
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomated glycan assembly of Streptococcus pneumoniae type 14 capsular polysaccharide fragments†
João Louçanoa,
Peter Bothb,
Andrea Marchesi b,
Linda del Binoc,
Roberto Adamoc,
Sabine Flitschb and
Mario Salwiczek*a
b,
Linda del Binoc,
Roberto Adamoc,
Sabine Flitschb and
Mario Salwiczek*a
aGlycoUniverse GmbH & Co KGaA, Am Mühlenberg 11, 14476 Potsdam, Germany. E-mail: m.salwiczek@glycouniverse.de
bSchool of Chemistry, University of Manchester, Manchester Institute of Biotechnology, 131 Princess Street, Manchester M1 7DN, UK
cGSK, Via Fiorentina 1, 53100 Siena, Italy
First published on 22nd June 2020
Abstract
S. pneumoniae is a major human pathogen with increasing antibiotic resistance. Pneumococcal vaccines consist of capsular polysaccharide (CPS) or their related fragments conjugated to a carrier protein. The repeating unit of S. pneumoniae type 14 CPS shares a core structure with the CPS of Group B Streptococcus (GBS) type III: the only difference is that the latter exhibits a sialic acid unit, with a α-2,3 linkage to galactose. Here, the automated glycan assembly (AGA) of two frameshifts of the repeating unit of S. pneumoniae type 14 is described. The same strategy is used to assemble dimers of the different repeating unit frameshifts. The four structures are assembled with only three commercially available monosaccharide building blocks. We also report an example of how enzymatic sialylation of the compounds obtained with AGA completes a synthetic route for GBS type III glycans. The synthesized structures were tested in competitive ELISA and further confirmed the branched tetrasaccharide Gal-Glc-(Gal-)GlcNAc to be the minimal epitope of S. pneumoniae type 14.
Introduction
Automated glycan assembly (AGA) is rising to the occasion as an efficient and growingly facile method to obtain chemically pure oligosaccharides1,2 that can be used to study the biological basis of their function in great molecular detail. The complexity of carbohydrate polymers prepared by AGA has been increasing continuously and structurally diverse antigens, including the Lewis antigens,3 oligosaccharides containing multiple cis-linkages,4 glycosaminoglycans,5 and long oligosaccharides (e.g. a mannose 50-mer6 and 150-mer7) have been reported. Success in the overall AGA process requires an optimized and highly efficient chemistry. The main challenge is to achieve highly efficient individual couplings to yield a specific stereochemistry for each of the emerging glycosidic linkages. The vast structural diversity of the monomeric building blocks, however, implicates equally diverse physicochemical properties and as such, the development of a reproducible synthetic protocol often requires a time-consuming iterative approach to optimize each coupling step. With the development of AGA and an increasing number of commercially available ready-to-use monomeric buildings blocks, optimizing a synthetic procedure can now be achieved within much smaller time frames. Given the demand for structurally defined synthetic carbohydrates in glycobiology and immunology, we engaged on an optimization study for the synthesis of S. pneumoniae capsular polysaccharide.S. pneumoniae is a leading cause of serious invasive diseases such as bacterial pneumonia, septicaemia, and meningitis in young children worldwide, and is listed in the WHO global priority list of antibiotic-resistant bacteria.8 Rising levels of antibiotic resistance reinforce the need for alternative treatments and prevention strategies such as vaccines. The capsular polysaccharide (CPS) is a major virulence factor and the basis of pneumococcal vaccines. Pneumococcal conjugate vaccines contain ten or thirteen CPS types, including type 14.9 The repeating unit of S. pneumoniae type 14 CPS (Sp14 CPS)10 is depicted in Fig. 1. This tetrasaccharide is the minimal immunogenic polysaccharide portion that elicits a protective response.11,12 A conjugate of this minimal epitope showed strong immunogenicity in mice, also as multivalent vaccine formulation combined with conjugates from other synthetic pneumococcal carbohydrate antigens.13
