by dissolving the adequate amount in deionized water. The potassium ferricyanide solution (5 mM) containing 0.1 M KCl was prepared in deionized water. CIP removal solution was prepared as follows: (a) methanol and deionized water in the ratio (1:1), (b) methanol and acetic acid (MeOH/AAc) in the ratio (9:1) (c) NaOH solution (0.1 M) was prepared in deionized water.
Preparation of the electrochemical sensor
The GCE was cleaned ultrasonically to get rid of the previous contamination followed by a rinse with ethanol/water solution and then followed by deionized water washing. Subsequently, 7.5 μL of Ch-AuNPs based NIP or Ch-AuNPs based adduct was drop cast onto the GCE surface followed by a drying step in an oven at 70 °C for 20 min. Then the electrode coated with adduct was immersed in a solution of MeOH/AAc (9:1) for 10 min under stirring conditions in order to remove the CIP from the adduct network followed by washing with PBS pH 7.4 (10 mM) to obtain Ch-AuMIPs. As mentioned in the preparation of MIPs and NIPs section above, UV-vis studies were performed on the eluted solvents to understand the effects of washing and the corresponding data is depicted in Fig. S2.†
Electrochemical analysis techniques
We performed CV measurements using the three electrodes system and by scanning the bias potential in the range −0.4 V to 0.9 V, with a potential step of 10 mV and 100 mV s−1 as scan rate. DPV measurements were performed by applying a sweep potential from 0.0 V to −0.9 V at a pulse amplitude of 20 mV and pulse width 0.1 s with a scan rate of 10 mV s−1. During the stability study, CV analysis was performed rigorously at a stretch and measurements were recorded for 100 cycles. A current corresponding to oxidation and reduction peaks during both the measurements was considered for further analysis with multiple analytes.Real samples preparation
Tap water samples were collected from the laboratory tap and CIP with 0.02 M PBS was directly added to it. Three different brands of mineral water samples were purchased from a local supermarket. Further, all the experimental procedures were performed as done before for the laboratory samples.The developed method was used for the determination of CIP in tablets (CIPROGEN®) purchased from a local drugstore in KAUST (Saudi Arabia). It contains hydrochloride as an extra ingredient along with CIP (as major constituent). The real sample solution was prepared as follows: 5 tablets were weighed and powdered, then an accurate weight of the powder equivalent to one tablet was mixed with deionized water in a 100 mL calibrated flask. Then the resultant solution is stirred for about 10 min before sonicating for 15 min and eventually strained through filter paper (from Whatmann) to separate any insoluble matter. Towards the end, the filtrate was collected in a clean flask. After dilution with PBS, an aliquot of sample solution was prepared in phosphate buffer pH 7.4. The amount of CIP per tablet was measured utilizing the developed sensing strategy and calculated from a standard calibration curve. The milk sample tested in this study was purchased from a local supermarket. To reduce the matrix effect, the milk sample was prepared by diluting 1 mL of the samples in 20 mL of PBS 10 mM (pH 7.4). The sample was fortified by 10 μM of CIP and tested using the developed sensor.
Results and discussion
The synthesized Ch-AuMIP and Ch-AuNIP were characterized using SEM, AFM and FT-IR. The following sections detail on different morphological characterizations of these materials.Fourier-transform infrared (FT-IR) spectroscopy characterization
The FT-IR spectra of NIP, MIP, and adduct are shown in Fig. S3a.† The obtained results showed the presence of a common absorbance band in all the three spectra. The broadband around 2400 cm−1 to 3400 cm−1 showed the presence of intramolecular hydrogen bonding between –NH2 and –OH group of chitosan. The absorbance band at 1650 cm−1 was allocated to the amide group in chitosan.35 A common band at 1375–1398 cm−1 is due to the stretching vibration of –CH2– group. A band at 2917–2926 cm−1 belongs to stretching vibration of –C–H group in adduct, MIP and NIP and a band at near about 1020–1080 cm−1 is due to the presence of C–O–C stretching vibration.36,37 In the case of the adduct, there are extra bands signifying the presence of CIP antibiotic as shown in Fig. S3a and Table S1.† They are absorbance bands at 1750 cm−1 and 1064 cm−1 which can be assigned to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and C–F stretching vibration, respectively.33 The absorption band at 1654 cm−1 is strong in adduct as compared to NIP and MIP which may be assigned to –NH bending of quinoline in CIP and –NH present in chitosan, whereas this band is weak in adduct and NIP due to chitosan. Similarly, Fig. S3b and Table S2† give FTIR information on the Ch-AuNPs alone.
O stretching vibration and C–F stretching vibration, respectively.33 The absorption band at 1654 cm−1 is strong in adduct as compared to NIP and MIP which may be assigned to –NH bending of quinoline in CIP and –NH present in chitosan, whereas this band is weak in adduct and NIP due to chitosan. Similarly, Fig. S3b and Table S2† give FTIR information on the Ch-AuNPs alone.
