
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKOH activation of coal-derived microporous carbons for oxygen reduction and supercapacitors†
Shaokui Guoab,
Beibei Guob,
Ruguang Ma b,
Yufang Zhu*ab and
Jiacheng Wang*b
b,
Yufang Zhu*ab and
Jiacheng Wang*b
aSchool of Materials Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, P. R. China
bState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P. R. China. E-mail: zjf2412@163.com; jiacheng.wang@mail.sic.ac.cn
First published on 21st April 2020
Abstract
Due to the dilemma of rapid consumption of fossil fuels and environmental pollution, development of clean, efficient and renewable energy conversion and storage technology has become an urgent need. Supercapacitors and hydrogen–oxygen fuel cells as typical representatives have become the focus of scientific research, in which the electrode materials are of much importance to their improved activity. In this work, a series of porous carbons (PCs) with high specific surface areas were prepared using natural coals as carbon precursors coupled with KOH activation. The effects of the mass ratio of coal and KOH as well as different activation temperatures on the microstructures of the PCs and electrochemical properties were studied in detail. The optimal PC4 (KOH: coal = 4) possessed a high specific surface area (SSA) of 2092 m2 g−1 and a well-developed microporous structure. As the electrocatalyst, it exhibited a positive onset potential of 0.88 V (vs. reversible hydrogen electrode (RHE)) and half-wave potential of 0.78 V (vs. RHE) towards the oxygen reduction reaction (ORR) in an alkaline solution. PC4 also showed the highest specific capacitance of 128 F g−1 at a current density of 0.5 A g−1 among all the samples in this work. The relatively good performance of PC4 resulted from its well-developed microporous structure and large SSA, enabling fast mass transfer of electrolytes.
1. Introduction
Nowadays, the energy crisis and environmental pollution are two major problems that restrict the development of this world. Due to the fast exhaustion of fossil resources, the development of clean energy conversion and storage technology is the only way to achieve sustainable development.1–4 Porous carbon (PC) has attracted enormous attention due to its great potential application in energy conversion and storage technology.5 For example, this functional carbon material has been considered as a potential electrocatalyst for oxygen reduction reactions (ORR) to overcome the inherent disadvantages of platinum-based catalysts (high cost, extremely scarce, poor stability and crossover deactivation). On the other hand, great capacitive performance has been verified because of its high electrical double-layer capacitance (EDLC).6 Most advantages of carbon materials, such as low price, large SSA, rich porosity, excellent conductivity, chemical stability, thermal stability and easy synthesis,7,8 result in their wide applications in fuel cells,9–11 supercapacitors (SCs),12–15 batteries,16 organic pollutant or pollution gas adsorption17,18 and supports for nanoparticulate catalysts.19,20It is necessary to select a suitable carbon precursor to prepare porous carbon materials with excellent properties. Coal as a kind of non-renewable energy is a fuel in industry. Coal mainly consists of carbon and many heteroelements (mainly S, O and N) and has the advantage of abundant, stable supply and low price. So coal is a good carbon precursor material.21,22 Some by-products in coal coking industry are studied generally,23,24 but coal is seldom reported. However, the performance of pure coal is poor when used in the devices of energy conversion and storage. So we need to further improve the structure through various physical and chemical strategies. As we know, there are various activating agents (such as CO2, ZnCl2, KOH, H3PO4, NaOH, etc.)25–29 and hard templates (SiO2, MgO, etc.)30 applied in the preparation of porous carbons with high SSA and different pore structures. KOH activation is one of the most common activating methods due to its mature technology and convenient operation.31,32 Compared with other activating reagents, the porous carbons (PCs) obtained from KOH activation commonly have a very high SSA with considerable micropores.
In this study, porous carbon (PC) materials were prepared by high temperature pyrolysis activation with coal as a carbon precursor and KOH as an activating agent. We studied the effects of different ratios of activating agent and reactant on the structures of the final PC materials in detail, as well as the ORR activity and the electric double-layer capacitor (EDLC) properties. The effects of the activation temperatures on the final PCs were also investigated. The optimal PC material (PC4) has the highest specific surface area of 2092 m2 g−1 and a mean micropore size of 2.2 nm. It shows a positive onset potential of 0.88 V (vs. RHE) and half-wave potential of 0.78 V (vs. RHE) towards ORR in an alkaline solution. Moreover, it exhibits an approximate four-electron dominant pathway as well as favorable stability and excellent methanol crossover resistance. As for supercapacitors, PC4 exhibits a higher specific capacitance of 128 F g−1 at a current density of 0.5 A g−1. When the current density increases from 0.5 to 10 A g−1, PC4 exhibits a specific capacitance of 91 F g−1 and 71% of original specific capacitance still remained to imply a good rate capacity.
2. Experiment section
2.1 Synthesis of porous carbons (PCs)
Firstly, commercially available bulky coals were broken and ground into powder in a mortar with a pestle. Further, the powder was ball milled in order to make the powder finer. 2 g coal powder and 8 g KOH were weighed and placed in a mortar. After grinding for 0.5 h, the mixed powder was placed in a curved crucible and placed in a metal tube furnace. The resultant mixture was heated to 750 °C at 5 °C min−1 and held for 120 min in Ar atmosphere, followed by cooling to room temperature to obtain black solid. Finally, the product was washed by 1 M HCl solution for 3 times in order to remove residual KOH and other impurities. Then the product was washed by deionized water for 5 times to remove redundant acid and adjust pH to 7. The as-made material was named as PC4. Besides, the PCs made from different ratio between KOH and coal were named as PCx (x = 1, 2, 3, 4 and 5).2.2 Structural characterization
The morphologies of samples were characterized by using scanning electron microscopy (SEM, SPM-9500J3) and the transmission electron microscopy (TEM, JEM-2100F) was carried out at an accelerating voltage of 200 kV. X-ray diffraction patterns were collected using high resolution powder X-ray diffractometer (Bruker D8 ADVANCE). Raman spectra were recorded by Raman spectrometer (LABRAM HR800). N2 adsorption/desorption isotherms of samples were studied by ASAP 2010 at 77 K. Before absorption, samples were degassed at 300 °C in vacuum for 12 h to remove volatile impurities. Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda method were studied to determine the specific surface area and pore size. The X-ray photoelectron spectroscopy (XPS) was measured using the X-ray photoelectron spectrometer (ESCALAB 250).2.3 Electrochemical measurements
The ORR performance was investigated on an electrochemical workstation (Pine instrument Co. LTD. USA) with platinum plate as the counter electrode and saturated calomel electrode (SCE) as the reference electrode in O2-saturated 0.1 M KOH aqueous solution. The working electrode is the glassy carbon electrode coated with catalyst ink. Typically, 5 mg sample and 20 μL Nafion (5 wt%) were dispersed into the solution containing 500 μL of deionized water and 500 μL ethanol followed by ultrasonic treatment for 2 h to form a homogenous ink. 20 μL of ink was dropped on the surface of the glassy carbon electrode and then dried at 50 °C to form the working electrode. Electrochemical measurements including cyclic voltammetry (CV), rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) were tested at a potential range from −1.0 to 0.1 V (vs. SCE).Supercapacitor (SC) performance was measured using a CHI760 electrochemical workstation (Shanghai Chenhua Instruments Co.) with Pt as the counter electrode and saturated calomel electrode (SCE) as the reference electrode. In the supercapacitor (SC) experiments, the working electrode was made by mixing an active material (80 wt%) with acetylene black (10 wt%) and poly(vinylidene difluoride) (PVDF) (10 wt%) in N-methylpyrrolidone on the Ni foam (1 cm × 1 cm) and then dried at 100 °C. The mass loading of activated material on the Ni form was ∼4.0 mg. Cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) and electrochemical impedance spectroscopy (EIS) methods were carried out in 6 M KOH aqueous solution to evaluate the SC performance.
3. Results and discussion
Fig. 1 illustrated the preparation process of PC4. The massive coal was ground evenly by ball mill to form fine pulverized coal. The one-step activation carbonization of the mixture of KOH and coal was completed at 750 °C for 2 h. After cooling at room temperature, the product was washed several times with dilute hydrochloric acid and water to remove the impurities. The mechanism of KOH activation was to react some of the carbon in the raw material with KOH to form some intermediate products such as K2CO3 and K2O.33,34 After acid pickling, these intermediate products and excess KOH were washed and then a large number of pores were formed in the etched position. The basic chemical reaction formula was listed as follows:| 4KOH + –CH2– → K2CO3 + K2O + 3H2 | (1) |
 | ||
| Fig. 1 Schematic illustration of preparing PCs by KOH activation of coals and their potential applications in oxygen reduction reaction (ORR) and supercapacitors. | ||
The morphology and microstructure of the prepared porous carbon with coal as the carbon source and KOH as activator were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Scanning electron microscopy (SEM) image of pure coal without KOH activation reveals a smooth surface on the surface (Fig. S1†). After subjecting the subsequent KOH activation (C/KOH = 1/4), the smooth surface became rough and porous (Fig. 2a). The increased density of small holes on the surface is ascribed to the combination of micropores. TEM image once again proves the porous structure of PC4 (Fig. 2c). And high-resolution TEM shows the amorphous carbon texture and there were existing large amount of micropores within the carbon matrix. Such a hierarchical porous structure is beneficial for increased exposure of active sites and mass transfer and during ORR35 and storage charge in SC,36 thereby improving the performance of PCs in the area of electrocatalysis.
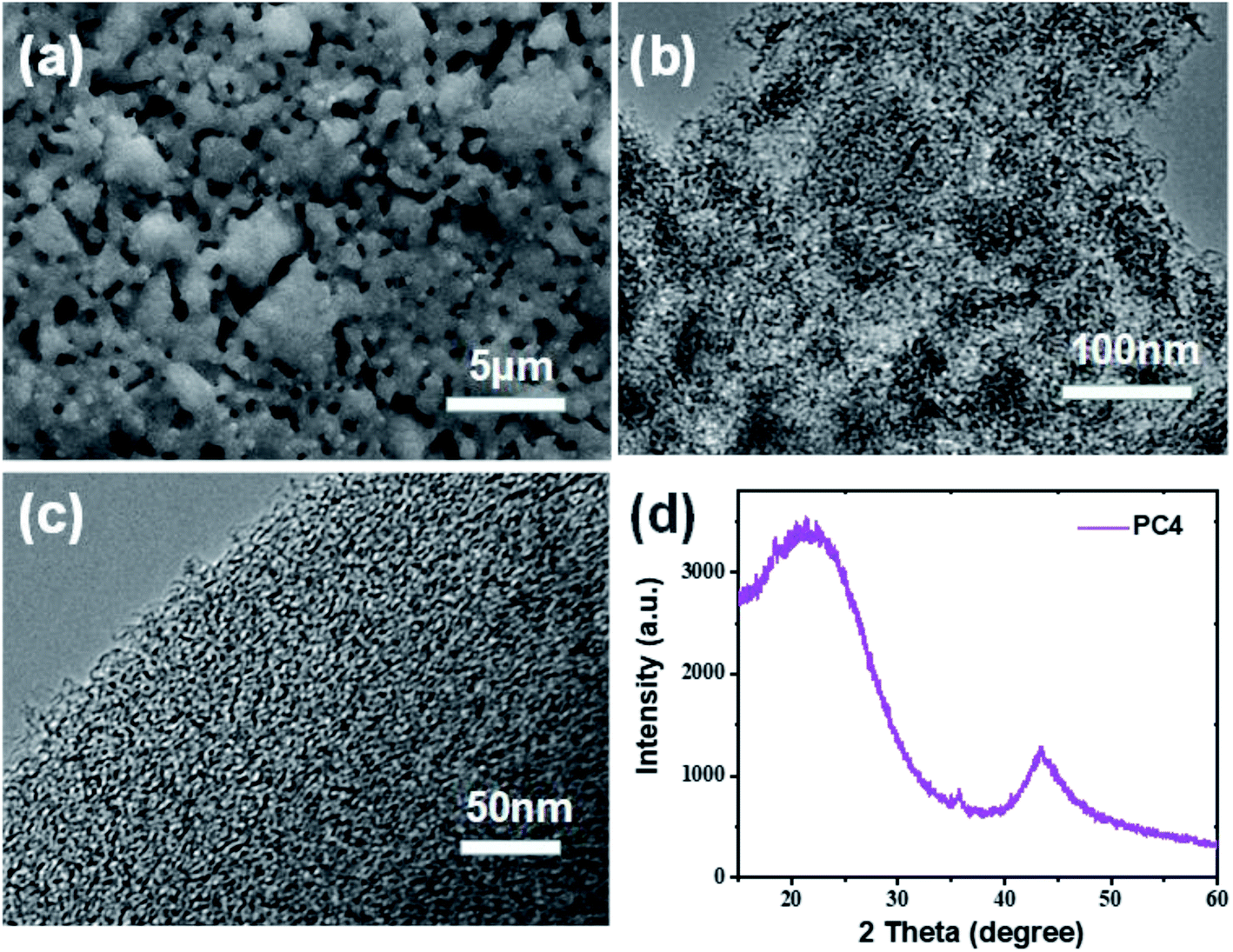 | ||
| Fig. 2 (a) SEM, (b and c) TEM, and (d) XRD pattern of PC4. | ||
Fig. 2d shows the XRD pattern of PC4. There are two broad diffraction peaks centered at around 22.6° and 43.5°, which are due to the (002) and (101) diffraction of the amorphous carbon structure.37 No other diffraction peaks prove that there are no other metal elements remaining. By X-ray photoelectron spectroscopy (XPS), the surface chemical performance of PC4 material is determined. In the full XPS spectrum (Fig. S2a†), there are two distinct C 1s peaks at 284 eV and O 1s peak at 532 eV. Very small amount (0.19 wt%) of N content was found to exist in PC4. The nitrogen functional groups are easy to oxidize and lose, especially in an alkaline environment.38–40 PC4 shows a high content of O heteroatoms (29.87 wt%).The high-resolution XPS spectra of C 1s and O 1s of PC4 are shown in Fig. S2b and c.† There are three individual peaks for the different types of C atoms in Fig. S1b,† namely, C–C (284.79 eV), C–O (286.26 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (294 eV), respectively.41 As presented in Fig. S1c,† the O 1s fitted peaks positions at 532.1 and 533.2 eV are assigned to CO (oxygen in quinone or carbonyl groups) and C–O (oxygen in hydroxyls and in esters), respectively.42,43 These abundant oxygen-containing groups provide supererogatory pseudo-capacitance and improve the wettability of the PCs electrodes in the aqueous electrolytes, which could enhance capacitance.44
O (294 eV), respectively.41 As presented in Fig. S1c,† the O 1s fitted peaks positions at 532.1 and 533.2 eV are assigned to CO (oxygen in quinone or carbonyl groups) and C–O (oxygen in hydroxyls and in esters), respectively.42,43 These abundant oxygen-containing groups provide supererogatory pseudo-capacitance and improve the wettability of the PCs electrodes in the aqueous electrolytes, which could enhance capacitance.44
The Raman spectra of all PCs show two strong peaks around 1339 cm−1 and 1600 cm−1 which could be assigned to D and G bands (Fig. 3a). The appearance of the D peak is attributed to the crystallite size effect referring to the defect and disorder induction. The G peak is produced by the stretching motion of all pairs of sp2 atoms in a carbon ring or a long chain.45 Generally, the intensity ratio of D and G peaks is used to measure the disorder and defect degree of carbon materials.46 As we all know, KOH activation could generate a lot of micropores and defects into the carbon matrix. The ratio of D to G band intensity was found to be the highest in PC4. The pure carbon has an ID/IG ratio of 1.16. As the ratio of alkali increases, the ratio of ID/IG becomes a trend of increasing first and then decreasing. The intensity ratios (ID/IG) of PC1, PC2, PC3, PC4 and PC5 were 1.2, 1.21, 1.26, 1.27 and 1.17, demonstrating that all the PCs had more defects than pure coal. The large specific surface area (SSA) and abundant micropores of PCs were verified by the nitrogen adsorption–desorption measurements. As showed in Fig. 3b, all the nitrogen adsorption–desorption isotherms were typical characteristics of I type based on the IUPAC classification. At low pressure (P/P0 = 0–0.2), the adsorbed volume of all PCs increases rapidly and reaches the saturation soon which was proved the existence of abundant micropores. At the medium pressure, all PCs did not show any hysteresis loops, indicating the absence of mesopores in all PCs. Fig. 3c shows the pore size distribution curves of PCs. Distinctly, most of the pore diameters of all PCs are below 2 nm and only a few pores exceed 2 nm. The pore structure parameters of all PCs samples are summarized in Table 1 and Fig. 3d. The pure coal has a specific surface area of 7.6 m2 g−1 and rarely no pore volume. After the activation with KOH, the specific surface area and pore volume are greatly improved. The specific surface area (SBET) of PC1, PC2, PC3, PC4 and PC5 was 332.9, 913.5, 1141.7, 2092.2 and 1801.8 m2 g−1 while the total pore volume (VTotal) was 0.19, 0.49, 0.64, 1.12 and 1.05 cm3 g−1, respectively. PC4 has the largest SSA and pore volume. Generation of such microporous feature could be attributed to surface activation by KOH at high temperature, where functionalization at cross linking carbon atoms broke the connected lamellar structure of coal and gave rise to interlayer voids, thereby resulting higher SSA and porosity. Beyond that, a higher pyrolysis temperature leads to the decomposition of carbon around some heteroatoms (found to be mainly oxygen), which in turn further increase the surface area of the doped carbon network.
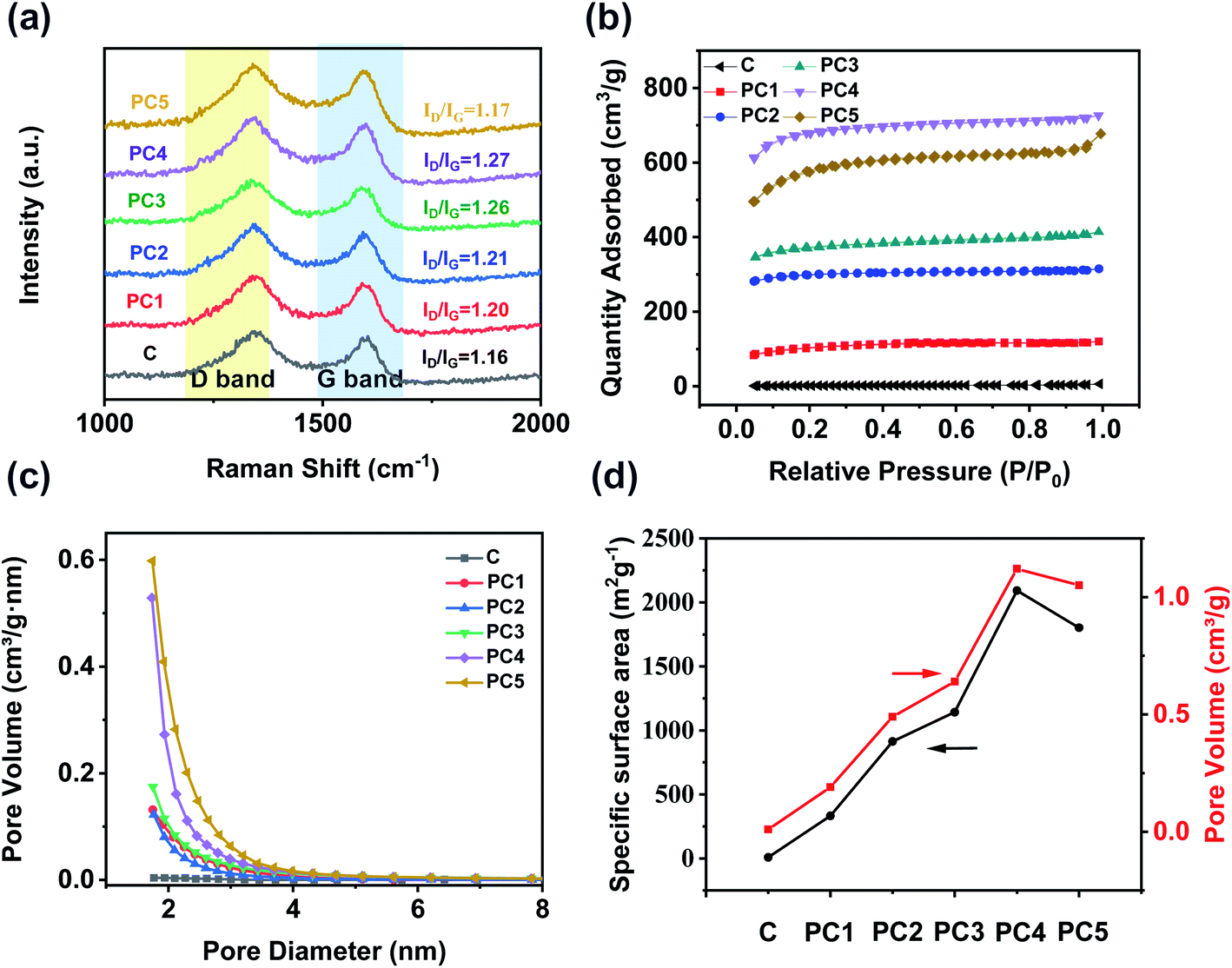 | ||
| Fig. 3 (a) Raman spectra, (b) nitrogen adsorption–desorption isotherms, and (c) pore size distribution curves of various PCs. (d) Trend of specific surface areas and pore volumes for different PCs. | ||
| Samples | SBET/(m2 g−1) | VTotal/(cm3 g−1) | Rpore/(nm) |
|---|---|---|---|
| Coal | 7.6 | 0.01 | 5.19 |
| PC1 | 332.9 | 0.19 | 2.23 |
| PC2 | 913.5 | 0.49 | 2.14 |
| PC3 | 1141.7 | 0.64 | 2.24 |
| PC4 | 2092.2 | 1.12 | 2.15 |
| PC5 | 1801.8 | 1.05 | 2.33 |
The effect of different temperatures on the porous carbon structure was also discussed in detail (Fig. S3†). Raman spectra show the intensity ratios (ID/IG) of PC4-700, PC4-750 and PC4-800 were 1.06, 1.27 and 0.99, respectively. When the carbonization temperature was raised from 700 to 750 °C, the defects of the porous carbon increased. When the temperature was 800 °C, the ID/IG ratio was 0.99. This means that the degree of graphitization of porous carbon was greatly enhanced. Besides, the width of the D band got reduced when the pyrolysis temperature increased, while the width of the G band almost remained the same. We inferred the lowering in the defect density and increasing in graphitic lattice formation in the amorphous PCs.47 The PC4-700 and PC4-800 samples show the specific surface area of 922 m2 g−1 with pore volume of 0.55 cm3 g−1 and 1229 m2 g−1 with pore volume of 0.74 cm3 g−1, respectively. The nitrogen adsorption–desorption isotherms of PC4-800 was typical characteristic of I and IV type. When the activation temperature exceeds the boiling point of metal potassium (762 °C), the generated potassium metal was sublimated into potassium vapor, which was diffused into the carbon layer structure of the carbon to activate it to form a rich pore structure. As the temperature increases, the degree of graphitization increases. Excessive temperatures cause the collapse of the microporous structure, resulting in a decrease in specific surface area. Therefore, an optimal carbon–KOH ratio and a suitable carbonization temperature have been explored to prepare the optimal porous carbon materials for energy conversion and storage.
The electrocatalytic ORR activity of all samples were measured in O2-saturated 0.1 M KOH aqueous solution at room temperature. Fig. 4a showed the CV curves of PC4 in O2 or N2 saturated 0.1 M KOH aqueous solution. An evident peak was obtained in O2 saturated electrolyte in 0.78 V (vs. RHE), while it disappears in N2 saturated electrolyte under the same condition. It suggests the obvious ORR occurs on the modified electrode. All samples were further studied using rotating disk electrode (RDE) at 1600 rpm. The pure C shows poor ORR activity as expected because of its low SSA and lacking of pores to expose the surface active sites. In contrast, after KOH activation, the specific surface area and pore size were larger in all PCs which significantly improve the ORR performance. Especially, PC4 (KOH/carbon = 4/1) exhibits the best ORR performance with onset potential of 0.88 V (vs. RHE) and half-wave potential of 0.78 V (vs. RHE) (Fig. 4b and c). The limited current density of PC4 also reaches 5.1 mA cm−2, indicating the best ORR performance. However it is still inferior to commercial Pt/C. We investigated the ORR rate determining step in PCs based on the Tafel plots analysis (Fig. 4d). PC4 has the smallest Tafel slope of 82 mV dec−1. This confirms that the rate determining step involves multiple electron transfer to adsorbed oxygen via an associative mechanism. Fig. 4e shows the LSV curves of PC4 at different rotating speeds from 400 to 2025 rpm. Obviously, the increased current density with the enhanced rotating speed was due to the shortened diffusion layer for electrode material. The Koutecky–Levich (K–L) plots at various potentials display good linearity (Fig. 4f). The slopes remain approximately constant over the potential range from −0.3 to −0.45 V (vs. RHE), implying similar electron transfer numbers for oxygen reduction at different potentials. From the K–L equation, the electron transfer numbers per oxygen molecule are 3.88, very close to 4, suggesting that the ORR catalyzed by PC4 mainly follows the 4e− reduction mechanism which is important for fuel cells, because the side-product of peroxides by a two-electron process could poison the cells via corroding the catalyst layer.48,49
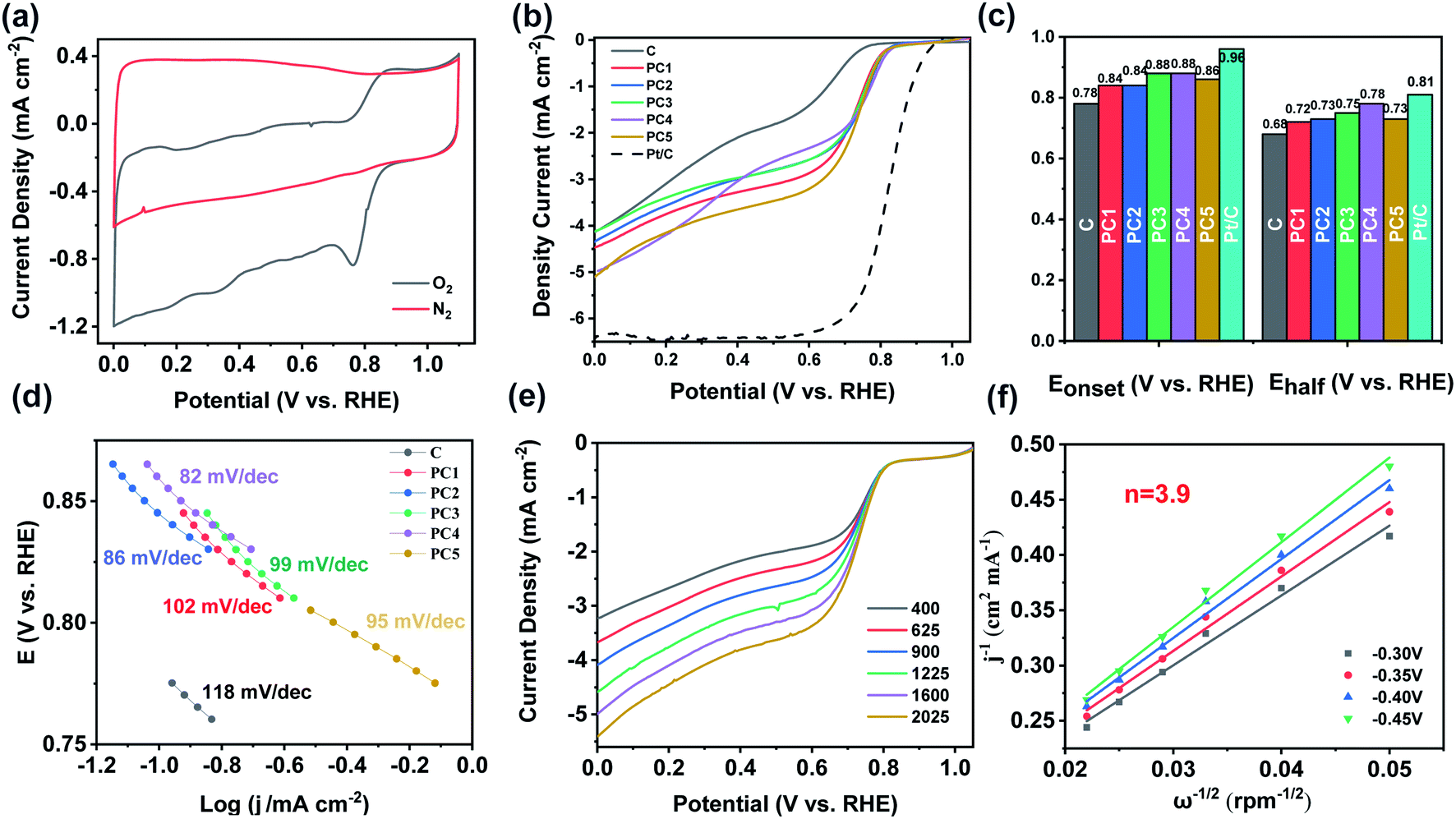 | ||
| Fig. 4 (a) CV curves of PC4 in N2 or O2-saturated electrolyte in 0.1 M KOH solution; (b) LSV curves at 1600 rpm, (c) bar graph of Eonset and Ehalf-wave, and (d) the corresponding Tafel plots for all samples. (e) LSV curves of PC4 at different rotating rates from 400 to 2025 rpm; (f) Koutecký–Levich plots at different potentials for PC4 (the number of transferred electrons is 3.9). | ||
Fig. 5a showed the ring and disk current densities for PCs at a rotating speed of 1600 rpm collected using a rotating ring-disc electrode (RRDE) measurement. And the electron transfer number (n) and peroxide yields were calculated from the RRDE data shown in Fig. 5b. The electron transfer number of PC4 is 3.3–3.4 at the potential range of 0 to 0.7 V (vs. RHE), indicating a four-electron dominant pathway in the ORR. The hydrogen peroxide yield is 19–29%. It suggests that the ORR process catalyzed by PC4 was mainly the four electrons process. This result is consistent with that calculated from the K–L equation. The methanol-tolerance capacity of PC4 was measured (Fig. 5c). Pt/C suffered a dramatic decline when 3 M methanol was added at 200 s during the chronoamperometric measurement. In contrast, a slight increase of the current existed in PC4. It indicates the excellent methanol-tolerance capacity which could satisfy the demands of the practical applications. The long-time stability was also a key factor for excellent ORR catalysts. After 3 h of ORR process, there was 84% of the initial current remained for PC4 (Fig. 5d), implying a long-term stability. As shown in Fig. S4a–c,† 750 °C was the best temperature for activating coal to obtain PC with the optimal ORR activity. The PC4-750 has the maximum onset potential of 0.88 V (vs. RHE) and the maximum half-wave potential of 0.78 V (vs. RHE).
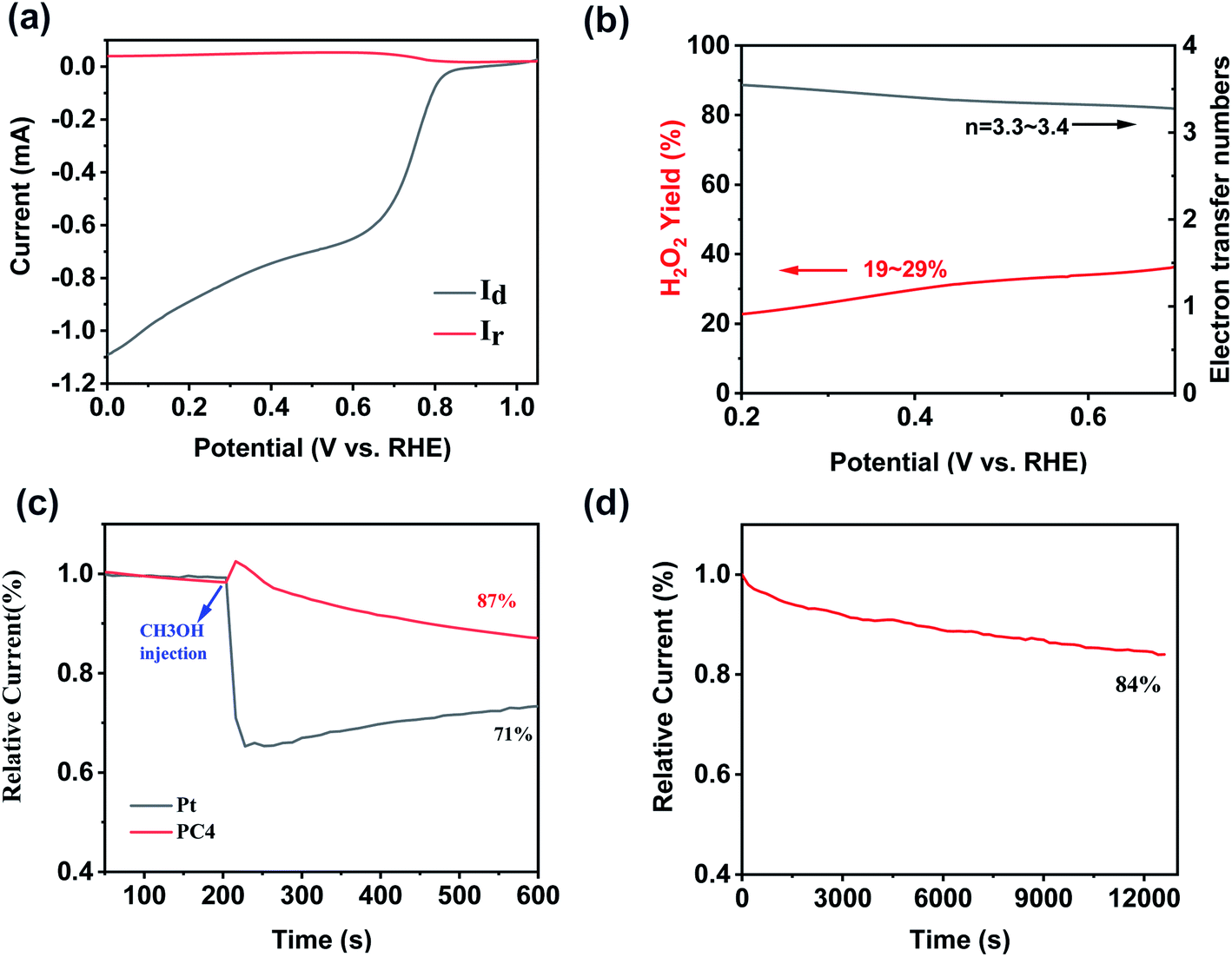 | ||
| Fig. 5 (a) RRDE polarization curves of PC4 at 1600 rpm; (b) the electron transfer number and hydrogen peroxide yield of PC4; (c) methanol-crossover from current versus time (i–t) and (d) chronoamperometric responses and stability of PC4. | ||
Besides potential application as oxygen reduction electrocatalyst, PCs also play an important role in energy storage. Supercapacitor behavior of all the samples was evaluated using a three-electrode system in 6 M KOH aqueous solution. Fig. 6a compared CV curves of all samples at a scan rate of 50 mV s−1. CV curves of all PCs exhibit approximately rectangular shapes, suggesting an ideal electric double-layer capacitive behavior.50 The calculated specific capacitance from GCD curves was shown in Fig. 6b. At the current density of 0.5 A g−1, the specific capacitance of PC1, PC2, PC3, PC4 and PC5 were 37, 42, 66, 128 and 114 F g−1, respectively. With enhancing the mass ratio of KOH/coal, the specific capacitance of PCs increases due to the enlarged specific surface area and pore volume. PC4 has the best performance because of its largest SSA and pore volume. When the current density increases to 10 A g−1, the remained specific capacitance of PC1, PC2, PC3, PC4 and PC5 is 16, 28, 28, 91 and 73 F g−1, respectively. It was worth mentioning that the specific capacitance of PC4 remains 71%. Fig. 6c and d showed CV curves of PC4 at different scan rates and GCD curves of PC4 electrode, respectively. All the CV curves of PC4 exhibit approximately rectangular shapes which proves the largest specific surface area (SSA) contributing to high electrical double layer capacitance (EDLC). The cyclic stability of PC4 was studied via galvanostatic charge/discharge at the current density of 10 A g−1. The specific capacitance kept steady at first and slightly decreases after 1000 cycles. After 2000 times charge/discharge cycles, the specific capacitance still remains 97.3% of the first cycle, which shows that PC4 possesses superior cyclic stability and excellent reversibility. Electrochemical impedance spectroscopy (EIS) measurement in Fig. S5a† was carried out to evaluate the capacitive properties of PCs.51 The semicircle at high-frequency region was on x-axis and the diameter of semicircle reflects solution resistance and charge transfer resistance. And PC4 has the smallest diameter of semicircle at a high-frequency region. The small values of Rs for PC4 indicate the ideal electric double-layer capacitive property. According to the electrical double-layer (EDLC) theory, carbon materials store energy by accumulating charges and ions on their surface. High SSA and developed pore structure could ensure its superior capacitive behavior because of the large SSA causing high specific capacitance and moderate pore width promoting fast ion diffusion. To explore the effect of different temperatures on the performance of the supercapacitor, the data was shown in Fig. S6.† At the current density of 0.5 A g−1, the specific capacitance of PC4-700 and PC4-800 is 111 and 99 F g−1, respectively. The specific capacitance of PC4-700 and PC4-800 remains 73.9% and 70.7%, respectively. So the best activation temperature was 750 °C. We also made a comparison with some other literature materials. The materials synthesized in this paper are simple in method, with high specific surface area and good electrocatalytic performance in Table S1.†
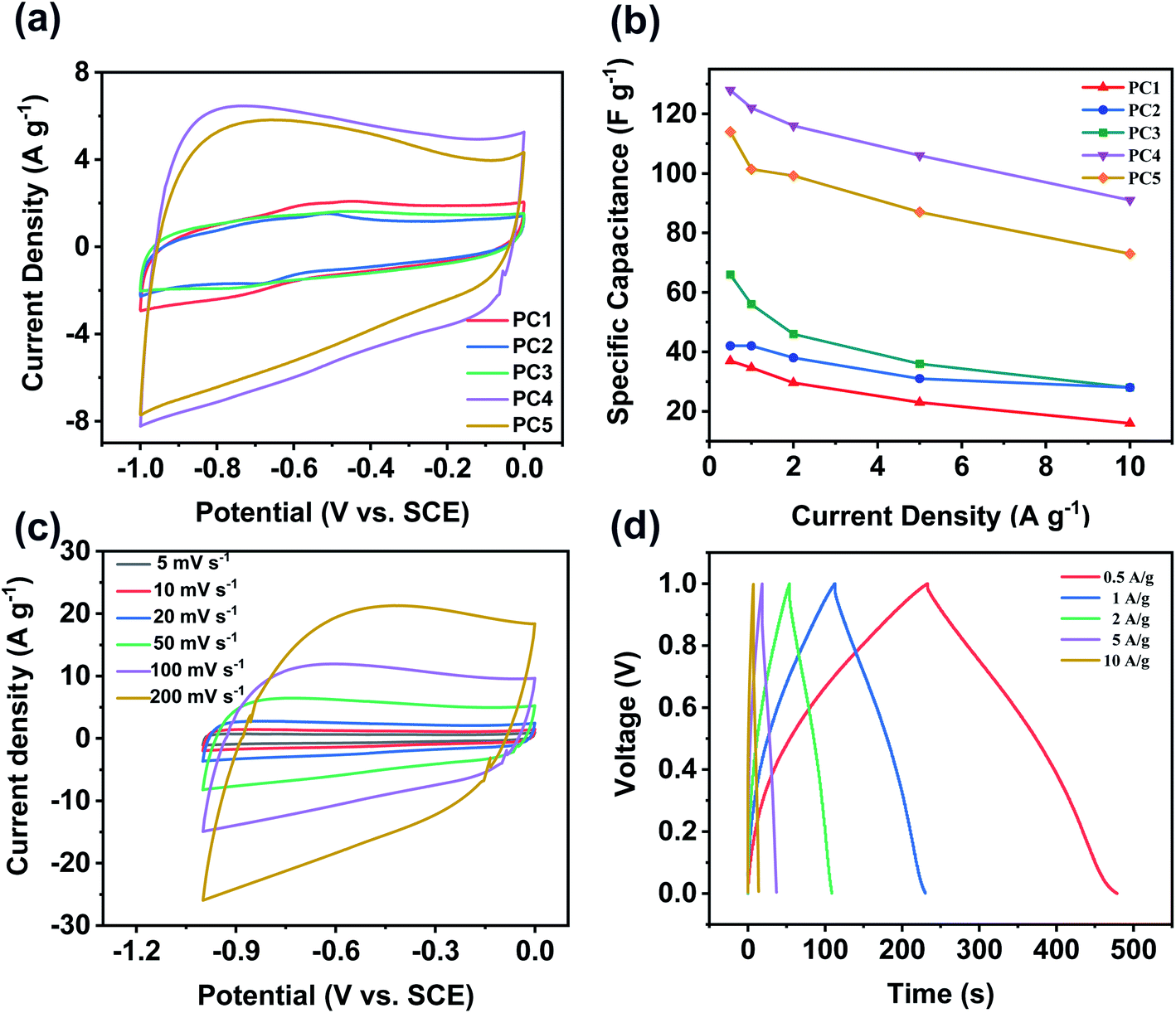 | ||
| Fig. 6 Supercapacitor performances were tested in 6 M KOH aqueous solution using a three-electrode system. (a) CV curves for different samples at 50 mV s−1; (b) specific capacitance at different current densities. (c) CV curves of PC4 at different scan rates; (d) GCD curves of PC4 electrode. | ||
4. Conclusions
Porous carbon materials (PCs) were synthesized by high temperature pyrolysis activation with coal as carbon precursor and KOH as activator. We studied the effects of different ratios of activator and reactants and different activation temperatures on the morphology and structure of porous carbon materials, as well as the ORR properties and the EDLC properties. The best ratio (KOH/coal) was 4 and the best activation temperature was 750 °C. The optimal PC4 possesses a high SSA of 2092 m2 g−1 and a developed microporous structure. It also exhibits a positive onset potential of 0.88 V (vs. RHE) and half-wave potential of 0.78 V (vs. RHE) towards ORR in alkaline solution. It also shows the highest specific capacitance of 128 F g−1 at a current density of 0.5 A g−1 among all the samples in this work. When the current density increases from 0.5 to 10 A g−1, PC4 exhibits a specific capacitance of 91 F g−1 and 71% of original specific capacitance still remains, therefore implying a good rate capacity. PC4 is rich in oxygen-containing functional groups, but contains less nitrogen. Subsequent studies could be continued to investigate the doping of N or other elements to enhance their oxygen reduction performance and supercapacitor performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the financial support from Technology Commission of Shanghai Municipality (16DZ2260603, 19ZR1465100, 19ZR1479500), and Equipment Research Program (6140721050215).References
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- G. Wan, C. Yang, W. Zhao, Q. Li, N. Wang, T. Li, H. Zhou, H. Chen and J. Shi, Adv. Mater., 2017, 29, 1703436 CrossRef PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- N. M. Markovic, Nat. Mater., 2013, 12, 101–102 CrossRef CAS PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed. Engl., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS.
- G. Lin, R. Ma, Y. Zhou, C. Hu, M. Yang, Q. Liu, S. Kaskel and J. Wang, J. Colloid Interface Sci., 2018, 527, 230–240 CrossRef CAS PubMed.
- H. W. Liang, W. Wei, Z. S. Wu, X. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- R. Sharma and K. K. Kar, Electrochim. Acta, 2016, 191, 876–886 CrossRef CAS.
- S.-M. Alatalo, K. Qiu, K. Preuss, A. Marinovic, M. Sevilla, M. Sillanpää, X. Guo and M.-M. Titirici, Carbon, 2016, 96, 622–630 CrossRef CAS.
- K. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2016, 19, 373–381 CrossRef CAS.
- C. Pean, B. Daffos, B. Rotenberg, P. Levitz, M. Haefele, P. L. Taberna, P. Simon and M. Salanne, J. Am. Chem. Soc., 2015, 137, 12627–12632 CrossRef CAS PubMed.
- X. Zhang, J. Luo, P. Tang, X. Ye, X. Peng, H. Tang, S.-G. Sun and J. Fransaer, Nano Energy, 2017, 31, 311–321 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- J. Xu, Z. Tan, W. Zeng, G. Chen, S. Wu, Y. Zhao, K. Ni, Z. Tao, M. Ikram, H. Ji and Y. Zhu, Adv. Mater., 2016, 28, 5222–5228 CrossRef CAS PubMed.
- J. Zhu, K. Sakaushi, G. Clavel, M. Shalom, M. Antonietti and T. P. Fellinger, J. Am. Chem. Soc., 2015, 137, 5480–5485 CrossRef CAS PubMed.
- D. K. Singh, K. S. Krishna, S. Harish, S. Sampath and M. Eswaramoorthy, Angew. Chem., Int. Ed. Engl., 2016, 55, 2032–2036 CrossRef CAS PubMed.
- M. Rana, R. B. Reddy, B. B. Rath and U. K. Gautam, Angew. Chem., Int. Ed. Engl., 2014, 53, 13523–13527 CrossRef CAS PubMed.
- X. Sun, L. Lu, Q. Zhu, C. Wu, D. Yang, C. Chen and B. Han, Angew. Chem., Int. Ed. Engl., 2018, 57, 2427–2431 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- V. C. Hoang, M. Hassan and V. G. Gomes, Appl. Mater. Today, 2018, 12, 342–358 CrossRef.
- M. Yu, C. Zhong, Y. Zhang, Q.-l. Chen, X. Ao, X. Lei and C. Li, J. Anal. Appl. Pyrol., 2018, 134, 293–300 CrossRef CAS.
- W. Geng, F. Ma, G. Wu, S. Song, J. Wan and D. Ma, Electrochim. Acta, 2016, 191, 854–863 CrossRef CAS.
- X. Xie, X. He, X. Shao, S. Dong, N. Xiao and J. Qiu, Electrochim. Acta, 2017, 246, 634–642 CrossRef CAS.
- M. Yu, Y. Han, J. Li and L. Wang, Chem. Eng. J., 2017, 317, 493–502 CrossRef CAS.
- G. J. F. Cruz, M. Pirilä, L. Matějová, K. Ainassaari, J. L. Solis, R. Fajgar, O. Šolcová and R. L. Keiski, Chem. Eng. Technol., 2018, 41, 1649–1659 CrossRef CAS.
- G. Lin, R. Ma, Y. Zhou, Q. Liu, X. Dong and J. Wang, Electrochim. Acta, 2018, 261, 49–57 CrossRef CAS.
- X. Xing, W. Jiang, S. Li, X. Zhang and W. Wang, Waste Manag., 2019, 89, 64–72 CrossRef CAS PubMed.
- A. C. Martins, O. Pezoti, A. L. Cazetta, K. C. Bedin, D. A. S. Yamazaki, G. F. G. Bandoch, T. Asefa, J. V. Visentainer and V. C. Almeida, Chem. Eng. J., 2015, 260, 291–299 CrossRef CAS.
- R. Xing, T. Zhou, Y. Zhou, R. Ma, Q. Liu, J. Luo and J. Wang, Nano-Micro Lett., 2018, 10, 3 CrossRef PubMed.
- W. He, R. Ma, Y. Zhu, M. Yang and J. Wang, J. Inorg. Mater., 2019, 34, 1115–1122 Search PubMed.
- A. Tyagi, S. Banerjee, S. Singh and K. K. Kar, Int. J. Hydrogen Energy, 2019, 06, 195 Search PubMed.
- Y. Huang, E. Ma and G. Zhao, Ind. Crop. Prod., 2015, 69, 447–455 CrossRef CAS.
- C. Ma, X. Chen, D. Long, J. Wang, W. Qiao and L. Ling, Carbon, 2017, 118, 699–708 CrossRef CAS.
- L. Yang, X. Zeng, W. Wang and D. Cao, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- X. Dong, H. Jin, R. Wang, J. Zhang, X. Feng, C. Yan, S. Chen, S. Wang, J. Wang and J. Lu, Adv. Energy Mater., 2018, 8, 1702695 CrossRef.
- X. Wang, Y. Zheng, Y. Xu, J. Ben, S. Gao, X. Zhu, Y. Zhuang, S. Yue, H. Bai, Y. Chen, L. Jiang, Y. Ji, Y. Xu, L. Fan, J. Sha, Z. He and Q. Chen, Biochim. Biophys. Acta, 2009, 1791, 76–83 CrossRef CAS PubMed.
- R. J. White, M. Antonietti and M.-M. Titirici, J. Mater. Chem., 2009, 19, 8645 RSC.
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed.
- H. Zhu, J. Yin, X. Wang, H. Wang and X. Yang, Adv. Funct. Mater., 2013, 23, 1305–1312 CrossRef CAS.
- C. Chen, B. Liang, A. Ogino, X. Wang and M. Nagatsu, J. Phys. Chem. C, 2009, 113, 7659–7665 CrossRef CAS.
- F. Ma, J. Wan, G. Wu and H. Zhao, RSC Adv., 2015, 5, 44416–44422 RSC.
- J. Cherusseri and K. K. Kar, J. Mater. Chem. A, 2016, 4, 9910–9922 RSC.
- X. He, N. Zhang, X. Shao, M. Wu, M. Yu and J. Qiu, Chem. Eng. J., 2016, 297, 121–127 CrossRef CAS.
- H. Zhou, J. Zhang, I. S. Amiinu, C. Zhang, X. Liu, W. Tu, M. Pan and S. Mu, Phys. Chem. Chem. Phys., 2016, 18, 10392–10399 RSC.
- F. Ma, H. Zhao, L. Sun, Q. Li, L. Huo, T. Xia, S. Gao, G. Pang, Z. Shi and S. Feng, J. Mater. Chem., 2012, 22, 13464 RSC.
- A. Jorio, E. H. M. Ferreira, M. V. O. Moutinho, F. Stavale, C. A. Achete and R. B. Capaz, Phys. Status Solidi B, 2010, 247, 2980–2982 CrossRef CAS.
- Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- B. Guo, R. Ma, Z. Li, S. Guo, J. Luo, M. Yang, Q. Liu, T. Thomas and J. Wang, Nano-Micro Lett., 2020, 12, 20 CrossRef.
- M. Zhou, F. Pu, Z. Wang and S. Guan, Carbon, 2014, 68, 185–194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01705a |
| This journal is © The Royal Society of Chemistry 2020 |