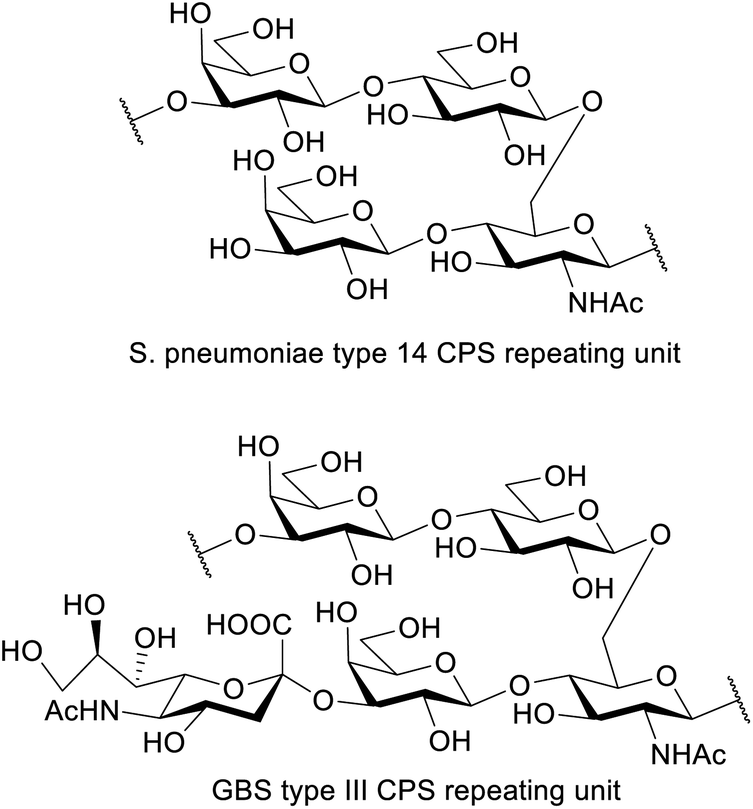 | ||
| Fig. 1 Repeating unit of the capsular polysaccharide of S. pneumoniae type 14 and GBS type III. | ||
Interestingly, the repeating unit of Sp14 CPS shares high structural identity with that of Group B Streptococcus (GBS) type III capsule, which exhibits an additional Neu5Ac residue α(2-3)-linked to the upstream Gal residue.14 GBS is a leading cause of sepsis and meningitis in newborns and causes an estimated 147![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 annual stillbirths and infant deaths globally.15 GBS capsule is a virulence factor and an important target for vaccine development.16 Ten different serotypes differentiated based on their structurally distinct sialylated polysaccharide capsules are known (Ia, Ib, II to IX) and all are associated with human disease. Serotype III is the most prevalent causing more than half of the infection cases.17
000 annual stillbirths and infant deaths globally.15 GBS capsule is a virulence factor and an important target for vaccine development.16 Ten different serotypes differentiated based on their structurally distinct sialylated polysaccharide capsules are known (Ia, Ib, II to IX) and all are associated with human disease. Serotype III is the most prevalent causing more than half of the infection cases.17
Pneumococcal antigens have been previously synthesized by solution-phase classical methods18,19 and AGA.20 One solution phase strategy to prepare Sp14 used a thioglycoside tetrasaccharide block, corresponding to the repeating unit of Sp14. The coupling of this block with a spacer, followed by removal of an isopropylidene acetal, yielded an acceptor that was elongated with the donor block to give a protected dimer of the repeating unit.21 Other strategies include the use of differentially protected lactose and lactosamine building blocks that were coupled to provide fragments up to a length of eight monosaccharides.22 For the synthesis of GBS structures that contain a terminal sialic acid, enzymatic methods offer an effective alternative to the usually low yielding chemical methods.23 The synthesis of the GBSIII repeating unit has been achieved by enzymatic introduction of the sialic acid in Sp14 structures.24,25 To avoid sialylation in the lactose arm of the structure, the corresponding galactose was blocked with a methyl ether in position 3 that could, however, not be removed in the end of the synthesis. Boons et al. described a fully chemical synthesis of a heptasaccharide fragment of GBS type III based on a convergent approach. The incorporation of sialic acid was based on α-NeuNAc-(2-3)-Galp methylthioglycoside disaccharide which had been assembled by coupling a methylthiosialoside and a galactose acceptor. Similarly, a series of GBSIII fragments have been recently assembled chemically to map the sialylated GBSIII structural epitope.26,27
Here, we take the repeating unit of Sp14 as a target structure and describe the optimization of AGA for this bacterial glycan. We proceed to show an example of enzymatic sialylation of one of the obtained structures to generate the GBS type III CPS repeating unit.
Results and discussion
AGA
The target structures for AGA (Scheme 1) represent two different frameshifts of the repeating unit (monomer) and the corresponding dimer of Sp14. In the branched frameshift (compounds 1 and 2), the reducing terminus is comprised of glucosamine. In the linear frameshift (compounds 3 and 4), glucose lies at the reducing terminus. Our retrosynthetic analysis of the target structures led to three thioglycoside building blocks that can be used to make both frameshifts. In our strategy, Fmoc served as the main temporary protecting group whereas Lev was used as an orthogonal temporary protecting to enable branching (glucosamine). Benzyl ether groups were used as permanent protecting groups. Benzoyl ester, and for the aminosugar, trichloroacetyl amide (NHTCA) at C2 were used as participating protecting groups to ensure selective trans-glycosylation. One galactose building block (6) was used both for terminal positions and where O3 elongation was required. In the first case, a capping step was used right after deprotecting the Fmoc group giving rise to a 3-O-acetylated galactose residue. This strategy allows time saving during the set-up of the synthesis and proves the versatility of the building block.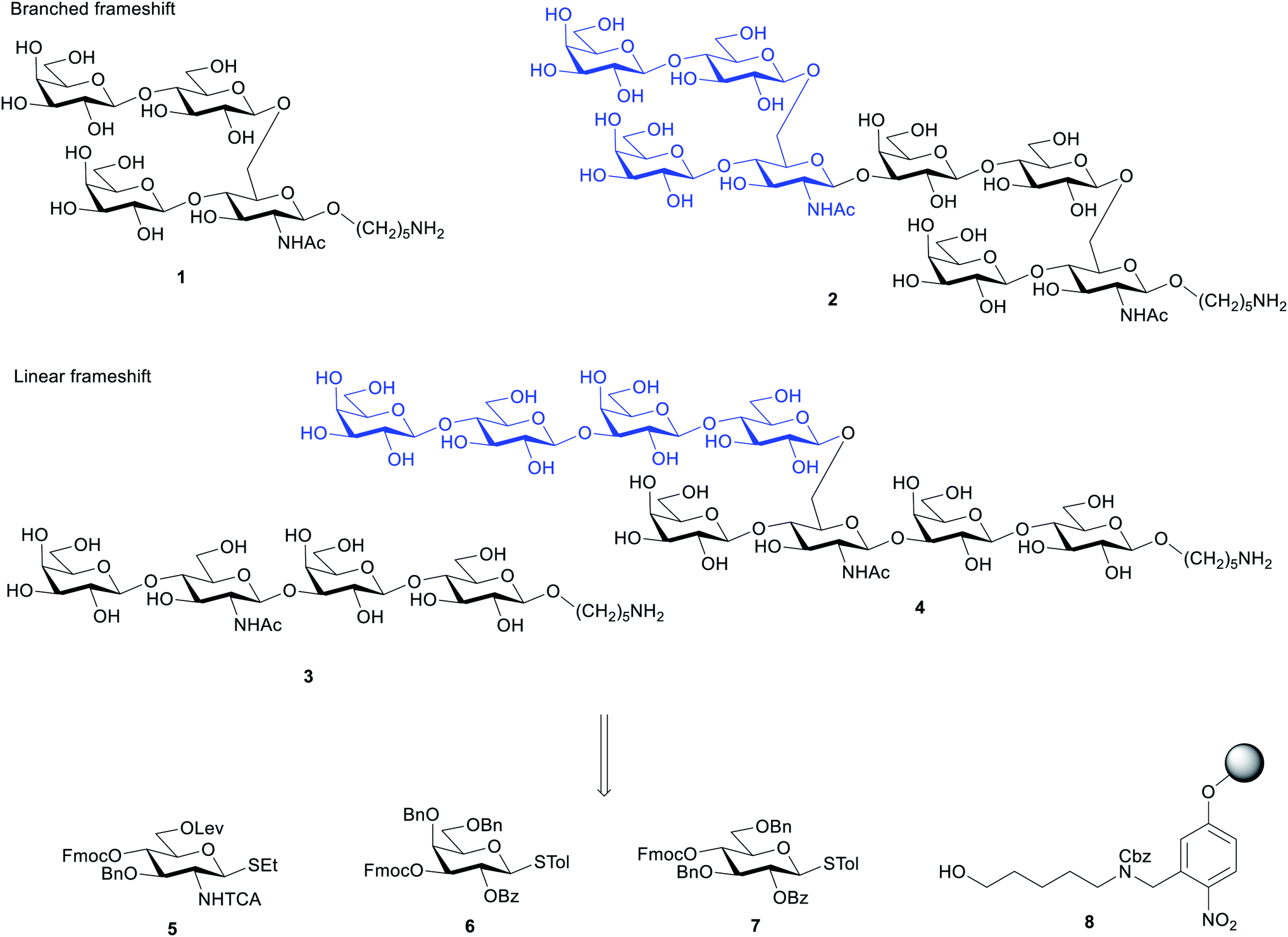 | ||
| Scheme 1 Retrosynthesis of oligosaccharides 1–4 using building blocks 5–7 and the functionalized Merrifield resin 8. | ||
The elongation cycle to incorporate each building block on the Glyconeer is comprised of four pre-programmed modules: acid wash, glycosylation, capping and deprotection (Table 1). The capping procedure prevents the growth of deletion sequences which facilitates purification and increases yield of the target product.28 Fmoc and Lev groups were cleaved as previously reported.28 The success of AGA depends largely on very high individual coupling yields which can be achieved by optimizing the key parameters related to the glycosylation module: temperature of glycosylation, number of coupling repetitions, and excess of donor.
| Module | Description |
|---|---|
| (1) Acid Wash | Resin is washed with a solution of TMSOTf in DCM |
| (2) Glycosylation | Building block and the activator solution (NIS/TfOH) are delivered to the reaction vessel at temperature T1 and incubated for 5 min. The temperature is then raised to T2 and an incubation time of 20 min is followed |
| (3) Capping | Resin is capped by acetylation with a solution of acetic anhydride and methanesulfonic acid in DCM preceded by a short wash with 10% pyridine in DMF |
| (4) Deprotection | Temporary protecting group is deprotected. Fmoc is cleaved with a solution of 20% (v/v) piperidine in DMF for 5 min. Lev is deprotected with a 0.15 M solution of hydrazine acetate in Py/AcOH/H2O (3 × 30 min) |
We defined the initial standard conditions as: 12.5 μmol scale (based on linker loading); glycosylations using 2× 6.5 equivalents (41 mM) of donor, T1 = −20 °C and T2 = 0 °C. After AGA and photocleavage to release the glycans from the solid support, the crude material was analysed by HPLC evaluate the success of the synthesis and the target product was purified by preparative HPLC. With these conditions, the target tetrasaccharide 9 was obtained in 12% yield (Table 2, entry 1). The HPLC analysis of the crude product revealed a significant contamination with the capped linker (see ESI†). When the first building block 5 was coupled using ten equivalents (62 mM), the signal for the capped linker was negligible and tetrasaccharide 9 was isolated in 20% yield (Table 2, entry 2). This finding suggests that the glucosamine building block 5 requires a more vigorous approach to achieve good coupling yield. Before proceeding to the assemble of the dimer 10, the synthesis of oligosaccharide 9 was repeated using ten equivalents of donor in one single coupling for building blocks 6 and 7. The crude material obtained after photocleavage showed negligible deletion sequences (see HPLC in ESI†) and the product was isolated in 31% yield (Table 2, entry 3). With these conditions, the synthesis time was reduced from 13.5 to 10.5 h and 2.5 equivalents per coupling of building block 6 and 7 were saved.