Surface morphology study
We performed AFM surface morphological study by taking 5 μm × 5 μm images at a frequency of 0.98 Hz and a sampling rate of 256. We observed that MIP has many big grooves over the surface as compared to NIP, which essentially confirms the presence of active frames inside them with embedded imprints. The overall RMS roughness for MIP is 43 nm and for NIP is 27 nm. The high roughness in the MIP sample corresponds to the presence of the active sites, which eventually helps transfer the adsorption effects of analytes into electrical changes in CV and DPV measurements. Thus, we observed highly selective changes during electrical characterization with the MIP than NIP. Furthermore, Fig. S4† shows the corresponding 2D AFM images of MIP and NIP and the height profiles shown in Fig. S4c and d,† confirming the varying pore depths in MIPs and NIPs. As per the image pores are small in size and more sporadic only in the case of MIPs. We did SEM imaging of both Ch-AuMIPs as prepared and Ch-AuNIPs samples in the Zeiss merlin instrument and presented in Fig. 2. The above images were taken at a scale of 2 μm each to show the surface coverage and to detail the minimum features of the MIP. It is evident from the Ch-AuNIPs (control) as shown in Fig. 2d that the surface is smoother as compared to Ch-AuMIP. The surface morphology of Ch-AuMIP obtained after removal of the template showed rough surfaces making it more porous (Fig. 2c). Ch-AuMIP had a better surface area with more cavities than that of Ch-AuNIP and thus Ch-AuMIP has more affinity towards the CIP molecule compared to much Ch-AuNIP.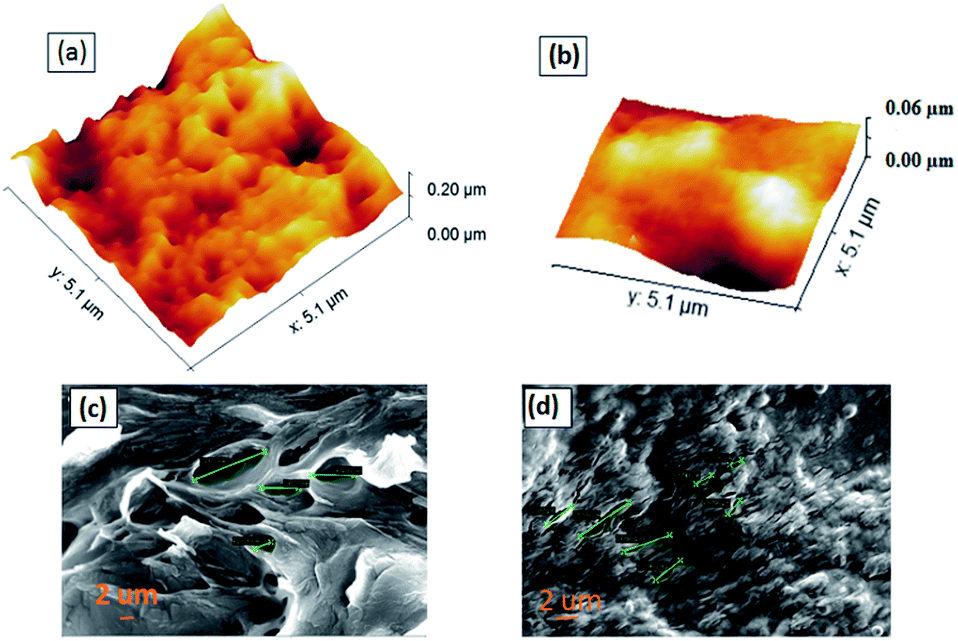 | ||
| Fig. 2 AFM images of (a) Ch-AuMIP and (b) Ch-AuNIP; SEM images of (c) Ch-AuMIP and (d) Ch-AuNIP. | ||
Electrochemical characterization of the sensing system
Different electrodes of GCE, Ch-AuNIP, Ch-AuMIP after removal of CIP and Ch-AuMIP after binding to CIP were characterized using CV measurements in 5 mM of potassium ferricyanide solution containing 0.1 M KCl. As shown in Fig. S5,† it can be observed clearly that the Ch-AuMIP/GCE after binding 10 μM of CIP shows a decreasing current intensity compared with GCE in the absence of CIP analyte. This decrease can be referred to as the resistance to electrons transfer due to the occupation of recognition sites (cavities) by the template CIP.17 However, after the removal of CIP from the polymer network the current intensity increases significantly (approx. 60%). Therefore, the sensing system based on Ch-AuMIP/GCE could have high sensitivity towards the detection of CIP molecules.The electrochemical sensors modified with MIP and NIP hybrid composites were characterized using 5 mM solution of ferricyanide containing 0.1 M KCl, after binding using 10 μM of CIP template molecule. The obtained results presented in Fig. S5b† show that the Ch-AuMIP/GCE sensor exhibited a high affinity towards the CIP compared to the Ch-AuNIP/GCE. This could be due to the effect of the recognition sites created on Ch-AuMIP after CIP removal from the adduct. The calculated imprinting factor is 1.57, thus confirming this Ch-AuMIP use for the detection of CIP molecules. The plausible reason for this could be attributed to the non-specific sorption of CIP in the NIP material. The following sections consist of the optimization of some crucial parameters that affect the selectivity and sensitivity of the proposed sensor.
Optimization of analytical parameters
:1 exhibits a high extraction/removal of the CIP template from the imprinted polymer leading to high current intensity response after removal compared to the other removal agents tested. Fig. 3c and d depicts the bar graphs with the error bars obtained for all the three removal agents. Hence, MeOH/AAc was selected to make free sites available in further experiments related to sensitivity and selectivity.
