
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriple-negative breast cancer suppressive activities, antioxidants and pharmacophore model of new acylated rhamnopyranoses from Premna odorata†
Abeer H. Elmaidomya,
Rabab Mohammeda,
Asmsaa I. Owisa,
Mona H. Hettab,
Asmaa M. AboulMagd c,
Abu Bakar Siddiqued,
Usama Ramadan Abdelmohsenef,
Mostafa E. Ratebag,
Khalid A. El Sayedd and
Hossam M. Hassan*a
c,
Abu Bakar Siddiqued,
Usama Ramadan Abdelmohsenef,
Mostafa E. Ratebag,
Khalid A. El Sayedd and
Hossam M. Hassan*a
aDepartment of Pharmacognosy, Faculty of Pharmacy, Beni-Suef University, Beni-Suef 62514, Egypt. E-mail: hossam.abdelazeem@pharm.bsu.edu.eg
bDepartment of Pharmacognosy, Faculty of Pharmacy, Fayoum University, Fayoum 63514, Egypt
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Nahda University, Beni-Suef 62514, Egypt
dSchool of Basic Pharmaceutical and Toxicological Sciences, College of Pharmacy, University of Louisiana at Monroe, Monroe, LA 71201, USA
eDepartment of Pharmacognosy, Faculty of Pharmacy, Minia University, Minia 61519, Egypt
fDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, Minia 61111, Egypt
gSchool of Computing, Engineering and Physical Sciences, University of the West of Scotland, PA1 2BE Paisley, UK
First published on 12th March 2020
Abstract
Phytochemical investigation of Premna odorata Blanco “Lamiaceae” young stems afforded four new acylated rhamnopyranoses 1–4, along with fourteen known compounds 5–19. The structures of the new compounds were confirmed using extensive 1D, 2D NMR, and HRESIMS analysis. The isolated compounds were tested for their cell proliferation and migration inhibition activities against the invasive human triple-negative breast cancer cells MDA-MB-231 and MCF-7, and the normal human breast cell line MCF-10A. In addition, free radical scavenging activities using 2,2′-diphenyl-1-picrylhydrazyl (DPPH) were studied. Compound 1 was the most active as an antiproliferative agent, showing a high to moderate antiproliferative effect with an IC50 value of 4.95 and 17.7 μM against MCF-7 and MDA-MB-231, respectively. The antiproliferative activities of compounds 1–5 against the normal breast cell line MCF-10A were moderate to low with IC50 values of 13.91 to 27.70 μM. On the other hand, compounds 1 and 10 suppressed MDA-MB-231 cell migration in the wound-healing assay at 10 μM concentration. Meanwhile, compounds 1–5 exhibited the highest value of DPPH radical scavenging activities with an IC50 value range of 17.5–20.43 ± 0.5 μg mL−1. The pharmacophore model generated using Molecular Operating Environment (MOE) for compounds 1–5 showed three hydrogen bond acceptors (HBAs), one hydrogen bond donor (HBD), one aromatic ring (Aro), and one hydrophobic (Hyd.) group. The central HBA feature lies at a distance of 4.36 Å and 6.38 Å from the remaining two HBA features. Also, the HBD feature maintains a distance of 2.74 Å from the aromatic feature. Acylated rhamnopyranoses can be considered good scaffolds for developing new anti-breast cancer and antioxidant compounds.
1. Introduction
Breast cancer (BC) is the most commonly diagnosed cancer and the second-leading cause of cancer-related death in women worldwide.1,2 BC is a heterogeneous disease that can be classified into four major molecular subtypes: luminal A, luminal B, human epidermal growth factor receptor (HER2)-overexpressing, and basal-like, based on clinical, histopathological, and microarray criteria.1,2 Most BC mortalities are mainly linked to metastatic or recurrent complications.3One of the key processes leading to metastasis of cancer is cancer cell migration.1 Cancer cells migrate within the primary tumor stroma after invasion to access the circulation and lymphatic system, and metastasize to secondary sites at distant organs. Due to the importance of cancer cell migration for metastasis and subsequent morbidity and mortality, targeting migration is critical for cancer control especially in highly metastatic and recurrent phenotypes (e.g., TNBC characterized by poor progression-free survival).3,4 Standard chemotherapeutics mainly induce programmed cell death in rapidly dividing cancer cells through mechanisms including the inhibition of cell division and interruption of deoxyribonucleic acid/ribonucleic acid (DNA/RNA) synthesis, but do not necessarily block cell motility. Therefore, residual surviving resistant tumor cells or dormant tumor cells can resist chemotherapy and/or radiotherapy, and can be activated and undergo invasion, migration and develop distant metastases.4
On the other hand, reactive oxygen species (ROS) are produced by living organisms due to their negative cellular metabolism.5 At low-to-moderate concentrations, ROS improved physiological cell processes. Conversely, they changed cell components such as lipids, proteins, and deoxyribonucleic acid at high concentrations.6 ROS are related to almost all aspects of cancer.6 Consequently, reducing ROS can be correlated to the anticancer effect.7 Natural antioxidants can eliminate free radicals such as single oxygen or peroxides by donating hydrogen and chelating metal ions. These activities decrease deoxyribonucleic acid damage, reduce lipid peroxidation, and inhibit cell proliferation related to cancer development.8
Phenolic natural compounds are distributed in plants, and have gained much attention in recent years because of their antioxidant activity, free radical scavenging ability, antitumor effects, and their proven efficacy and safety.8,9 Recently, natural products have begun to play an important role in the discovery of anticancer antioxidant lead compounds. About 50% of today's anticancer drugs in clinical practice are either natural products or modeled after parent natural product compounds such as taxanes, doxorubicin, epothilones, vincristine, camptothecin, and several others.6–9
Plants of the genus Premna, Lamiaceae, are rich resources for the production of diverse bioactive secondary metabolites that have cytotoxic antioxidant activities. For example, the diterpenes latifolionol and dihydrolatifolionol isolated from the bark of P. latifolia showed potent cytotoxic activity against HT-29 and the hepatocellular carcinoma (HepG2) cell lines.10 Also, the leaf extract from P. odorata afforded multiple acylated iridoid glycosides and rhamnopyranoses with good antiproliferative and anti-migratory activities against triple-negative breast cancer cell lines by targeting the c-Met kinase domain.11 The flavone glycosides 4′-hydroxy-8,3′-dimethoxy-6-acroleinylflavan-3,4-diol and naringenin isolated from P. latifolia leaves significantly inhibited the oxidation of DPPH with a half maximal inhibitory concentration (IC50) of 22.5 and 16.0 μg mL−1, respectively.12 The furofuran lignans (4-β-hydroxyasarinin-1-O-β-D-glucopyranoside, premnadimer) and iridoid glycosides (premnosidic acid, 10-O-trans-p-coumaroylcatalpol, 10-O-trans-p-coumaroyl-6-O-α-L rhamnopyranosyl catalpol, 4′′-hydroxy-E-globularinin) were found to contribute to the antioxidant activity of the stem bark of P. integrifolia when evaluated with radical scavenging DPPH, nitric oxide (NO) and ferric reducing antioxidant power (FRAP) assays.13 A recent study also identified the aromatic diterpene 11,12,16-trihydroxy-2-oxo-5-methyl-10-demethyl-abieta-1,10 and 6,8,11,13-pentene as an antioxidant constituent from P. serratifolia with an IC50 of 20.4 ± 1.3 μg mL−1 towards a DPPH assay.14 The diterpenoids, icetexanes I and III, and the dimeric diterpene premnalatifolin A isolated from the bark of P. tomentosa showed selective inhibitory activity for the growth of the human estrogen-dependent breast cancer Michigan Cancer Foundation-7 (MCF-7) cells and the colorectal cancer human adenocarcinoma colorectal cell line (HT-29) cells.15,16
This study reports the isolation of four new acylated rhamnopyranoside esters 1–4, along with several known secondary metabolites isolated from the young stems of P. odorata. A previous successful biological investigation on the acylated rhamnopyranose 1-O-trans-p-coumaroyl-2-O-trans-caffeoyl-α-L-rhamnopyranose isolated from the leaves of P. odorata showed antiproliferative activity, with an IC50 value of 13.3 μM against MDA-MB-231.11 This previous report encouraged further biological investigations on the isolated acylated rhamnopyranoside esters from the young stems of P. odorata as antiproliferative and antimigratory agents against the invasive human triple-negative breast cancer cells MDA-MB-231, MCF-7, as well as the normal cell line MCF-10A and antioxidant activities.
2. Experimental
2.1. Plant material
Young stems from P. odorata were collected on May 2018 from the Giza Zoo garden, Egypt and identified by Dr Abd El-Halim A. Mohammed, Horticultural Research Institute, Department of Flora and Phytotaxonomy Researches, Dokki, Cairo, Egypt. A voucher specimen (2018-BuPD 46) was deposited at the Department of Pharmacognosy, Faculty of Pharmacy, Beni-Suef University, Egypt.2.2. Chemicals and reagents
The solvents used in this work included n-hexane (n-hex., boiling point b.p. 60–80 °C), dichloromethane (DCM), ethyl acetate (EtOAC), n-butanol (n-but.), ethanol (EtOH), methanol (MeOH), formic acid (F.A.), glacial acetic acid (G.A.A.) and sulfuric acid purchased from El-Nasr Company for Pharmaceuticals and Chemicals (Egypt). High Performance Liquid Chromatography (HPLC) and deuterated solvents used for chromatographic and spectroscopic analyses were purchased from Sigma-Aldrich (Saint Louis, Missouri, USA), including HPLC-methanol, HPLC-water, chloroform (CDCl3), methanol (CD3OD), and dimethyl sulfoxide (DMSO-d6). Column chromatography (CC) was performed using silica gel 60 (63–200 μm, E. Merck, Sigma-Aldrich), and polyamide-6 (50–160 μm), while silica gel GF254 for Thin-layer chromatography (TLC) (El-Nasr Company for Pharmaceuticals and Chemicals, Egypt) was employed for vacuum liquid chromatography (VLC). Thin-layer chromatography (TLC) was carried out using pre-coated silica gel 60 GF254 plates (E. Merck, Darmstadt, Germany; 20 × 20 cm, 0.25 mm in thickness). Spots were visualized by spraying with para-anisaldehyde (PAA) reagent (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:10:0.5 absolute EtOH:sulfuric acid:G.A.A.:para-anisaldehyde), followed by heating at 110 °C.17 For the biological study, DPPH and L-ascorbic acid were purchased from (Sigma-Aldrich, Saint Louis, Missouri, USA).