| Entry | BB | equiv. | T1 | T2 | Isolated yield |
|---|---|---|---|---|---|
| 1 | 5 | 2× 6.5 | −20 | 0 | 12% |
| 6 | 2× 6.5 | −20 | 0 | ||
| 7 | 2× 6.5 | −20 | 0 | ||
| 6 | 2× 6.5 | −20 | 0 | ||
| 2 | 5 | 2× 10 | −20 | 0 | 20% |
| 6 | 2× 6.5 | −20 | 0 | ||
| 7 | 2× 6.5 | −20 | 0 | ||
| 6 | 2× 6.5 | −20 | 0 | ||
| 3 | 5 | 2× 10 | −20 | 0 | 36% |
| 6 | 1× 10 | −20 | 0 | ||
| 7 | 1× 10 | −20 | 0 | ||
| 6 | 1× 10 | −20 | 0 |
These conditions were then applied for the synthesis of the dimer 10 (Table 3, entry 1). Unfortunately, the major product was the deletion sequence tetrasaccharide 14 (Fig. 2). The formation of 14 can be explained by a very low coupling efficiency of glucosamine building block 5 to the O3 position of galactose. In an attempt to improve this step, the effect of the glycosylation temperature for building block 5 was probed: the temperatures T1 = −10 °C and T2 = 0 °C led to an increase in the formation of the target octasaccharide; T1 = 0 °C and T2 = 20 °C led to a decrease in the formation of the target octasaccharide. However, even with the best temperature set (Table 3, entry 2) the deletion sequence 14 prevailed as the major product. When glucosamine 5 was coupled at −10 °C to 10 °C and repeated in a quadruple coupling, the synthesis proceeded with the formation of 10 as the major product (Table 3, entry 3, 8% yield). Building block 7 was used again in a double coupling to prevent the formation of deletion sequence 13 and the amount of building block 6 was reduced to 6.5 equiv. without formation of deletion sequences.
| Entry | BB | equiv. | T1 | T2 | Isolated yield |
|---|---|---|---|---|---|
| 1 | 5 | 2× 10 | −20 | 0 | 1% |
| 6 | 1× 10 | −20 | 0 | ||
| 7 | 1× 10 | −20 | 0 | ||
| 6 | 1× 10 | −20 | 0 | ||
| (2×) | |||||
| 2 | 5 | 2× 10 | (i) −20 (ii) −10 | (i) 0 (ii) 10 | 4% |
| 6 | 1× 6.5 | −20 | 0 | ||
| 7 | 2× 6.5 | −20 | 0 | ||
| 6 | 1× 6.5 | −20 | 0 | ||
| (2×) | |||||
| 3 | 5 | (i) 2× (ii) 4× 10 | (i) −20 (ii) 0 | (i) 0 (ii) 20 | 8% |
| 6 | 1× 6.5 | −20 | 0 | ||
| 7 | 2× 6.5 | −20 | 0 | ||
| 6 | 1× 6.5 | −20 | 0 | ||
| (2×) |
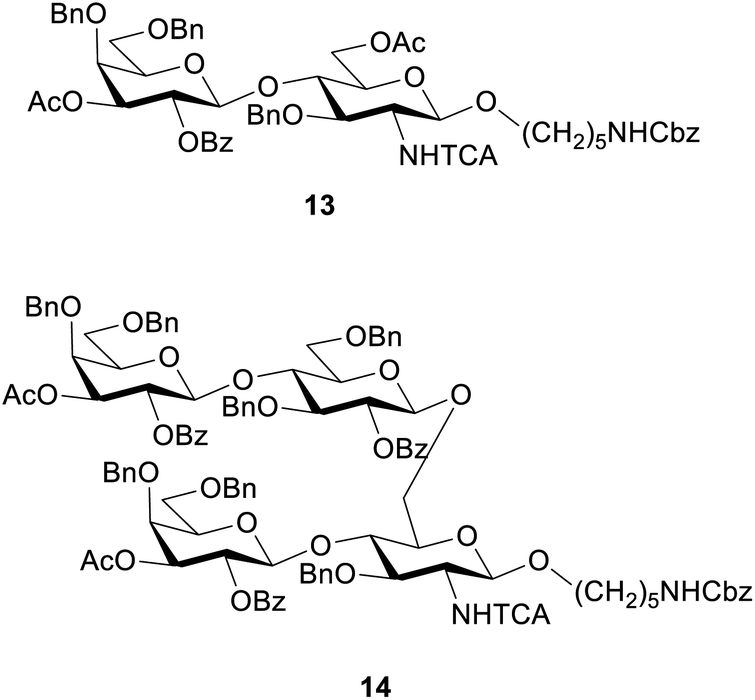 | ||
| Fig. 2 Deletion sequences identified in the synthesis of octasaccharide 10. | ||
To generate the fragments of the linear frameshift, the best conditions for the synthesis of dimer 10 were used. The synthesis of tetrasaccharide 11 (Table 4, entry 1) was successful and led to 23% yield. The synthesis of 12 was performed by repeating the elongation cycles. An unexpected deletion sequence was identified as a tetrasaccharide with the Lev still present at the branching point at C6 of glucosamine indicating incomplete cleavage of this protecting group. Nevertheless, the octasaccharide could still be isolated in 8% yield (Scheme 2).