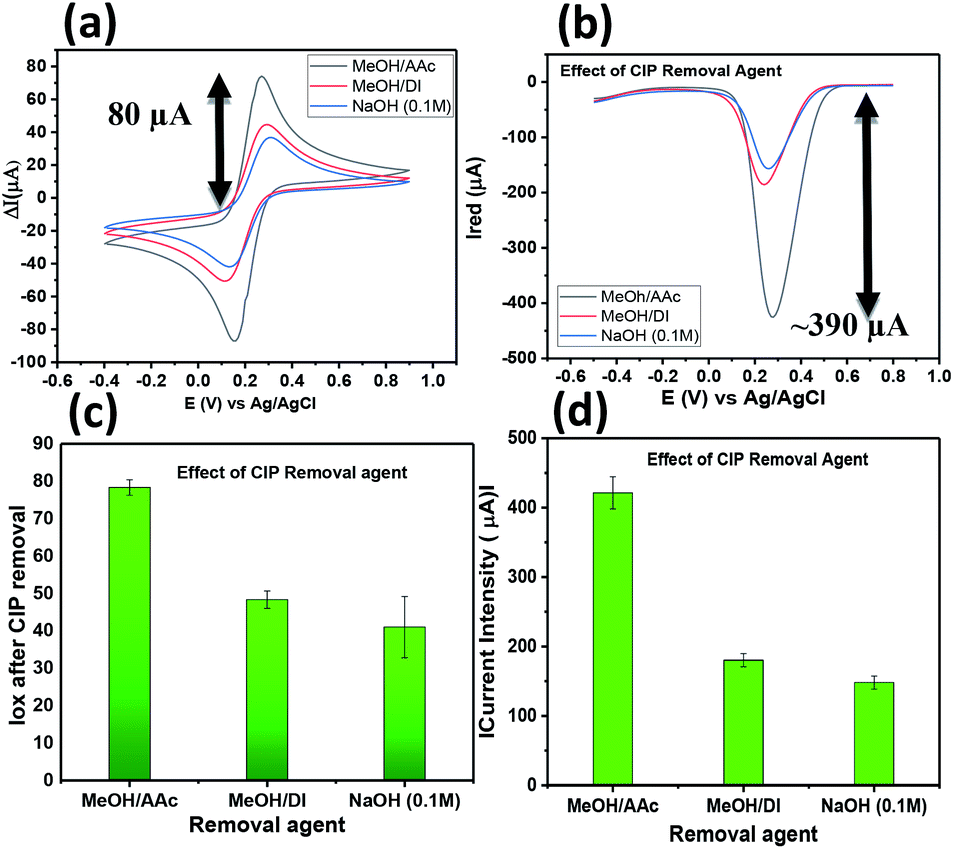 | ||
| Fig. 3 Effect of CIP removal agent NaOH 0.1 M (blue line), MeOH/DI (red line), MeOH/AAc (gray line) using (a) cyclic voltammetry (b) differential pulse voltammetry; corresponding histograms of effect of removal agent used for (c) CV and (d) DPV. | ||
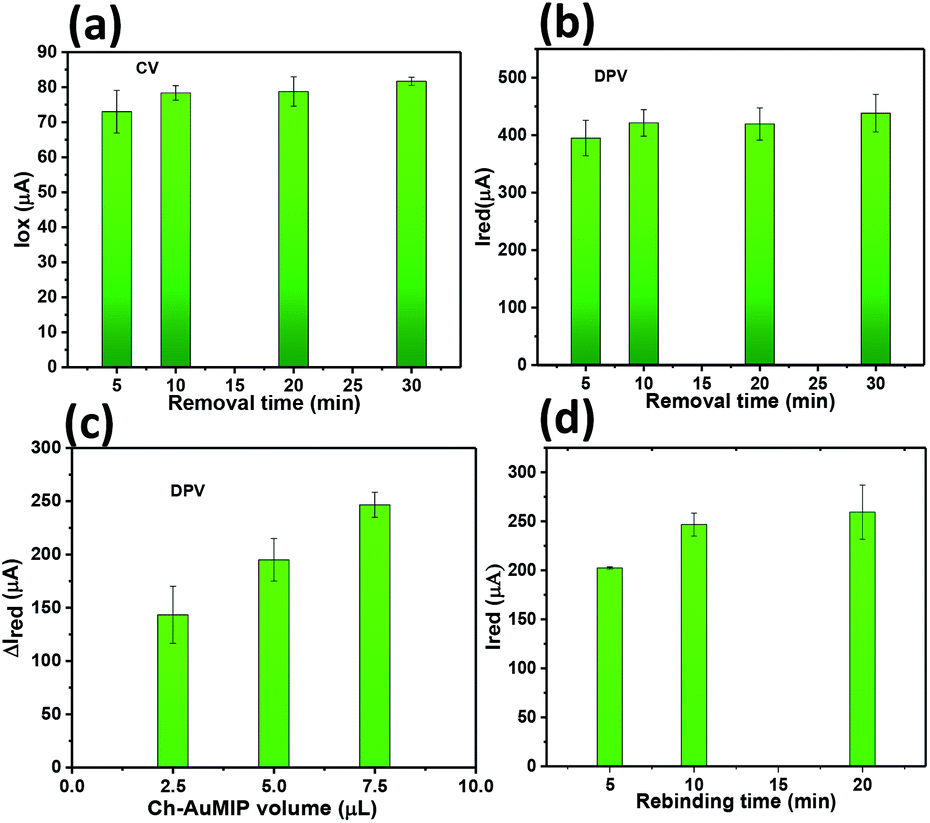 | ||
| Fig. 4 (a) CV and (b) DPV response of Ch-AuMIP in a solution of 5 mM ferricyanide containing 0.1 M KCl after CIP removal using MeOH/AAc for different times. (c) CV and (d) DPV responses of Ch-AuMIP/GCE obtained for different amounts of Ch-AuMIP in a solution of 5 mM ferricyanide containing 0.1 M KCl (d) CIP rebinding time effect. | ||
Thus, we discarded volumes beyond 7.5 μL as this may lead to the erroneous data, which corresponds to the unknown concentration. The rebinding time for CIP has been optimized by varying the CIP contact time with the Ch-AuMIP starting from 5 to 20 min and is shown in Fig. 4d. The results obtained showed that after 10 min, the Ch-AuMIP tends the bind the maximum of the analyte showing a high current intensity. Thus, 10 min was chosen as an optimal value of the rebinding time for the rest of the experiments.