:5:10:0.5 absolute EtOH:sulfuric acid:G.A.A.:para-anisaldehyde), followed by heating at 110 °C.17 For the biological study, DPPH and L-ascorbic acid were purchased from (Sigma-Aldrich, Saint Louis, Missouri, USA).
2.3. Spectral analyses
Proton 1H and Distortionless Enhancement by Polarization Transfer-Q (DEPT-Q) 13C NMR spectra were recorded at 400 and 100 MHz, respectively. Tetramethylsilane (TMS) was used as an internal standard in chloroform (CDCl3), methanol (CD3OD), and dimethyl sulfoxide (DMSO-d6), using the residual solvent peak (δH = 7.26 and δC = 77.0), (δH = 3.34, 4.78 and δC = 49.9) and (δH = 2.50 and δC = 39.5) as references, respectively. Measurements were performed on a Bruker Advance III 400 MHz with BBFO Smart Probe and a Bruker 400 MHz EON Nitrogen-Free Magnet (Bruker AG, Billerica, MA, USA). Carbon multiplicities were determined using a DEPT-Q experiment. The optical rotation in methanol was obtained using a Perkin-Elmer 343 polarimeter (PerkinElmer Inc., Waltham, MA, USA). The ultraviolet radiation (UV) spectrum in methanol was obtained using a Shimadzu UV 2401PC spectrophotometer (Shimadzu Corporation – UV-2401PC/UV-2501PC, Kyoto, Japan). Infrared (IR) spectra were measured using a Jasco FTIR 300E infrared spectrophotometer. HRESIMS data were obtained using an Acquity Ultra Performance Liquid Chromatography system coupled to a Synapt G2 HDMS quadrupole time-of-flight hybrid mass spectrometer (Waters, Milford, MA, USA). HPLC chromatographic separations were conducted using an Agilent 1260 Infinity preparative pump (G1361A), Agilent 1260 diode array detector VL (G1315 D), Agilent 1260 Infinity Thermostand column compartment (G1361 A), Agilent 1260 Infinity preparative autosampler (G2260A) and a YMC-Pack ODS-A A-324 column (i.d. 10 × 300 mm, YMC, Kyoto, Japan).2.4. Extraction and fractionation of plant material
The young stems (5 kg) were collected and air-dried in the shade for one month. After drying, the young stems were finely powdered using an OC-60B/60B set small corn bark grinding machine for herbs (60–120 mesh, Henan, Mainland China). The finely powdered young stems were extracted by maceration without agitation using 70% ethanol (5 L, 3×, seven days each) at room temperature, and concentrated under vacuum at 45 °C using a rotary evaporator (Buchi Rotavapor R-300, Cole-Parmer, Vernon Hills, IL, USA) to afford 150 g crude extract. The dry extract was suspended in 300 mL distilled water (H2O), and successively portioned with solvents of different polarities (n-Hex., DCM, EtOAC, and n-but.). The organic phase in each step separately evaporated under reduced pressure to afford the corresponding fractions I (8.0 g), II (8.0 g), III (10.0 g) and IV (50.0 g), respectively, while the remaining mother liquor was then concentrated down to give the aqueous fraction (V). All resulting fractions were kept at 4 °C for biological and phytochemical investigations.2.5. Isolation and purification of compounds
A part of fraction I (5 g) was subjected to VLC fractionation using silica gel GF254 (column 6 × 30 cm, 50 g). Elution was performed using n-hex.:EtOAC gradient mixtures in order of increasing polarities (0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 80 and 100%, 1 L each, flow rate (FR) 3 mL min−1), then with DCM:EtOAC (50, 50, 1 L, FR 3 mL min−1), and finally with EtOAC. The effluents from the column were collected in fractions (100 mL each); and each collected fraction was concentrated and monitored by TLC using the system n-hex.:EtOAC 8:2 and PAA reagent. Similar fractions were grouped and concentrated under reduced pressure to provide three sub-fractions (I1–I3). Crystallization of sub-fractions I1 and I3 was performed separately using MeOH, and afforded compounds 10 (16 mg) and 18 (30 mg), respectively. Subfraction I2 (1.0 g) was further fractionated on a silica gel 60 column. An elution was performed using n-hex.:EtOAC gradient mixtures in order of increasing polarities (0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 80 and 100%, 250 mL each, FR 3 mL min−1). The effluents were collected in fractions (20 mL each); each fraction was concentrated and monitored by TLC to afford compounds 13 (10 mg), 14 (40 mg), and 16 (10 mg).
Fraction II (8 g) was subjected to VLC fractionation on a silica gel (column 6 × 30 cm, 50 g). Elution was performed using DCM:EtOAC gradient mixtures in the order of increasing polarities (0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 80 and 100%, 1 L each, FR 3 mL min−1), then with EtOAC:MeOH (50, 50, 1 L, FR 3 mL min−1), and finally with MeOH. The effluents were collected in fractions (100 mL each); each fraction was concentrated and monitored by TLC using the system DCM:EtOAC 8:2 and PAA reagent. Similar fractions were grouped and concentrated under reduced pressure to provide three sub-fractions (II1–II3). Subfraction II1 (1 g) was further fractionated on a silica gel column (65 × 1.5 cm, 50 g), and an elution was performed using n-hex.:EtOAC gradient mixtures in order of increasing polarities (0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 80 and 100%, 250 mL each, FR 3 mL min−1). The effluents were collected in fractions (20 mL each); each fraction was concentrated and monitored by TLC to afford compounds 11 (16 mg) and 17 (7 mg). Subfraction II2 (0.5 g) was further fractionated on silica gel 60 (100 × 1 cm, 25 g). Elution was performed using DCM:EtOAC isocratic mixture (20%, 500 mL, FR 3 mL min−1) to afford compound 15 (16 mg). Finally, the crystallization of subfraction II3 from MeOH afforded compound 12 (6 mg).
Moreover, part of the EtOAC fraction III (8 g) was fractionated on a polyamide-6 (column 50–160 μm, 1000 × 5 cm, 100 g) using gradient elution, starting with water (H2O) and ending with MeOH in the order of increasing polarities (0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 80 and 100%, 1000 mL each, FR 5 mL min−1). The effluents were collected in fractions (200 mL each); each fraction was concentrated and monitored by TLC using system EtOAC:G.A.A.:F.A.:H2O 10:1:1:2 and PAA reagent. Similar fractions were grouped and concentrated under reduced pressure to also provide three sub-fractions (III1–III3).
Sub-fractions III1-3 (1.0 g) were separately further fractionated on a silica gel (column 63–200 μm, 65 × 1.5 cm, 50 g). Elution was performed using DCM:MEOH gradient mixtures in order of increasing polarities (0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 80 and 100%, 250 mL each, FR 3 mL min−1). The effluents were collected in fractions (20 mL each); each fraction was concentrated and monitored by TLC using system EtOAC:G.A.A.:F.A.:H2O 10:1:1:2 and PAA reagent to afford one promising sub-subfraction III1-3 (80, 30, 50 mg, respectively), which was further purified in preparative TLC eluted with system EtOAC:G.A.A:F.A.:H2O 10:1:1:2 to give compounds 1 (6 mg), 2 (7 mg), 3 (6 mg), 4 (7 mg), 5 (7 mg), 6 (5 mg), 7 (6 mg), 8 (6 mg), and 9 (7 mg).
| Moiety | Position | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| a In CD3OD, 400 MHz for 1H and 100 MHz for 13C NMR; J in Hz. Carbon multiplicities were determined by DEPT-Q experiments. qC = quaternary, CH = methine, CH2 = methylene, CH3 = methyl carbons, α-L-Rhamn.: α-L-rhamnopyranoside, trans-p-Coum.: trans-p-coumaroyl moiety, trans-Caff.: trans-caffoyl moiety. | |||||||||
| α-L-Rhamn. | 1, CH | 91.9 | 5.18, d (1.6) | 94.4 | 5.13, d (1.6) | 91.9 | 5.18, d (1.6) | 94.4 | 5.13, d (1.6) |
| 2, CH | 73.7 | 5.19, m | 69.7 | 4.12, dd (2.0, 3.6) | 73.7 | 5.18, m | 69.6 | 4.12, dd (2.0, 3.6) | |
| 3, CH | 69.1 | 4.11, dd (3.6, 9.4) | 74.1 | 5.24, dd (3.6, 9.4) | 69.0 | 4.11, dd (3.6, 9.4) | 73.9 | 5.24, dd (3.6, 9.4) | |
| 4, CH | 73.2 | 3.62, t (9.4) | 70.4 | 3.76, t (9.4) | 73.1 | 3.62, t (9.4) | 70.4 | 3.76, t (9.4) | |
| 5, CH | 68.2 | 3.99, m | 68.1 | 3.95, m | 68.1 | 3.99, m | 68.0 | 3.95, m | |
| 6, CH3 | 17.0 | 1.34, d (6.0) | 17.0 | 1.33, d (6.0) | 17.0 | 1.34, d (6.0) | 17.0 | 1.33, d (6.0) | |
| trans-p-Coum. | 1′, qC | 125.9 | 125.9 | ||||||
| 2′/6′, CH | 130.0 | 7.41, d (8.0) | 130.0 | 7.39, d (8.0) | |||||
| 3′/5′, CH | 115.6 | 6.83, d (8.0) | 115.6 | 6.81, d (8.0) | |||||
| 4′, qC | 159.6 | 159.6 | |||||||
| 7′, CH | 145.6 | 7.41, d (16.0) | 145.9 | 7.56, d (16.0) | |||||
| 8′, CH | 113.7 | 6.37, d (16.0) | 113.7 | 6.28, d (16.0) | |||||
| 9′, qC | 167.7 | 167.7 | |||||||
| trans-p-Coum. | 1′, qC | 126.0 | 126.0 | ||||||
| 2′/6′, CH | 130.1 | 7.69, d (8.0) | 130.1 | 7.39, d (8.0) | |||||
| 3′/5′, CH | 115.6 | 6.83, d (8.0) | 115.6 | 6.81, d (8.0) | |||||
| 4′, qC | 159.7 | 159.7 | |||||||
| 7′, CH | 145.9 | 7.64, d (16.0) | 146.2 | 7.66, d (16.0) | |||||
| 8′, CH | 114.1 | 6.44, d (16.0) | 114.2 | 6.37, d (16.0) | |||||
| 9′, qC | 167.9 | 167.9 | |||||||
| trans-Caff. | 1′′, qC | 126.3 | 126.3 | ||||||
| 2′′, CH | 114.0 | 7.08, d (2.4) | 114.0 | 7.08, d (2.4) | |||||
| 3′′, qC | 148.3 | 148.3 | |||||||
| 4′′, qC | 145.4 | 145.4 | |||||||
| 5′′, CH | 115.1 | 6.81, d (8.0) | 115.1 | 6.81, d (8.0) | |||||
| 6′′, CH | 121.6 | 6.91, dd (2.4, 8.0) | 121.6 | 6.91, dd (2.4, 8.0) | |||||
| 7′′, CH | 145.7 | 7.57, d (16.0) | 145.7 | 7.58, d (16.0) | |||||
| 8′′, CH | 113.6 | 6.31, d (16.0) | 113.6 | 6.35, d (16.0) | |||||
| 9′′, qC | 167.6 | 167.6 | |||||||
| trans-Caff. | 1′′, qC | 126.3 | 126.3 | ||||||
| 2′′, CH | 114.0 | 7.08, d (2.4) | 114.0 | 7.08, d (2.4) | |||||
| 3′′, qC | 148.3 | 148.3 | |||||||
| 4′′, qC | 146.0 | 146.0 | |||||||
| 5′′, CH | 115.1 | 6.81, d (8.0) | 115.1 | 6.81, d (8.0) | |||||
| 6′′, CH | 121.7 | 6.91, dd (2.4, 8.0) | 121.7 | 6.91, dd (2.4, 8.0) | |||||
| 7′′, CH | 145.7 | 7.57, d (16.0) | 145.7 | 7.58, d (16.0) | |||||
| 8′′, CH | 113.8 | 6.33, d (16.0) | 113.8 | 6.42, d (16.0) | |||||
| 9′′, qC | 167.7 | 167.7 | |||||||
2.6. Cell lines, culture conditions, and cell viability assessments
The human triple-negative breast cancer cell line MDA-MB-231 was purchased from the American Type Culture Collection (ATCC, Manassas, VA). This cancer cell line was maintained in RPMI-1640 (Gibco® by Life Technologies, Grand Island, NY), which was supplemented with 10% fetal bovine serum (Gemini Bio-Products), 100 U mL−1 penicillin G, 100 μM mL−1 streptomycin, and 2 mmol L−1 glutamine. MCF-7 and MCF-10A cell lines were obtained from American Type Culture Collection, and the cells were cultured using DMEM (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 μg mL−1 of insulin (Sigma), and 1% penicillin–streptomycin.The cells were subcultured upon attaining 80% confluence. The cells were maintained at 37 °C in a humidified incubator under 5% CO2. A stock solution was prepared by dissolving each tested compound in sterilized DMSO solvent at concentration of 10 mM for all assays. The working solutions at their final concentrations for each assay was prepared in an appropriate culture medium immediately before use. The vehicle DMSO control was prepared by adding the maximum volume of DMSO, which was used in preparing test compounds to the media type such that the final DMSO concentration maintained as the same in the all treatment groups within an experiment and never exceeded 0.1%. (−)-Oleocanthal used in all experiments as a positive control based on earlier studies.18 A viable cell count was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. The optical density of each sample was measured at 570 nm on a Synergy 2-micro-plate reader (BioTek, VT, USA). The number of cells per well was calculated against a standard curve, which was prepared at the start of each experiment by plating various concentrations of cells (1000–60000 cells per well), as determined using a hemocytometer.
