
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic degradation of an organophosphorus pesticide using a ZnO/rGO composite†
Zihan
Zhu
,
Feng
Guo
 *,
Zhonghao
Xu
,
Xiaoxuan
Di
and
Qian
Zhang
*,
Zhonghao
Xu
,
Xiaoxuan
Di
and
Qian
Zhang
Key Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education), School of Ocean Science and Technology, Dalian University of Technology, Panjin 124221, China. E-mail: 0411guofeng@dlut.edu.cn; Tel: +86-427-2631892
First published on 24th March 2020
Abstract
A zinc oxide (ZnO)/reduced graphene oxide (rGO) nanocomposite was synthesized via a hydrothermal synthesis method and used for the photocatalytic degradation of dimethoate. In the synthesis process of the ZnO/rGO nanocomposite, hexamethylenetetramine (HMT) was used as both a mineralizer and reducing agent. When the ZnO nanoparticles formed on the surfaces of graphene oxide sheets, the sheets were simultaneously reduced by HMT to form rGO. The photodegradation rate and photodegradation efficiency of dimethoate by the ZnO/rGO nanocomposite were 4 and 1.5 times, respectively, higher than those of bare ZnO. The ZnO/rGO nanocomposite possessed a high surface area of 41.0 m2 g−1 and pore volume of 4.72 × 10−3 cm3 g−1, which were conducive to the adsorption and mass transfer of pesticides and oxygen. The enhanced photocatalytic performance of the ZnO/rGO nanocomposite was attributed to the decrease in electron–hole recombination rate and effective carrier transport caused by the presence of rGO. Photoelectrochemical measurements confirmed that the nanocomposite exhibited a high charge transfer rate at the ZnO/rGO interface. These results indicate that ZnO/rGO nanocomposites have great application potential in pollutant degradation.
1. Introduction
Organophosphorus pesticides (OPPs) are widely used to control major diseases, insect pests, and weeds.1,2 However, because of their persistence and accumulation, OPPs gradually migrate to freshwater sources such as rivers, lakes, wells, and underground water from the soil, which is the primary pollution source.3–5 Effective removal of OPPs from aquatic systems is an essential issue to solve. Dimethoate is a typical OPP that has high toxicity and a wide insecticidal range to many kinds of pests, especially to chewing and piercing-sucking insects. Exposure to dimethoate may have many different adverse effects on humans, such as carcinogenic,6 neurological,7 reproductive,8 and endocrine effects.9The removal of OPPs by various techniques including photocatalysis,10,11 adsorption,12–14 biodegradation,15–17 chemical oxidation,18,19 and electrochemical oxidation20 has been investigated. Among these methods, semiconductor photocatalysis is regarded as a green and efficient technology for the removal of organic pollutants from water without high-cost equipment and catalysts.21,22 Although various semiconductor materials including TiO2, ZnO, CdS, ZnS, and Fe2O3 have been used for photocatalysis, much photocatalysis research still focuses on TiO2 and its nanocomposites. ZnO is considered an attractive alternative semiconductor material to replace TiO2 because of its higher exciton binding energy (60 meV compared to 4 meV for TiO2) and carrier transmission efficiency.23 ZnO boasts the advantages of high quantum efficiency, easy synthesis, low cost, and environmental friendliness, which have led to its extensive use in photodegradation. However, the rapid recombination of photogenerated electron–hole pairs greatly limits the photodegradation efficiency of ZnO under ultraviolet (UV) light.
In general, the photocatalytic performance of ZnO is improved by promoting both the separation of photogenerated electron–hole pairs on the ZnO surface and carrier transport.24 To achieve this, ZnO is usually doped with carbon materials such as carbon nanotubes, graphitic carbon nitrude, graphene, and graphene oxide (GO).25–27 Graphene is a two-dimensional hexagonal lattice of sp2-hybridized carbon atoms. Because of its unique electronic structure, graphene has a large specific surface area and high electron mobility.24 GO is a chemically modified derivative of graphene. The abundant oxygen-containing groups on the surface of GO enable it to combine with many inorganic materials through covalent and/or ionic bonds to form functional composites. Coupling CdS and TiO2 with GO can markedly improve their photocatalytic performance.28,29 Therefore, loading ZnO nanoparticles on GO sheets should lead to better pollutant degradation performance. Recombination of photogenerated electron–hole pairs can be suppressed by transferring electrons from ZnO to GO, which is an excellent electron acceptor.30
In the present work, ZnO nanoparticles are grown uniformly on few-layer GO via a hydrothermal synthesis process that uses hexamethylenetetramine (HMT) as both a mineralizer and reducing agent. The crystal structure, morphology, specific surface area, light absorption properties, and photoelectrical properties of the prepared ZnO/reduced graphene oxide (rGO) composite are characterized. The photocatalytic performance of the ZnO/rGO nanocomposite and its mechanism are also systematically investigated.
2. Material and methods
2.1 Materials
Graphite powder (Sinopharm Chemical, China) was used as a starting material. Sodium hydroxide (NaOH), HMT, and zinc sulfate heptahydrate (ZnSO4) were used as raw chemicals (Damao Chemicals, China). Deionized water was used to prepare all solutions. All raw chemicals were used without further purification.2.2 Preparation of ZnO/rGO composites
GO was prepared from natural graphite using a reported method.31 The incorporation of the prepared GO with ZnO was performed using a simple hydrothermal method (Fig. S1†). In a typical preparation process, GO (0.04 g) was dispersed in water by ultrasonication for 30 min to obtain a stable GO suspension. ZnSO4 (99%, 1.44 g, 5 mmol) was added to form a ZnSO4/GO suspension. NaOH (99%, 1.6 g, 40 mmol) was dissolved in deionized water and then added dropwise to the ZnSO4/GO solution. After constant stirring for 30 min, HMT (99%, 0.7 g, 5 mmol) was added. The mixture was vigorously stirred for 30 min. The suspension was transferred to a Teflon-lined stainless-steel autoclave (100 mL) and then heated at 110 °C for 9 h. The ZnO/rGO nanocomposite with a mass ratio of 10% of rGO to ZnO (labeled as ZG2) was purified by centrifugation, washed, and then dried in an oven at 60 °C overnight. The mass ratio of rGO was changed to 0%, 5%, 15%, and 20% to prepare ZG0, ZG1, ZG3, and ZG4 nanocomposites, respectively, using the same procedure.2.3 Characterization