| Ipa = 8.16(v)1/2 − 3.73 R2 = 0.999 | (1) |
| Ipc = −7.95(v)1/2 − 6.74 R2 = 0.999 | (2) |
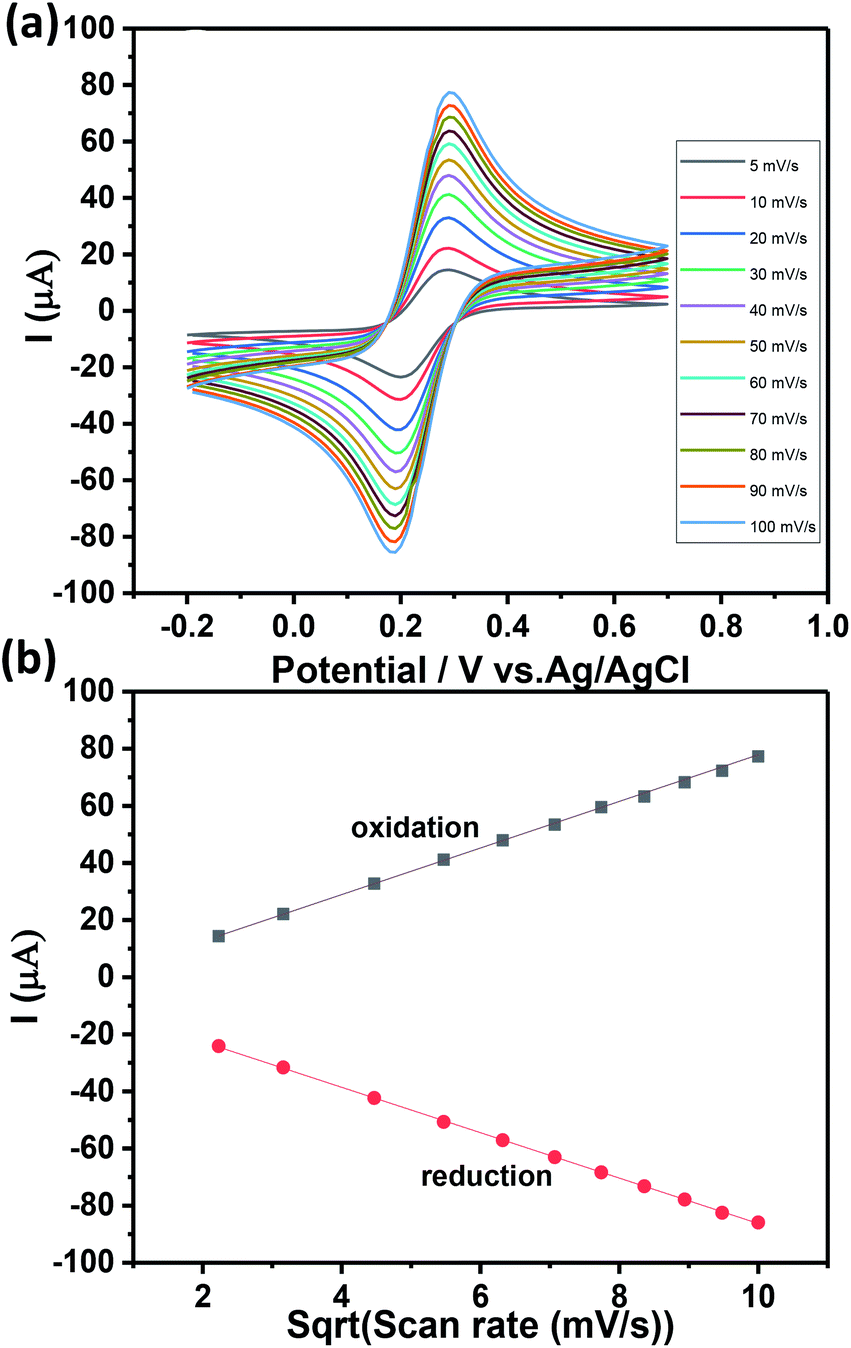 | ||
| Fig. 5 (a) CV analysis of the Ch-AuMIP/GCE as a function of scan rates (from 5 to 100 mV s−1) in PBS (10 mM, pH 7.4) and (b) linear plot of the oxidation and reduction peak currents vs. square root of scan rates. | ||
Fig. 5b shows a well-defined ferricyanide redox peaks suggesting that surface controlled electrochemistry and the ratio of redox peak current value was close to 1 which indicates there is no side reaction.38,39 The active surface area of the Ch-AuMIP/GCE was estimated based on Randles–Sevick equation as presented below:
|
Ipa = 268600n3/2AD1/2Cv1/2
| (3) |
Selectivity studies
The selectivity of the Ch-AuMIP/GCE was investigated by comparing the CV responses in the presence of some possible interferents such as dopamine (DA), uric acid (UA), glucose (Glu) and ascorbic acid (AA) as shown in Fig. S6a.† A similar study was performed by investigating the sensor response in the presence of NFX and OFX, which belongs to the same drug family as CIP. The obtained results are presented in Fig. S6b as shown in the ESI.† The obtained results presented in Fig. S6a and b† showed that the developed sensing system based on Ch-AuMIP exhibits a high affinity towards CIP compared to other structural analogue molecules. These results manifest that this sensing system could be useful for the detection of CIP with good sensitivity. Along with Ch-AuMIP coated GCEs, we also checked the binding capability of Ch-AuNIP towards different possible interferents. As can be seen from Fig. S6a and b,† Ch-AuNIP exhibits a small response compared to Ch-AuMIP which can be attributed to the physical adsorption of the interferents on the polymer network. Moreover, all ΔIox values obtained for the tested interferences were lower than the one obtained for the Ch-AuMIP which is due to the presence of the MIP cavities at Ch-AuMIP. We also checked the cross-selectivity, which we discussed along with stability in the later part.Sensitivity of Ch-AuMIP