2.7. Cell proliferation assay
Proliferation inhibition activities of the free hydroxyl phenolic and/or carboxylic acid groups containing compounds (acylated rhamnopyranoses 1–4, verbascoside 5, premnoside E–F 6–7, oleanolic acid 10, and maslinic acid 12) were tested against the invasive human triple-negative breast cancer cells MDA-MB-231, MCF-7 and normal MCF-10A cell lines.19,20 Cell proliferation was evaluated in the celllines by the MTT assay in triplicate. In brief, breast cells in exponential growth were seeded at a density of 1 × 104 cells per well (6 wells per group) in 96-well culture plates and maintained in RPMI-1640 media supplemented with 10% fetal bovine serum. These cells were treated for 24 h with isolated compounds using 10 μM MTT, and allowed to adhere overnight at 37 °C under 5% CO2 in a humidified incubator.19 One day later, cells were washed with phosphate-buffered saline, followed by division into different treatment groups. The cells were then fed with serum-free defined RPMI-1640 media and experimental treatments containing designated concentrations of each tested compound or vehicle-treated control media, and incubation resumed at 37 °C under 5% CO2 for 48 h. The control and treatment media were then removed and replaced with fresh media, and 50 μL of fresh MTT solution (1 mg mL−1) was added to each well, and the plates were re-incubated for 4 h at 37 °C. The color reaction was stopped by removing the media and adding 100 μL DMSO in each well to dissolve the formed formazan crystals. The incubation at 37 °C was resumed for up to 20 min to ensure complete dissolution of crystals. The absorbance was determined at λ570 (nm) using an ELISA plate microreader (BioTek, Winooski, VT, USA). The % of the cell survival was calculated as follows: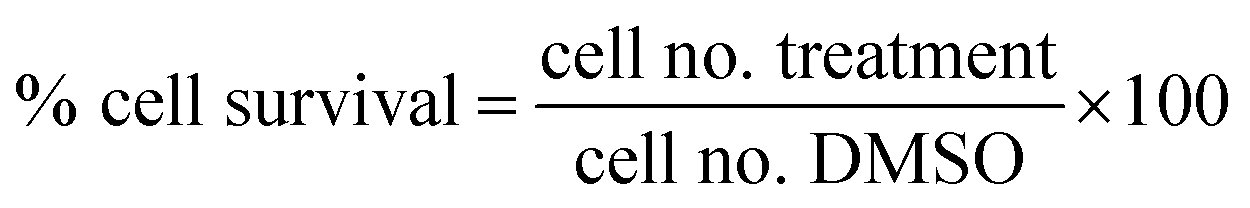
2.8. Cell migration assay
Migration inhibition activities of the free hydroxyl phenolic and/or carboxylic acid groups containing compounds (acylated rhamnopyranoses 1–4, verbascoside 5, premnoside E–F 6–7, oleanolic acid 10, and maslinic acid 12) were tested against the invasive human triple-negative breast cancer cells MDA-MB-231, MCF-7 and normal MCF-10A.19,20 Breast cells were plated in sterile 24-well plates and allowed to form a confluent monolayer/well overnight.19,20 The wounds were then inflicted in each cell monolayer using a sterile 200 μL pipette tip, the media was then removed, and the cells were washed twice using phosphate-buffered saline followed by fresh RPMI medium to remove cell debris. Each test compound concentration (10 μM) prepared in fresh serum-free defined media contained 40 μg mL−1 Hepatocyte growth factor (HGF) as a mitogen source was added to the wells in triplicate. The cells were incubated for 24 h and then the medium was removed. The cells were washed, fixed, and stained using Giemsa stain. The wound healing was visualized at 0 and 24 h by a Nikon ECLIPSE TE200-U microscope, and the digital images were captured using Nikon NIS Elements software (Nikon Instruments Inc., Melville, NY). The distance traveled by the cells was determined by measuring the wound width after 24 h. The percentages of the cell migration were calculated using the following formula:where T0 was the wound thickness at time zero, TDMSO was the wound thickness in the DMSO-treated control wells, while Tt was the wound thickness in the treated wells.
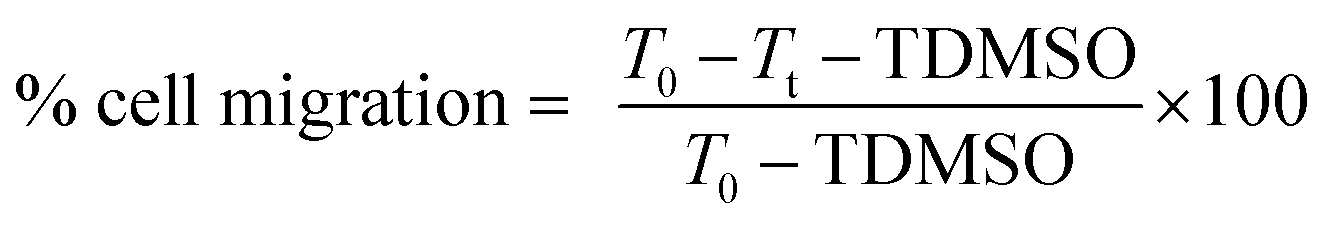
2.9. DPPH radical scavenging activity assay
The radical scavenging activity of the free hydroxyl phenolic and/or carboxylic acid groups containing compounds (acylated rhamnopyranoses 1–4, verbascoside 5, premnoside E–F 6–7, oleanolic acid 10, and maslinic acid 12) were tested using the stable radical DPPH assay.21 In a brief manner, a 1 mL solution of the tested compounds at different concentrations (2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 40, 60, 80, 100 μg mL−1 in absolute ethanol) was mixed with 2 mL of the freshly prepared DPPH solution (20 μg mL−1 in absolute ethanol), followed by incubation at room temperature in the dark for 30 min. The absorbance was measured at λ517 (nm) using a UV-Vis Jenway 6003 spectrophotometer. Ascorbic acid was used as a positive control, and absolute ethanol was used as a blank. The DPPH radical scavenging activity was calculated according to the following equation:where A0 is the absorbance of the blank and A1 is the absorbance in the presence of the tested compound or standard.