The surface areas of the samples were determined by N2 adsorption measurements using an automatic physical adsorption apparatus (Autosorb-iQ-C, Quantachrome Instruments, USA) and the data were evaluated by the Brunauer–Emmett–Teller method. Pore volumes were obtained from adsorption data at a relative pressure of 0.995. The particle sizes of samples were obtained using particle size analyzer (Zeta sizer Nano ZS, Malvern, UK). Scanning electron microscopy (SEM) investigations were performed using a scanning electron microscope (Nova NanoSEM; FEI; USA) with an acceleration voltage of 30 kV. Elemental analysis of the samples was conducted using an energy-dispersive X-ray detector (Vario EL cube; Elementar Analysensysteme GmbH; Germany). The photoluminescence (PL) spectra of the samples were measured using a fluorescence spectrophotometer (FL-7000; Hitachi; Japan) with an excitation wavelength of 325 nm. The functional groups of catalysts were analyzed by using a Fourier transform infrared spectroscope (Nicolet iN10 MX & iS10; ThermoFisher, USA) operated in the range of 4000–650 cm−1. UV-vis reflectance spectra of the samples were recorded using a spectrometer (Lambda 950, PerkinElmer, USA). Raman spectra were obtained by a Raman spectrometer (inVia, Renishaw, UK) with an excitation wavelength of 532 nm. X-ray diffraction (XRD) patterns were recorded by an X-ray diffractometer (XRD-7000S, Shimadzu, Japan) with Cu Kα radiation (λ = 0.15406 nm) using a scan rate of 5° min−1. X-ray photoelectron spectroscopy (XPS) patterns were recorded by an X-ray photoelectron spectrometer (ESCALAB™ 250Xi, ThermoFisher, USA).2.4 Photocatalytic test
The obtained ZnO and ZnO/rGO nanocomposites were used as photocatalysts for the degradation of dimethoate under UV irradiation at room temperature. A 250 W UV lamp (λ = 254 nm) with an average light intensity of 2.45 mW cm−2 was used as a light source. Photocatalyst (50 mg) was suspended in an aqueous solution of dimethoate (5 mg L−1, 100 mL) with vigorous stirring. At certain time intervals, aliquots of the suspension (1.5 mL) were sampled and then filtered through a 0.22 μm filter membrane to remove the photocatalyst from the solution. The degradation efficiency of dimethoate was analyzed by high-performance liquid chromatography (Agilent 1260) at 210 nm.2.5 Photoelectrochemical measurements
ZnO and ZnO/rGO electrodes were prepared by a dip-coating method. In general, acetone and FTO glass are commonly used as dip-coating solution and substrate in photoelectric test. In this study, Nafion and stainless-steel substrate (500 mesh) were chosen as candidate materials, which does not affect the comparison of photocurrent differences in different photocatalytic material. Each nanocomposite (25 mg) was dispersed in Nafion (20 mL) by ultrasonication for 60 min. Each suspension was spray coated on a stainless-steel substrate (500 mesh) and then the prepared films were dried at 60 °C for 30 min. A three-electrode cell system was used for electrochemical property measurements. Ag/AgCl (saturated KCl) and Pt electrodes were immersed in the electrolytic cell as reference and counter electrodes, respectively. In all measurements, 0.5 M Na2SO4 aqueous solution was used as the electrolyte. The variations of the photocurrent responses over time (I–t curves) of the ZnO and ZnO/rGO samples were also measured under 0.95 mW cm−2 light illumination at a bias of 1.03 V (vs. Ag/AgCl). In electrochemical impedance spectroscopy (EIS) measurements, the frequency range was from 10−1 to 105 Hz with an alternating-current (AC) amplitude of 10 mV at the open circuit potential. To obtain Mott–Schottky (MS) plots, the potential was cycled in the range from −0.8 to 0.5 V (vs. Ag/AgCl) at a frequency of 1000 Hz and AC amplitude of 10 mV.3. Results and discussion
The ZnO/rGO nanocomposites were fabricated by a hydrothermal method. First, GO was obtained by the oxidation of graphite, which led to the introduction of various organic moieties on graphene, such as hydroxyl, carboxyl, and epoxy groups. Therefore, GO will be negatively charged when dissolved in aqueous solution, which gives it the ability to adsorb a large amount of Zn2+ through electrostatic interactions. The decomposition of HTM can produce ammonia (NH3) and formaldehyde (HCHO); NH3 releases hydroxide ions (OH−) and HCHO promotes the deoxygenation reaction of GO. In addition, the nucleation and growth of ZnO nanocrystals are affected by pH, so the presence of OH− could enhance the nucleation rate of nanocrystals. When the OH−/Zn2+ molar ratio exceeded 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, the excess OH− dissolved and regenerated ZnO, leading to ZnO nanocrystallites with a larger grain diameter. ZnO crystals nucleated in situ on the GO sheets to form the ZnO/rGO nanocomposite after addition of OH−. The main reactions in the synthesis process are as follows (eqn (1)–(6)):
:2, the excess OH− dissolved and regenerated ZnO, leading to ZnO nanocrystallites with a larger grain diameter. ZnO crystals nucleated in situ on the GO sheets to form the ZnO/rGO nanocomposite after addition of OH−. The main reactions in the synthesis process are as follows (eqn (1)–(6)):| Zn2+ + 2OH− → Zn(OH)2 | (1) |
| (CH2)6N4 + 10H2O → 6HCHO + 4NH3·H2O | (2) |
| NH3·H2O ↔ NH4+ + OH− | (3) |
| Zn2+ + 4OH− → Zn(OH)42− | (4) |
| Zn(OH)2 → ZnO + H2O | (5) |
| Zn(OH)42− → ZnO + H2O + 2OH− | (6) |
3.1 Characterization
XRD patterns of GO, ZnO, and ZnO/rGO nanocomposite samples with different GO contents are displayed in Fig. 1. The XRD pattern of GO showed a diffraction peak at 9.86° corresponding to the (001) plane of GO. The XRD pattern of ZnO in Fig. 1(a) exhibited six diffraction peaks at 31.77°, 34.42°, 36.25°, 47.54°, 56.60°, and 62.86° that corresponded to the (100), (002), (101), (102), (110), and (103) planes, respectively, of the hexagonal wurtzite crystal structure (JCPDS card no. 36-1451). The XRD patterns of the ZnO/rGO nanocomposites were similar to that of pure ZnO. No diffraction peak from GO was detected, which indicated that the addition of up to 20 wt% GO to ZnO did not change the crystallinity or preferred orientation of ZnO.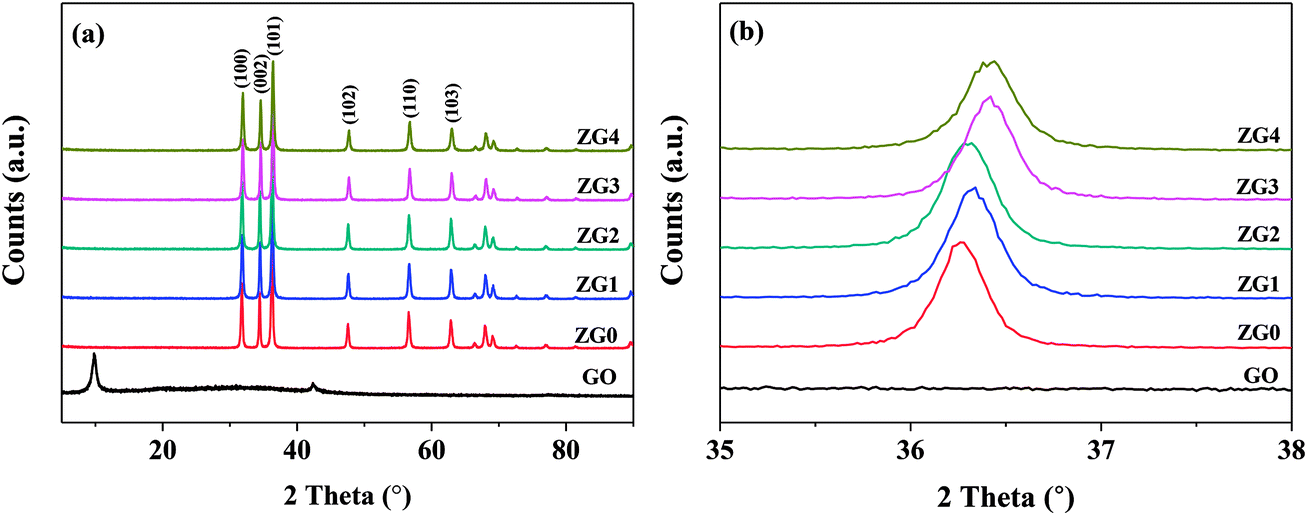 | ||
| Fig. 1 XRD spectra of ZnO and ZnO/rGO nanocomposite in the range (a) 5–90° and (b) 35–38°. | ||
The lattice constants a and c of ZnO determined from the XRD patterns were 0.325 and 0.521 nm, respectively, which were almost identical to those of the ZnO/rGO thin films. This result indicated that GO doping did not affect the lattice parameters or unit cell volume of ZnO; therefore, the C atoms were not doped into the lattice of ZnO through substitution of O or Zn atoms. Moreover, because of its low surface energy, ZnO grew preferentially along with the (101) direction. Fig. 1(b) shows that the (101) diffraction peak shifted to slightly higher angle in the range of 35°–38° as the doping content of GO increased. The mean grain size (D) of the ZnO and ZnO/rGO nanocomposite samples calculated using the Scherrer formula decreased from 34.70 to 29.05 nm (Table S1†):24
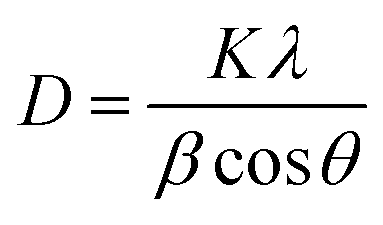 | (7) |
SEM images of the ZnO/rGO nanocomposites fabricated by the hydrothermal method are shown in Fig. 2(a–d). The ZnO/rGO nanocomposites consisted of agglomerations of ZnO nanosheets that were well anchored on the rGO film. The TEM images in Fig. 2(e) and (f) clearly show the lamellar structure of ZnO with a thickness of ∼2.3 nm and particle size of 14–36 nm, which are consistent with the result calculated from the XRD patterns (∼34 nm). Moreover, the lattice spacing determined by high-resolution TEM analysis was 0.262 nm, which is consistent with the lattice spacing of the [0001] crystal plane, confirming that ZnO grew along the [0001] direction to form nanosheets.32 These results demonstrated that HMT played an important role in controlling the morphology and structure of ZnO nanocrystals.