The electrochemical response of the Ch-AuMIP/GCE has been studied as a function of CIP concentration as shown in Fig. 6a using DPV at a scan rate of 20 mV s−1 in 5 mM ferricyanide solution containing 0.1 M KCl. It can be seen from the inset of Fig. 6a that the magnitude of the electrochemical response current of the Ch-AuMIP/GCE was observed to decrease with an increase in the CIP concentrations. This was attributed to the successful binding of the CIP template to the created imprinted sites in the polymer network leading to a selective interaction. A linear plot drawn between changes in current and the log of concentrations of CIP is almost linear (Fig. 6b), which is expressed in the following equation (eqn (4))|
ΔIred = −47.6logCCIP + 479.7
| (4) |
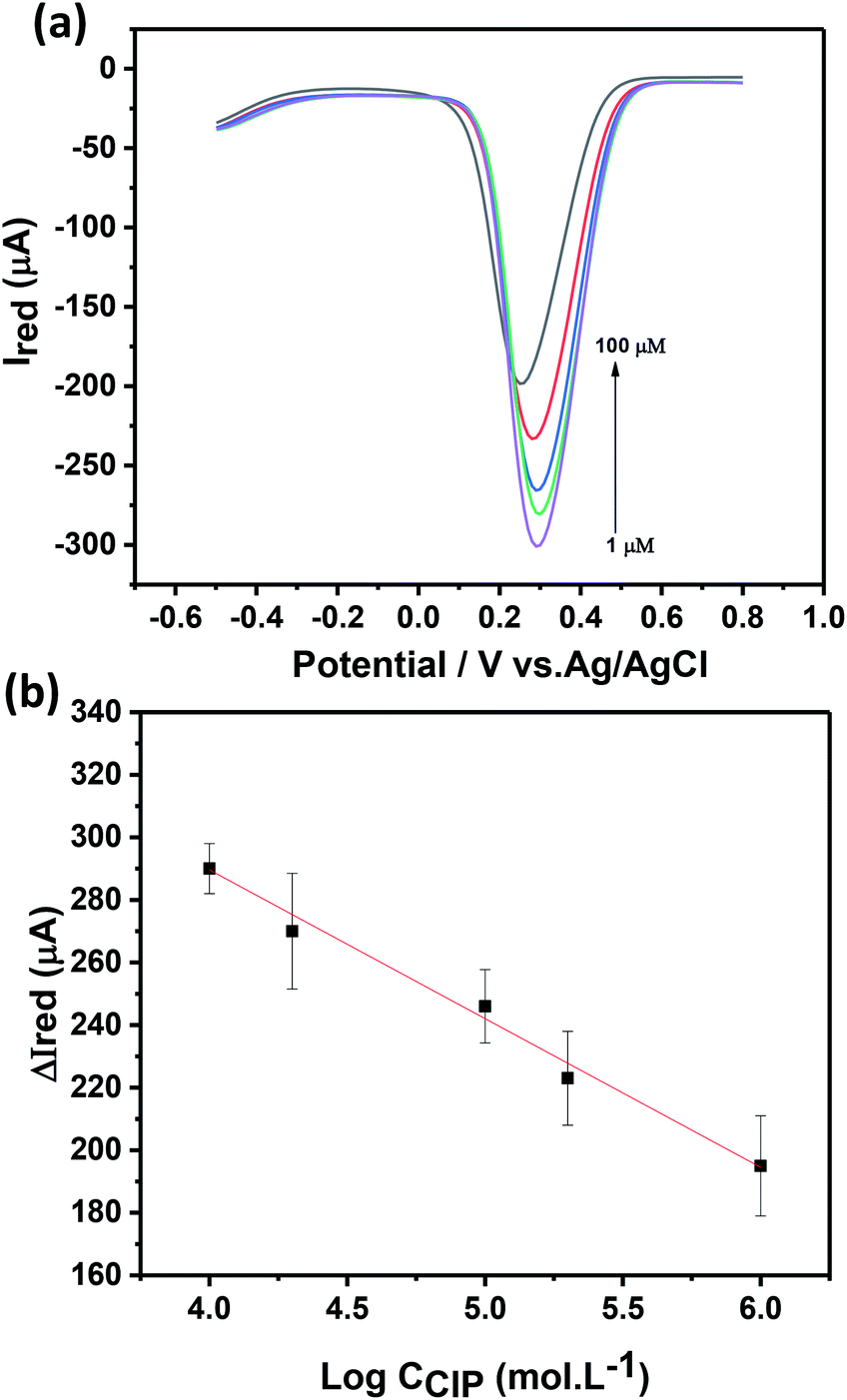 | ||
| Fig. 6 (a) Electrochemical studies of Ch-AuMIP/GCE as a function of CIP concentration varied from 1–100 μM in PBS 10 mM, pH 7.4, (b) linear plot between electrochemical peak current responses and logarithm of CIP concentrations. | ||
DPV studies show that the prepared sensor showed a logarithmic linear response to CIP concentrations ranging from 1–100 μM with a correlation coefficient of 0.989. The detection limit of the developed sensor has been estimated as 0.21 μM. The following comparison (Table 1) covers all the details on sensors developed for the detection of CIP. In terms of limit of detection, we are on par with most of the recent reports but as per our knowledge, this is the first report using Au nanoparticles in MIP to detect CIP. Though C. Yan et al. successfully demonstrated a MIP based CIP detector, they confined the experiments only to one application.49 Similarly, H. Bagheri et al., used a complex hybrid such as magnetic multi-walled carbon nanotube with MIPs to obtain a sensor material matrix.50 The linearity range of this hybrid material is low as compared to many of the materials in the table and the limit of detection was on par with most of the sensors. Due to individual compatibilities and fortes, a combination of multi-array sensors with the suggested materials in Table 1 would yield a good platform for CIP detection in multiple ranges and environments.