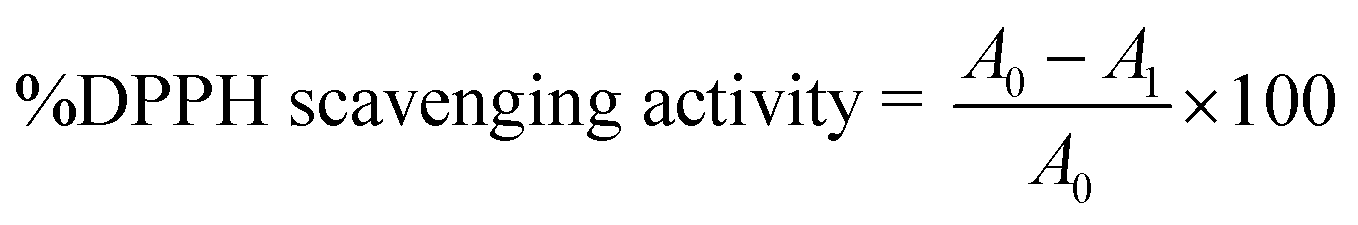
2.10. Pharmacophore model generation
From the previous DPPH results, ChemBioDraw Ultra 12.0 (CambridgeSoft, 100 Cambridge Park Drive, Cambridge, MA 02140) was used to sketch the 2D chemical structures of compounds 1–5 (acylated rhamnopyranoses 1–4, and verbascoside 5), saved in the MDL-molfile format and a conformational training dataset of compounds 1–5 was implemented in Molecular Operating Environment (MOE) (Chemical Computing Group software, Canada) to generate the pharmacophore hypotheses. These were converted to 3D structures and their energies were minimized with the Merck Molecular Force Field (MMFF), using an energy threshold value of 15 kcal mol−1 above the global energy minimum until a local energy minimum was reached. MOE 2019 was used to generate the pharmacophore models for the aligned molecules using common features for the pharmacophore generation.22 The 3D space of the pharmacophore sites of a ligand are demonstrated by a set of points. During the pharmacophore hypothesis generation, all compounds were selected with common features such as HBDs, HBAs, ring aromatics (Aro), and hydrophobic groups (Hyd). A scoring function was employed for examining the different pharmacophore hypotheses produced so that the best alignment of the ligands would be produced. Finally, molecular sequence and lowest Root Mean Square Deviation (RMSD) were used as a base to select the developed pharmacophore model (see Table 2).23| Compound no. | RMSD |
|---|---|
| a RMSD: lowest root mean square deviation. | |
| 1 | 0.416 |
| 2 | 0.417 |
| 3 | 0.432 |
| 4 | 0.474 |
| 5 | 0.365 |
2.11. Statistical analysis
All in vitro experiments were performed in triplicate. Pooled data were presented as the mean ± standard error of the mean (SEM) of at least three independent experiments. The differences among various treatment groups were determined by ANOVA, followed by Dunnett's test using PASW Statistics® version 18 (Quarry Bay, Hong Kong). A difference of p < 0.05 was considered statistically significant compared with a vehicle-treated control group and shown by a * symbol. The IC50 values were determined using a nonlinear regression curve fitting analysis using GraphPad Prism software version 6 (La Jolla, CA, USA).3. Results and discussion
3.1. Phytochemical investigation of P. odorata young stems
Based on the physicochemical and chromatographic properties, the spectral analyses from UV, 1H, and DEPT-Q NMR, as well as comparisons with the literature and some authentic samples, the crude ethanolic extract of P. odorata young stems afforded the new acylated rhamnopyranoside esters 1–4, along with the known verbascoside 5 (ref. 24) and premnoside E–H 6–9.11 Additionally, oleanolic acid 10,25 oleanolic acid methyl ester 11,26 maslinic acid 12,25 β-amyrin 13,27 β-sitosterol 14,28 daucosterol 15,29 citrostadienol 16,30 palmitic acid 17,31 and tricosane-1-ol 18,32 were identified (Fig. 1). All characterized compounds (11, 12, and 16) were isolated herein for the first time from the genus Premna.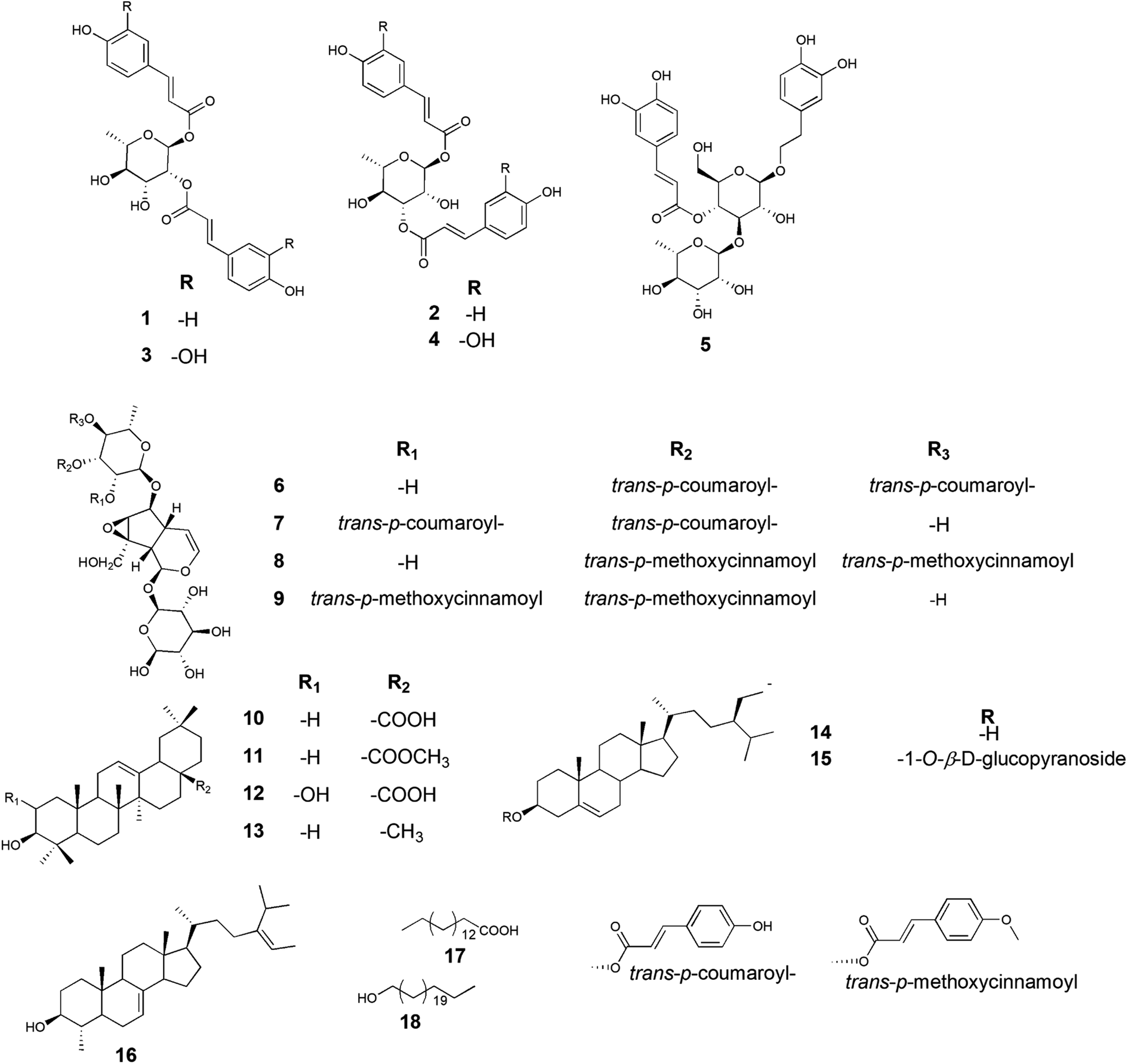 | ||
| Fig. 1 Chemical structure of compounds 1–18 isolated from Premna odorata young stems extract. | ||
Analysis of the HRESIMS, 1D and 2D NMR data of compounds 1–4 suggested a possible acylated rhamnopyranoside core scaffold.11 The HRESIMS data for compound 1 showed an adduct pseudo-molecular ion peak at m/z 457.1494 [M + H]+, (calc. for C24H25O9, 457.1499), suggesting 13 degrees of unsaturation. The 1H and DEPT-Q 13C NMR data (Table 1, Fig. S2–S3†), along with the Heteronuclear Single Quantum Correlation Experiment (HSQC) data (Fig. S4†), suggested that six characteristic resonances appeared: five oxymethine groups at δH 5.18 (1H, d, J = 1.6) δC 91.1, δH 5.18 (1H, m) δC 73.7, δH 4.11 (1H, dd, J = 3.6, 9.4) δC 69.1, δH 3.62 (1H, t, J = 9.4) δC 73.2, and δH 3.99 (1H, m) δC 68.2, and one methyl group at δH 1.34 (1H, d, J = 6) δC 69.1, suggesting the characteristic core structure for a disubstituted α-L-ramnopyranoside unit.11 NMR data also showed eight aromatic methine groups at δH 7.41 (2H, d, J = 8) δC 130.0, δH 6.83 (2H, d, J = 8) δC 115.6, δH 7.69 (2H, d, J = 8) δC 130.1, δH 6.83 (2H, d, J = 8) δC 115.6, four trans-olefinic methine groups at δH 7.41 (1H, d, J = 16) δC 145.6, δH 6.37 (1H, d, J = 16) δC 113.7, δH 7.64 (1H, d, J = 16) δC 145.9, δH 6.44 (1H, d, J = 16) δC 114.1, and six quaternary carbons at δC 125.9, 126.0, 159.6, 159.7, 167.7 and 167.9. These signals are suggestive characteristics for two trans-p-coumaroyl moieties.11 The Homonuclear Correlation Spectroscopy (1H–1H COSY) data of 1 (Fig. 2) confirmed the expected coupling of the oxymethines and methyl H-1-H3-6. Moreover, the Heteronuclear Multiple-Bond Correlation (HMBC) experiment showed the 3J-HMBC correlation of the proton H-1 δH 5.18 (δC 91.1) with the quaternary carbonyl carbon C-9′ (δC 167.7) and the 3J-HMBC correlations of the proton H-2 δH 5.18 (δC 73.7) with the quaternary carbonyl carbon C-9′′ (δC 167.9), confirming the connections of the two trans-p-coumaroyl moieties at C-1 and C-2 of the α-L-rhamnopyranosyl moiety (Fig. 2). The structure of compound 1 was very similar to the known related 1-O-trans-p-coumaroyl-2-O-trans-caffeoyl-α-L-rhamnopyranose, previously isolated from P. odorata leaves,11 with the replacement of the C-2 caffeoyl ester by a C-2 p-coumaroyl ester moiety. This change induced an upfield chemical shift of carbons C-2′′, C-4′′ and C-6′′ in the known C-2 caffeoyl α-L-rhamnopyranose by ΔδC −15.9, −14.6 and −8, respectively, while carbon C-3′′ was downfield shifted by ΔδC +32.4, as compared to those of compound 1. Accordingly, compound 1 was identified as 1,2-di-O-trans-p-coumaroyl-α-L-rhamnopyranose.
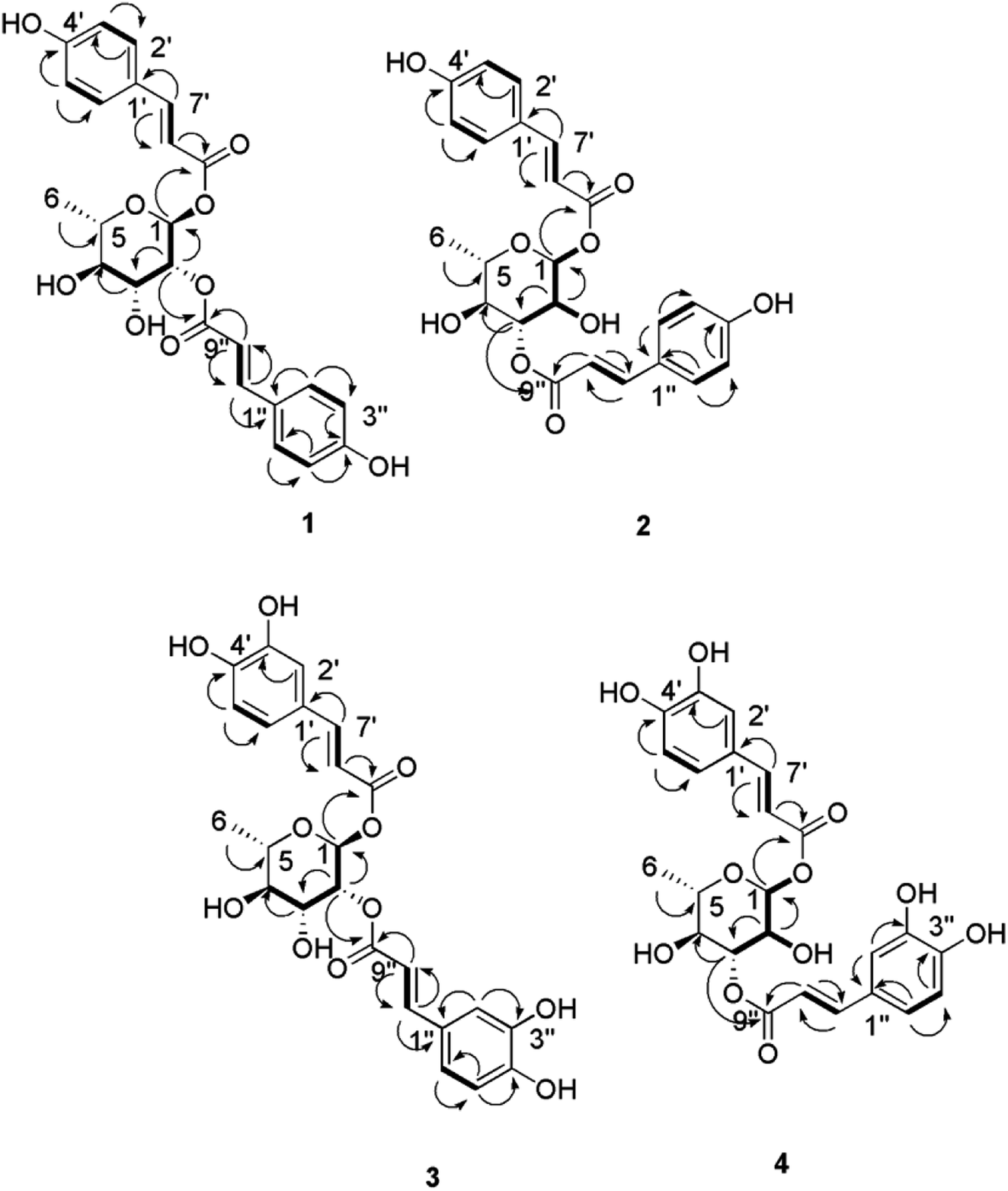 | ||
Fig. 2 Selected HMBC ( ) and 1H–1H COSY ( ) and 1H–1H COSY ( ) correlations of compounds 1–4. ) correlations of compounds 1–4. | ||
The molecular formula of compound 2 was identical to that of 1 based on HRESIMS (C24H25O9). The 1H and 13C NMR data was also very close to those of compound 1 for the two trans-p-coumaric acid moieties, but differed in the resonated chemical shifts of the oxymethine groups of the core α-L-rhamnopyranosyl moiety. This suggested a positional difference of the location of the two trans-p-coumaroyl moieties in the α-L-rhamnopyranosyl moiety versus 1 (Table 1, Fig. 2, and S8–S9†). The assignment of the location of the two trans-p-coumaric acid moieties in 2 was aided by the HMBC experiment. A 3J-HMBC correlation of the compound 2 proton H-1 δH 5.13 (δC 94.4) with the quaternary carbonyl carbon C-9′ (δC 167.7) and a 3J-HMBC correlation of the proton H-3 δH 5.24 (δC 74.1) with the carbonyl carbon C-9′′ (δC 167.9) confirmed the location of the two trans-p-coumaric moieties at carbons C-1 and C-3 of the α-L-rhamnopyranosyl moiety. The 1H–1H COSY data of 2 (Fig. 2) confirmed the coupling of the oxymethine H-1 (δH 5.13) with H-2 (δH 4.12), which in turn coupled with H-3 (δH 5.24). This data further validated the 1,3-disubstituted trans-p-coumaroyl-α-L-rhamnopyranose identity of 2. Comparing the DEPT-Q 13C NMR data of compound 2 with those of 1 showed a downfield shifting of carbons C-1 and C-3 (ΔδC +2.5 and +4.9, respectively), while carbons C-2 and C-4 were upfield shifted (ΔδC +4.0 and 2.8, respectively) compared with those of compound 1 (Table 1, Fig. 1). Accordingly, compound 2 was identified as 1,3-di-O-trans-p-coumaroyl-α-L-rhamnopyranose.
Compound 3 showed an adduct molecular ion peak at m/z 489.1395 [M + H]+ (calc. for C24H25O11, 489.1391), which was 32 mass units higher than compounds 1 and 2 and also suggested 13 degrees of unsaturation. The 1H and 13C NMR data (Table 1, Fig. S14–S15†) suggested six characteristic resonances for a disubstituted α-L-rhamnopyranoside unit (Table 1, Fig. 2). NMR data also showed six aromatic methine groups at δH 7.08 (2H, d, J = 2.4) δC 114.0, δH 6.81 (2H, d, J = 8) δC 115.1, δH 6.91 (2H, dd, J = 2.4, 8) δC 121.6, 121.7, four trans-olefinic methine groups at δH 7.57 (2H, d, J = 16) δC 145.7, δH 6.31, 6.33 (2H, d, J = 16) δC 113.6, 113.8, and eight quaternary carbons at δC 126.3, 126.3, 148.3, 145.4, 146, 148.3, 176.6 and 167.7, which suggested two possible trans-caffeic acid moieties.16 The additional oxygen in each caffeic acid moiety justified the additional 32 mass units increase observed for compound 3 versus compounds 1 and 2. The 1H–1H COSY data of 3 (Fig. 2), was similar to that of 1 for the 1,2-disubstituted α-L-rhamnopyranoside unit. Moreover, the 3J-HMBC correlation of H-1 δH 5.18 (δC 91.1) with the quaternary carbonyl carbon C-9′ (δC 167.6) and the 3J-HMBC correlation of H-2 δH 5.18 (δC 73.7) with the carbonyl carbon C-9′′ (δC 167.7) confirmed the location of the two trans-caffeic moieties at carbons C-1 and C-2 of the α-L-rhamnopyranosyl moiety. Accordingly, compound 3 was identified as 1,2-di-O-trans-p-caffeoyl-α-L-rhamnopyranose.
The molecular formula of compound 4 was identical to that of compound 3 based on HRESIMS (C24H25O11). Similar to compound 2, the 1H and 13C NMR data of 4 showed two trans-caffeic acid moieties attached at C-1 and C-3 of the core α-L-rhamnopyranose (Table 1, Fig. 2, and S18–S19†). This was further confirmed through the HMBC experiment, which showed the 3J-HMBC correlation of the proton H-1 δH 5.13 (δC 94.4) with the quaternary carbonyl carbon C-9′ (δC 167.6) and the 3J-HMBC correlation of the proton H-3 δH 5.24 (δC 74.1) with the carbonyl carbon C-9′′ (δC 167.7) (Fig. 2). Accordingly, compound 4 was identified as 1,3-di-O-trans-p-caffeoyl-α-L-rhamnopyranose.
3.2. Biological activity
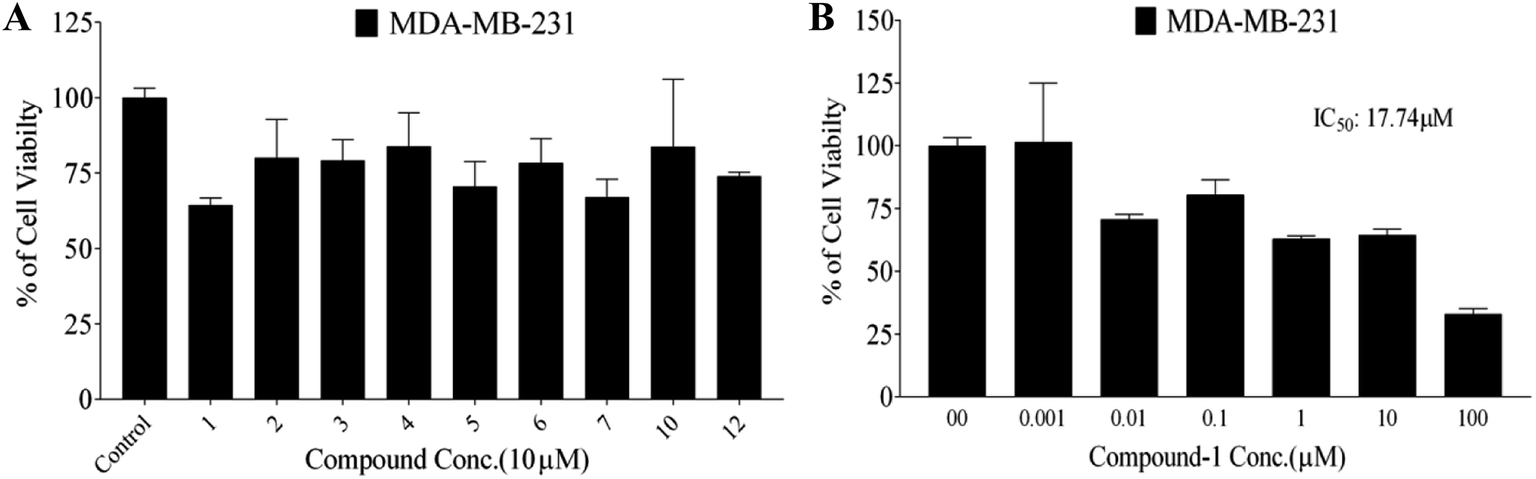 | ||
| Fig. 3 (a) Antiproliferative activity of compounds 1–7, 10, and 12 against the human TNBC MDA-MB-231 cells at 10 μM screening doses using the MTT assay. (b) Dose–response effects of various compound 1 treatments on the viability of the TNBC MDA-MB-231 cells compared with the DMSO vehicle control. The viable cell count was determined using the MTT assay. Error bars show the SEM of N = 3/dose, *p < 0.05 as compared with the vehicle-treated control groups. (−) Oleocanthal was used as a positive control at 10 μM. #p < 0.05 as compared with the (−)-oleocanthal treated control group. The IC50 values are included for activity comparison. | ||
The isolated acylated rhamnopyranoses 1–4, and verbascoside 5 were further screened for their antiproliferative activities against the breast cancer cell line MCF-7 and the normal cell line MCF-10A at 10 μM using the MTT assay. Results showed that compound 1 had the highest activity against MCF-7 with an IC50 value of 4.95 μM (similar to the standard saturosporine), with low activity on MCF-10A with an IC50 value of 17.35 μM (Table 3). The other compounds 2–4 showed moderate activities against MCF-7 with IC50 values in the range of 11.09–20.04 μM and moderate activities against MCF-10A.
| Compound | Cytotoxicity (IC50 μM) | |
|---|---|---|
| MCF-7 | MCF-10A | |
| a IC50 = the concentration (μM) that caused a 50% inhibition of cell growth in vitro. | ||
| 1 | 4.95 ± 0.24 | 17.35 ± 0.86 |
| 2 | 11.09 ± 0.70 | 20.01 ± 0.34 |
| 3 | 17.07 ± 0.85 | 25.76 ± 1.28 |
| 4 | 20.04 ± 0.90 | 27.70 ± 1.10 |
| 5 | 10.45 ± 0.52 | 13.91 ± 0.69 |
| Staurosporine | 3.44 ± 0.17 | 11.42 ± 0.57 |
Data were expressed as means ± standard error of the mean (SEM) of at least three independent experiments. The differences among various treatment groups were determined by ANOVA followed by Dunnett's test using PASW Statistics® version 18 (Quarry Bay, Hong Kong). A difference of p < 0.05 was considered statistically significant compared with a vehicle-treated control group. Staurosporine was used as a positive control.
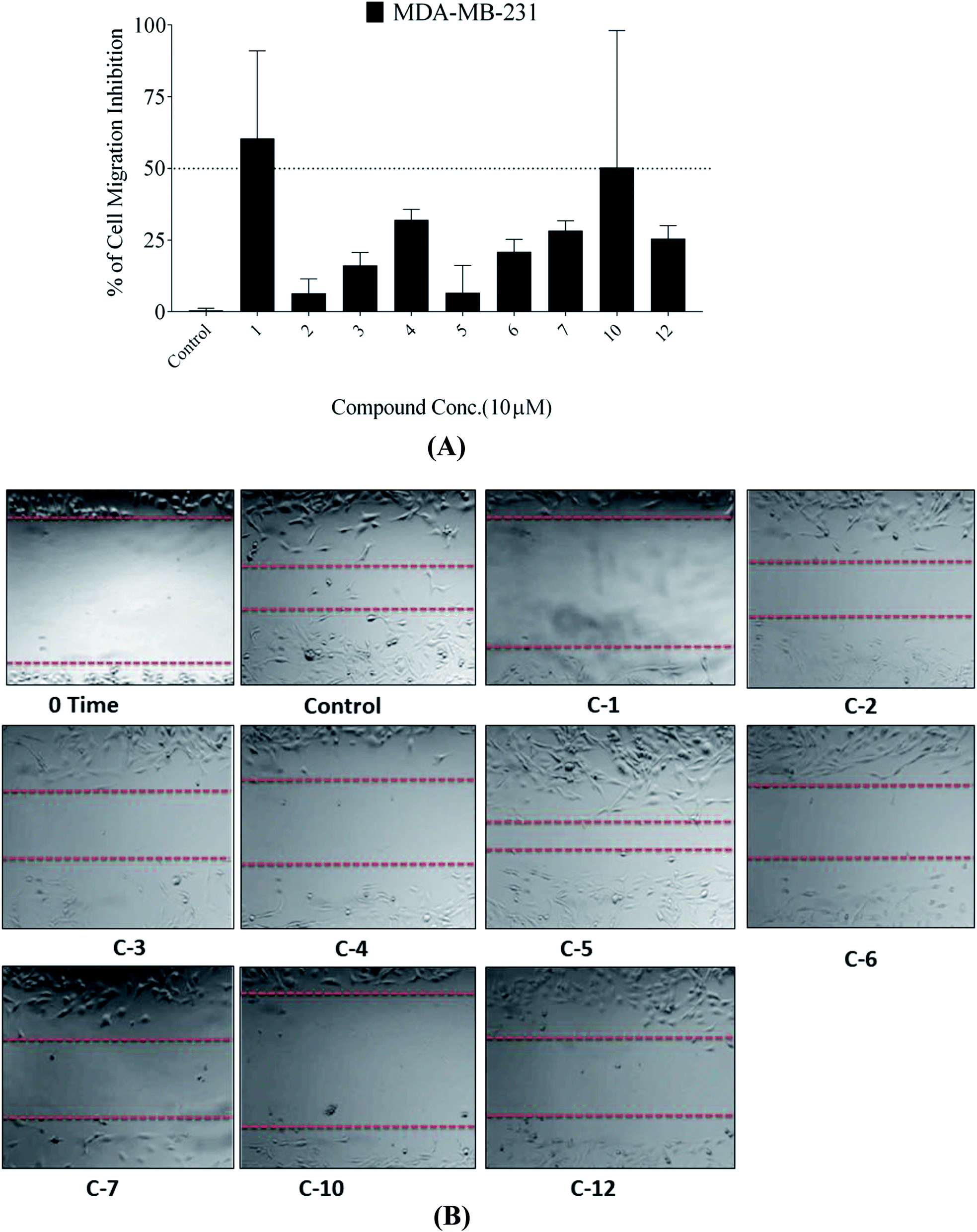 | ||
| Fig. 4 (a) Anti-migratory activity of compounds 1–7, 10 and 12 at 10 μM against the human TNBC MDA-MB-231 cells using the wound-healing scratch assay. (b) Photomicrographs showing the migration inhibitory activity of compounds 1–7, 10 and 12 against the TNBC MDA-MB-231 cells in the wound healing assay, compared with DMSO as a vehicle control. | ||
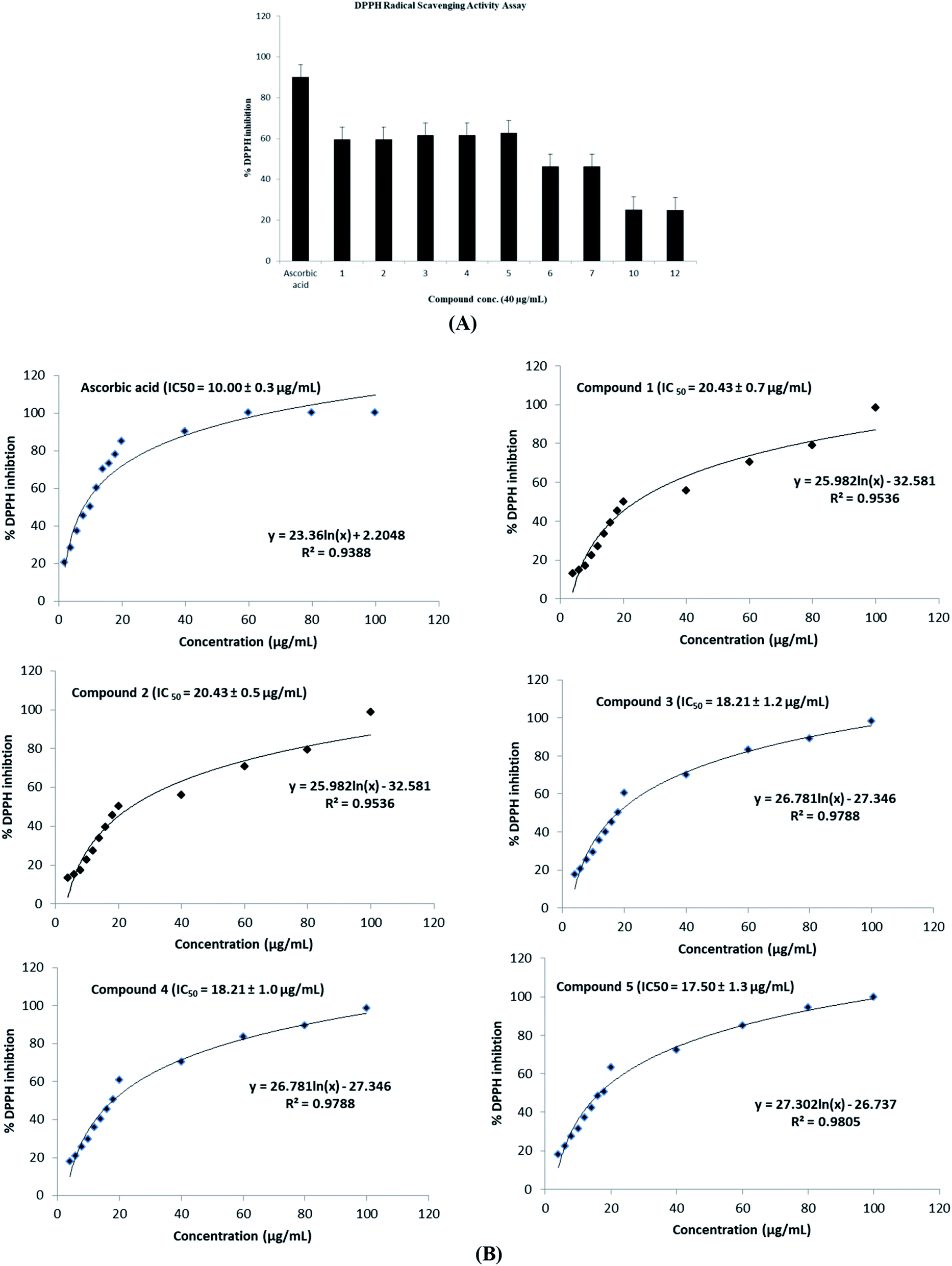 | ||
| Fig. 5 (a) Antioxidant activity of compounds 1–7, 10, and 12 against DPPH at 40 μg mL−1 screening doses using the DPPH radical scavenging activity assay. (b) Dose–response effects of various concentrations of compounds 1–5 treatments on the ability of the compounds to scavenge DPPH using the DPPH scavenging activities assay. (−) Ascorbic acid used as a positive control. The IC50 values were included for activity comparison. | ||
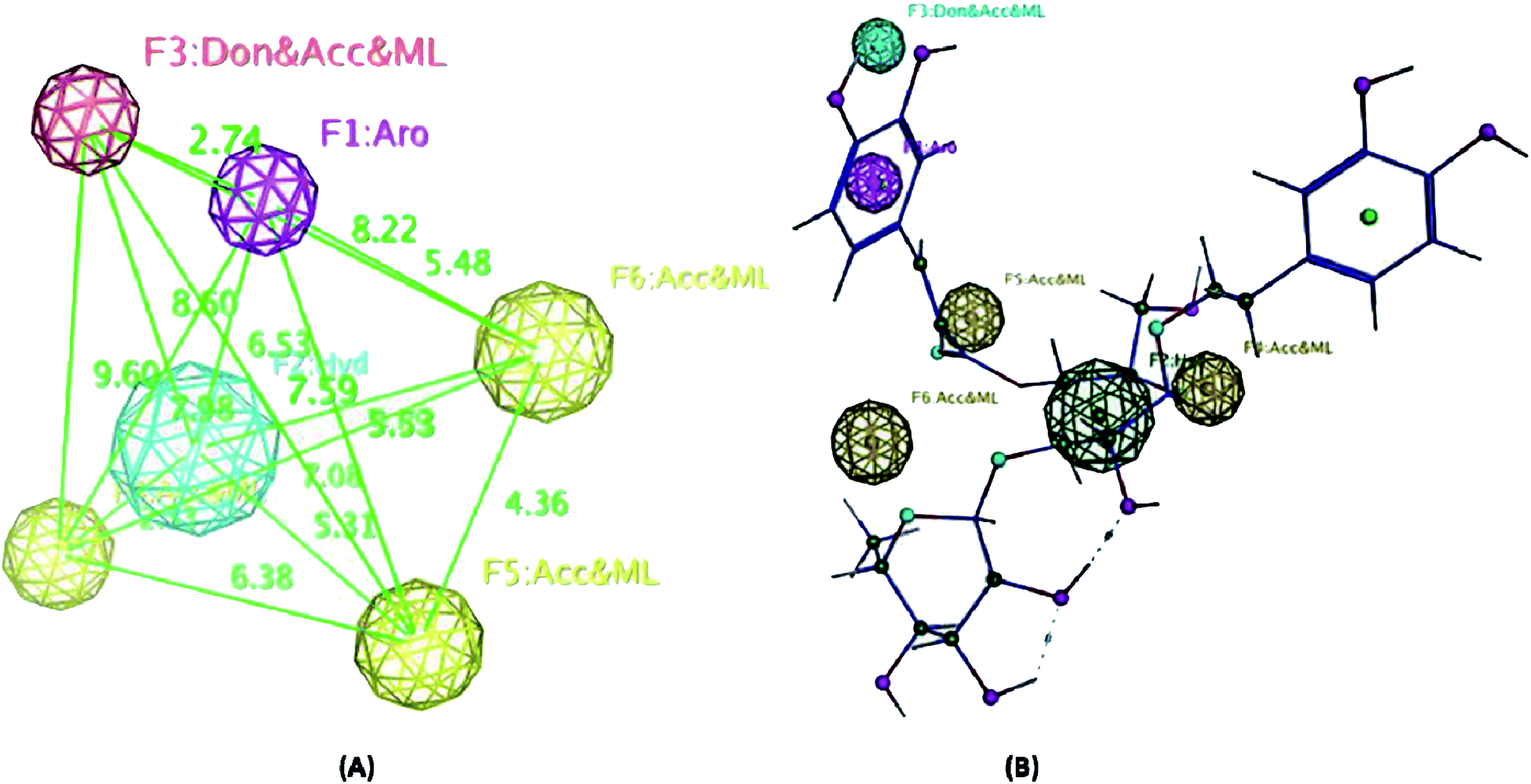 | ||
| Fig. 6 (A) The highest-ranked pharmacophore model generated for compounds 1–5 showing the distances among the different features. Hydrogen bond acceptors (HBAs) are represented as yellow-vectored spheres, the hydrogen bond donor (HBD) is represented as a red-vectored sphere, ring aromatic features are represented as a violet-vectored sphere, and the hydrophobic group as a blue sphere. (B) Structure of the most active compound 5 and how it aligns the pharmacophore map. Exclusion spheres were hidden for clarity. | ||
According to literature, and in general, plant-derived phenolic compounds have been shown to inhibit the initiation and progression of cancers by modulating the genes that regulate key processes, such as the oncogenic transformation of normal cells, growth and development of cancer, angiogenesis and metastasis.34 Recent studies focused on identifying the molecular basis for plant phenolic-induced cancer cell death have demonstrated the down regulation of oncogenic survival kinases such as phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt); cell proliferation regulators that include extracellular signal-regulated kinase Erk1/2, D-type cyclins, and cyclin-dependent kinases (CDKs); transcription factors such as nuclear factor kappa beta (NF-kβ), and nuclear factor erythroid 2 (NRF2); histone deacetylases HDAC1 and HDAC2; and angiogenic factors vascular endothelial growth factor (VEGF), and fibroblast growth factor receptor 1 (FGFR1). Furthermore, while inhibiting oncogenic proteins, the phenolic compounds elevate the expression of tumor suppressor proteins P53, P21, and P27.34 In addition, plant phenolic compounds modulate the levels of reactive oxygen species (ROS) in cells, thereby regulating cell proliferation, survival and apoptosis.34
Even though the phenolic compounds have been shown to exhibit anticancer activity, the efficacy varies from one compound to another due to the variations in their structures as well as their molecular targets. For example, the structure–activity-relationship (SAR) studies delineating the key functional groups required for exhibiting potent anticancer effects of phenolic compounds have identified the involvement of an aromatic ring and hydroxyl groups,35 where compounds with a greater number of hydroxyl groups exhibited better activity compared to those with no hydroxyl groups or compounds with methoxy moieties. In addition, the presence of an unsaturated short fatty acid side chain also makes the phenolic compounds more potent.34 Esters of aromatic acids are more potent when compared to the free versions.34–36
In this study, the antiproliferative and antimigratory activities for free hydroxyl phenolic and/or carboxylic acid groups containing compounds (acylated rhamnopyranoses 1–4, verbascoside 5, premnoside E–F 6–7, oleanolic acid 10, and maslinic acid 12) were tested against the invasive human triple-negative breast cancer cells MDA-MB-231, MCF-7, and normal MCF-10A. Results showed that 1,2-di-O-trans-p-coumaroyl-α-L-rhamnopyranose 1 was able to inhibit the HGF-mediated cell proliferation in a dose-dependent manner, with an IC50 value of 17.74 μM, MCF-7 with an IC50 value of 4.95 μM, with low activity against MCF-10A with an IC50 value of 17.35 μM using the MTT assay. On the other hand, compound 1 showed an antimigratory effect against the same cell line (Table 3, Fig. 3 and 4).
In response to the above biochemical parameters, the acylated rhamnopyranoses 1–4 contained two aromatic acid moieties attached to the α-L-rhamnopyranoside in the ester form containing free phenolic hydroxyl groups and two unsaturated short fatty acid side chains. These features increased the antiproliferative and antimigratory activities of the acylated rhamnopyranoses compared to the other tested compounds (verbascoside 5, premnoside E–F 6–7, oleanolic acid 10, and maslinic acid 12) (Fig. 1).
Moreover, this study showed that the acylated rhamnopyranoside 1,2-di-O-trans-p-coumaroyl-α-L-rhamnopyranose 1 was the most active as an antiproliferative, antimigratory, and cytotoxic agent against MDA-MB-231 and MCF-7 compared to the other acylated rhamnopyranosides. Comparing this result with the previously reported acylated rhamnopyranose (1-O-trans-p-coumaroyl-2-O-trans-caffeoyl-α-L-rhamnopyranose) isolated from P. odorata leaves (IC50 12.9 μM)11 and the isolated tested hits 2, 3, 4 in the present work, a preliminary structure–activity relationship conclusion can be drawn by assuming the preference of the 1,2-di-aromatic acid substitution over the 1,3-disubstitution. Also, the p-coumaroyl with one C-4 OH group conferred better activity than that with the caffeoyl moiety (with the C-3 and C-4 dihydroxy/catechol system), showing that the lower number of –OH groups in the attached aromatic acids seemed better for the activity.
On the other hand, the radical scavenging activity of the free hydroxyl phenolic and/or carboxylic acid groups containing compounds (acylated rhamnopyranoses 1–4, verbascoside 5, premnoside E–F 6–7, oleanolic acid 10, and maslinic acid 12) were tested using the stable radical DPPH assay. Results showed that the acylated rhamnopyranoses 1–4 and verbascoside 5 had the highest value of DPPH radical scavenging activities with an IC50 value range of 17.5–20.0 ± 0.5 μg mL−1, where verbascoside 5 was the most active compound. The pharmacophore model of the most active antioxidant compounds 1–5 was generated using MOE software, and showed three HBAs, one HBD, one aromatic ring, and one Hyd. group. The central HBA feature lies at a distance of 4.36 Å and 6.38 Å from the remaining two HBA features. Also, the HBD feature maintains a distance of 2.74 Å from the aromatic feature. The significant activity of compounds 1–5 can be explained by the importance of the HBA groups that function by the mechanism of single electron transfer followed by de-protonation. The mapping of verbascoside 5, which has the most potent antioxidant activity with the developed pharmacophore, indicated that the hydroxyl group present in the aromatic ring and the phenyl ring constituting the HBDs and the aromatic features, respectively, are critical for the antioxidant activity profile of the known natural antioxidant derivatives,23 as the interaction of the electron-rich free radicals is captured by the hydrophobic nature of the phenyl ring. Additionally, the carbonyl linked to the caffeoyl nucleus constitutes an essential fragment, facilitating another HBA feature of the developed 3D pharmacophore model (Fig. 5–6).
As a result of the antiproliferative, antimigratory, cytotoxic, and antioxidants activities for the acylated 1,2-disubstituted α-L-rhamnopyranoses, it could be considered another novel hit appropriate for further optimization for users to control breast cancer.
4. Conclusion
A phytochemical investigation of P. odorata young stems offered four new acylated rhamnopyranoses 1–4, along with fourteen known compounds. This study confirmed the antiproliferative, antimigratory and cytotoxic potential of the acylated rhamnopyranoses against the human TNBC MDA-MB-231, MCF-7, and MCF-10A. 1,2-di-O-trans-p-coumaroyl-α-L-rhamnopyranose 1 was able to inhibit the HGF-mediated cell proliferation in a dose-dependent manner, with an IC50 value of 17.74 μM compared with (−)-oleocanthal as the positive control (IC50 15 μM). On the other hand, compound 1 showed an antimigratory effect against the same cell line at 10 μM concentrations. Furthermore, compound 1 showed cytotoxic activity against MCF-7 with an IC50 value of 4.95 μM, and showed low cytotoxic activity on the MCF-10A cell line with an IC50 value of 17.35 μM using the MTT assay. Additionally, acylated rhamnopyranoses 1–4 and verbascoside 5 showed the highest potential of DPPH radical scavenging activities with an IC50 value range of 17.5–20.0 ± 0.5 μg mL−1. The pharmacophore model generated using Molecular Operating Environment (MOE) for compounds 1–5 showed three hydrogen bond acceptors (HBAs), one hydrogen bond donor (HBDs), one aromatic ring (Aro), and one hydrophobic (Hyd.) group. The central HBA feature lies at a distance of 4.36 Å and 6.38 Å from the remaining two HBA features. Also, the HBD feature maintains a distance of 2.74 Å from the aromatic feature. Acylated rhamnopyranose 1 can be considered a good scaffold for developing new anti-breast cancer compounds. In the light of the previous findings, it is worth mentioning that P. odorata produced a minor amount of the acylated rhamnopyranose. On the other hand, the organic solvent consumption does not favor the commercialized production of the rhamnopyranoses. Consequently, further synthesis and/or compound optimization for the active hits can be considered a great way to commercialize this class of compounds.Conflicts of interest
The authors declare no conflict of interest.References
- J. Ferlay, M. Colombet, I. Soerjomataram, C. Mathers, D. Parkin, M. Piñeros, A. Znaor and F. Bray, Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods, Int. J. Cancer, 2019, 144, 1941–1953, DOI:10.1002/ijc.31937
.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2018, 68, 394–424, DOI:10.3322/caac.21492
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2019, Ca-Cancer J. Clin., 2019, 69, 7–34, DOI:10.3322/caac.21551
- E. Giovannucci, D. M. Harlan, M. C. Archer, R. M. Bergenstal, S. M. Gapstur, L. A. Habel, M. Pollak, J. G. Regensteiner and D. Yee, Diabetes and cancer: a consensus report, Ca-Cancer J. Clin., 2010, 60, 207–221, DOI:10.3322/caac.20078
- E. Birben, U. M. Sahiner, C. Sackesen, S. Erzurum and O. Kalayci, Oxidative stress and antioxidant defense, World Allergy Organ. J., 2012, 5, 9–19 CrossRef CAS PubMed
- N. Noda and H. Wakasugi, Cancer and oxidative stress, Japan Med. Assoc. J., 2001, 44, 535–539 Search PubMed
- M. Leporini, G. Catinella, M. Bruno, T. Falco, R. Tundis and M. R. Loizzo, Investigating the Antiproliferative and Antioxidant Properties of Pancratium maritimum L. (Amaryllidaceae) Stems, Flowers, Bulbs, and Fruits Extracts, J. Evidence-Based Complementary Altern. Med., 2018, 2018, 1–8 CrossRef PubMed
- N. Sharma, A. Sharma, G. Bhatia, M. Landi, M. Brestic, B. Singh, J. Singh, S. Kaur and R. Bhardwaj, Isolation of phytochemicals from Bauhinia variegata l. Bark and their in vitro antioxidant and cytotoxic potential, Antioxidants, 2019, 8, 492 CrossRef CAS PubMed
- A. Puttisri, A. Pamarapa, M. Jayanton Patumanond, M. Apichat Tantraworasin and C. Charoentum, Recurrence and death from breast cancer after complete treatments: An experience from hospitals in Northern Thailand, J. Med. Assoc. Thailand, 2014, 97, 932–938 Search PubMed
- G. Suresh, K. S. Babu, V. R. S. Rao, M. S. A. Rao, V. L. Nayak and S. Ramakrishna, Novel cytotoxic icetexane diterpenes from Premna latifolia Roxb, Tetrahedron Lett., 2011, 52, 1273–1276 CrossRef CAS
- A. H. Elmaidomy, M. M. Mohyeldin, M. M. Ibrahim, H. M. Hassan, E. Amin, M. E. Rateb, M. H. Hetta and K. A. El Sayed, Acylated Iridoids and Rhamnopyranoses from Premna odorata (Lamiaceae) as Novel Mesenchymal–Epithelial Transition Factor Receptor Inhibitors for the Control of Breast Cancer, Phytother. Res., 2017, 31, 1546–1556, DOI:10.1002/ptr.5882
- P. S. Ghosh, N. Das and B. Dinda, Antioxidant flavone glycosides and other constituents from Premna latifolia leaves, Indian J. Chem., 2014, 53B, 746–749 CAS
- D. Yadav, N. Masood, S. Luqman, P. Brindha and M. M. Gupta, Antioxidant furofuran lignans from Premna integrifolia, Ind. Crops Prod., 2013, 41, 397–402 CrossRef CAS
- S. Habtemariam and G. K. Varghese, A Novel Diterpene Skeleton: Identification of a highly aromatic, cytotoxic and antioxidant 5-methyl-10-demethyl-abietane-type diterpene from Premna serratifolia, Phytother. Res., 2015, 29, 80–85, DOI:10.1002/ptr.5229
- A. Hymavathi, K. S. Babu, V. Naidu, S. R. Krishna, P. V. Diwan and J. M. Rao, Bioactivity-guided isolation of cytotoxic constituents from stem–bark of Premna tomentosa, Bioorg. Med. Chem. Lett., 2009, 19, 5727–5731 CrossRef CAS PubMed
- G. Suresh, K. S. Babu, M. S. A. Rao, V. R. S. Rao, P. A. Yadav, V. L. Nayak and S. Ramakrishna, Premnalatifolin A, a novel dimeric diterpene from Premna latifolia Roxb, Tetrahedron Lett., 2011, 52, 5016–5019 CrossRef CAS
- E. Stahl, Thin Layer Chromatography, Springer Verlage, Berlin, Heidelberg, New York, 2nd, 1970, p. 810, DOI:10.1002/star.19700221110
- D. L. Holliday and V. Speirs, Choosing the right cell line for breast cancer research, Breast Cancer Res., 2011, 13, 215 CrossRef PubMed
- M. M. Mohyeldin, M. R. Akl, H. Y. Ebrahim, A. M. Dragoi, S. Dykes, J. A. Cardelli and K. A. El Sayed, The oleocanthal-based homovanillyl sinapate as a novel c-Met inhibitor, Oncotarget, 2016, 7, 32247 CrossRef PubMed
- A. H. Elmaidomy, R. Mohammed, H. M. Hassan, A. I. Owis, M. E. Rateb, M. A. Khanfar, M. Krischke, M. J. Mueller and U. R. Abdelmohsen, Metabolomic Profiling and Cytotoxic Tetrahydrofurofuran Lignans Investigations from Premna odorata Blanco, Metabolites, 2019, 9, 223 CrossRef CAS PubMed
- M. R. Loizzo, M. B. Jemia, F. Senatore, M. Bruno, F. Menichini and R. Tundis, Chemistry and functional properties in prevention of neurodegenerative disorders of five Cistus species essential oils, Food Chem. Toxicol., 2013, 59, 586–594 CrossRef CAS PubMed
- M. A. Khanfar, M. M. AbuKhader, S. Alqtaishat and M. O. Taha, Pharmacophore modeling, homology modeling, and in silico screening reveal mammalian target of rapamycin inhibitory activities for sotalol, glyburide, metipranolol, sulfamethizole, glipizide, and pioglitazone, J. Mol. Graphics Modell., 2013, 42, 39–49 CrossRef CAS PubMed
- I. Mitra, A. Saha and K. Roy, Predictive modeling of antioxidant coumarin derivatives using multiple approaches: descriptor-based QSAR, 3D-pharmacophore mapping, and HQSAR, Sci. Pharm., 2013, 81, 57–80 CrossRef CAS PubMed
- T. Kanchanapoom, R. Kasai and K. Yamasaki, Phenolic glycosides from Barnettia kerrii, Phytoch, 2002, 59, 565–570 CrossRef CAS
- M. A. Hossain and Z. Ismail, Isolation and characterization of triterpenes from the leaves of Orthosiphon stamineus, Arabian J. Chem., 2013, 6, 295–298 CrossRef CAS
- T. B. Ayeleso, M. G. Matumba and E. Mukwevho, Oleanolic acid and its derivatives: biological activities and therapeutic potential in chronic diseases, Molecules, 2017, 22, 1915 CrossRef PubMed
- L. H. Vázquez, J. Palazon and A. Navarro-Ocaña, The Pentacyclic Triterpenes α-, β-amyrins: A Review of Sources and Biological Activities, Phytochem, 2012, 5, 1–5 CrossRef
- L. L. Pierre and M. N. Moses, Isolation and characterisation of stigmasterol and β-sitosterol from Odontonema strictum (acanthaceae), J. Innovations Pharm. Biol. Sci., 2015, 2, 88–96 CAS
- S. Moradkhani, F. Kobarfard and S. A. M. Ayatollahi, Phytochemical investigations on chemical constituents of Achillea tenuifolia Lam, Iran J. Pharm. Res., 2014, 13, 1049 CAS
- I. S. Lee, S. R. Oh, K. Y. Jung, D. S. Kim, J. H. Kim and H. K. Lee, Anticomplementary activity and complete 13C NMR assignment of citrostadienol from Schizandra chinensis, Intern. J. Pharmacogn., 1997, 35, 358–363, DOI:10.1080/09251619708951283
- J. Bulama, S. Dangoggo, M. Halilu, A. Tsaf and S. Hassan, Isolation and characterization of palmitic acid from ethyl acetate extract of root bark of Terminalia glaucescens, J. Chem. Mater. Res., 2014, 6, 140–143 Search PubMed
- M. Wen, Chemical analysis and biosynthesis of secondary alcohols in plant cuticular waxes, University of British Columbia, 2009 Search PubMed
- M. R. Akl, N. M. Ayoub, M. M. Mohyeldin, B. A. Busnena, A. I. Foudah, Y.-Y. Liu and K. A. E. Sayed, Olive phenolics as c-Met inhibitors:(-)-Oleocanthal attenuates cell proliferation, invasiveness, and tumor growth in breast cancer models, PLoS One, 2014, 9, e97622 CrossRef PubMed
- P. G. Anantharaju, P. C. Gowda, M. G. Vimalambike and S. V. Madhunapantula, An overview on the role of dietary phenolics for the treatment of cancers, Nutr. J., 2016, 15, 99 CrossRef PubMed
- Y.-J. Lee, P.-H. Liao, W.-K. Chen and C.-C. Yang, Preferential cytotoxicity of caffeic acid phenethyl ester analogues on oral cancer cells, Cancer Lett., 2000, 153, 51–56 CrossRef CAS PubMed
- M. Chen, H. Meng, Y. Zhao, F. Chen and S. Yu, Antioxidant and in vitro anticancer activities of phenolics isolated from sugar beet molasses, BMC Complementary Altern. Med., 2015, 15, 313 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01697g |
| This journal is © The Royal Society of Chemistry 2020 |