 | ||
| Fig. 2 SEM images (a–d) of ZnO/rGO nanocomposite, TEM images (e–g) and corresponding size distribution (h) of ZG2 catalysts. | ||
The nucleation and growth processes of ZnO nanocrystals in the presence of GO and HMT were further studied. The morphology of ZnO nanocrystals is influenced by the relative growth rates of various crystal surfaces, which can be controlled by the hydrothermal conditions. Generally, the Zn2+-terminated [0001] plane and O2−-terminated [000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] plane have high surface energy, which makes the crystal growth occur preferentially along the c-axis.33 Because HMT is a nonpolar chelating agent, it can anchor onto the nonpolar surface of ZnO crystals to impede the access of Zn2+, leaving only the polar [0001] plane available for continued growth.34,35 As a result, a 2-D ZnO nanosheet structure is formed because the growth of ZnO crystals along the c-axis is blocked. It has also been reported that GO inhibits crystal growth in the [0001] plane.36 The negatively charged oxygen-containing functional groups (e.g., COOH, OH) on the GO sheets can form strong charge interactions with the Zn2+-terminated [0001] plane, which impedes its c-axis growth.37 Thus, the intimate interaction between GO and ZnO inhibited the growth of ZnO in the [0001] plane. This is a reason why the grain size of ZnO decreased after the introduction of GO. Moreover, during the hydrothermal process, most of the HMT decomposed into NH3 and HCHO rather than being adsorbed on the nonpolar surface of ZnO, resulting in the formation of ZnO nanosheets. Therefore, the inhibition effects of GO and HMT coexisted during the ZnO synthesis process, and the competition between these two effects determined the final morphology and structure of ZnO.34
] plane have high surface energy, which makes the crystal growth occur preferentially along the c-axis.33 Because HMT is a nonpolar chelating agent, it can anchor onto the nonpolar surface of ZnO crystals to impede the access of Zn2+, leaving only the polar [0001] plane available for continued growth.34,35 As a result, a 2-D ZnO nanosheet structure is formed because the growth of ZnO crystals along the c-axis is blocked. It has also been reported that GO inhibits crystal growth in the [0001] plane.36 The negatively charged oxygen-containing functional groups (e.g., COOH, OH) on the GO sheets can form strong charge interactions with the Zn2+-terminated [0001] plane, which impedes its c-axis growth.37 Thus, the intimate interaction between GO and ZnO inhibited the growth of ZnO in the [0001] plane. This is a reason why the grain size of ZnO decreased after the introduction of GO. Moreover, during the hydrothermal process, most of the HMT decomposed into NH3 and HCHO rather than being adsorbed on the nonpolar surface of ZnO, resulting in the formation of ZnO nanosheets. Therefore, the inhibition effects of GO and HMT coexisted during the ZnO synthesis process, and the competition between these two effects determined the final morphology and structure of ZnO.34
The surface area, pore volume, pore size, and particle size of the ZnO/rGO nanocomposites are summarized in Table S2.† An appropriate content of GO increased the surface area of ZnO/rGO the nanocomposite to 41.0 m2 g−1, which was higher that of pure ZnO. (30.9 m2 g−1). Further increase of GO content caused a decrease of specific surface area because of the partial agglomeration of GO and resulted in a decrease of photocatalytic activity (vide infra) The pore volume and pore size of the samples showed the same trend as that of the specific surface area whereas the particle size gradually decreased with increasing GO content.
XPS measurements were conducted to analyze the chemical compositions and states of the ZnO and ZnO/rGO nanocomposites [Fig. 3(a–d)]. The XPS survey scans of ZG0 and ZG2 showed the characteristic peaks of Zn 2p, C 1s, and O 1s [Fig. 3(a)]. The presence of a weak C 1s peak position in the spectrum of ZG0 was ascribed to adventitious carbon originating from air pollution [Fig. 3(b)]. Three peaks centered at 284.9, 286.1, and 289.0 eV were observed in the C 1s spectrum of ZG2. The peak overlap between the carbon species in different functional groups led to the asymmetry of the main peak. The main C 1s peak of ZG2 was deconvoluted into two peaks that corresponded to oxygen-containing groups on the GO surface: sp2-hybridized C–C bonds and C–OH bonds (284.9 eV) and C–O bonds of epoxy and hydroxyl groups (286.1 eV).38 Moreover, the satellite peaks located at 289.0 eV for ZG0 and ZG2 were attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of GO or HCHO. The XPS results confirmed that GO was reduced to rGO in the nanocomposites. In Fig. 3(c), the O 1s peaks for ZG0 and ZG2 were ascribed to lattice oxygen and adsorbed oxygen: Zn–O bonds in wurtzite ZnO crystals at 530.1 eV and OH− groups chemically adsorbed on the ZnO surface at 531.5 eV.39,40Fig. 3(d) reveals that the Zn 2p signals of ZG2 moved to higher binding energy relative to those of ZG0 by 0.6 eV. The Zn 2p peaks at binding energies between 1021.1 and 1044.2 eV for ZG0 and 1021.85 and 1044.86 eV for ZG2 (originating from Zn 2p3/2 and Zn 2p1/2, respectively) confirmed that Zn was in the Zn2+ state in both samples.41
O bonds of GO or HCHO. The XPS results confirmed that GO was reduced to rGO in the nanocomposites. In Fig. 3(c), the O 1s peaks for ZG0 and ZG2 were ascribed to lattice oxygen and adsorbed oxygen: Zn–O bonds in wurtzite ZnO crystals at 530.1 eV and OH− groups chemically adsorbed on the ZnO surface at 531.5 eV.39,40Fig. 3(d) reveals that the Zn 2p signals of ZG2 moved to higher binding energy relative to those of ZG0 by 0.6 eV. The Zn 2p peaks at binding energies between 1021.1 and 1044.2 eV for ZG0 and 1021.85 and 1044.86 eV for ZG2 (originating from Zn 2p3/2 and Zn 2p1/2, respectively) confirmed that Zn was in the Zn2+ state in both samples.41
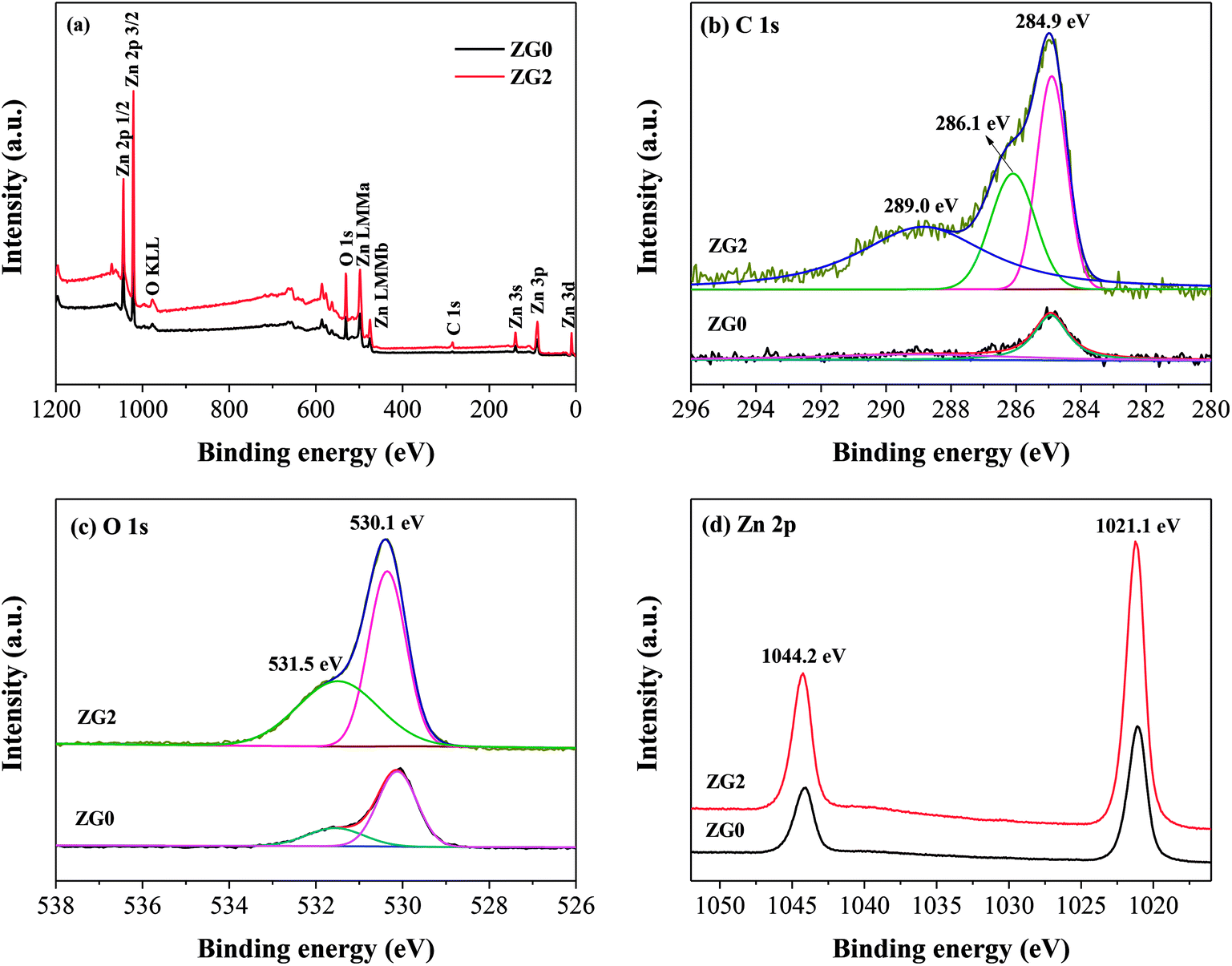 | ||
| Fig. 3 (a) XPS total survey spectra, (b) C 1s, (c) O 1s, (d) Zn 2p XPS core level spectra of ZG0 and ZG2 catalysts. | ||
Fourier transform infrared (FT-IR) spectra of GO, ZnO, and ZnO/rGO samples were further collected and are displayed in Fig. 4(a). Peaks observed at 1670, 1555, 1240, 1110, and 1000 cm−1 were assigned to CO stretching vibrations, skeleton vibrations of unoxidized sp2-hybridized graphitic domains, C–OH stretching vibrations, and C–O–C and C–O stretching vibrations, respectively.24,42 A new peak appeared at 1415 cm−1 that was attributed to the typical transverse optical stretching modes of ZnO.24 The FT-IR results indicated that some oxygen-containing functional groups remained in rGO. Moreover, all the peaks corresponding to the oxygenated functional groups in the ZnO/rGO nanocomposites gradually weakened as the rZnO content increased because of deoxygenation/reduction. In addition, no characteristic peaks of HMT were observed, indicating that HMT was completely decomposed during the hydrothermal synthesis of the ZnO/rGO nanocomposites.
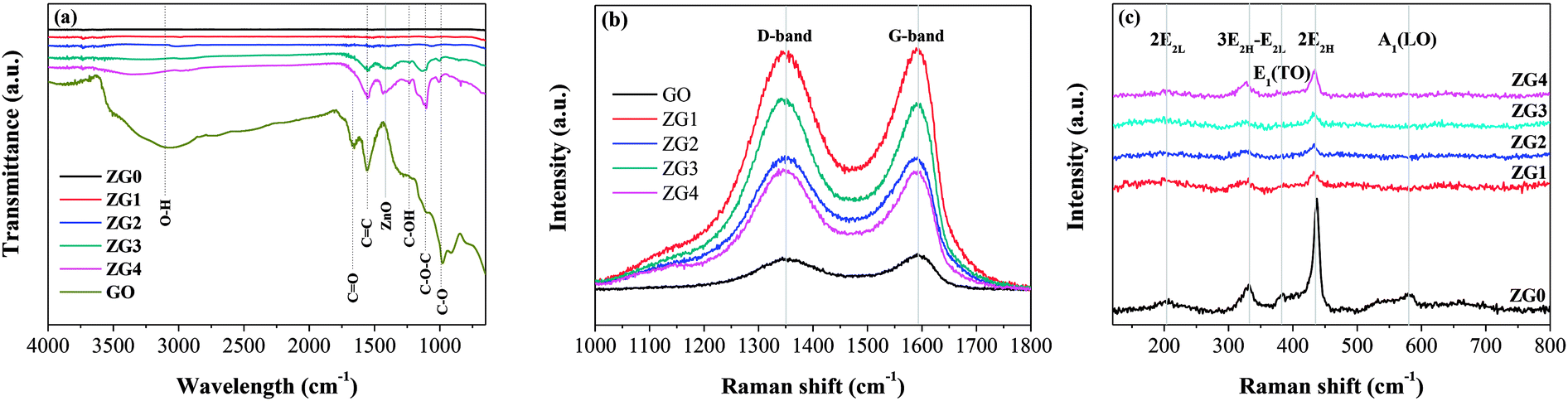 | ||
| Fig. 4 (a) FT-IR spectra of GO, ZG0, ZG1, ZG2, ZG3 and ZG4 catalysts. Raman spectra of (b) GO and ZnO/rGO nanocomposite in the range 1000–1800 cm−1 and (c) ZnO and ZnO/rGO nanocomposite in the range 120–800 cm−1. | ||
Fig. 4(b) shows Raman spectra of GO and the ZnO/rGO nanocomposites, which contained two vibrations bands at 1350 cm−1 (D band) and 1595 cm−1 (G band) in the range of 1000–1800 cm−1. The intensity ratio of the D to the G band (ID/IG) is a measure of the graphitization degree and sp2-hybridized regions in a graphene-based sample. As seen in Table S3,† the ID/IG values of the ZnO/rGO nanocomposites were higher than that of GO, indicating the decrease in the size of the sp2 domains, which led to the formation of more defects in rGO.43 To confirm the reduction of GO, ZnO in the nanocomposites was dissolved with acid and then the remaining GO was tested by elemental analysis (Table S3†). The results showed that the C content, C/H ratio, and C/O ratio all increased with the amount of GO in the nanocomposite samples. This may be partially because the reduction of HCHO to GO was hindered by the growth of more ZnO on the GO template when a small amount of GO was present in the nanocomposite. Fig. 4(c) shows the Raman spectra of ZnO and the ZnO/rGO nanocomposites in the range of 120–800 cm−1. The spectra displayed the typical active modes of ZnO with bands at 207, 331, 386, 437, and 583 cm−1. The bands at 437 and 583 cm−1 were attributed to the E2H and A1(LO) primary phonon vibrational modes of ZnO hexagonal structure, respectively.44 The E2H mode is caused by the oxygen sublattice, whereas the A1 (LO) mode is related to the amount of the defects and zinc-free carriers (i.e., Zn interstitials and oxygen vacancies). The bands at 207, 331, and 386 cm−1 were attributed to the 2 E2L, 3E2H–E2L, and E1 (TO) multiphoton scattering modes, respectively. The decrease of the band intensity of E2L and A1(LO) modes with increasing GO content might be caused by the decrease in Zn vacancies resulting from GO doping. Moreover, bands characteristic of both GO and ZnO appeared simultaneously in the Raman spectra of the ZnO/rGO nanocomposites, confirming that ZnO was well incorporated into GO.
The optical properties of ZnO and the ZnO/rGO nanocomposite were evaluated by UV-vis diffuse reflectance spectroscopy (DRS). Fig. 5(a) reveals that the maximum absorption of all samples occurred at ∼450 nm. The absorption band of the ZnO/rGO samples showed a red shift after the addition of GO, which was because the presence of GO decreased the reflectivity of the ZnO/rGO samples to light. The band-gap energy (Eg) of the samples was calculated using the Kubelka–Munk function:
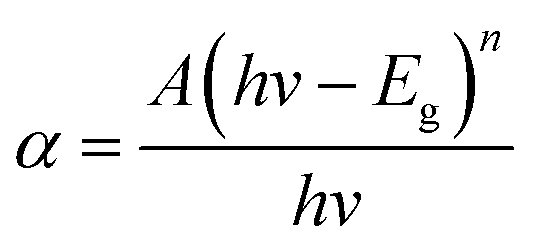 | (8) |
 | ||
| Fig. 5 (a) UV-vis diffuse reflectance spectra of modified ZnO and ZnO/rGO nanocomposite. (b) A plot of (αhv)2versus the band gap energy (eV) for ZG0 and ZG2 catalysts. (c) PL spectra of ZnO and ZnO/rGO nanocomposite. | ||
The migration, separation, and recombination of photogenerated electron–hole pairs were further evaluated by PL spectroscopy. PL spectra of ZnO and ZnO/rGO nanocomposites are shown in Fig. 5(c). The PL spectrum of rZnO contained three peaks located at 384.8, 421.7, and 439.6 nm corresponding to the recombination of free excitons, Zn interstitials, and oxygen vacancies, respectively.45–47 The addition of GO led to the fluorescence quenching of the ZnO nanosheets. It can be speculated that the carrier transfer from the ZnO nanoparticles to the rGO surface suppressed the recombination of electron–hole pairs, which weakened the fluorescence of ZnO.48 Therefore, introduction of GO decreased the defect content of ZnO, resulting in the improved photocatalytic activity of the ZnO/rGO composites. In particular, ZG2 showed the weakest PL signal, indicating that it had the highest electron transfer rate of the nanocomposites. These results suggest that the photocatalytic activity of ZG2 should be higher than that of other ZnO/rGO nanocomposites.
3.2 Photocatalytic performance
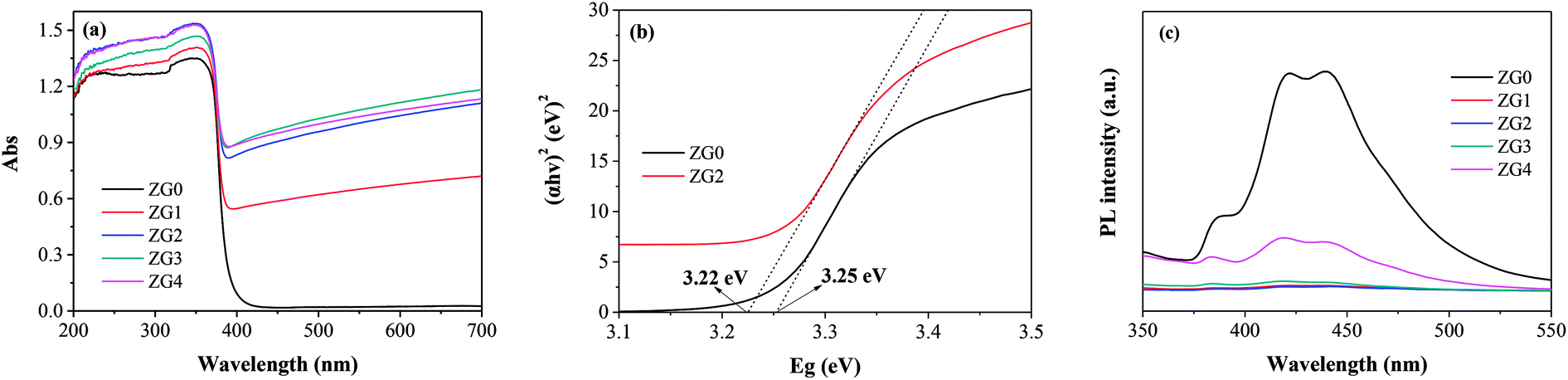
The photodegradation ability of ZnO and the ZnO/rGO nanocomposites was examined by assessing the photocatalytic decomposition of dimethoate under UV light at room temperature. As shown in Fig. 6(a), the degradation capacities of the ZnO/rGO catalysts were much better than that of the ZnO catalyst. rGO performed high adsorption capacity due to its large specific surface area, unique pore structure and electrostatic attraction of surface functional groups. During the preparation of ZG material, a large number of functional groups on rGO are loosed. Moreover, the surface of rGO was covered by ZnO, which also reduced the surface area and pore volume. On the other hand, the composite process makes the dispersion of ZnO on GO get better and cause the recombination rate of hole electron pairs decreased, which improves the degradation efficiency of the composites. The rate of dimethoate adsorption by ZG2 catalyst under dark conditions after 180 min was lower than 5%. The photocatalytic efficiency of ZG2 in dimethoate degradation reached to approximately 99% after 180 min of UV light irradiation.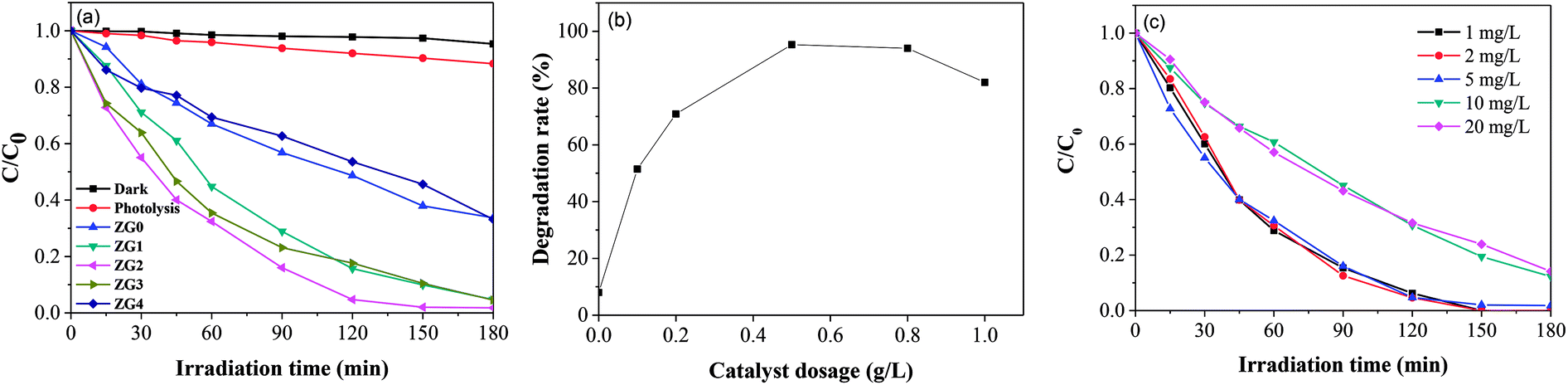 | ||
| Fig. 6 Effect of photocatalytic parameters on degradation rate of dimethoate: (a) rGO dosage (catalyst dosage, 0.5 g L−1; dimethoate, 5 mg L−1); (b) catalyst dosage (photocatalyst, ZG2; dimethoate, 5 mg L−1; 2 h). (c) dimethoate concentration (photocatalyst, ZG2; catalyst dosage, 0.5 g L−1). | ||
In this work, the kinetic rate constants of dimethoate degradation were obtained by the following pseudo-first-order and second-order kinetic models:46
| ln(C0/C) = k1t | (9) |
| (1/C) − (1/C0) = k2t | (10) |
The effect of ZG2 dosage (0.1 to 1.0 g L−1) on dimethoate degradation was then evaluated; the results are illustrated in Fig. 6(b). Increasing the concentration of ZG2 from 0 to 0.5 g L−1 led to a remarkable increase in the photocatalytic efficiency of dimethoate degradation. A higher dose of catalyst provided more active sites, which promoted the generation of electron–hole pairs and accelerated the production of reactive radicals that induced dimethoate degradation. However, further increasing the catalyst dose (from 0.5 to 1.0 g L−1) decreased the light penetration through the reaction system, which led to a decrease of photocatalytic activity. Subsequent reactions were carried out using the optimum catalyst dosage of 0.5 g L−1.
The initial concentration of dimethoate in the photocatalytic system is a very important parameter. The initial dimethoate concentration (1.0 to 20 mg L−1) in photodegradation experiments was thus investigated. Fig. 6(c) shows that increasing the dimethoate concentration from 1.0 to 20 mg L−1 resulted in a decrease of the degradation rate after 3 h from 100% to 85.9%. It is presumed that the dimethoate adsorbed on the surface of the catalyst blocked the active sites. Moreover, a high concentration of dimethoate will compete with the photocatalyst to absorb light energy, decreasing the light absorbed by the photocatalyst.47 To maintain a high degradation rate, 5 mg L−1 was selected as the optimum initial dimethoate concentration.
3.3 Photocatalytic mechanism
Fig. 7(a) illustrates I–t curves of the prepared photoelectrodes in the dark and under 0.95 mW cm−2 illumination. Both the ZG2 and ZG0 electrodes showed sensitive and stable photoinduced current responses for each switch-on and switch-off event. The I–t curves of the electrodes were almost identical and I rapidly decreased to zero when the light was switched off. The photoinduced current density of the ZG2 photoelectrode was 2.91 × 10−9 A cm−2 (vs. Ag/AgCl), which was 1.33 times higher than that of ZG0 (2.18 × 10−9 A cm−2vs. Ag/AgCl). The photocurrent enhancement of the ZG2 electrode indicated the separation efficiency of photogenerated electron–hole pairs and better carrier transmission efficiency in the ZnO/rGO nanocomposite because of the presence of rGO. | ||
| Fig. 7 (a) The photoinduced current density curves of ZG0 and ZG2 electrodes (V vs. Ag/AgCl). (b) EIS spectra of ZG0 and ZG2 photoanodes. (c) Mott–Schottky plots of ZG0 and ZG2 electrodes (V vs. Ag/AgCl). (d) Trapping experiments of active species during the photocatalytic reaction. | ||
Nyquist plots of the ZG0 and ZG2 electrodes revealed the electron transfer process at the electrolyte–electrode interface [Fig. 7(b)]. For each electrode, only one arc was observed in the Nyquist plot, indicating that only surface charge transfer occurred in the photocatalytic degradation process. The arc in the Nyquist plot of the ZG2 electrode showed a smaller radius than was the case for ZG0, indicating lower resistance and faster reaction rate on its electrode surface. That is to say, the introduction of rGO accelerated the transfer of interfacial electrons and promoted the separation of photogenerated charge carriers.
MS measurements were performed to characterize the flat-band potentials (Efb) and charge carrier densities (Nd) at the photoanode/electrolyte interface. Fig. 7(c) shows the MS plots of the ZG0 and ZG2 electrodes, which had positive slopes consistent with the typical behavior of n-type semiconductors. Efb and Nd of the electrodes were calculated using the MS equation:
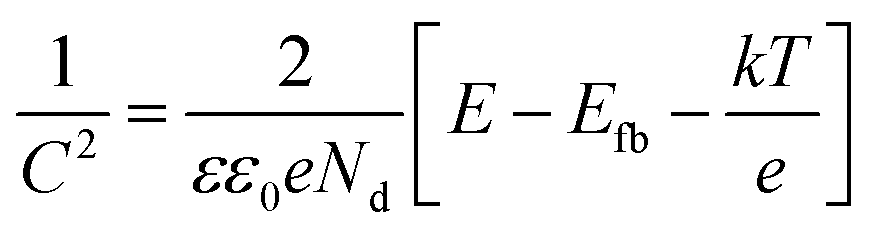 | (11) |
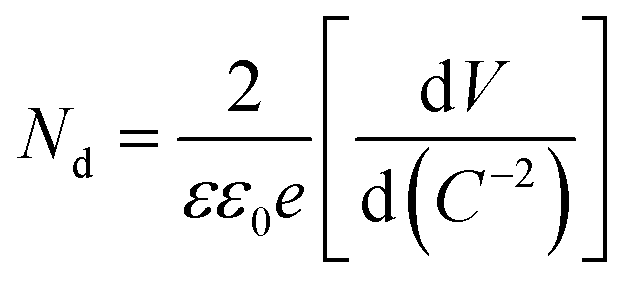 | (12) |
To evaluate the photocatalytic degradation pathway of dimethoate, AgNO3 (1.0 mmol L−1), disodium ethylenediaminetetraacetate (EDTA–2Na), isopropyl alcohol (IPA), and benzoquinone (BQ) were used as scavengers of electrons, holes, superoxide radicals (˙O2−), and hydroxyl radicals (˙OH), respectively. As shown in Fig. 7(d), dimethoate displayed resistance to degradation under UV irradiation in the presence of AgNO3. Addition of IPA caused the degradation efficiency of dimethoate by ZG2 to decrease slightly, confirming that electrons and ˙OH did not have a strong effect of the degradation of dimethoate, which is in agreement with a previous report.48 In contrast, the addition of BQ and EDTA–2Na led to considerable decreases of the degradation rate constant, indicating that ˙O2− and holes play major roles in the photocatalytic degradation process of dimethoate.
The proposed photodegradation mechanism of dimethoate by the ZnO/rGO nanocomposite is shown in Fig. S2.† The delocalized conjugated π structure and superior electrical conductivity of graphene improve the transmission efficiency of photogenerated electrons.49 Therefore, the recombination of electron–hole pairs generated on ZnO was suppressed by electron transfer to rGO. Photogenerated electrons can easily jump from the VB to the CB under UV light. Because the work function of ZnO (5.2 eV) is higher than that of GO (4.6 eV), electrons could transfer into the composite and reach rGO.50,51 Therefore, the increased separation of electron–hole pairs leads to higher photodegradation efficiency. The photogenerated electrons then react with oxygen adsorbed on the ZnO surface to generate reactive species such as ˙O2− that decompose dimethoate. Moreover, dimethoate can act as an electron donor to generate excited electrons and form free radical cations.52 The electrons are transferred to the rGO sheets, where they react with surface-adsorbed O2 to form free radicals.
3.4 Recycling of ZnO/rGO
In the photocatalyst recycling test, the ZG2 powder obtained after one photocatalytic cycle was filtered, washed with distilled water and ethanol, dried, and then reused for four consecutive runs. Because of the inevitable loss of catalyst powder during the washing and recycling process, fresh catalyst was used to maintain the catalyst content. Even though the photodegradation efficiency decreased slightly with increasing runs [Fig. S3(a)†], the degradation of dimethoate by ZG2 remained above 95% even in the fourth run.XPS measurements were used to analyze the ZG2 catalyst before and after use to better investigate the change in the elements present and their electronic states after the recycling test. After reuse four times, small peaks corresponding to P 2p from dimethoate were detected in the XPS survey scan, indicating the presence of dimethoate on the catalyst [Fig. S3(b)†]. The binding energy of Zn did not change after recycling. However, compared with that of the Zn species of the fresh catalyst, the peak area of the Zn species decreased for the used catalyst [Fig. S3(c)†], indicating that dimethoate was adsorbed onto the ZG2 catalyst. In addition, the intensity of peaks from C–O and CO groups at binding energies of 286.1 and 288.9 eV, respectively, increased for the used ZG2 catalyst [Fig. S3(d)†]. Therefore, trace dimethoxy molecules remained adsorbed on the ZnO/rGO nanocomposite and covered some its active sites, which weakened the characteristic peaks of Zn and strengthened those of C in the XPS profiles. These results confirmed that the ZnO/rGO nanocomposite can be conveniently recovered for reuse, which provides a potentially effective and environmentally friendly pathway to treat environmental pollutants.
4. Conclusions
A ZnO/rGO nanocomposite photocatalyst was synthesized by a hydrothermal method using HMT as a mineralizer and reducing agent. Compared with those of bare ZnO, the ZnO/rGO nanocomposite exhibited a photodegradation rate and photodegradation efficiency of dimethoate that were four and 1.5 times higher, respectively. GO increased surface area and pore volume of the composite, which were conducive for the adsorption and mass transfer of pesticides and oxygen. The improved photocatalytic performance of the nanocomposite was attributed to the decreased electron–hole recombination rate and more effective carrier transport in the presence of rGO. A high rate of interfacial charge transfer was confirmed by photoelectrochemical testing. Overall, the results showed that ZnO/rGO nanocomposites have broad application prospects in optics, electrical, and optoelectronics applications.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors wish to acknowledge the financial support from National Natural Science Foundation of China (No. 31270620), Fundamental Research Funds for the Central Universities (No. DUT19JC13) and Dalian Scientific and Technological Innovation Foundation (No. 2018J12SN072).References
- M. R. Bonner and M. C. R. Alavanja, Food Energy Secur., 2017, 6, 89–93, DOI:10.1002/fes3.112.
- J. Cooper and H. Dobson, Crop Protect., 2007, 26, 1337–1348, DOI:10.1016/j.cropro.2007.03.022.
- J. Li, F. Li and Q. Liu, Mar. Pollut. Bull., 2015, 100, 516–522, DOI:10.1016/j.marpolbul.2015.09.003.
- H. Yuan, E. Liu, E. Zhang, W. Luo, L. Chen, C. Wang and Q. Lin, Chemosphere, 2017, 173, 78–88, DOI:10.1016/j.chemosphere.2017.01.047.
- S. H. Douglas, B. Dixon and D. Griffin, Phys. Geogr., 2018, 1–19, DOI:10.1080/02723646.2017.1406300.
- S. A. Mansour, Toxicology, 2004, 198, 91–115, DOI:10.1016/j.tox.2004.01.036.
- U. Hoffmann and T. Papendorf, Intensive Care Med., 2006, 32, 464–468, DOI:10.1007/s00134-005-0051-z.
- I. Jaabiri Kamoun, O. O. Jegede, O. J. Owojori, J. Bouzid, R. Gargouri and J. Rombke, Integr. Environ. Assess. Manage., 2018, 14, 92–104, DOI:10.1002/ieam.1966.
- J. Zhang, X. Huang, H. Liu, W. Liu and J. Liu, Toxicol. Sci., 2018, 162, 53–63, DOI:10.1093/toxsci/kfx244.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027, DOI:10.1016/j.watres.2010.02.039.
- D. Ayodhya and G. Veerabhadram, Photochem. Photobiol. Sci., 2018, 17, 1429–1442, 10.1039/c8pp00220g.
- M. Yuan, X. Liu, C. Li, J. Yu, B. Zhang and Y. Ma, Int. J. Biol. Macromol., 2019, 126, 857–866, DOI:10.1016/j.ijbiomac.2018.12.157.
- H. Zheng, Q. Zhang, G. Liu, X. Luo, F. Li, Y. Zhang and Z. Wang, Sci. Total Environ., 2019, 657, 953–962, DOI:10.1016/j.scitotenv.2018.12.018.
- J.-B. Huo, L. Xu, X. Chen, Y. Zhang, J.-C. E. Yang, B. Yuan and M.-L. Fu, Microporous Mesoporous Mater., 2019, 276, 68–75, DOI:10.1016/j.micromeso.2018.09.017.
- S. Deng, Y. Chen, D. Wang, T. Shi, X. Wu, X. Ma, X. Li, R. Hua, X. Tang and Q. X. Li, J. Hazard. Mater., 2015, 297, 17–24, DOI:10.1016/j.jhazmat.2015.04.052.
- D. Meng, W. Jiang, J. Li, L. Huang, L. Zhai, L. Zhang, Z. Guan, Y. Cai and X. Liao, J. Environ. Sci. Health, Part B, 2019, 1–8, DOI:10.1080/03601234.2019.1571363.
- T. Liu, S. Xu, S. Lu, P. Qin, B. Bi, H. Ding, Y. Liu, X. Guo and X. Liu, Sci. Total Environ., 2019, 651, 2247–2268, DOI:10.1016/j.scitotenv.2018.10.087.
- H. Liu, J. Chen, N. Wu, X. Xu, Y. Qi, L. Jiang, X. Wang and Z. Wang, Ecotoxicol. Environ. Saf., 2019, 170, 259–266, DOI:10.1016/j.ecoenv.2018.11.132.
- R. Saini and P. Kumar, J. Environ. Chem. Eng., 2016, 4, 2952–2963, DOI:10.1016/j.jece.2016.06.003.
- H. Rubí-Juárez, S. Cotillas, C. Sáez, P. Cañizares, C. Barrera-Díaz and M. A. Rodrigo, Appl. Catal., B, 2016, 188, 305–312, DOI:10.1016/j.apcatb.2016.02.006.
- C. Dong, J. Yang, Y. Xiao, M. Xing and J. Zhang, Chin. Chem. Lett., 2019, 853–862, DOI:10.1016/j.cclet.2019.03.020.
- I. Nabi, Z. Fu, K. Li, H. Cheng and L. Zhang, Chin. Chem. Lett., 2019, 30, 2225–2230, DOI:10.1016/j.cclet.2019.07.058.
- D. Chaudhary, S. Singh, V. D. Vankar and N. Khare, J. Photochem. Photobiol., A, 2018, 351, 154–161, DOI:10.1016/j.jphotochem.2017.10.018.
- R. Paul, R. N. Gayen, S. Biswas, S. V. Bhat and R. Bhunia, RSC Adv., 2016, 6, 61661–61672, 10.1039/c6ra05039e.
- M. Sun, Z. Chen, X. Jiang, C. Feng and R. Zeng, J. Alloys Compd., 2019, 780, 540–551, DOI:10.1016/j.jallcom.2018.12.019.
- L. Wang, Z. Li, J. Chen, Y. Huang, H. Zhang and H. Qiu, Environ. Pollut., 2019, 249, 801–811, DOI:10.1016/j.envpol.2019.03.071.
- Y. Rambabu, U. Kumar, N. Singhal, M. Kaushal, M. Jaiswal, S. L. Jain and S. C. Roy, Appl. Surf. Sci., 2019, 485, 48–55, DOI:10.1016/j.apsusc.2019.04.041.
- B. Zeng, W. Liu and W. Zeng, J. Mater. Sci. Mater. Electron., 2019, 30, 3753–3759 CrossRef CAS.
- H. Li, P. Wang, X. Yi and H. Yu, Appl. Catal., B, 2020, 264, 118504, DOI:10.1016/j.apcatb.2019.118504.
- D. Xu, B. Cheng, S. Cao and J. Yu, Appl. Catal., B, 2015, 164, 380–388 CrossRef CAS.
- J. Chen, Y. Li, L. Huang, C. Li and G. Shi, Carbon, 2015, 81, 826–834 CrossRef CAS.
- S. Zhu, X. Chen, F. Zuo, M. Jiang, Z. Zhou and D. Hui, J. Solid State Chem., 2013, 197, 69–74, DOI:10.1016/j.jssc.2012.09.001.
- V. R. Shinde, T. P. Gujar, T. Noda, D. Fujita, A. Vinu, M. Grandcolas and J. Ye, Chemistry, 2010, 16, 10569–10575, DOI:10.1002/chem.200903370.
- H. Li, S. Jiao, H. Li and L. Li, J. Mater. Sci.: Mater. Electron., 2014, 25, 2569–2573, DOI:10.1007/s10854-014-1911-5.
- S. Baruah and J. Dutta, Sci. Technol. Adv. Mater., 2009, 10, 013001, DOI:10.1088/1468-6996/10/1/013001.
- X. Pan, M. Q. Yang and Y. J. Xu, Phys. Chem. Chem. Phys., 2014, 16, 5589–5599, 10.1039/c3cp55038a.
- J. Maryam, B. Qiaoliang, Y. Jia-Xiang and L. Kian Ping, J. Am. Chem. Soc., 2010, 132, 14487–14495 CrossRef PubMed.
- B. Li, T. Liu, Y. Wang and Z. Wang, J. Colloid Interface Sci., 2012, 377, 114–121, DOI:10.1016/j.jcis.2012.03.060.
- H. Chen, W. Liu and Z. Qin, Catal. Sci. Technol., 2017, 7, 2236–2244 RSC.
- Z.-W. Wu, S.-L. Tyan, H.-H. Chen, Y.-C. Huang, C.-R. Lee and T.-S. Mo, Superlattices Microstruct., 2017, 107, 38–43 CrossRef CAS.
- A. S. Alshammari, L. Chi, X. Chen, A. Bagabas, D. Kramer, A. Alromaeh and Z. Jiang, RSC Adv., 2015, 5, 27690–27698, 10.1039/c4ra17227b.
- A. M. Rocco, D. P. Moreira and R. P. Pereira, Eur. Polym. J., 2003, 39, 1925–1934 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565, DOI:10.1016/j.carbon.2007.02.034.
- I. Kazeminezhad, S. Saadatmand and R. Yousefi, Bull. Mater. Sci., 2016, 39, 719–724, DOI:10.1007/s12034-016-1206-y.
- S. Alamdari, M. S. Ghamsari, H. Afarideh, A. Mohammadi, S. Geranmayeh, M. J. Tafreshi and M. H. Ehsani, Opt. Mater., 2019, 92, 243–250 CrossRef CAS.
- C. Worathitanon, K. Jangyubol, P. Ruengrung, W. Donphai, W. Klysubun, N. Chanlek, P. Prasitchoke and M. Chareonpanich, Appl. Catal., B, 2019, 241, 359–366, DOI:10.1016/j.apcatb.2018.09.048.
- M. Muruganandham, N. Shobana and M. Swaminathan, J. Mol. Catal. A: Chem., 2006, 246, 154–161, DOI:10.1016/j.molcata.2005.09.052.
- A. Habibi-Yangjeh and M. Shekofteh-Gohari, Sep. Purif. Technol., 2017, 184, 334–346, DOI:10.1016/j.seppur.2017.05.007.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387, DOI:10.1016/j.apcatb.2010.10.007.
- H. Kou, L. Jia and C. Wang, Carbon, 2012, 50, 3522–3529, DOI:10.1016/j.carbon.2012.03.020.
- J. Liu, Y. Xue, Y. Gao, D. Yu, M. Durstock and L. Dai, Adv. Mater., 2012, 24, 2228–2233, DOI:10.1002/adma.201104945.
- M. K. Sabullah, M. F. Rahman, S. A. Ahmad, M. R. Sulaiman, M. S. Shukor, N. A. Shamaan and M. Y. Shukor, Journal of Mathematical and Fundamental Sciences, 2017, 49, 193–210, DOI:10.5614/j.math.fund.sci.2017.49.2.8.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01741h |
| This journal is © The Royal Society of Chemistry 2020 |