| Sensor | Transducer | Conc. range (μmol L−1) | LOD (μmol L−1) | Sample | Ref. |
|---|---|---|---|---|---|
| DNA sensor | CCP, DPV | 40–80 | 24 | Salmon sperm | 40 |
| MWCNTs/GCE | LSV | 3–1200 | 0.9 | Human urine, plasma and in pharmaceutical samples | 41 |
| Ds-DNA-BDD | SWV, DPV | 0.5–60 | 0.44 | Urine, calf thymus in aqueous solution | 42 |
| MWCNTs/GCE | Amperometry | 40–1000 | 6 | Urine, serum, spiked sample | 43 |
| Ba0.5Co0.5Fe2O4 nanoparticle ferrites/GCE | CV | 0.01–1000 | 0.0058 | Spiked water, tablet | 44 |
| Au/C3N4/GN/GCE | CV, SWV | 0.6–120 | 0.42 | Milk | 45 |
| CPE with cetyltrimethylammonium bromide | DPV | 20.0 | 0.05 | Spiked water | 46 |
| MIP silica NPs | Fluorescent | — | 0.13 | Urine | 47 |
| MIPs@SiO2–FITC | Fluorescent | 4–250 (nM) | 4 nM | Aquaculture water | 48 |
| MIP | SWV | 0.001–1000 | 75 pM | Pharmaceuticals | 49 |
| MMWCNT/CPE | DPV | 0.005–0.85 | 0.0017 | Urine, serum, pharmaceuticals | 50 |
| Ch-AuMIP/GCE | DPV | 1–100 | 0.21 | Mineral and tap water, milk and pharmaceuticals | This work |
Stability of the electrochemical sensor
In any sensor system, stability of the device is a crucial step and needs to be calibrated properly. The stability of Ch-AuMIP/GCE towards the electrochemical scanning was evaluated by measuring the current response of the sensing system for 100 cycles in 5 mM ferricyanide solution containing 0.1 M KCl using CV as shown in Fig. 7a.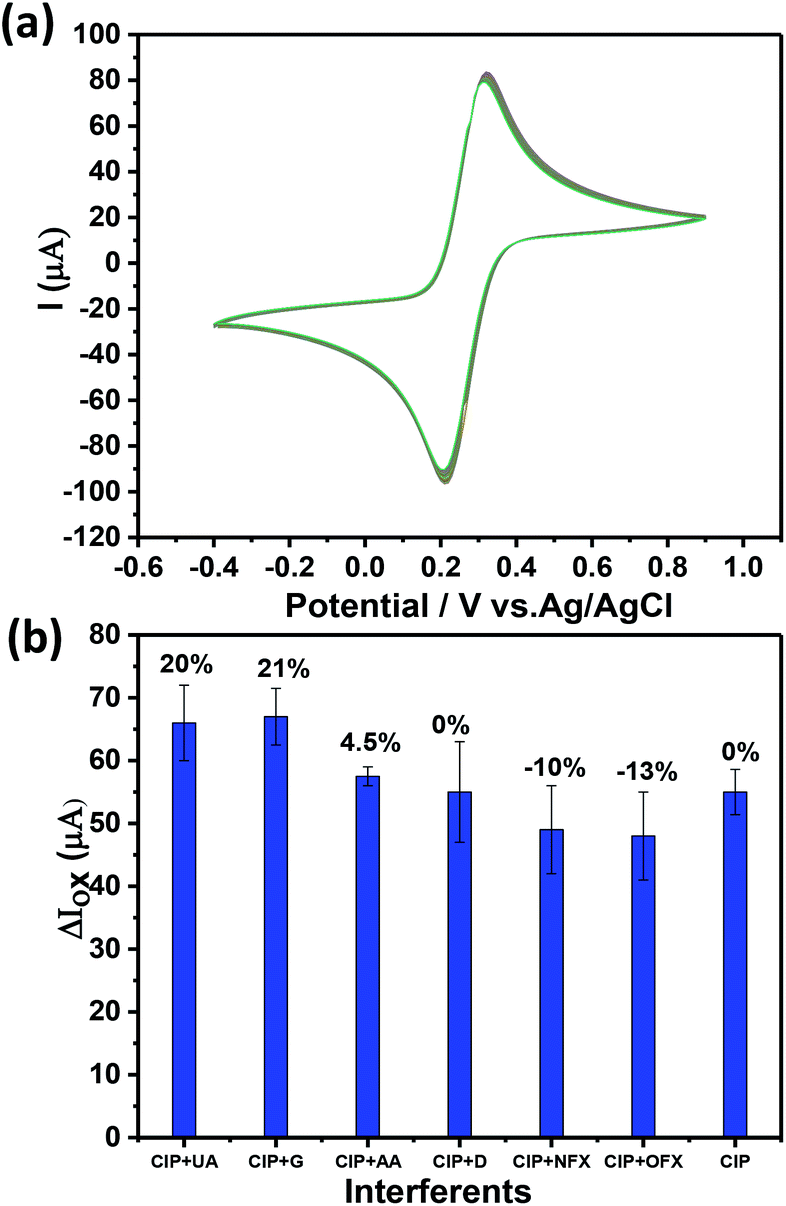 | ||
| Fig. 7 (a) The stability analysis of Ch-AuMIP for 100 cycles in 5 mM ferricyanide solution containing 0.1 M KCl. (b) The competitive study by mixing CIP and other interferents in equal ratios. | ||
The relative standard deviation (RSD) values were calculated from the current responses obtained for Ch-AuMIP was of ±5, which further assured good stability of the electrode. Thus it implies that even after 100 cycles of applied potential the electrode doesn't face any bias stability issues and continues to perform well in the CIP detection. The device coated with a single run of Ch-AuMIP performs well in the detection process for about 4 cycles with an average value of 46.75 μA with a standard deviation of 3.5.
To extend the selectivity study of the developed Ch-AuMIP/GCE, we added different interferents in the presence of CIP analyte and the corresponding results are plotted in Fig. 7b. The results indicate a relative standard deviation related to each possible interferent in the presence of CIP as shown in Fig. 7b. Further, one can notice that there was a change of −13% to +21% for different interferent combinations along with CIP. Apart from glucose and uric acid, the other interferents such as OFX, NFX, ascorbic acid, and dopamine exhibited a small change in the current intensity compared to CIP alone. In the case of dopamine alone as interferent, there was no interference effect observed. Overall, this data shows the good selectivity of the developed Ch-AuMIP/GCE sensing system.