| Entry | BB | equiv. | T1 | T2 | Isolated yield |
|---|---|---|---|---|---|
| 1 | 7 | 2× 6.5 | −20 | 0 | 23% (11) |
| 6 | 1× 6.5 | −20 | 0 | ||
| 5 | 4× 10 | −10 | 10 | ||
| 6 | 1× 6.5 | −20 | 0 | ||
| 2 | 7 | 2× 6.5 | −20 | 0 | 8% (12) |
| 6 | 1× 6.5 | −20 | 0 | ||
| 5 | 4× 10 | −10 | 10 | ||
| 6 | 1× 6.5 | −20 | 0 | ||
| (2×) |
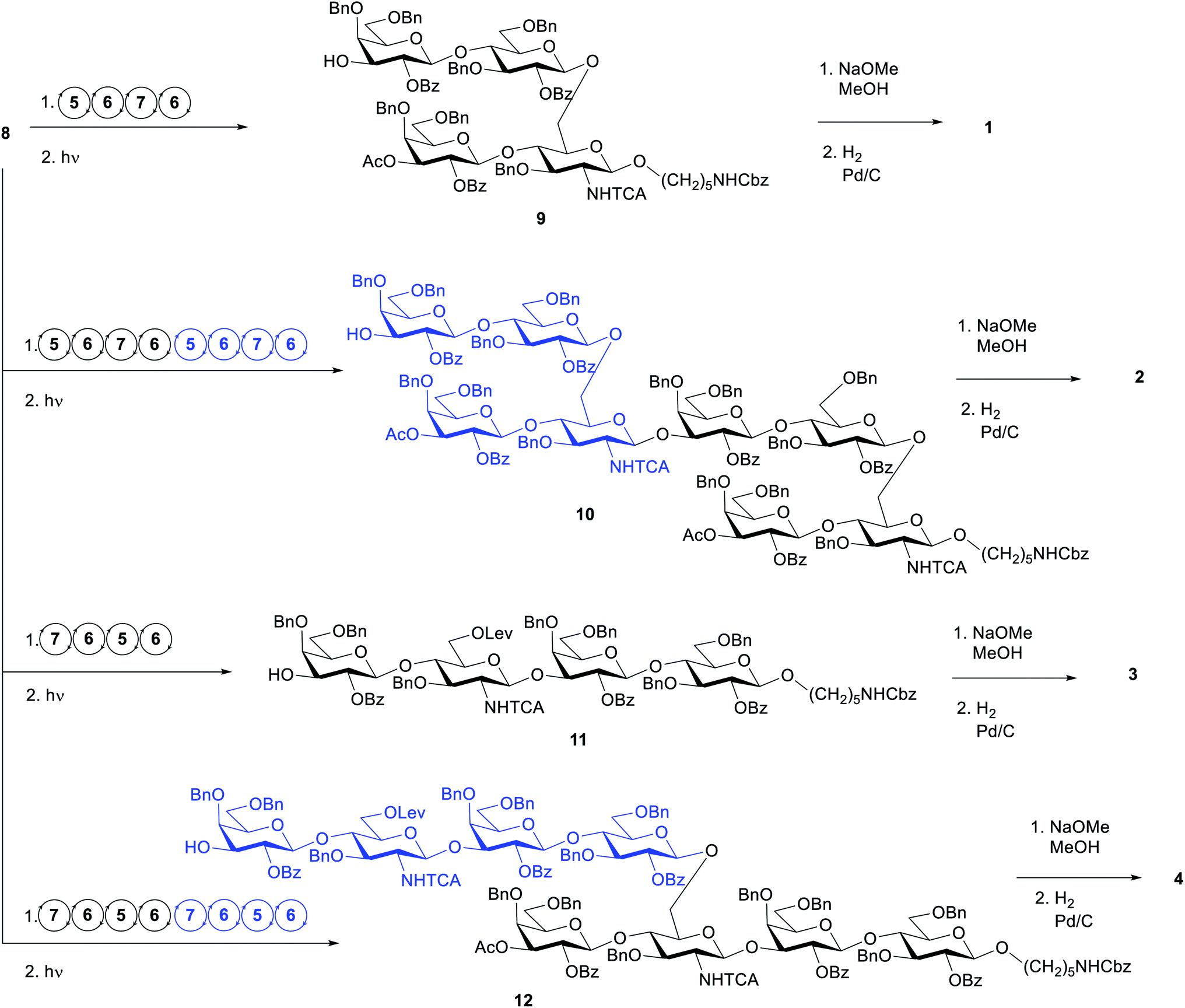 | ||
| Scheme 2 AGA of oligosaccharides 9–12, and global deprotection to generate 1–4. Circular arrows represent the elongation cycle for each BB (Table 1). | ||
Upscaling on the Glyconeer
The AGA synthesis described so far refer to a 12.5 μmol scale. A linear scale-up to 25 μmol was performed with the conditions in Table 2, entry 2 and the target tetrasaccharide 9 was isolated in 28%. The synthesis of the tetrasaccharide 11 was also linearly scaled up to 25 μmol using the conditions in Table 4, entry 1, leading to an isolated yield of 21%. These are comparable to the lower scale experiments (20% and 23%, respectively). The synthesis of tetrasaccharide 9 was further called up to 50 μmol. In this case, due to volume limitations, the amount of BBs was reduced to 2 × 5 equivalents per coupling, keeping the concentration at 41 mM. Interestingly, this time, tetrasaccharide 9 was isolated in 33%.Deprotection
Following AGA, cleavage from the solid support, and HPLC purification, compounds 9, 10, 11 and 12 were deprotected in two steps. Ester groups (benzoyl, acetyl and levaloyl) were cleaved with sodium methoxide in 1:1 MeOH/DCM. In the second step, benzyl ethers, NHTCA and the carboxybenzyl group at the amino linker were removed by hydrogenation catalyzed by Pd/C. Methanolysis of tetrasaccharide 9 was complete after 24 h using 25 mM NaOMe (by MALDI). The crude product was then hydrogenated and purified using a C18 cartridge to give 1 in 93% overall deprotection yield. The linear tetrasaccharide 11 was submitted to the same methanolysis procedure. However, after 24 h, mass analysis showed partially cleaved intermediates and when left to react for 4 days the sample degraded. When performed with an increased concentration of NaOMe (50 mM), the reaction showed similar results. Upon addition of another portion of NaOMe (final concentration 0.1 M) and after another 24 h, the conversion was complete. The crude product was then subjected to hydrogenation. Purification using a C18 cartridge was not sufficient and tetrasaccharide 3 was obtained after HPLC purification as the formate salt (71% overall deprotection yield).
Deprotection of octasaccharides 10 and 12 proved to be cumbersome as the methanolysis could not be followed by LC-MS or MALDI. For branched structure 10, the procedure used for methanolysis was the same as for the branched tetrasaccharide 9. After hydrogenation, the crude product was subjected to RP-HPLC but was isolated with a triethylamine contamination, originating from the work up procedure. The linear structure 12 was deprotected using the same conditions as for linear tetrasaccharide 11. After hydrogenation and RP-HPLC purification, the product was isolated in 22% yield (overall deprotection yield).
Enzymatic sialylation
To convert the S. pneumoniae type 14 repeating unit (oligosaccharide 3) into the GBS type III repeating unit (compound 15), an α(2,3)-sialyltransferase from Pasteurella multocida (PmST1)29 was used. The reaction was performed using three equivalents of CMP-Neu5Ac (3 mM) as donor in the presence of an alkaline phosphatase to dephosphorylate the CMP by-product. The enzyme was used in a concentration of 6 μg mL−1 and after incubation at 37 °C with a Tris–HCl buffer (pH 9.3), the sialoside was filtered by a polyethersulfone membrane (MWCO 10 kDa) to remove the protein. A final purification by RP-HPLC gave 15 in 30% yield (Scheme 3).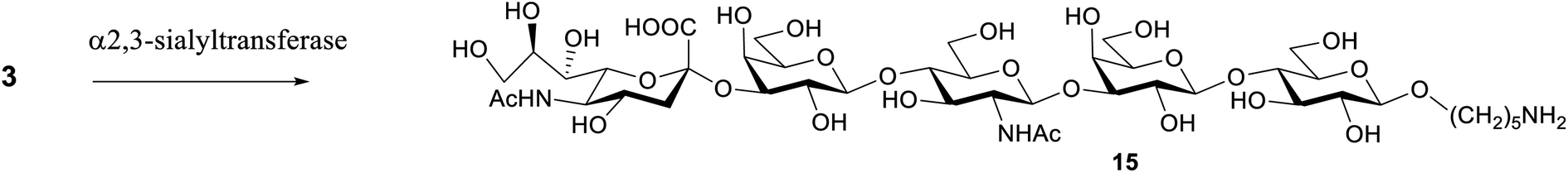 | ||
| Scheme 3 Enzymatic synthesis of sialoside 14. α-(2,3)-Sialyltransferase PmST1, CMP-Neu5Ac, alkaline phosphatase, Tris–HCl pH 9.3, MgCl2. | ||
Antigenic evaluation of the synthesized pneumococcal glycans
The antigenicity of the synthesized pneumococcal serotype 14 fragments was assessed by competitive ELISA (Fig. 3). To this end, plates were coated with Sp14 polysaccharide and binding to the corresponding murine anti serum was inhibited with serial dilutions of the synthesized oligosaccharides 1–3 and 15. The natural polysaccharide Sp14 was the positive control, while the GBSIII capsular polysaccharide was the negative control. The branched structure 1 and its dimer 2 were able to fully inhibit the binding of the Sp14 serum to the polysaccharide. In contrast, the linear glycan 3 and its sialylated form 15 resembling the GBSIII polysaccharide linear frameshift failed to inhibit the binding. These results further support the theory whereby the minimal immunogenic epitope of Sp14 is the branched tetrasaccharide 1.11 The lack of specificity of the fragments 1 and 2 towards anti GBSIII antibodies was also confirmed by inhibiting the binding of a rabbit GBSIII mAb (Fig. S1, ESI†), that has been shown to recognize a sialylated GBSIII portion comprised of two repeating units.27 While GBSIII polysaccharide at the tested dilutions showed to deplete the mAb recognition, the two pneumococcal fragments did not. This result agrees with the previous finding that anti-GBSIII human antibodies recognize a shared linear structure in the backbone of both Sp14 and GBSIII, represented by the -Glc-GlcNAc-Gal-motif.30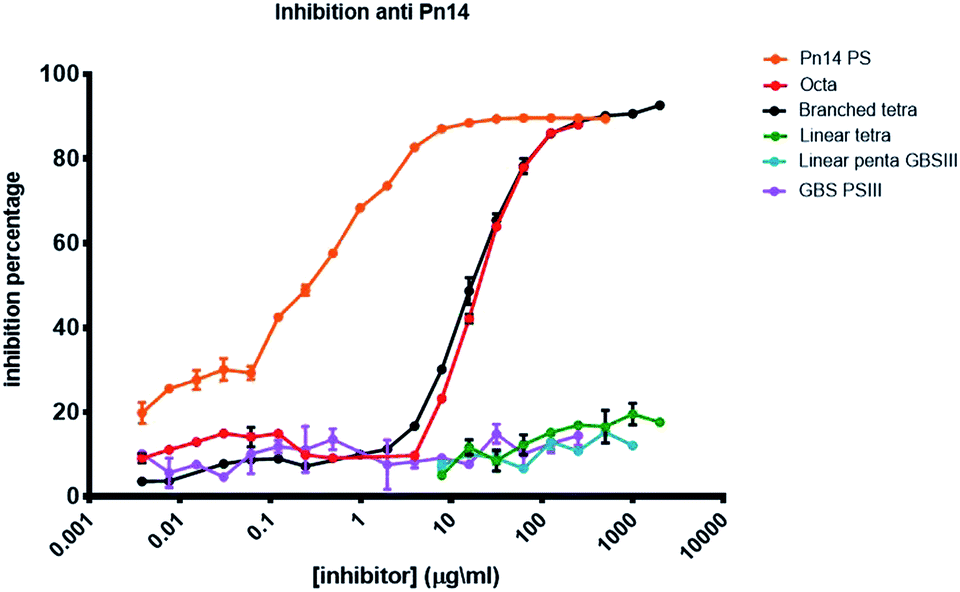 | ||
| Fig. 3 Inhibition percentage of Sp14 capsular polysaccharide using a specific murin serum (saccharide concentration expressed as μg mL−1). | ||
Conclusions
The success of AGA relies on general and standardized procedures. The coupling of BBs 5–7 was explored with the best conditions leading to reproducible good coupling yields in two different frameshifts of Sp14 CPS oligosaccharides. This showcases the importance of coupling optimization and proves the versatility the monosaccharides building blocks, setting the scene for their use in different structures. Using only three building blocks, four different antigens 1–4 were prepared. The optimized yields after AGA ranged from 36% to 8%, the lower values corresponding to the longer structures. Exploring BBs with different protecting groups or leaving groups (e.g. phosphate donors) can lead to higher yields at this stage. The larger structures also showed lower deprotection yields, which reinforces the need for more efficient hydrogenation methods. The combination of enzymatic sialylation with AGA proved to be an expeditious approach to the preparation of complex glycans for vaccine development. Compounds 1–3 and 15 were used in competitive ELISA experiments. Structure 1 and its dimer 2 showed to inhibit the binding of Sp14 polysaccharide with specific murine serum, providing further evidence for the assignment of the branched tetrasaccharide 1 as the minimal epitope. At the same time, 1 and 2 did not deplete the recognition of GBSIII polysaccharide with a mAb, supporting the need for the Glc-GlcNAc-Gal-motif in for binding. Vaccine development can greatly advance with the increased accessibility to the glycans prepared in this work as the elucidation of minimal epitopes opens the possibility to generate analogues in the pursuit of safer and more efficient active ingredients as well as analogues with easier production routines.Conflicts of interest
J. L. and M. S. work at GlycoUniverse GmbH & Co. KGaA, the company that commercializes the synthesis instrument and building blocks. L. del B. and R. A. are employees of GSK group of companies. R. A. is owner of GKS stocks.Acknowledgements
This project has received funding from the European Union's Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie Sklodowska-Curie Grant Agreement No. 675671. We acknowledge Tanistha Gupta for fruitful discussions and Prof. Peter H. Seeberger for valuable feedback.Notes and references
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS PubMed.
- M. Guberman, M. Bräutigam and P. H. Seeberger, Chem. Sci., 2019, 1–96 Search PubMed.
- H. S. Hahm, M. Hurevich and P. H. Seeberger, Nat. Commun., 2016, 7, 1–8 RSC.
- C. F. Liang, H. S. Hahm and P. H. Seeberger, Methods Mol. Biol., 2014, 1229, 3–10 CrossRef PubMed.
- K. Naresh, F. Schumacher, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2017, 53, 9085–9088 RSC.
- A. A. Joseph, A. Pardo-Vargas and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 8561–8564 CrossRef PubMed.
