
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination environment evolution of Co(II) during dehydration and re-crystallization processes of KCoPO4·H2O towards enhanced electrocatalytic oxygen evolution reaction†
Quande Che *a,
Xiaobin Xieb,
Qian Maa,
Junpeng Wanga,
Yuanna Zhua,
Ruixia Shia and
Ping Yang*a
*a,
Xiaobin Xieb,
Qian Maa,
Junpeng Wanga,
Yuanna Zhua,
Ruixia Shia and
Ping Yang*a
aSchool of Materials Science and Engineering, University of Jinan, Jinan 250022, P. R. China. E-mail: mse_cheqd@ujn.edu.cn; mse_yangp@ujn.edu.cn; Fax: +86 531 87974453; Tel: +86 531 89736225
bSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China
First published on 16th April 2020
Abstract
Development of efficient and stable electrodes for electrocatalytic oxygen evolution reaction (OER) is essential for energy storage and conversion applications, such as hydrogen generation from water splitting, rechargeable metal–air batteries and renewable fuel cells. Alkali metal cobalt phosphates show great potential as OER electrocatalysts. Herein, an original electrode design strategy is reported to realize an efficient OER electrocatalyst through engineering the coordination geometry of Co(II) in KCoPO4·H2O by a facile dehydration process. Experimental result indicated that the dehydration treatment is accompanied by a structural transformation from orthorhombic KCoPO4·H2O to hexagonal KCoPO4, involving a concomitant coordination geometry evolution of Co(II) from octahedral to tetrahedral configuration. More significantly, the local structural evolution leads to an advantageous electronic effect, i.e. increased Co–O covalency, resulting in an enhanced intrinsic OER activity. To be specific, the as-produced KCoPO4 can deliver a current density of 10 mA cm−2 at a low overpotential of 319 mV with a small Tafel slope of 61.8 mV dec−1 in alkaline electrolyte. Thus, this present research provides a new way of developing alkali metal transition-metal phosphates for efficient and stable electrocatalytic oxygen evolution reaction.
1. Introduction
The oxygen evolution reaction (OER) is one of the crucial steps of energy storage and conversion applications, such as hydrogen generation from water splitting, rechargeable metal–air batteries and renewable fuel cells.1–5 However, the intrinsic sluggish kinetics of the OER involving four-electron transfer requires an overpotential to drive the process.6–8 Therefore, efficient electrocatalysts with high activity and high stability are desirable to realize its practical application.9,10 To date, noble-metal oxides, such as RuO2 and IrO2 are considered to be the state-of-the-art electrocatalysts for the OER,11,12 but their high cost, limited reserves and stability problem prevent their large-scale industrial application.13 Currently, extensive research efforts have been focused towards the development of cost-effective transition-metal based OER catalysts.14–20In particular, alkali metal cobalt phosphates have gained increasing attention due to their high activity and favorable kinetics, such as orthophosphate (LiCoPO4),21 pyrophosphate (NaCoP2O7),22 and metaphosphate (NaCo(PO3)3).23 The diverse orientations of phosphate ligands lead to various crystal structures, which are usually beneficial for the structural stability during OER process.24–26 As illustrated in Fig. S2,† phosphate/pyrophosphate/metaphosphate-containing compounds have various cobalt(II) geometries including octahedral (Oh), tetrahedral (Td) and trigonal bipyramidal (TBP), with diverse connections and low-symmetry crystal structures due to the bulky phosphate ligands. Thus, alkali metal Co phosphates with various structural configurations provide an ideal platform to design efficient and stable OER electrocatalyst. Previous research has demonstrated that the Co geometric coordination has a great impact on the OER activity of Co based phosphates. Kim et al. selected four Co based phosphates with various Co coordination environment, including Na2CoP2O7 (Td), LiCoPO4 (Oh), NaCoPO4 (Oh), Li2CoP2O7 (Oh/TBP), as a platform to explore the relationship between the local coordination and the OER activity.27 The result demonstrated that the Na2CoP2O7 shows enhanced activity relative to NaCoPO4 due to a highly distorted tetrahedral geometry. Nevertheless, Wan et al. subsequently reported that the NaCo4(PO4)3 containing a rare 5 coordinated Co(II) outperformed the Na2CoP2O7 (Td).28 Besides, some other reported alkali metal cobalt phosphates, such as LiCoPO4 (Oh)21 and NaCo(PO3)3 (Oh)23 exhibited acceptable OER activity.
Admittedly, local coordination geometry in alkali metal cobalt phosphates plays a significant role in improving their OER activity. However, there is still a lack of deep understanding of the origin of this relationship. It is well accepted that OER activity are highly dependent on both geometric configuration and electronic structure of electrocatalysts. Thus, to address this issue, there is the need for reveal the dual effect of coordination geometry of Co(II) in alkali metal cobalt phosphates. In this work, we found that the dehydration treatment of KCoPO4·H2O is accompanied by a structural transformation from orthorhombic to hexagonal phase, involving a concomitant coordination geometry evolution of Co(II) from octahedral to tetrahedral configuration. By this, special consideration was paid to the influence of this local structural evolution on electronic structure. Experimental result and theoretical analysis indicated that the co coordination evolution increases the Co–O covalency, resulting in an enhanced OER activity. The as-resulted KCoPO4 exhibited an enhanced OER activity relative to the pristine KCoPO4·H2O. Specifically, it can deliver a current density of 10 mA cm−2 at a low overpotential of 319 mV with a small Tafel slope of 61.8 mV dec−1 in alkaline electrolyte. Moreover, stability test demonstrated it can hold its catalytic OER activity for 50 h at least. Besides, to the best of our knowledge, this work for the first time demonstrated the OER performance of the hexagonal KCoPO4 containing Co(II)-Td local coordination environment. Overall, this uniqueness of this material design not only lies in an effective strategy for local coordination evolution, but more significantly it provides an ideal platform to better understand the local coordination–activity relationship for alkali metal cobalt phosphates electrocatalysts.
2. Experimental
KCoPO4·H2O nanoplates were synthesized via a simple hydrothermal process. In a typical step, an aqueous solution of CoCl2·6H2O (0.004 mol, 30 ml) was completely added into an aqueous solution of K2HPO4 (0.04 mol, 30 ml) under continuous stirring. After stirred for 2 h at room temperature (25 °C), the mixture was transferred into a Teflon-lined stainless autoclave and kept at 140 °C in an electric drying box for 20 h. The resulting pink product was collected by centrifugation, rinsed with the DI water and absolute ethanol, dried at 50 °C for 12 h to obtain the KCoPO4·H2O nanoplates.The coordinated water molecule in KCoPO4·H2O can be removed by heating at 300 °C under N2 atmosphere for 3 h to obtain the dehydrated phase, i.e. KCoPO4. The step involved in the dehydration process is illustrated in Fig. 1(a). The characterization techniques and the electrochemical measurements were explained in the ESI.†
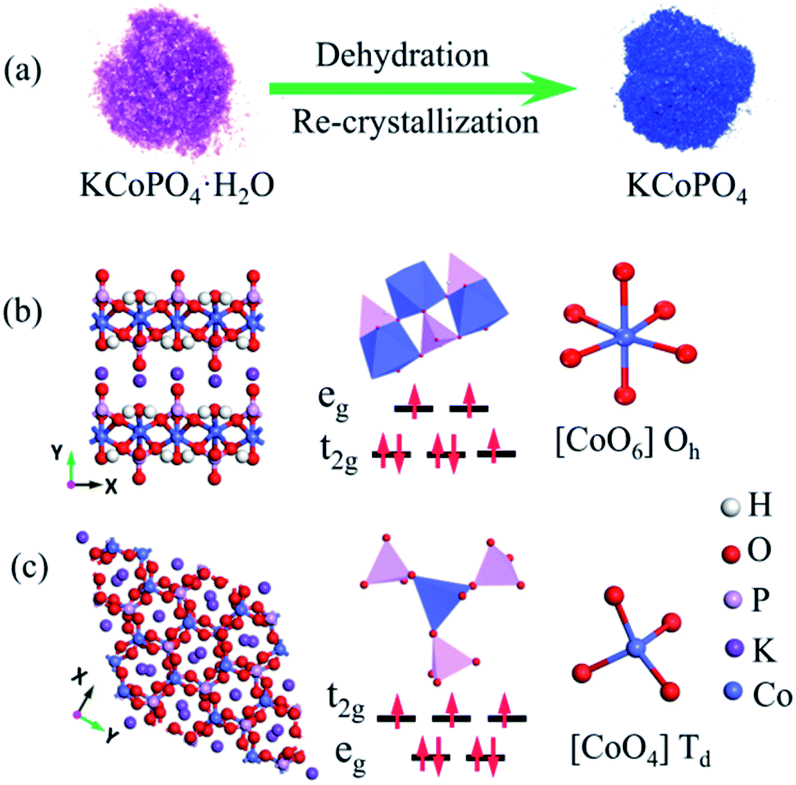 | ||
| Fig. 1 (a) Schematic illustration for the dehydration and re-crystallization processes of KCoPO4·H2O. Crystal structures of (b) KCoPO4·H2O, and (c) KCoPO4. The inset shows the local environment around the Co subunit (blue) and the spin state of the Co(II). | ||
3. Results and discussion
The XRD pattern (Fig. 2(a)) indicates the successful preparation of the KCoPO4·H2O. The diffraction peaks of the obtained powders perfectly matched to the orthorhombic-phase potassium cobalt phosphate monohydrate KCoPO4·H2O (JCPDS no. 86-0590), while the dehydrated phase shows different crystalline structure. Upon dehydration, the KCoPO4·H2O was transformed into hexagonal-phase KCoPO4 (JCPDS no. 82-0762), as indicated in Fig. 2(b). Fig. 1(b and c) shows the crystal structures of KCoPO4·H2O and KCoPO4, respectively. It is clearly illustrated that the dehydration is accompanied by a structural transformation and re-crystalline process. Observations by the SEM (Fig. 3(a and b)) and TEM (Fig. 3(c and e)) indicate that the as-synthesized KCoPO4·H2O and dehydrated KCoPO4 exhibit plate-like nanostructures with an average size tunable from 2 to 5 μm. Furthermore, the HRTEM images show the interlayer d spacing of 0.41 nm (Fig. 3(d)) and 0.31 nm (Fig. 3(f)) nm of the two phases, which are consistent with the XRD data.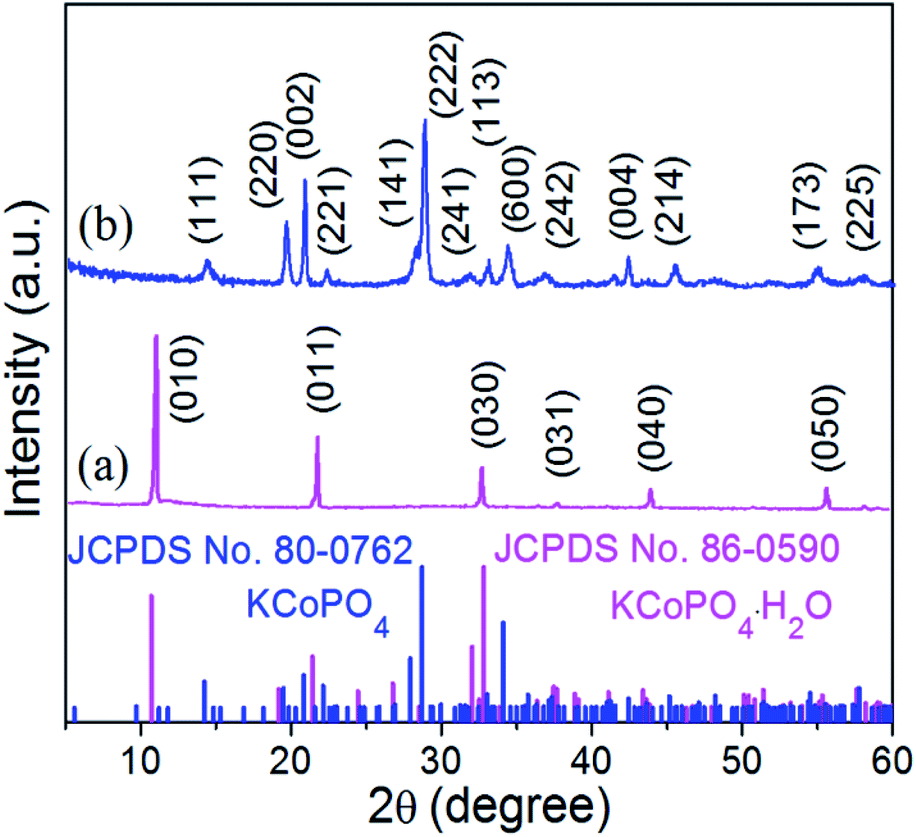 | ||
| Fig. 2 XRD patterns of the as-prepared (a) KCoPO4·H2O (JCPDS no. 86-0590), and (b) KCoPO4 (JCPDS no. 82-0762), respectively. | ||
 | ||
| Fig. 3 SEM images of the as-prepared (a) KCoPO4·H2O, and (b) KCoPO4. TEM (c and e) and HRTEM (d and f) images of KCoPO4·H2O and KCoPO4, respectively. | ||
Thermogravimetric analysis (TGA) was carried out under flowing N2 to further demonstrate the dehydration and re-crystalline process of KCoPO4·H2O, as shown in Fig. 4(a). The TG curve shows a sharp weight loss of 8.61%, and is characterized by a strong endothermic DTG peak at 195.7 °C, which corresponds to the loss of coordinated water molecule in KCoPO4·H2O (calcd: 8.53%). There is no weight after that, indicating it transforms to be a stable KCoPO4.
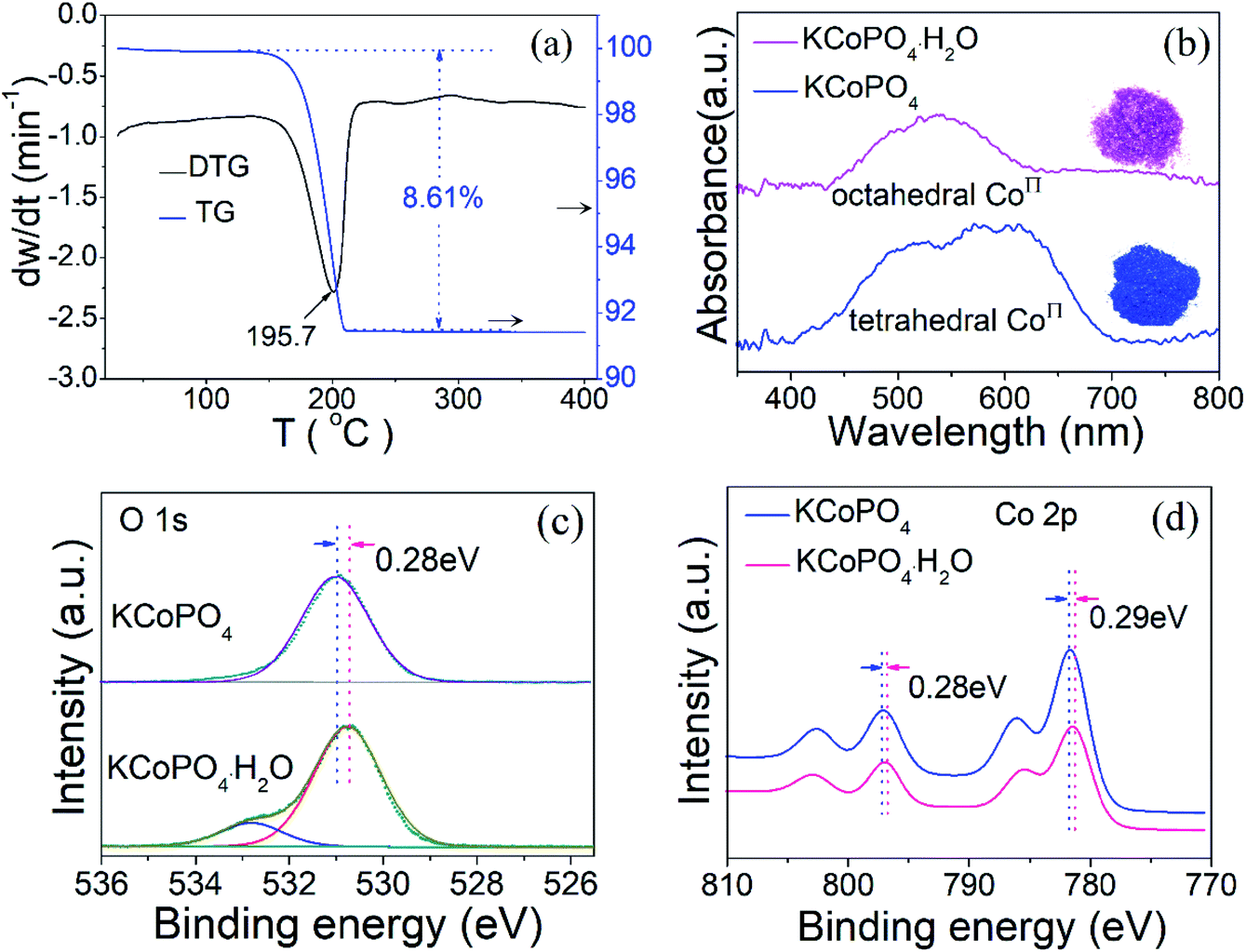 | ||
| Fig. 4 (a) TGA/DTG curves of the KCoPO4·H2O nanoplates. (b) UV-vis spectra of KCoPO4·H2O and KCoPO4. High-resolution XPS spectra for (c) O 1s, and (d) Co 2p of the KCoPO4·H2O and KCoPO4, respectively. | ||
Significantly, upon dehydration, the coordination environment around Co(II) changed from octahedral to tetrahedral configuration, which is demonstrated by a concomitant color change from pink to blue. In other words, The Co–O bond from the oxygen in the water molecule coordinated with Co(II) is lost during dehydration, leading to the coordination of the Co(II) becomes to tetrahedral. The diffuse-reflectance solid-state UV-vis spectra of the two phases was shown in Fig. 4(b). The spectrum of KCoPO4·H2O shows a broad band located at approximately 539 nm, which correspond to the 4T1g (F) 4A2g (P) transition (ν3 band) of high-spin Co(II) in octahedral [CoO5–(H2O)]2+,29 while for KCoPO4 the bands located at about 526 nm, 575 nm, and 615 nm are assigned to the 4A2g (F) 4T1 (P) of high-spin tetrahedral Co(II).30 Thus, the UV-vis spectra suggests a structural conversion from octahedral to tetrahedral coordination. Fig. 1(b and c) shows the local coordination environment and the spin state of Co(II) in the two phases, respectively. As mentioned above, the Co geometric coordination has a great impact on the OER activity of Co based phosphates. Therefore, the coordination environment evolution of Co(II) in the two phases motivate us to explore the corresponding change of their OER performance and the original relationship between coordination configuration with OER activity of Co phosphates. However, just knowing the structural transformation is far from enough, there is the need for further efforts to reveal the corresponding electronic effect.
The chemical compositions and bonding states in the as-prepared KCoPO4·H2O and dehydrated KCoPO4 were examined by XPS, which contributes to estimate the electron states of Co in the two phases, and in turn is conductive to reveal the electronic effect of coordination environment evolution of Co(II) on OER activity. First, the full survey spectra (Fig. S3(a and b)†) indicates the presence of cobalt (Co 2p), phosphorus (P 2p), oxygen (O 1s), potassium (K 2p) from both of the two compounds. Furthermore, the O 1s spectrum of KCoPO4·H2O (Fig. 4(c)) fitted into two peaks situated at 530.7 eV and 532.8 eV can be ascribed to the P–O bonds and coordinated water molecule, respectively,31 while for KCoPO4 only lattice oxygen can be tested, as illustrated by the peak located at about 531.0 eV. More significantly, it is found that there is a positive shift (about 0.3 eV) of binding energy for oxygen after the dehydration process, which indicates the increased valence state of oxygen in KCoPO4. Similarly, the valence state of cobalt increases on dehydration, as confirmed by the positive shift (about 0.3 eV) of binding energy, as shown in (Fig. 4(d)). Specifically, the Co 2p spectrum of KCoPO4·H2O fitted into two peaks located at 796.8and 781.3 eV, while for KCoPO4 the peaks situated at 797.1 and 781.6 eV, respectively. Besides, the Co 2p spectra of the two compounds indicate the Co2+ oxidation state of cobalt in both of them.32 In a word, the XPS result suggests the increased degree of Co–O hybridization, i.e. Co–O covalency in KCoPO4.
Based on the above characterizations including crystal structure and chemical bonding states, it is clearly indicated that the dehydration process of KCoPO4·H2O is accompanied by a structural transformation from orthorhombic-phase to hexagonal-phase. More significantly, the local coordination geometry evolution of Co(II) from octahedral to tetrahedral configuration increases the Co–O covalency. Previous research demonstrated that the geometric configuration and electronic structure of cobalt phosphates can largely influence its OER catalytic activity.33,34 Therefore, the local Co geometry evolution with increased Co–O covalency may imply an enhanced OER activity.
The OER performance of the as-prepared two compounds was evaluated via linear scanning voltammetry (LSV) using a conventional three-electrode system at a scan rate of 5 mV s−1 in 1.0 M KOH (electrochemical measurements, see details in ESI†). Fig. 5(a) shows the LSV curves of the as-prepared KCoPO4, together with the KCoPO4·H2O, and RuO2 for comparison. As illustrated in Fig. 5(a), the KCoPO4·H2O requires 387 mV overpotential to reach a current density of 10 mA cm−2. On KCoPO4, the overpotential greatly decreased to 319 mV, which is even comparable to that of the reference sample (309 mV for noble RuO2). Not only that, the catalytic performance of KCoPO4 is comparable to or even superior to many recently reported transition-metal phosphate based electrocatalysts (the detailed comparison can be seen in Table S3, ESI†). Besides, a smaller Tafel slope of 61.8 mV dec−1 of KCoPO4 than that of KCoPO4·H2O (66.2 mV dec−1) indicates the favorable reaction kinetic for OER, as shown in Fig. 5(b). It could also be inferred from the electrochemical impedance spectroscopy (EIS) result (Fig. 5(c)). An equivalent circuit model was suggested, as shown in inset of Fig. 5(c). The charge transfer resistance (Rct), was tested to be 36.1 Ω for KCoPO4, much lower than 139.9 Ω for KCoPO4·H2O. Furthermore, CV curves with different scan rates were recorded to evaluate electrochemically active surface area (ECSA) of the two compounds, as shown in Fig. 6(a and b). It can be evaluated with the electrochemical double-layer capacitance (Cdl), which is determined by current density differences plotted against various scan rates. Specifically, the Cdl of KCoPO4 was calculated to be 14.15 mF cm−2, which is very close to that of KCoPO4·H2O (13.55 mF cm−2), indicating an equivalent ECSA value of them. Therefore, it is proposed that the transformation of KCoPO4·H2O to KCoPO4 lead to the enhanced intrinsic OER activity. In order to estimate the intrinsic activity, turnover frequency (TOF) was calculated for them. The TOF value of KCoPO4, KCoPO4·H2O and RuO2 were 0.0635 s−1, 0.0413 s−1, 0.015 s−1, respectively, which are comparable with the previously reported values.35 Such a high TOF value means a very high intrinsic activity of KCoPO4.
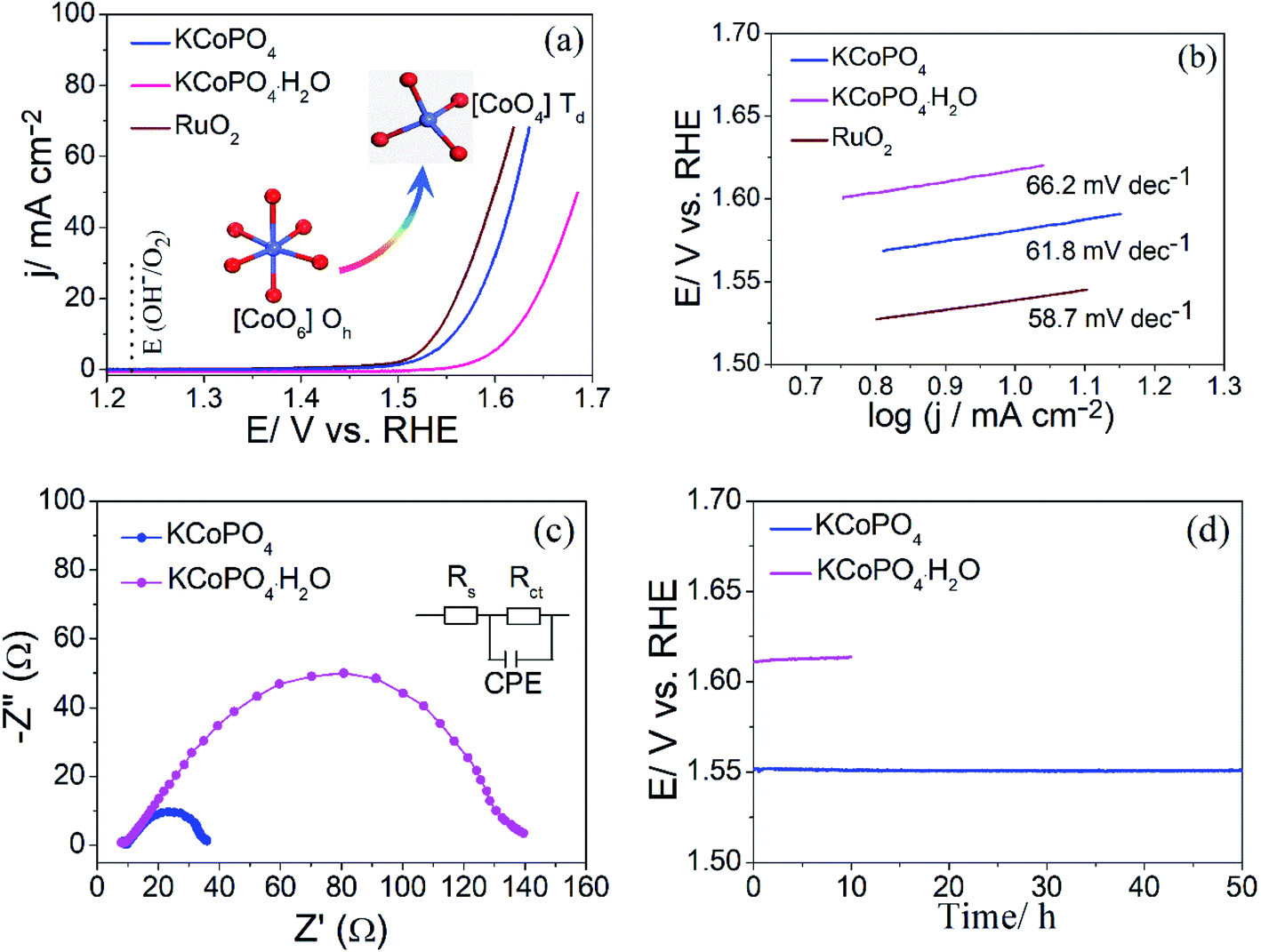 | ||
| Fig. 5 OER performance of the as-prepared KCoPO4·H2O, KCoPO4, and RuO2. (a) LSV curves, (b) corresponding Tafel plots, (c) EIS spectra, and (d) chronopotentiometric curves at 10 mA cm−2. | ||
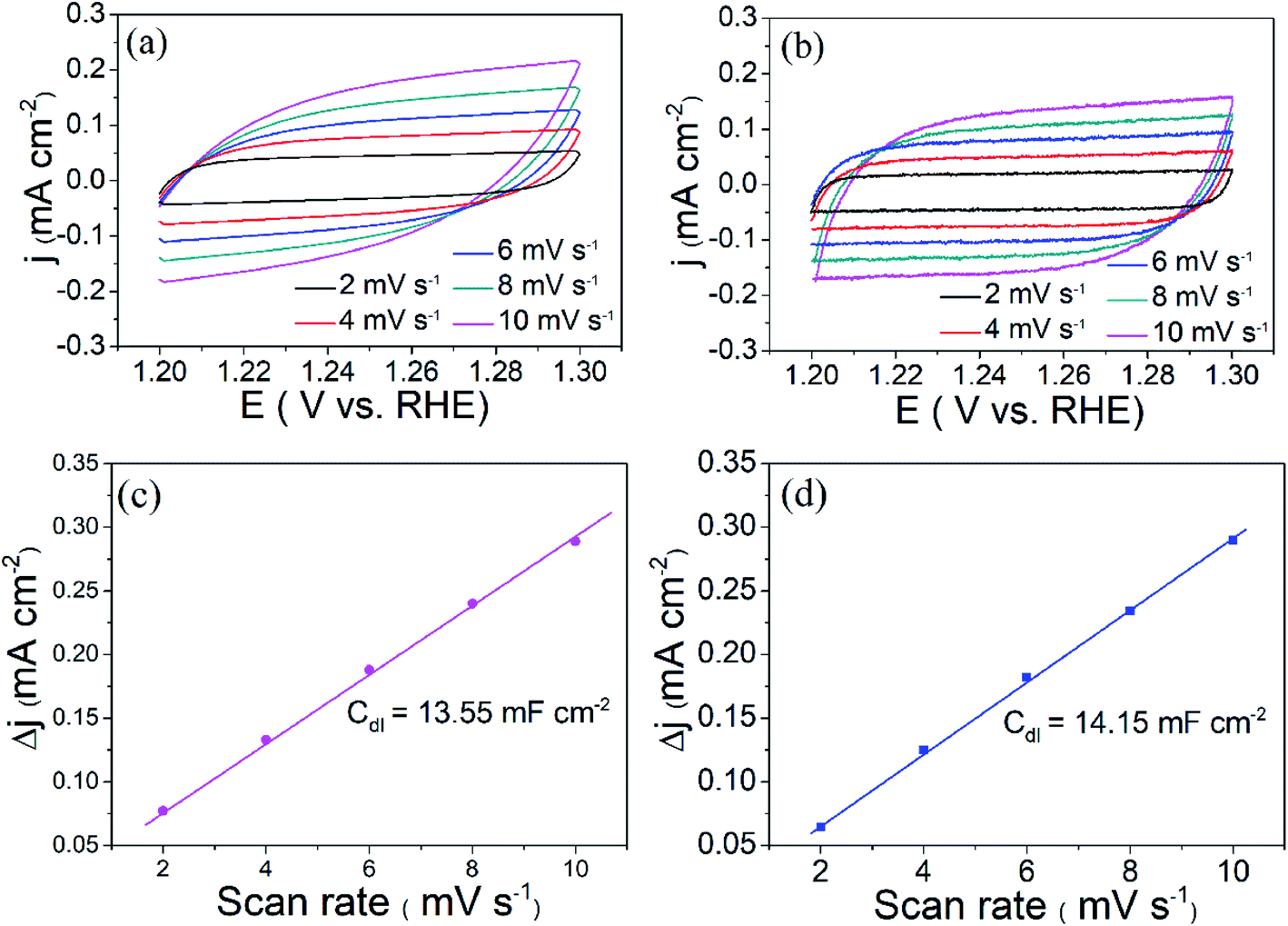 | ||
| Fig. 6 Cyclic voltammetry curves of (a) KCoPO4·H2O, and (b) KCoPO4 in 1 M KOH with different scan rates. Δj of as-above electrodes plotted against scan rates, (c) KCoPO4·H2O, and (d) KCoPO4. | ||
Based on the above comparison study on structural transformation and OER performance evaluation, it is suggested that the enhanced OER activity is mainly derived from the electronic effect of local Co(II) geometry, i.e. the increased Co–O covalency.
In addition, the stability measurement for the as-prepared KCoPO4 manifests that the long-term durability requirement can be met. The chronopotentiometry result of KCoPO4 at a constant 10 mA cm−2, as shown in Fig. 5(d), indicating the potential kept stable at ∼1.55 V in an OER process up to 50 h. The LSV curve after 50 h test exhibited a negligible shift, as shown in Fig. S4.† Besides, the structural characterizations, as illustrated in Fig. S5,† further demonstrated the stability.
Based on the above analysis, it is suggested that the distinct OER activity is associated with the local electronic structure modulation by coordination environment evolution of Co(II). In other words, the local Co geometry evolution from octahedral to tetrahedral configuration with increased Co–O covalency result in an enhanced OER activity. This analysis is consistent with the previous research. Recent studies demonstrated that the increased Co–O covalency promotes the interfacial charge transfer between Co and oxygen intermediates, resulting in an enhanced OER kinetics.36–39 To further confirm the electronic effect, first-principles density functional theory plus Hubbard U (DFT+U) calculations were performed on the two compounds (computational models and methods can be seen in the ESI†). The calculated partial densities of the states (pDOS) of Co 3d and O 2p band are shown in Fig. 7(a and b). As a feasible descriptor for OER activity, the electronic parameter, i.e., O 2p band center (εO2p) can be extracted out from the calculated pDOS,40–42 which also indicates the degree of Co–O hybridization, as illustrated in Fig. 7(c and d). In specific, a larger εO2p signify greater covalency and better OER activity. It is observed that the calculated εO2p of KCoPO4 (−2.95 eV) is more positive than that of KCoPO4·H2O (−3.24 eV), indicating the increased Co–O covalency, which is confirmed by the XPS analysis.
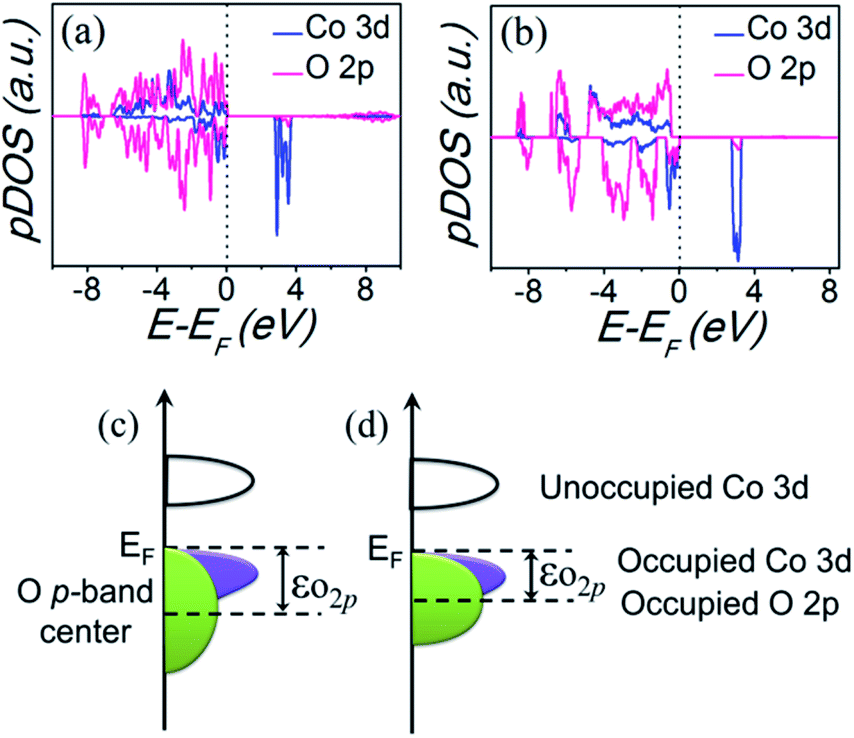 | ||
| Fig. 7 Computed partial electronic density of states (pDOS) of Co 3d and O 2p band of (a) KCoPO4·H2O, and (b) KCoPO4. Illustration of the relationships between Co 3d and O 2p band of (c) KCoPO4·H2O, and (d) KCoPO4, respectively. | ||
4. Conclusions
In summary, KCoPO4·H2O nanoplates were successfully synthesized via a hydrothermal process. The coordination water molecule in KCoPO4·H2O can be removed by heating at 300 °C under N2 atmosphere to obtain the dehydrated KCoPO4. Significantly, the dehydration process is accompanied by a phase transformation, and a local coordination environment evolution of Co(II) from octahedral to tetrahedral configuration, which increases the Co–O covalency, resulting in an enhanced OER activity. In specific, the dehydrated KCoPO4 can deliver a current density of 10 mA cm−2 at a low overpotential of 319 mV with a small Tafel slope of 61.8 mV dec−1 in alkaline electrolyte. Thus, this present research enlightens a new way of developing alkali metal transition-metal phosphates for efficient and stable water oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (51602128), the Research Foundation from University of Jinan (XKY1401, XBS1508, XBH1504).Notes and references
- T. Fujimura, W. Hikima, Y. Fukunaka and T. Homma, Electrochem. Commun., 2019, 101, 43 CrossRef CAS.
- W. Yang, K.-Y. Kim, P. E. Saikaly and B. E. Logan, Energy Environ. Sci., 2017, 10, 1025 RSC.
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989 CrossRef CAS PubMed.
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G.-D. Li, X. Zou and J.-S. Chen, Nat. Commun., 2018, 9, 2609 CrossRef PubMed.
- A. Pirkarami, S. Rasouli and E. Ghasemi, Appl. Catal., B, 2019, 241, 28 CrossRef CAS.
- S. Fukuzumi, Y. M. Lee and W. Nam, Dalton Trans., 2019, 48, 779 RSC.
- Y. Q. Li, C. Wang, M. Cui, S. R. Chen and T. L. Ma, Catal. Commun., 2019, 131, 105800 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399 CrossRef CAS PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430 CrossRef.
- Y. Zhang, X. Wang, F. Luo, Y. Tan, L. Zeng, B. Fang and A. Liu, Appl. Catal., B, 2019, 256, 117852 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011 CrossRef CAS PubMed.
- J. Yi, W. H. Lee, C. H. Choi, Y. Lee, K. S. Park, B. K. Min, Y. J. Hwang and H. S. Oh, Electrochem. Commun., 2019, 104, 106469 CrossRef CAS.
- J. Jiang, Q. Liu, C. Zeng and L. Ai, J. Mater. Chem. A, 2017, 5, 16929 RSC.
- X. F. Zhang, A. X. Shan, S. B. Duan, H. F. Zhao, R. M. Wang and W.-M. Lau, RSC Adv., 2019, 9, 40811 RSC.
- I. V. Odynets, N. Y. Strutynska, J. Z. Li, W. Han, I. V. Zatovsky and N. I. Klyui, Dalton Trans., 2018, 47, 15703 RSC.
- S. S. Zhu, J. L. Lei, Y. H. Qin, L. N. Zhang and L. J. Lu, RSC Adv., 2019, 9, 13269 RSC.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- Y. W. Liu, C. Xiao, M. J. Lyu, Y. Lin, W. Z. Cai, P. C. Huang, W. Tong, Y. M. Zou and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 1123 Search PubMed.
- M. Huynh, C. Y. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887 CrossRef CAS PubMed.
- J. Y. Wang, X. Teng, Y. L. Niu, L. X. Guo, J. F. Kong, X. M. He and Z. F. Chen, RSC Adv., 2019, 9, 21679 RSC.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959 CrossRef CAS PubMed.
- L. Q. Gui, X. Y. Miao, C. J. Lei, K. L. Wang, W. Zhou, B. B. He, Q. Wang and L. Zhao, Chem.–Eur. J., 2019, 25, 11007 CrossRef CAS PubMed.
- R. Gond, D. K. Singh, M. Eswaramoorthy and P. Barpanda, Angew. Chem., Int. Ed., 2019, 131, 8418 CrossRef.
- T. H. Zhou, Y. H. Du, D. P. Wang, S. M. Yin, W. G. Tu, Z. Chen, A. Borgna and R. Xu, ACS Catal., 2017, 7, 6000 CrossRef CAS.
- X. L. Hu, S. Piccinin, A. Laio and S. Fabris, ACS Nano, 2012, 6, 10497 CrossRef CAS PubMed.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109 RSC.
- H. Kim, J. Park, I. Park, K. Jin, S. E. Jerng, S. H. Kim, K. T. Nam and K. Kang, Nat. Commun., 2015, 6, 8253 CrossRef CAS PubMed.
- H. Wan, R. Z. Ma, X. H. Liu, J. L. Pan, H. D. Wang, S. Q. Liang, G. Z. Qiu and T. Sasaki, ACS Energy Lett., 2018, 3, 1254 CrossRef CAS.
- B. M. Weckhuysen, D. Baetens and R. A. Schoonheydt, Angew. Chem., Int. Ed., 2000, 39, 3419 CrossRef CAS PubMed.
- D. Grandjean, A. M. Beale, A. V. Petukhov and B. M. Weckhuysen, J. Am. Chem. Soc., 2005, 127, 14454 CrossRef CAS PubMed.
- C. Z. Wei, C. Cheng, S. S. Wang, Y. Z. Xu, J. D. Wang and H. Pang, Chem.–Asian J., 2015, 10, 1731 CrossRef CAS PubMed.
- B. J. Tan, K. J. Klabunde and P. M. Sherwood, J. Am. Chem. Soc., 1991, 113, 855 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501 CrossRef CAS PubMed.
- H. F. Du, W. Ai, Z. L. Zhao, Y. Chen, X. Xu, C. J. Zou, L. S. Wu, L. Su, K. K. Nan, T. Yu and C. M. Li, Small, 2018, 14, 1801068 CrossRef PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- Y. Duan, S. N. Sun, S. B. Xi, X. Ren, Y. Zhou, G. L. Zhang, H. T. Yang, Y. H. Du and Z. C. Xu, Chem. Mater., 2019, 29, 10534 CrossRef.
- J. G. Lee, J. Hwang, H. J. Hwang, O. S. Jeon, J. Jang, O. Kwon, Y. Lee, B. Han and Y. G. Shul, J. Am. Chem. Soc., 2016, 138, 3541 CrossRef CAS PubMed.
- W. T. Hong, K. A. Stoerzinger, Y. L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. L. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190 RSC.
- Y. Zhou, S. N. Sun, J. J. Song, S. B. Xi, B. Chen, Y. H. Du, A. C. Fisher, F. Y. Cheng, X. Wang, H. Zhang and Z. C. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, XPS, XRD, SEM, TEM and computational models and methods. See DOI: 10.1039/d0ra01813a |
| This journal is © The Royal Society of Chemistry 2020 |