Analytical application of the electrochemical sensor in real samples
In order to evaluate the performance of the Ch-AuMIP/GCE sensor in real analytical applications, the voltammetric procedure was carried out for the determination of CIP in real water samples (tap and mineral water samples), commercial milk, and as well as in a pharmaceutical formulation. The samples were first spiked with 10 μM of CIP and analyzed following the above procedure described in the experimental section.Table 2 summarized the obtained results for the different tested real samples. The recoveries obtained were in the range of 93.9–108.4% with RSD values comprised between 2% and 7%. A commercial pharmaceutical product containing CIP as an active compound was analyzed using DPV measurements following the above described electroanalytical strategy. The obtained results for three consecutive determinations of CIP were 518.33 ± 10.69 which is almost equivalent to the labeled value (500 mg). RSD obtained for three determinations was lower than 2%. Subsequently, we tested a commercial milk sample fortified with 10 μM of CIP and analyzed using the proposed sensor. The results obtained show satisfactory recovery of CIP almost equivalent to 94%. Thus, the results obtained for the analysis of CIP in real samples show the efficiency of the sensing method for multiple applications. To determine the CIP presence in water samples and in milk by applying a new metrological alternative to demonstrate accuracy. As we have only one type of sensor and different water sources, a single factor ANOVA was sufficient to analyze the data. The obtained p-value for this method was 0.018836569 and is lower than 0.05, which means that the differences observed are not due to random sampling (Table. S3†). The population from different water sources does not have identical means and at least one mean differs in the population from the rest. But even such a difference is not huge as p ∼ 0.02 and not less by an order or more. Thus at least one mean data in the population is significantly different (p < 0.05) by ANOVA and Tukey test.
| Real sample | CIP amount added (μM) | CIP amount founda (μM) | Recovery% | RSD% |
|---|---|---|---|---|
| a Data followed by the same superscript are negligibly different (p = 0.018) by ANOVA and Tukey test. Where we performed 4 repetitive tests. | ||||
| Tap water | 10 | 9.98 ± 0.02 | 99.8 | 5 |
| Milk | 10 | 9.39 ± 0.17 | 93.9 | 2 |
| M1 | 10 | 9.99 ± 0.72 | 99.9 | 7 |
| M2 | 10 | 10.8 ± 0.45 | 108.4 | 4 |
| M3 | 10 | 10.6 ± 0.3 | 106.3 | 3 |
Conclusions
In this work, we presented the synthesis of chitosan gold nanoparticles decorated MIP and its application for the development of a sensitive and selective electrochemical sensor for CIP detection. The developed sensing system based on MIP Ch-AuNPs composite could specifically recognize CIP, which resulted in a decrease of the peak current intensity of ferricyanide using the modified electrode. The proposed sensor was characterized and used for the electrochemical determination of CIP in a concentration range between 1 to 100 μmol L−1 with a detection limit of 210 nmol L−1. We achieved 66% sensitivity for the target molecule CIP and sensitivities of 40%, 40%, 36%, 45%, 44% and 45% for DA, AA, Glu, UA, NFX and OFX, respectively. The sensing system showed easiness of preparation, good reproducibility of measurement as well as good selectivity towards CIP analyte. The developed sensing strategy was successfully applied for CIP determination in real samples of milk, mineral and tap water as well as in a pharmaceutical formulation. The proposed sensing system showed several advantages such as easiness of electrode preparation, a low-cost and good application for real samples.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors would like to acknowledge the Department of Science and Technology (DST), Government of India (Project INT/RUS/RFBR/P-292 dated 20/10/2017). We also thank Tutku Beduk Ashirova for her help with the ANOVA calculations.Notes and references
- M. Cruciani and D. Bassetti, J. Antimicrob. Chemother., 1994, 33, 403–417 CrossRef CAS PubMed.
- F. Kamali, S. H. L. Thomas and C. Edwards, Eur. J. Clin. Pharmacol., 1993, 44, 365–367 CrossRef CAS PubMed.
- I. Sanseverino, A. C. Navarro, R. Loos, D. Marinov, and T. Lettieri, State of the Art on the Contribution of Water to Antimicrobial Resistance, Publications Office of the European Union, 2018 Search PubMed.
- B. L. Wise, C. Peloquin, H. Choi, N. E. Lane and Y. Zhang, Am. J. Med., 2012, 125, 1223–1228 CrossRef PubMed.
- M. Assouad, R. J. Willcourt and P. H. Godman, Ann. Intern. Med., 1995, 122, 396–397 CrossRef CAS PubMed.
- D. G. J. Larsson, Upsala J. Med. Sci., 2014, 119, 108–112 CrossRef PubMed.
- G. Klecak, F. Urbach and H. Urwyler, J. Photochem. Photobiol., B, 1997, 37, 174–181 CrossRef CAS.
- C. Pierfitte, P. Gillet and R. J. Royer, N. Engl. J. Med., 1995, 332, 193 CrossRef CAS PubMed.
- R. Wise, J. Andrews and L. Edwards, Antimicrob. Agents Chemother., 1983, 23, 559–564 CrossRef CAS PubMed.
- N. Tarannum, O. D. Hendrickson, S. Khatoon, A. V. Zherdev and B. B. Dzantiev, Crit. Rev. Anal. Chem., 2019, 1–20 CrossRef PubMed.
- A. A. Lahcen and A. Amine, Electroanalysis, 2019, 31, 188–201 CrossRef CAS.
- R. Ahmad, N. Griffete, A. Lamouri, N. Felidj, M. M. Chehimi and C. Mangeney, Chem. Mater., 2015, 27, 5464–5478 CrossRef CAS.
- W. Zhao, T. FangKang, L. Lu, F. Shen and S. Cheng, J. Electroanal. Chem., 2017, 786, 102–111 CrossRef CAS.
- S. Ansari and M. Karimi, Talanta, 2017, 164, 612–625 CrossRef CAS.
- A. Speltini, A. Scalabrini, F. Maraschi, M. Sturini and A. Profumo, Anal. Chim. Acta, 2017, 974, 1–26 CrossRef CAS.
- X. Sun, F. Li, G. Shen, J. Huang and X. Wang, Analyst, 2014, 139, 299–308 RSC.
- A. A. Lahcen, A. Baleg, P. Baker, E. Iwuoha and A. Amine, Sens. Actuators, B, 2018, 276, 114–120 CrossRef.
- A. Zamora-Galvez, A. Ait-Lahcen, L. A. Mercante, E. Morales-Narvaez, A. Amine and A. Merkoci, Anal. Chem., 2016, 88, 3578–3584 CrossRef CAS PubMed.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- L. Uzun and A. P. F. Turner, Biosens. Bioelectron., 2016, 76, 131–144 CrossRef CAS PubMed.