- E. Balsells, L. Guillot, H. Nair and M. H. Kyaw, PLoS One, 2017, 12, 1–20 CrossRef PubMed.
- J. N. Weiser, D. M. Ferreira and J. C. Paton, Nat. Rev. Microbiol., 2019, 16, 355–367 CrossRef PubMed.
- B. Lindberg, J. Lonngren and D. A. Powell, Carbohydr. Res., 1977, 58, 177–186 CrossRef CAS PubMed.
- D. Safari, H. A. T. Dekker, J. A. F. Joosten, D. Michalik, A. C. De Souza, R. Adamo, M. Lahmann, A. Sundgren, S. Oscarson, J. P. Kamerling and H. Snippe, Infect. Immun., 2008, 76, 4615–4623 CrossRef CAS PubMed.
- F. Mawas, J. Niggemann, C. Jones, M. J. Corbel, J. P. Kamerling and J. F. G. Vliegenthart, Infect. Immun., 2002, 70, 5107–5114 CrossRef CAS PubMed.
- P. Kaplonek, N. Khan, K. Reppe, B. Schumann, M. Emmadi, M. P. Lisboa, F.-F. Xu, A. D. J. Calow, S. G. Parameswarappa, M. Witzenrath, C. L. Pereira and P. H. Seeberger, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 13353–13358 CrossRef CAS PubMed.
- H. J. Jennings, K.-G. Rosell and D. L. Kasper, Can. J. Biochem., 1980, 58, 112–120 CrossRef CAS PubMed.
- A. C. Seale, H. Blencowe, F. Bianchi-Jassir, N. Embleton, Q. Bassat, J. Ordi, C. MenCrossed, C. Cutland, C. Briner, J. A. Berkley, J. E. Lawn, C. J. Baker, L. Bartlett, M. G. Gravett, P. T. Heath, M. Ip, K. Le Doare, C. E. Rubens, S. K. Saha, S. Schrag, A. S. Ter Meulen, J. Vekemans and S. A. Madhi, Clin. Infect. Dis., 2017, 65, S125–S132 CrossRef PubMed.
- A. Nuccitelli, C. D. Rinaudo and D. Maione, Ther. Adv. Vaccines, 2015, 3, 76–90 CrossRef CAS PubMed.
- A. M. Weisner, A. P. Johnson, T. L. Lamagni, E. Arnold, M. Warner, P. T. Heath and A. Efstratiou, Clin. Infect. Dis., 2004, 38, 1203–1208 CrossRef PubMed.
- M. Alpe and S. Oscarson, Carbohydr. Res., 2002, 337, 1715–1722 CrossRef CAS PubMed.
- C. L. Pereira, A. Geissner, C. Anish and P. H. Seeberger, Angew. Chem., Int. Ed., 2015, 54, 10016–10019 CrossRef CAS PubMed.
- B. Schumann, H. S. Hahm, S. G. Parameswarappa, K. Reppe, A. Wahlbrink, S. Govindan, P. Kaplonek, L. A. Pirofski, M. Witzenrath, C. Anish, C. L. Pereira and P. H. Seeberger, Sci. Transl. Med., 2017, 9, eaaf5347 CrossRef PubMed.
- A. Sundgren, M. Lahmann and S. Oscarson, J. Carbohydr. Chem., 2005, 24, 379–391 CrossRef CAS.
- E. V. Sukhova, D. V. Yashunsky, Y. E. Tsvetkov, E. A. Kurbatova and N. E. Nifantiev, Russ. Chem. Bull., 2014, 63, 511–521 CrossRef CAS.
- C.-C. Yu and S. G. Withers, Adv. Synth. Catal., 2015, 357, 1633–1654 CrossRef CAS.
- V. Pozsgay, J. R. Brisson, H. J. Jennings, S. Allen and J. C. Paulson, J. Org. Chem., 1991, 56, 3377–3385 CrossRef CAS.
- V. Pozsgay, J. Gaudino, J. C. Paulson and H. J. Jennings, Bioorg. Med. Chem. Lett., 1991, 1, 391–394 CrossRef CAS.
- V. Cattaneo, F. Carboni, D. Oldrini, R. De Ricco, N. Donadio, I. M. Y. Ros, F. Berti and R. Adamo, Pure Appl. Chem., 2017, 89, 855–875 CAS.
- F. Carboni, R. Adamo, M. Fabbrini, R. De Ricco, V. Cattaneo, B. Brogioni, D. Veggi, V. Pinto, I. Passalacqua and D. Oldrini, et al., Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5017–5022 CrossRef CAS PubMed.
- Y. Yu, A. Kononov, M. Delbianco and P. H. Seeberger, Chem.–Eur. J., 2018, 24, 6075–6078 CrossRef CAS PubMed.
- H. Yu, H. Chokhawala, R. Karpel, H. Yu, B. Wu, J. Zhang, Y. Zhang, Q. Jia and X. Chen, J. Am. Chem. Soc., 2005, 127, 17618–17619 CrossRef CAS PubMed.
- D. Safari, H. A. T. Dekker, G. T. Rijkers, A. van der Ende, J. P. Kamerling and H. Snippe, Glycoconjugate J., 2011, 28, 557–562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01803a |
| This journal is © The Royal Society of Chemistry 2020 |