
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic-scale synthesis of nanoporous gallium–zinc oxynitride-reduced graphene oxide photocatalyst with tailored carrier transport mechanism†
Babak Adeli a and
Fariborz Taghipour*ab
a and
Fariborz Taghipour*ab
aDepartment of Chemical and Biological Engineering, University of British Columbia, Vancouver, Canada. E-mail: fariborz.taghipour@ubc.ca
bClean Energy Research Center (CERC), University of British Columbia, Vancouver, Canada
First published on 15th April 2020
Abstract
Surface modified gallium–zinc oxynitride solid solution exhibited outstanding stability and visible-light activity for water splitting. However, the considerable rate of photo-induced charge recombination and the low surface area of the bulk photocatalyst limited its performance. Here, an efficient technique is proposed for the synthesis of a nanoporous oxynitride photocatalyst and its graphene-hybridized material. The nanoporous oxynitride photocatalyst was prepared via a nanoscale solid-state route, using microwave irradiation as an intermolecular-state activation method, Ga3+/Zn2+ layered double hydroxide as an atomic-level uniform mixed-metal precursor, and urea as a non-toxic ammonolysis soft-template. The graphene-hybridized photocatalyst was fabricated using a facile electrostatic self-assembly technique. The photocatalytic activity of the synthesized graphene hybridized nanoporous oxynitride photocatalyst was systematically improved through shortening the majority-carrier diffusion length and enhancing the density of active hydrogen evolution sites within the quasi-three-dimensional nanostructure, reaching 7.5-fold sacrificial photocatalytic hydrogen evolution, compared to the conventional 1 wt% Rh-loaded oxynitride photocatalyst.
Introduction
The development of novel photocatalyst materials for the direct conversion of sunlight to hydrogen has been the focus of intensive studies in the last decade. In particular, oxynitride semiconductors have garnered considerable attention due to their photo-electronic characteristics, which enable visible-light applications.1,2 However, the photocatalytic hydrogen evolution reaction (HER) scarcely proceeded on the surface of oxynitride photocatalysts, as the surface of metal nitrides and oxynitrides are not active for the HER without surface decoration with catalytic sites.3 Therefore, the development of a highly active visible-light responsive oxynitride photocatalyst requires innovative morphologies to enhance the structural capacity for deposition of HER active sites.It is known that highly active HER catalysts also catalyzed the undesired water formation from evolved hydrogen and oxygen, the so-called water splitting reverse reaction.4 Moreover, the high cost associated with the utilization of highly active HER catalysts, typically noble metals such as Pt and Rh, reduces their commercial feasibility. Recent advances in nanoscale fabrication and photochemistry have led to various strategies to suppress the undesirable water splitting reverse reaction, as well as utilization of Earth abundant HER catalysts, those include surface modifications with highly efficient HER catalysts,4,5 and photosensitization.6–8 For instance, Sayama and Arakawa and Abe et al. showed that the rate of reverse reaction on a Pt catalyst surface could be suppressed by adding Na2CO3 or NaI to the water splitting solution,9,10 while Domen and co-workers engineered a metal–Cr2O3 core–shell co-catalytic system that suppresses the undesired reaction of H2 and O2, achieving mmol h−1 scale hydrogen evolution and quantum efficiencies as high as 5.9% in overall water splitting under visible light irradiation in acidic pHs.3,11,12 Our group has reported the facile synthesis of GaZnON photocatalyst through soft-chemistry methods, followed by rapid solid-state reaction within urea soft-template.13 The as prepared photocatalyst exhibited overall water splitting activity with up to 2.5% apparent quantum efficiency. Recently, Li et al. reported comparable photocatalytic performance, once the conventional Rh active sites are replaced by less content of Pd, reaching to 1.6% overall water splitting apparent quantum efficiency at pH of 4.5.12 Moreover, The development of highly active HER catalysts from Earth abundant compounds is at the frontier of nanotechnology and electrochemistry. Recent efforts, benefited from atomic-scale fabrication routes, resulted in the development of durable and cost-effective HER catalysts with performances favorably comparable to those reported for the noble metal state-of-the-art catalysts.14–17 However, activating the surface of semiconductors with a high density of HER active sites has remained a challenge,18 due to the low specific surface area of the photocatalyst materials. It is well-documented that excessive loading of co-catalyst active sites causes degradation in the photocatalytic activity by shading the surface of the photocatalyst from incident photons.19 To simultaneously enhance the rate of photo-generated carrier transport and the density of catalytic sites, the diffusion length of majority charge carriers must be shortened significantly, while the active surface area of the photocatalyst must be increased substantially.
Among oxynitride semiconductors, the crystalline solid alloy of gallium nitride and zinc oxide (GaN:ZnO) has exhibited outstanding stability and overall water splitting performance under visible-light irradiation.20 It is evident that the visible light activity of GaN:ZnO is attributed to large electron–hole spatial separation due to the preferential flow of holes to the GaN-rich surface and efficient electron extraction by the cocatalyst.21 However, the bulk structure of the GaN:ZnO photocatalyst burdens high quantum efficiency realization, as the majority of the photo-induced carriers are recombined at structural defects and grain boundaries.22 Moreover, the loading of HER-active sites is significantly hindered (typically less than 1.5 wt%) by the low specific surface area of the bulk GaN:ZnO (3.7–8.1 m2 g−1 (ref. 23)). Thus, the development of a morphologically tailored GaN:ZnO nanostructure with a large specific surface area for the high–density loading of HER sites, as well as a short majority charge carrier diffusion length for effective transportation of charges are of great importance.
In the past decade, the crystalline structure,20,24–26 optical and electronic characteristics,27–29 synthesis method,13,30–35 and surface modification3,11,36,37 of GaN:ZnO have been investigated in detail. In particular, considerable attention has been dedicated to the preparation of GaN:ZnO solid solution nanostructures with tailored morphologies and structures. Yan and co-workers reported ion-exchange-mediated synthesis of mesoporous GaZnON photocatalyst via ammonolysis of mesoporous ZnGa2O4.35,38 Yang and colleagues studied the one-pot preparation of nanoporous GaZnON solid solution with surface areas up to 31.1 m2 g−1.39 Lee et al. developed a multi-step route for the preparation of Zn-rich GaN:ZnO nanoparticles (dp ∼ 18 nm) using synthetic ZnGa2O4 and ZnO nanocrystallites as the starting materials.31 Our group proposed a facile route for the fabrication of multi-crystal GaN:ZnO solid solution (GaZnON) photocatalyst with a wide range of Zn-content, high surface areas, and encouraging overall water splitting activities.13 Recently, Katagiri and co-workers studied the formation of wurtzite oxynitride phase via urea-mediated ammonolysis of uniform oxide precursor and suggested a kinetically stable kinetics via Zn(CN2)-like intermediate.40 Further, Godin et al. highlighted the importance of charge carriers transport mechanism in visible light activity of GaN:ZnO.21
The fabrication of advanced photocatalytic systems based on GaZnON has received considerable attention.22,41–45 Graphene-hybridized GaZnON is acknowledged to be an excellent photocatalyst, owing to the fascinating characteristics of composite photocatalysts in promoting rapid charge transport, compare to its bare photocatalytic counterpart.46 Authors of this article for the first time reported the ex situ electrostatic self-assembly fabrication of GaN:ZnO-reduced graphene oxide (rGO) with over 70% improvement in apparent quantum efficiency.41 Li et al. reported a 4.5-fold photocatalytic activity improvement for the graphene-based composite of GaN:ZnO, prepared through nitridation of a hydrothermally synthesized ZnGa2O4–GO composite.42 In spite of the efforts to fabricate nanostructure composite oxynitride solid solutions, the utilization of the prepared photocatalysts to improve the deposition capacity of HER active sites and the engineering of their structures to shorten the carrier diffusion length have not been studied.
Here, we developed a systematic approach for enhancing the density of HER-catalytic sites, as well as reducing the rate of photo-recombination losses through engineering a three-dimensional (3D) framework, consisting of interconnected oxynitride photocatalyst nanostructure, catalytic sites, and two-dimensional conductive scaffold. We propose an efficient fabrication technique for an organic–inorganic hybrid nanostructure photocatalyst, consisting of nanoporous GaZnON and reduced graphene oxide, which to our knowledge is reported for the first time. For the synthesis of the nanoporous GaZnON photocatalyst microwave irradiation was employed as the intermolecular solid-state kinetics activation source, Ga3+/Zn2+ layered double hydroxide (LDHs), which can also be expressed as [(Zn2+)1−m(Ga3+)m(OH)2]m+(CO32−)m/2·nH2O, was used as the atomic-level uniform mixed-metal precursor, and urea was applied as the non-toxic ammonolysis soft-template. The reduced graphene oxide hybridized nanoporous GaZnON composite was prepared through a facile electrostatic self-assembly route. By utilizing the surface modified structural nanopores and graphene as the active sites of hydrogen evolution reaction, the redox active sites were positioned at distinct areas within the well-defined 3D structure; thus the photocatalytic activity was increased by 7.5-fold, compared to the typical 1 wt% Rh-loaded oxynitride photocatalyst. The present study highlights the effectiveness of morphology engineering in addressing the key water splitting challenges, including limited hydrogen and oxygen evolution catalytic sites density and bulk recombination of photo-induced charges. Further, the methodology developed and discussed in this article can be further adapted and applied to photocatalysts from a rich variety of materials.
Experimental
The following materials have been used for processing of various samples as received, without further purifications:Ga2O3 (99.99+%, Sigma-Aldrich), ZnO (99+%, Sigma-Aldrich), Zn (99+%, Sigma-Aldrich), urea (Sigma-Aldrich), NaOH (≥97%, Fisher Scientific), Na2CO3 (≥99.50%, Fisher Scientific), Na3RhCl6 (Sigma-Aldrich), H2PtCl6·6H2O (≥37.50% Pt basis, Sigma-Aldrich), Ni(NO3)2·6H2O (99.999%, Sigma-Aldrich), KMnO4 (99%, EMD Millipore), NaNO2 (≥99%, Sigma-Aldrich), natural graphite flakes (Sigma-Aldrich), H2SO4 (98%, Sigma-Aldrich), NaNO3 (99%, Sigma-Aldrich), and H2O2 (30–32 wt% in water, Sigma-Aldrich).
Nanoporous GaZnON synthesis
Layered double hydroxides (LDHs) of Ga3+ and Zn2+ were prepared via the method described previously.13 In a typical synthesis, Ga2O3 and ZnO powder ([Zn]/[Zn + Ga] = 0.5) were dissolved in 8 M HCl solution. The pH of the solution was adjusted to 9, with a mixed aqueous solution of 2 M NaOH and 1 M Na2CO3 added drop-wise to the acidic solution under vigorous stirring. The obtained slurry was aged in the mother liquor for 18–24 h at 80 °C. The precipitates were collected and washed thoroughly with pure water and ethanol and subsequently dried overnight in a laboratory oven at 80 °C. The prepared LDHs powder was mixed with Zn ([Zn]/[Ga] = 1.0) and urea powders in a stainless-steel ball-mill jar for 10 min to obtain a uniform precursor.The nanoporous GaZnON solid solution was synthesized through MW treatment of the prepared organic–inorganic hybrid precursor, using a Panasonic NN-SD671S MW oven. The prepaid hybrid precursor was loaded inside a semi-sealed alumina chamber, which was placed inside an alumina container. The space between the two containers was filled with CuO heated medium, as described previously.39 The precursor were thereafter treated at 720 W for 12 min, reaching to nearly 900 °C in temperature. The synthesized photocatalyst was thoroughly washed with water and ethanol aqueous solution for several times, dried at 80 °C overnight, and annealed in air at 600 °C for 1 h.
GaZnON–rGO composite synthesis
A solution of graphene oxide was prepared through the modified Hummers' method, using pre-exfoliated graphite flakes according to the method we have reported previously.41 A total of 1 mg mL−1 of GO solution was prepared by ultra-sonication of an appropriate amount of the prepared GO in pure water for 1–2 h. The GaZnON–GO composite was prepared by 1 h sonication and several hours mixing of the nanoporous GaZnON photocatalyst uniform suspension and GO solution. The prepared suspension was then dispersed in 10 V% ethanol solution, purged with ultra-pure nitrogen, and exposed to visible-light irradiation for 4–6 h to obtain the GaZnON–rGO composite. The prepared rGO composite was collected, washed with pure water, and dried overnight.Surface modification with co-catalyst
Na3RhCl6, H2PtCl6·6H2O, and Ni(NO3)2·6H2O salts were used for the deposition of Rh, Pt, and NiOx hydrogen evolution catalysts, respectively. The co-catalyst loading (wt%) stated in this article represents the metallic contents of the salts in the deposition solution.The co-catalyst nanoparticles were deposited through photo-deposition, as described previously,11 with modifications. The synthesized photocatalyst was stirred in a 10 V% ethanol solution and purged with ultra-pure nitrogen for several hours to obtain an oxygen-free suspension. An oxygen-free solution containing an appropriate amount of co-catalyst salt was introduced to the photocatalyst suspension in the dark and purged with ultra-pure nitrogen for another 2 h, when the suspension was irradiated with visible light for 4–6 h. The surface modified samples were collected, washed with pure water, dried overnight, and subsequently annealed in air at 350–400 °C for 1 h.
Material characterization
The surface morphology of the samples and their bulk compositions were studied through scanning electron microscopy (SEM) using an FEI Helios NanoLab 650 dual beam scanning electron microscope equipped with an EDAX TEAM Pegasus energy dispersive X-ray spectrometer. For imaging, the samples were coated with 4 nm Ir using a high-vacuum Leica EM MED 020 sputter coater. The edges of the samples were painted with silver suspension to ensure adequate conductivity.The surface area (SBET) measurement of the samples was obtained from the single-point Brunauer–Emmett–Teller (BET) analysis, using a Micrometrics FlowSorbII 2300. An appropriate amount of sample was loaded in a Pyrex BET tube and degassed for several hours prior to the test.
The studies of the surface chemical states and compositions of the samples were conducted using a Leybold MAX200 X-ray photoelectron spectrometer with Al K-alpha and Mg K-alpha X-ray sources. High-resolution XPS data were analyzed with XPSPEAK 4.1 software.
The structure and elemental distribution of the synthesized samples were investigated through transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) using an FEI Tecnai Osiris S/TEM system equipped with an Analytical TWIN (A-TWIN) objective lens integrated with the Super-X EDX detection system, Gatan Ultrascan CCD camera, high angle annular dark field (HAADF), and BF STEM detectors, capable of STEM/EDX mapping (Esprit), and S/TEM tomography. The MW-treated sample exhibited variations in chemical composition, likely due to the random irradiation pattern of the MW oven; thus, the S/TEM characterizations were performed for a similar sample prepared inside a furnace. The HAADF tomography was conducted by acquiring the STEM image of an individual particle in ±70° rotation at 2° intervals.
Photocatalytic reaction
Photocatalytic hydrogen production reactions were conducted in a 900 mL (reaction volume) borosilicate glass reactor, where a USHIO 100 W high pressure UV-vis Hg lamp was located at the center, in a quartz sleeve. The UV portion (λ ≤ 420 nm) of the lamp's output was filtered by 2 M NaNO2 solution. A total of 400 mg of the photocatalyst decorated with an appropriate amount of co-catalyst materials was dispersed in a 10 V% methanol aqueous solution inside the photo-reactor. The photo-reactor was purged with ultra-pure nitrogen until oxygen-free suspension was obtained. Analysis of the evolved hydrogen was carried out using an Agilent 7890A gas chromatograph equipped with a thermal conductivity detector (TCD). The reported photocatalytic hydrogen evolution refers to average evolution rate after 3 hours of reaction.Results and discussion
Nanoporous gallium–zinc oxynitride photocatalyst
The kinetics of phase transformation from the oxide starting materials to oxynitride solid solution was studied in detail by Rodriguez and co-workers, who highlighted the importance of spinel ZnGa2O4 formation and its effect on the Zn-content of the final oxynitride phase.47,48 At relatively low temperatures (<700 °C), the chemical transformation from spinel ZnGa2O4 to the wurtzite oxynitride phase is the rate-governing step, while this phase transition occurs in time orders of minutes at 850 °C (ref. 47 and 49) or at temperatures as low as 650 °C when nanostructure starting materials are used.31,35In order to enhance the rate of spinel and oxynitride phase formation, the solid-state reaction surface must be increased significantly. The atomic-scale solid-state synthesis of GaN:ZnO photocatalyst was realized by (1) maintaining the intimacy between the solid–solid starting materials through inter-atomic reaction design, and (2) introducing the ammonolysis reagent to the solid reactants at the molecular-level.34,35 To accomplish the aforementioned conditions, Ga3+/Zn2+ LDHs are used as the starting materials, urea is employed as both a synthetic soft template and ammonolysis reagent, and microwave (MW) irradiation is utilized as an effective solid-state reaction activation strategy.
As demonstrated in Fig. 1(a), at a high MW-assisted heating, urea pyrolysis forms a quasi-homogenous foam with a massive surface area, which was employed as the nanostructured soft-template to help the synthesis of oxynitride photocatalyst with high specific surface areas. Upon the formation of the urea-mediated nano-template, the mixed-metal precursor is distributed within this structure, as displayed in Fig. 1(b), and exposed to urea pyrolysis products, dominated with NH3 and oxygen, resulting in improved gaseous-solid reactant interactions. The solid-state diffusion rate is further enhanced by the utilization of Ga3+/Zn2+ LDHs ([(Zn2+)(Ga3+)(OH)4]1+(CO22−)·yH2O), where the atomically uniform mixed-metal oxyhydroxide precursor comes into contact with reductive NH3 via LDHs inter-galleries (0.7–1.2 nm basal spacing50), as illustrated in Fig. 1(c). Such interaction within at interatomic-level between the solid–solid and gaseous-solid reactants at high temperature is sufficient for both the formation of the spinel ZnGa2O4, and its phase transition to the wurtzite oxynitride. Thus, the synthesis of oxynitride phase was realized in order of minutes. The optimal synthesis conditions (i.e., MW energy and treatment duration) were obtained through a combinatorial experiment, where the mixed-metal organic–inorganic precursor was treated at P = 600–960 W and t = 6–20 min. The photocatalytic performances of the prepared oxynitride samples suggested an MW power of 720 W and treatment duration of 12 min as an optimal synthesis condition.
 | ||
| Fig. 1 Urea-LDHs mediated microwave synthesis of GaZnON photocatalyst. (a) The formation of nano-template during urea pyrolysis; (b) magnified display of the rectangular region indicated in (a), showing the distribution of mixed-metal LDHs precursor within the urea nano-template, and (c) effective ammonolysis through interaction of urea pyrolysis products (mostly NH3) with the Ga3+/Zn2+ LDHs precursor within interlayer galleries. | ||
Fig. 2(a) shows the MW-treated sample, containing particles up to 4 μm in size. Wide size and shape distributions are attributable to the high-temperature sintering and formation of aggregates. Decomposition of these clusters is advantageous; since the particles in the inter-core of clusters are not exposed to the incident photons and as the photo-generated charges are trapped in the grain boundaries, they do not participate in the redox reactions.
 | ||
| Fig. 2 (a) SEM image of the as synthesized GaZnON photocatalyst and (b) high resolution SEM image of the nanoporosity on the surface of the as prepared GaZnON photocatalyst. | ||
The urea thermal-decomposition gaseous off-products cause local nano- and micro-disruptions in the precursor mixture and create random structures in the bulk and on the surface of the synthesized oxynitride photocatalyst. The high-resolution SEM micrograph in Fig. 2(b) shows the surface nanoporosity formed as the result of such disruption. The as synthesized GaZnON photocatalyst possesses macroporous morphology, containing hierarchical porosity with diameters mainly between 50 nm and 200 nm, but in some cases as small as 20 nm. The presence of pores with smaller diameters (less than 50 nm) suggests a combined three-dimensional nanostructure of mesopores and macropores. Such three-dimensional structures are suitable for photocatalytic applications, owing to their significantly higher specific surface area for photocatlytic reaction,51 compared to their bulk counterpart, as well as short diffusion path of the photo-excited charge carriers to catalytic active sites,52 improved light harvesting efficiency due to scattering/reflection,53,54 and enhanced mass transport within the hierarchical porosity composed of mesopores connected with macropores.55–57
The surface of the prepared samples contains a uniform nanoporous morphology with a narrow pore size distribution, resulting in a high specific surface areas up to 61.2 m2 g−1, significantly greater than that reported for the traditional GaN:ZnO (SBET = 3.7–8.1 m2 g−1 (ref. 23)), as well as the improved values reported.39
The surface and bulk composition of the prepared nanoporous GaZnON were examined through XPS and EDX spectroscopy, indicating [Zn]/[Zn + Ga] = 0.13 and 0.19, respectively. The surface and bulk zinc-contents of the prepared GaZnON were smaller than those in the precursor, which is similar to a previously reported observation.23 Comparing the chemistry of the microwave-treated sample with that of the solid solution (Ga1−xZnx)(N1−xOx) suggests sub-stoichiometric Zn-content ([Zn]/[O] < 1) as well as N-deficiency ([Ga]/[N] > 1). This deviation indicates that the rapid microwave-assisted synthesis causes incomplete solid-state nitridation, which is attributed to the evaporation of the rapid surface Zn evaporation at high synthesis temperature and the partial collapse of the Ga–N bonds due to oxidation. Thus, although oxynitride phase can be formed within the short period of time, it is unlikely to maintain a single wurtzite phase formation.
STEM images of the prepared GaZnON sample, along with a compositional map of the key solid solution elements are displayed in Fig. 3. The STEM micrographs in Fig. 3(a and b) display the nanoporous structure of the synthesized GaZnON photocatalyst. The HAADF tomography presented in the ESI† also confirms that the nanopores are distributed uniformly within the surface and bulk of the photocatalyst particles, with no sign of aggregation. The EDX elemental maps in Fig. 3(c–f) reveals the homogenous distribution of Ga, N, Zn, and O, respectively, with no sign of aggregation, isolated phases, or elemental islands.
 | ||
| Fig. 3 Bright (a) and dark-field (b) high resolution STEM micrographs of the prepared GaZnON photocatalyst, accompanied by the EDX elemental map of Ga (c), N (d), Zn (e), and O (f). | ||
GaZnON-reduced graphene oxide composite photocatalyst
Graphene oxide was prepared through the modified Hummer's method using pre-exfoliated graphite. The majority of the prepared GO nanosheets were characterized with sizes over 10 μm monolayer nanosheets with wrinkled surface textures. The SEM and TEM images of the prepared GO are displayed in the ESI.†Through XPS analysis, the C/O atomic ratios of 12.36, 2.05, and 4.11 were estimated for the graphite, prepared GO, and GaZnON–rGO composite, respectively, suggesting the effectiveness of the Hummer's oxidization of pre-exfoliated graphite, as well as the in situ photo-assisted removal of oxygen functionalities from the GO surface. The high degree of oxidation-reduction is further evidenced by the high-resolution C 1s band characteristics of the samples demonstrated in Fig. 4. The C 1s peak of graphite in Fig. 4(a) is transformed into complex spectra, showing two maxima for GO in Fig. 4(b), consisting of sp2 and sp3 networks: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C (284.5 eV), C–O (286.5 eV), and O–CO (287.3 eV). The large C–O and O–CO peaks, which were deconvoluted from the high-resolution C 1s spectra of GO, indicate the occupation of the nanosheet surface with a high density of oxygen-containing functional groups. As displayed in Fig. 4(c), after photo-reduction, the densities of the oxygen functionalities are decreased significantly, thus the broken sp2 network is healed, and the epoxide and carbonyl groups are removed from the surface of the nanosheets.
C and C–C (284.5 eV), C–O (286.5 eV), and O–CO (287.3 eV). The large C–O and O–CO peaks, which were deconvoluted from the high-resolution C 1s spectra of GO, indicate the occupation of the nanosheet surface with a high density of oxygen-containing functional groups. As displayed in Fig. 4(c), after photo-reduction, the densities of the oxygen functionalities are decreased significantly, thus the broken sp2 network is healed, and the epoxide and carbonyl groups are removed from the surface of the nanosheets.
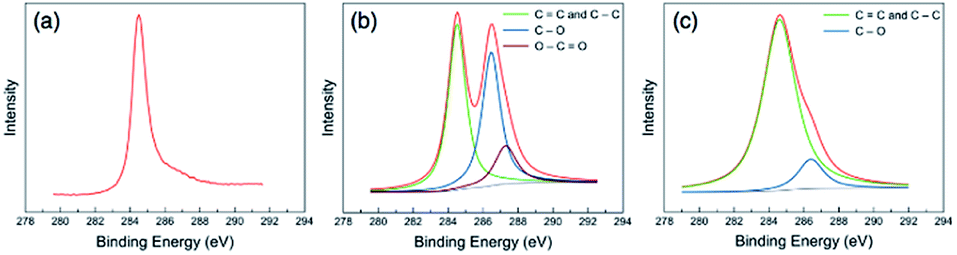 | ||
| Fig. 4 High-resolution XPS C 1s spectra of (a) graphite, (b) the as prepared GO, and (c) the synthesized GaZnON–rGO composite, showing phase transformation by introducing and removing the oxygen functional groups through exfoliation and photo-induced reduction, respectively. | ||
The remaining chemical oxygen groups in the structure of the rGO composite are believed to favor the composite's photocatalytic activity, as they maintain the solubility of the composite for most applications and stabilize the effective interfacial contact between the composite components. This is important, knowing that the removal of oxygen functionalities transfers the characteristics of GO towards those of pristine graphene, which is hydrophobic with negligible solubility in most solvents. In the course of reduction, the hydrophilic functionalities are removed; therefore, the electrostatic repulsion between negatively charged nanosheets is weakened, leading to van der Waals restacking. Excessive reduction results in the formation of aggregates with wrinkled and crumpled morphologies, which is irreversible and results in the loss of much of the graphene's surface area.
The high-resolution STEM image of the rGO in the composite sample in Fig. 5(a) shows the surface texture of the nanosheets, along with ultra-fine Rh nanoparticles as bright spots. The Rh compositional map in Fig. 5(b) can be overlapped to the bright spots in the green rectangle indicated in Fig. 5(a). The size distribution of the Rh nanoparticles were estimated between 1.0–5.3 nm (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) = 2.0 nm) from the area indicated by a white rectangle in Fig. 5(a), as demonstrated in the inset of the Fig. 5(b). The surface texture of the rGO nanosheets is further detectable in the high-resolution STEM image in Fig. 5(c), where the wrinkles are clearly distinct from the co-catalyst nanoparticles, suggesting a very uniform deposition of Rh nanoparticles on the rGO surface, with no sign of aggregation. The Rh compositional map of the area indicated by a green rectangle in Fig. 5(c) is presented in Fig. 5(d), confirming that the bright dots can be overlapped with the Rh dispersive signals. The presence of Rh nanoparticles on the surface of the rGO nanosheets testifies to the effective charge transfer from the bulk of nanoporous oxynitride photocatalyst to the rGO nanosheets. More importantly, it confirms that the density of electrons can reach to a sufficient level, required to commence an effective electrochemical interaction between electrolyte and nanosheets, hence drive photo-assisted reactions such as the photo-reduction of Rh salt to zero-valent metallic Rh nanoparticles.
= 2.0 nm) from the area indicated by a white rectangle in Fig. 5(a), as demonstrated in the inset of the Fig. 5(b). The surface texture of the rGO nanosheets is further detectable in the high-resolution STEM image in Fig. 5(c), where the wrinkles are clearly distinct from the co-catalyst nanoparticles, suggesting a very uniform deposition of Rh nanoparticles on the rGO surface, with no sign of aggregation. The Rh compositional map of the area indicated by a green rectangle in Fig. 5(c) is presented in Fig. 5(d), confirming that the bright dots can be overlapped with the Rh dispersive signals. The presence of Rh nanoparticles on the surface of the rGO nanosheets testifies to the effective charge transfer from the bulk of nanoporous oxynitride photocatalyst to the rGO nanosheets. More importantly, it confirms that the density of electrons can reach to a sufficient level, required to commence an effective electrochemical interaction between electrolyte and nanosheets, hence drive photo-assisted reactions such as the photo-reduction of Rh salt to zero-valent metallic Rh nanoparticles.
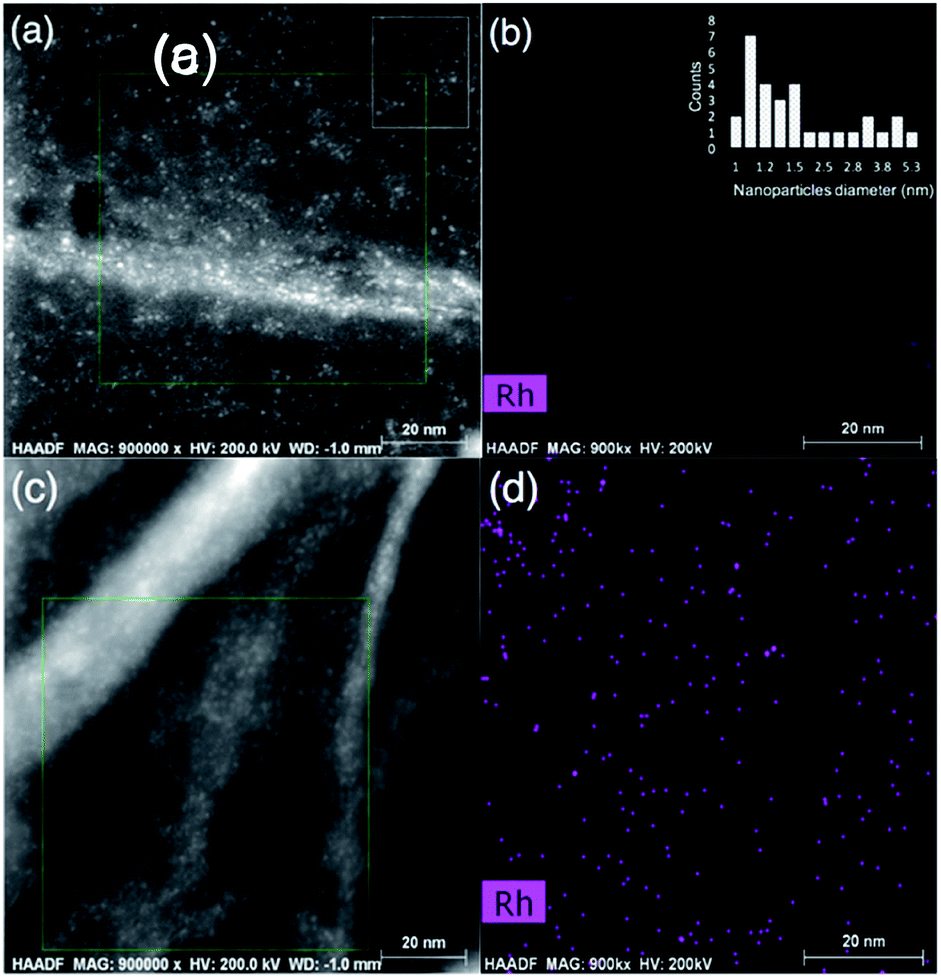 | ||
| Fig. 5 High-resolution STEM image of rGO surface loaded with Rh nanoparticles in the synthesized graphene GaZnON composite (a and c), and Rh elemental map of the area indicated with the green rectangle in (a and c) (b and d). The inset in (b) displays the estimated Rh particle size distribution, obtained from the area indicated with a white rectangle in (a). | ||
Photocatalytic activity
The rate of hydrogen evolution of the bulk GaN:ZnO solid solution is limited to the low surface capacity of the oxynitride photocatalyst for the deposition of HER active sites (typically less than 2 wt%), as most co-catalyst materials are optically opaque, so they absorb the incident light that is meant to be the driving force of water splitting. It is documented that the heavy loading of co-catalyst active sites reduces the photocatalytic activity by shading the surface.19 Unlike water electrolysis, in which the functional electrodes are typically loaded heavily with the HER and OER catalysts, co-catalyst deposition for photocatalytic water splitting cannot exceed a few percent of the utilized photocatalyst's weight. Therefore, the photocatalytic performance of nitride and oxynitride semiconductors is significantly limited by the insufficient density of HER-active sites.Fig. 6(a) shows the photocatalytic performance of the nanoporous GaZnON samples decorated with various amounts of NiOx, Pt and Rh HER co-catalysts against the metallic content of the HER catalyst precursor in photo-deposition solution. As expected, the NiOx-loaded photocatalysts showed the lowest photocatalytic activity, compared to those decorated with Pt and Rh co-catalysts, due to its lower work function (4.7–5.0 eV),58 compared to noble metal HER catalysts. The improved rate of photocatalytic hydrogen evolution in respect to increases in loading amount of the co-catalyst indicates that the metallic nanoparticles neither blocked the surface of the photocatalyst from incident photons, nor disrupted the nanoporous structure of the photocatalyst. The nanoporous GaZnON photocatalyst loaded with up to 9 wt% of Pt and 7 wt% of Rh exhibited higher photocatalytic activity, compared to the same samples loaded with less co-catalyst. The average hydrogen evolution of the 9 wt% Pt- and 7 wt% Rh-loaded photocatalysts increased by 3.5 and 3.2 times, compared to those loaded with the typical amounts of 1 wt% Pt and Rh, respectively. Such improvements suggest the effectiveness of the structural nanopores in extensive deposition of HER-active sites (>2 wt%) within the entire nanoporous structure of the synthesized GaZnON photocatalyst—not only on the photocatalyst surface. The nanoporous structure of the prepared GaZnON enhances the loading limit of the co-catalyst materials without shading the surface from the incident photons, and thus the structural nanopores can be utilized as the active surface for hydrogen evolution reaction. As schematically illustrated in Fig. 6(b), it is believed that the majority of the co-catalyst nanoparticles are deposited inside the structural nanopores of the photocatalyst, where the density of majority carriers is substantially greater.
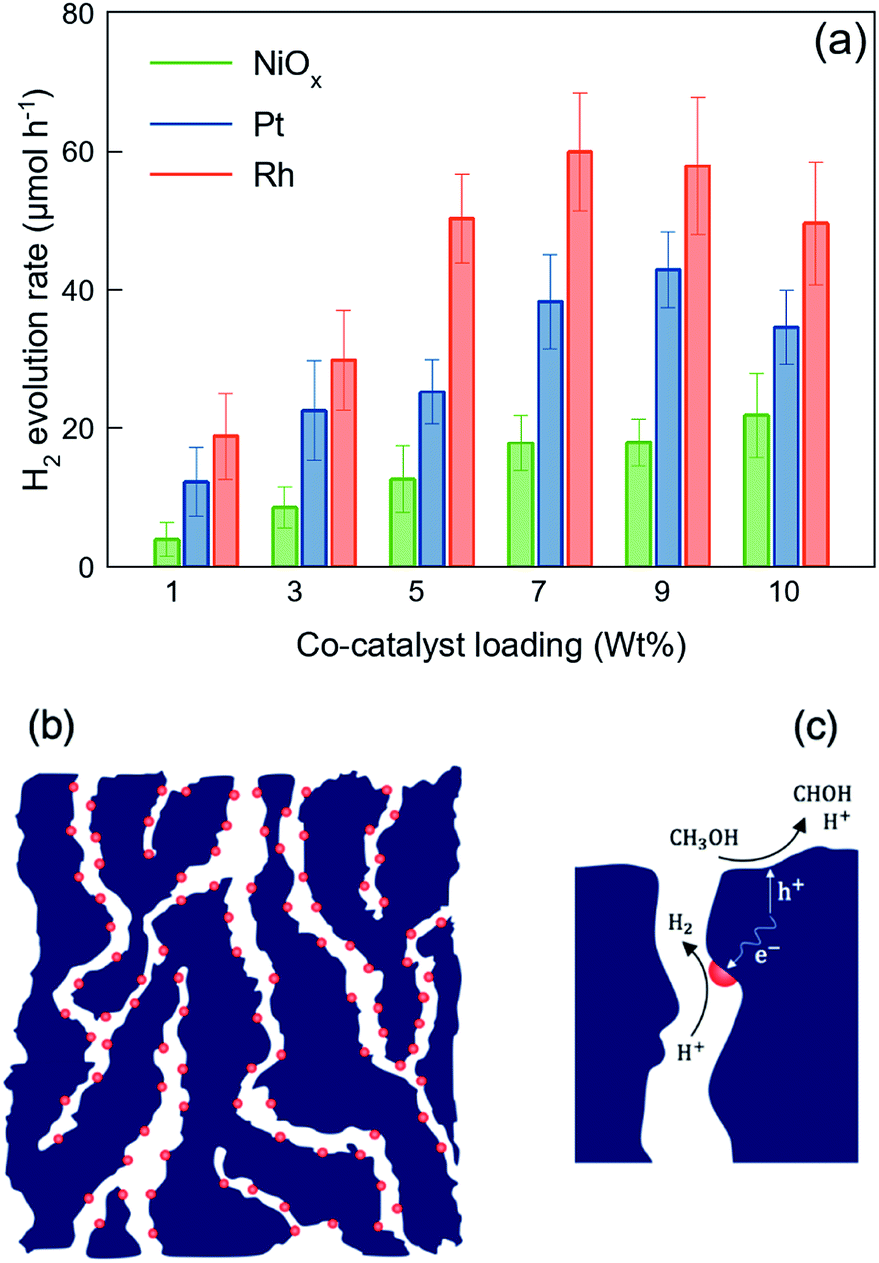 | ||
| Fig. 6 (a) Visible-light hydrogen evolution of the nanoporous GaZnON photocatalyst decorated with various amounts of NiOx, Pt, and Rh co-catalysts in 10 V% methanol aqueous solution. The error bars represent 90% confident intervals. (b) Utilization of structural nanopores for the deposition of co-catalyst nanoparticles, due to altered charge transfer mechanism, and (c) separation and placement of redox reactions active sites through employing the structural nanopores in reducing the photogenerated electrons (e−) diffusion length to the active site–electrolyte interface, and photocatalyst surface for hole (h+) methanol-assisted scavenging reaction. | ||
In addition to its role in enhancing the HER-active site deposition capacity, as illustrated in Fig. 6(c), the presence of catalyst nanoparticles inside the structural nanopores effectively contributes to increasing the likelihood that charge carriers will reach the redox reaction sites. In the quasi-3D framework of the photocatalyst, the transport path of majority (with micron-scale diffusivity) and minority (with nano-scale diffusivity) carriers to the HER catalyst-electrolyte and photocatalyst–electrolyte interfaces are decreases substantially. Despite the fact that photon absorption occurs on the near-surface region of the bulk particles, the resulting photo-excited charges can be transferred inside the bulk in response to variations in the charge densities. Particularly for n-type semiconductors, the majority of photo-induced electrons are transported to the bulk and recombine at the crystal defects and grain boundaries. The presence of co-catalyst nanoparticles within the quasi-3D structure (not only on the surface) significantly reduces the recombination losses by shortening the charge carrier's diffusion length. Therefore, even though the nanopores are not exposed to the incident photons, they can be utilized as the redox reaction active regions.
Fig. 7(a) demonstrates the hydrogen evolution activity of the 7 wt% Rh-loaded GaZnON–rGO composite photocatalyst with various GO compositions. The hydrogen evolution rate was increased significantly from the 7 wt% Rh–GaZnON photocatalyst to its corresponding rGO composite by over 2.2 and 2.4-times, upon incorporation of 1 and 2 wt% of GO, respectively, reaching to maximum of 5.1% apparent quantum yield. Adding further conductive nanosheets (over 2 wt%) reduces the photocatalytic performance of the composite photocatalyst due to inefficient light absorption by the multi-layer/encapsulated rGO hybridized. As displayed in Fig. 7(b), this improvement is attributable to the rapid injection of photo-induced electrons from the nanoporous photocatalyst to the conductive rGO nanosheets. This efficient charge transfer reduces the rate of photo-recombination losses and results in higher photocatalytic activities. It is known that the surface of rGO is readily active for HER,59,60 so, as shown in Fig. 7(b), the electron density on the surface of rGO directly interacts with electrolytes and contribute in hydrogen production. Thus, the incorporation of graphene not only increases the rate of photo-induced charge separation and the specific surface area for the excessive deposition of HER-active sites, but also could be utilized as a strategy for maintaining high photocatalytic activities at low metallic catalyst content. Fig. 7(c) illustrates the schematic energy band structure and charge transport path of the HER catalyst-decorated GaZnON–rGO composite. Under band gap excitation, the photo-induced holes directly participate in methanol oxidation reaction. The photo-generated electrons, however, cannot react with protons due to the lack of HER-active sites on the surface of the GaZnON photocatalyst. Therefore, electrons are either attracted to co-catalyst nanoparticles within the structure (on the surface and inside nanopores) of the photocatalyst or transported to the rGO nanosheets. Due to more negative conduction band potential of semi-metal rGO (∼−0.71 eV vs. Ag/AgCl60) compared to metal catalyst's Fermi levels, electrons can be readily transported to co-catalyst nanoparticles on the surface of rGO or directly engage in the hydrogen evolution reaction.
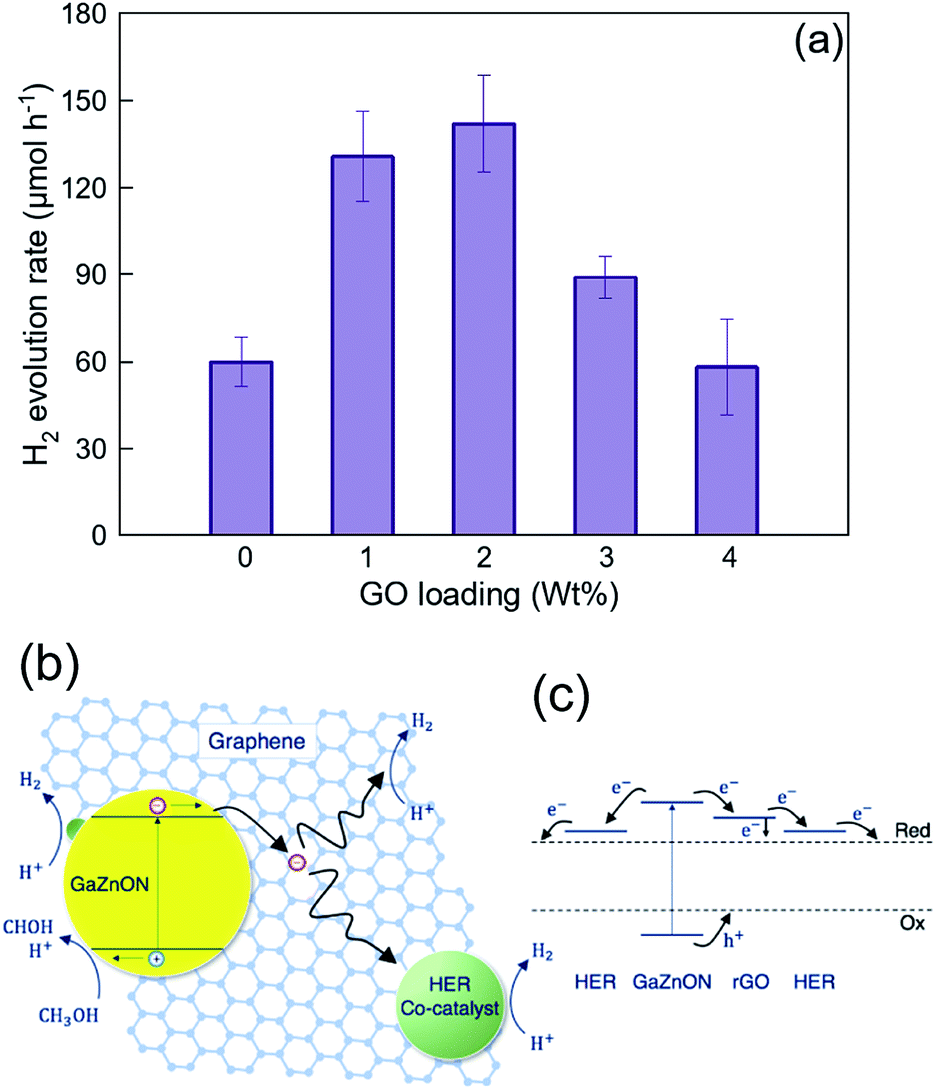 | ||
| Fig. 7 (a) Effect of graphene oxide hybridization on the photocatalytic hydrogen evolution of 7 wt% Rh-loaded nanoporous GaZnON–rGO composite photocatalyst. The error bars represent 90% confident intervals. (b) Schematic mechanism, describing the effect of reduced graphene oxide as the electron accepting medium in enhancing the charge separation through the rapid injection of photo-induced electrons to graphene nanosheets and the subsequent reduction reactions on catalytic active sites, and (c) the energy band diagram and charge transport path of the HER co-catalyst decorated GaZnON–rGO composite photocatalyst. | ||
The photocatalytic hydrogen evolution results presented in Fig. 6(a) and 7(a) shows that the rate of H2 evolution of the Rh–GaZnON–rGO (7 wt% Rh and 2 wt% GO) photocatalyst was enhanced significantly (7.5-fold) compared to the typical 1 wt% Rh–GaZnON sample. This improvement is attributed to engineering the morphology of the photocatalyst (nanoporous structure) and the role of graphene nanosheets as charge acceptors scaffold. The control over the morphology, which was presented here resulted in: (1) high capacity of nanoporous photocatalyst for deposition of HER-active sites without blocking the surface from incident photons; (2) availability of Rh nanoparticles within the structural nanopores, which effectively increases the density of the photo-induced charges available for photo-induced redox reactions and reduces the rate of photo-recombination rate; (3) high surface area of rGO nanosheets for hydrogen evolution reaction and deposition of Rh catalyst nanoparticles; and (4) enhanced charge transport from the photocatalyst particles to rGO nanosheets and further reduced photo-recombination losses. The advanced nanoporous and graphene-hybridized photocatalytic system studied in this research is of interest, considering that, for practical applications, devices with low densities and high active surface areas are highly desired. This approach could be further adapted and optimized for a wide spectrum of photocatalytic systems to increase the number of redox active sites and decrease the rate of photo-recombination losses.
Conclusion
In this article, the effect of photocatalyst morphology on the co-catalyst loading capacity and photo-induced carriers diffusion lengths was investigated. A facile MW-assisted route for the synthesis of nanoporous oxynitride solid solution of GaN and ZnO was designed through the utilization of an atomic-scale uniform layered double hydroxide precursor and abundant urea ammonolysis reagent, which resulted in effective inter-molecular interactions between solid–solid and solid-gaseous reactants at the nanoscale. The high surface area of the newly synthesized photocatalyst was increased up to 7.5-times, compared to the traditional GaN:ZnO. Such improved specific surface area is attributed to the quasi-three-dimensional framework of photocatalyst, consisting of uniform structural nanopores formed as the result of nano/micro-scale disruption occurring during urea thermal decomposition. An efficient self-assembly method was employed for the fabrication of a graphene-hybridized nanoporous oxynitride photocatalyst, and the characterization results indicated effective electrochemical interaction between the nanoporous photocatalyst particles and conductive nanosheets. The nanoporous photocatalyst and its graphene-based composite demonstrated outstanding capacity for loading hydrogen evolution active sites up to 7–9 wt%, as well as short charge diffusion lengths toward the catalyst-electrolyte interface. By utilizing the structural nanopores as the reduction reaction active sites, the 9 wt% Pt- and 7 wt% Rh-loaded photocatalysts achieved average hydrogen evolution rates 3.5- and 3.2 times higher, respectively, compared to the 1 wt% Pt- and Rh-loaded samples. The improved charge transport characteristics of the graphene-incorporated samples were observed through their visible-light hydrogen evolution, where the 2 wt% GO-loaded photocatalyst exhibited enhanced activity (2.4-fold), compared to the bare surface modified photocatalyst with same Rh-content. The optimized graphene composite photocatalyst decorated with 7 wt% Rh achieved 7.5-fold higher photocatalytic activity compared to the typical 1 wt% Rh-loaded oxynitride photocatalyst. The recently reported strategies for suppressing the water splitting reverse reaction and the development of Earth-abundant HER catalysts enabled heavy loading of the co-catalyst materials, to the extent that the photon absorption of the photocatalyst is not disrupted. The systematic engineering of photocatalyst morphology proposed in this article provides a general framework for effectively enhancing the density of HER-active sites and promoting the charge transport process. This approach can be further adapted and applied for other photocatalytic systems.Conflicts of interest
There are no conflicts to declare.References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G. J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- Y. J. Wang, G. Chang, Q. Chen, G. J. Yang, S. Q. Fan and B. Fang, Chem. Commun., 2015, 51, 685–688 RSC.
- Y. Corre, W. Iali, M. Hamdaoui, X. Trivelli, J. P. Djukic, F. Agbossou-Niedercorn and C. Michon, Catal. Sci. Technol., 2015, 5, 1452–1458 RSC.
- D. Xu, Q. Chu, Z. Wu, Q. Chen, S. Q. Fan, G. J. Yang and B. Fang, J. Catal., 2015, 325, 118–127 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Photochem. Photobiol., A, 1996, 94, 67–76 CrossRef CAS.
- R. Abe, K. Sayama and H. Arakawa, Chem. Phys. Lett., 2003, 371, 360–364 CrossRef CAS.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem.–Eur. J., 2010, 16, 7750–7759 CrossRef CAS PubMed.
- Z. Li, F. Zhang, J. Han, J. Zhu, M. Li, B. Zhang, W. Fan, J. Lu and C. Li, Catal. Lett., 2018, 148, 933–939 CrossRef CAS.
- B. Adeli and F. Taghipour, Appl. Catal., A, 2016, 521, 250–258 CrossRef CAS.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- H. Yan, C. Tian, L. Wang, A. Wu, M. Meng, L. Zhao and H. Fu, Angew. Chem., 2015, 127, 6423–6427 CrossRef.
- K. Edalati, R. Uehiro, S. Takechi, Q. Wang, M. Arita, M. Watanabe, T. Ishihara and Z. Horita, Acta Mater., 2020, 185, 149–156 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- K. Maeda, K. Teramura and K. Domen, J. Catal., 2008, 254, 198–204 CrossRef CAS.
- R. Godin, T. Hisatomi, K. Domen and J. R. Durrant, Chem. Sci., 2018, 9, 7546–7555 RSC.
- C. Hahn, M. A. Fardy, C. Nguyen, M. Natera-Comte, S. C. Andrews and P. D. Yang, Isr. J. Chem., 2012, 52, 1111–1117 CrossRef CAS.
- K. Maeda and K. Domen, Chem. Mater., 2010, 22, 612–623 CrossRef CAS.
- M. Yashima, K. Maeda, K. Teramura, T. Takata and K. Domen, Chem. Phys. Lett., 2005, 416, 225–228 CrossRef CAS.
- M. Yashima, K. Maeda, K. Teramura, T. Takata and K. Domen, Mater. Trans., 2006, 47, 295–297 CrossRef CAS.
- M. J. Ward, W.-Q. Han and T.-K. Sham, J. Phys. Chem. C, 2013, 117, 20332–20342 CrossRef CAS.
- M. Yashima, H. Yamada, K. Maeda and K. Domen, Chem. Commun., 2010, 46, 141–145 Search PubMed.
- E. J. McDermott, E. Z. Kurmaev, T. D. Boyko, L. D. Finkelstein, R. J. Green, K. Maeda, K. Domen and A. Moewes, J. Phys. Chem. C, 2012, 116, 7694–7700 CrossRef CAS.
- T. Hirai, K. Maeda, M. Yoshida, J. Kubota, S. Ikeda, M. Matsumura and K. Domen, J. Phys. Chem. C, 2007, 111, 18853–18855 CrossRef CAS.
- X. Sun, K. Maeda, M. Le Faucheur, K. Teramura and K. Domen, Appl. Catal., A, 2007, 327, 114–121 CrossRef CAS.
- K. Lee, B. M. Tienes, M. B. Wilker, K. J. Schnitzenbaumer and G. Dukovic, Nano Lett., 2012, 12, 3268–3272 CrossRef CAS PubMed.
- B. Adeli and F. Taghipour, ECS J. Solid State Sci. Technol., 2013, 2, Q118–Q126 CrossRef CAS.
- H. Hagiwara, R. Kakigi, S. Takechi, M. Watanabe, S. Hinokuma, S. Ida and T. Ishihara, Surf. Coat. Technol., 2016, 324, 601–606 CrossRef.
- J. Wang, B. Huang, Z. Wang, P. Wang, H. Cheng, Z. Zheng, X. Qin, X. Zhang, Y. Dai and M.-H. Whangbo, J. Mater. Chem., 2011, 21, 4562 RSC.
- S. Yan, Z. Wang, Z. Li and Z. Zou, J. Mater. Chem., 2011, 21, 5682 RSC.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753–13758 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, J. Catal., 2006, 243, 303–308 CrossRef CAS.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400–6404 CrossRef CAS PubMed.
- M. Yang, Q. Huang and X. Jin, Solid State Sci., 2012, 14, 465–470 CrossRef CAS.
- K. Katagiri, Y. Hayashi, R. Yoshiyuki, K. Inumaru, T. Uchiyama, N. Nagata, Y. Uchimoto, A. Miyoshi and K. Maeda, Inorg. Chem., 2018, 57, 13953–13962 CrossRef CAS PubMed.
- B. Adeli and F. Taghipour, Chem. Eng. Technol., 2016, 39, 142–148 CrossRef CAS.
- X. Li, Q. Zhang, H. Wang and Y. Li, Appl. Surf. Sci., 2015, 358, 57–62 CrossRef CAS.
- W. Q. Han, Y. Zhang, C. Y. Nam, C. T. Black and E. E. Mendez, Appl. Phys. Lett., 2010, 97, 083108 CrossRef.
- M. Zhong, Y. Ma, P. Oleynikov, K. Domen and J.-J. Delaunay, Energy Environ. Sci., 2014, 7, 1693 RSC.
- J. Li, B. Liu, W. Yang, Y. Cho, X. Zhang, B. Dierre, T. Sekiguchi, A. Wu and X. Jiang, Nanoscale, 2016, 8, 3694–3703 RSC.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- H. Chen, W. Wen, Q. Wang, J. C. Hanson, J. T. Muckerman, E. Fujita, A. I. Frenkel and J. A. Rodriguez, J. Phys. Chem. C, 2009, 113, 3650–3659 CrossRef CAS.
- H. Chen, L. Wang, J. Bai, J. C. Hanson, J. B. Warren, J. T. Muckerman, E. Fujita and J. A. Rodriguez, J. Phys. Chem. C, 2010, 114, 1809–1814 CrossRef CAS.
- H. Chen, L. Wang, J. Bai, J. C. Hanson, J. B. Warren, J. T. Muckerman, E. Fujita and J. A. Rodriguez, J. Phys. Chem. C, 2010, 114, 1809–1814 CrossRef CAS.
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 RSC.
- H. Yang, Y. Zhou, Y. Wang, S. Hu, B. Wang, Q. Liao, H. Li, J. Bao, G. Ge and S. Jia, J. Mater. Chem. A, 2018, 6, 16485–16494 RSC.
- W. Zhan, L. Sun and X. Han, Nano-Micro Lett., 2019, 11, 1–28 CrossRef CAS PubMed.
- B. Fang, Y. Xing, A. Bonakdarpour, S. Zhang and D. P. Wilkinson, ACS Sustainable Chem. Eng., 2015, 3, 2381–2388 CrossRef CAS.
- B. Fang, A. Bonakdarpour, K. Reilly, Y. Xing, F. Taghipour and D. P. Wilkinson, ACS Appl. Mater. Interfaces, 2014, 6, 15488–15498 CrossRef CAS PubMed.
- B. Fang, J. H. Kim, M. S. Kim and J. S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- W. B. Hua, X. D. Guo, Z. Zheng, Y. J. Wang, B. H. Zhong, B. Fang, J. Z. Wang, S. L. Chou and H. Liu, J. Power Sources, 2015, 275, 200–206 CrossRef CAS.
- J. H. Kim, B. Fang, M. Kim and J. S. Yu, Catal. Today, 2009, 146, 25–30 CrossRef CAS.
- B. Mustafa, J. Griffin, A. S. Alsulami, D. G. Lidzey and A. R. Buckley, Appl. Phys. Lett., 2014, 104, 063302 CrossRef.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- T. F. Yeh, F. F. Chan, C. Te Hsieh and H. Teng, J. Phys. Chem. C, 2011, 115, 22587–22597 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) High-angle annular dark field (HAADF) tomography video, showing the nanoporous structure of the prepared GaZnON photocatalyst. (2) Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the prepared graphene oxide (GO) nanosheets. See DOI: 10.1039/d0ra01725f |
| This journal is © The Royal Society of Chemistry 2020 |