- X. W. Kan, Y. Zhao, Z. R. Geng, Z. L. Wang and J. J. Zhu, J. Phys. Chem., 2008, 112, 4849–4854 CrossRef CAS PubMed.
- D. Lakshmi, A. Bossi, M. J. Whitcombe, I. Chianella, S. A. Fowler, S. Subrahmanyam, E. V. Piletska and S. A. Piletsky, Anal. Chem., 2009, 81, 3576–3584 CrossRef CAS PubMed.
- W. Guo, F. Pi, H. Zhang, J. Sun, Y. Zhang and X. Sun, Biosens. Bioelectron., 2017, 98, 299–304 CrossRef CAS PubMed.
- C. G. Xie, H. F. Li, S. Q. Li, J. Wu and Z. P. Zhang, Anal. Chem., 2010, 82, 241–249 CrossRef CAS PubMed.
- X. Shan, J. D. Habimana, J. Ji, J. Sun, F. Pi, Y. Zhang and X. Sun, J. Solid State Electrochem., 2019, 23, 1211–1220 CrossRef CAS.
- R. Singh, R. Verma, A. Kaushik, G. Sumana, S. Sood, R. K. Gupta and B. D. Malhotra, Biosens. Bioelectron., 2011, 26, 2967–2974 CrossRef CAS PubMed.
- X. Xuan, H. S. Yoon and J. Y. Park, Biosens. Bioelectron., 2018, 109, 75–82 CrossRef CAS PubMed.
- S. L. Wei, Y. Liu, T. Hua, L. Liu and H. W. Wang, J. Appl. Polym. Sci., 2014, 131, 1–8 CrossRef.
- W. J. Lian, S. Liu, J. H. Yu, X. R. Xing, J. Li, M. Cui and J. D. Huang, Biosens. Bioelectron., 2012, 38, 163–169 CrossRef CAS PubMed.
- W. J. Lian, S. Liu, J. H. Yu, J. Li, M. Cui, W. Xu and J. D. Huang, Anal. Lett., 2013, 46, 1117–1131 CrossRef CAS.
- X. L. Xu, S. Zhang, H. Chen and J. L. Kong, Talanta, 2009, 80, 8–18 CrossRef CAS PubMed.
- A. Regiel-Futyra, M. Kus-Liskiewicz, V. Sebastian, S. Irusta, M. Arruebo, G. Stochel and A. Kyziol, ACS Appl. Mater. Interfaces, 2015, 7, 1087–1099 CrossRef CAS PubMed.
- J. Wang, Y. X. Sang, W. H. Liu, N. Liang and X. H. Wang, Anal. Methods, 2017, 9, 6682–6688 RSC.
- Y. D. Wang, E. L. Wang, H. Dong, F. Liu, Z. M. Wu, H. A. Li and Y. Wang, Adsorpt. Sci. Technol., 2014, 32, 321–330 CrossRef CAS.
- J. Brugnerotto, J. Lizardi, F. M. Goycoolea, W. Arguelles-Monal, J. Desbrieres and M. Rinaudo, Polymer, 2001, 42, 3569–3580 CrossRef CAS.
- C. Z. Fan, K. Li, Y. L. Wang, X. F. Qian and J. P. Jia, RSC Adv., 2016, 6, 2678–2686 RSC.
- S. S. Pati, L. H. Singh, E. M. Guimaraes, J. Mantilla, J. A. H. Coaquira, A. C. Oliveira, V. K. Sharma and V. K. Garg, J. Alloys Compd., 2016, 684, 68–74 CrossRef CAS.
- C. Fenzl, P. Nayak, T. Hirsch, O. S. Wolfbeis, H. N. Alshareef and A. J. Baeumner, ACS Sens., 2017, 2(5), 616–620 CrossRef CAS PubMed.
- M. Elfiky, N. Salahuddin, A. Hassanein, A. Matsuda and T. Hattori, Microchem. J., 2019, 146, 170–177 CrossRef CAS.
- H. Nawaz, S. Rauf, K. Akhtar and A. M. Khalid, Anal. Biochem., 2006, 354, 28–34 CrossRef CAS PubMed.
- A. A. Ensafi, M. Taei, T. Khayamian and F. Hasanpour, Anal. Sci., 2010, 26, 803–808 CrossRef CAS PubMed.
- G. S. Garbellini, R. C. Rocha and O. Fatibello, Anal. Methods, 2015, 7, 3411–3418 RSC.
- L. Fotouhi and M. Alahyari, Colloids Surf., B, 2011, 81, 110–114 CrossRef PubMed.
- N. S. E. Osman, N. Thapliyal, W. S. Alwan, R. Karpoormath and T. Moyo, J. Mater. Sci.: Mater. Electron., 2015, 26, 5097–5105 CrossRef CAS.
- Y. H. Yuan, F. F. Zhang, H. Y. Wang, L. N. Gao and Z. H. Wang, ECS J. Solid State Sci. Technol., 2018, 7, 201–208 CrossRef.
- H. Yi and C. Li, Russ. J. Electrochem., 2007, 43, 1377–1381 CrossRef CAS.
- G. Bo, X. P. He, Y. Jiang, J. T. Wei, H. Suo and C. Zhao, J. Sep. Sci., 2014, 37, 3753–3759 CrossRef PubMed.
- W. Chunxia, R. Cheng, J. Wang, Y. Wang, X. Jing, R. Chen, L. Sun and Y. Yan, J. Sep. Sci., 2018, 41, 3782–3790 CrossRef PubMed.
- C. Yan, J. Li, T. Meng, X. Liu, R. Zhang, Y. Chen and G. Wang, Int. J. Electrochem. Sci., 2016, 11(8), 6466–6476 CrossRef CAS.
- H. Bagheri, H. Khoshsafar, S. Amidic and Y. H. Ardakanid, Anal. Methods, 2016, 8, 3383–3390 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01838d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |