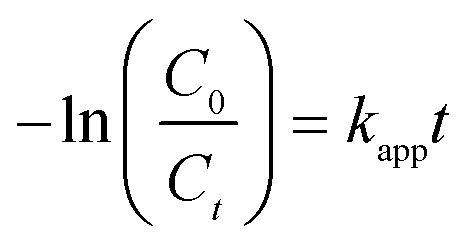DOI:
10.1039/D0RA01668C
(Paper)
RSC Adv., 2020,
10, 19961-19973
Novel recyclable BiOBr/Fe3O4/RGO composites with remarkable visible-light photocatalytic activity†
Received
21st February 2020
, Accepted 16th April 2020
First published on 27th May 2020
Abstract
Magnetic BiOBr/Fe3O4/RGO composites with remarkable photocatalytic capability were prepared by a simple hydrothermal method to load 3D flower-like microspherical BiOBr onto the surface of Fe3O4/RGO. Under visible-light irradiation (λ > 420 nm), the BiOBr/Fe3O4/RGO composite with 56% mass percentage of Fe3O4/RGO shows the optimal removal ability for Rhodamine B, and the total removal efficiency is 96%. The coupling of Fe3O4/RGO and BiOBr elevates the conduction band of BiOBr, which enhances the reduction level of BiOBr/Fe3O4/RGO composites. Ultimately, based on experiments and theoretical calculations, an n-type Schottky contact formed at the heterojunction interface between RGO doped with Fe3O4 and BiOBr is proposed for photoexcited charge transfer. The RGO with great adsorptivity plays a major role in the photocatalysts composed of BiOBr, RGO and Fe3O4. Further, BiOBr/Fe3O4/RGO composites with permanent-magnetism can be recovered and reused easily by external magnetic field and maintain a total removal efficiency of 90% after four cycles.
Introduction
Semiconductor-based photocatalysis is identified as an appealing advanced method for environmental contaminant treatment.1 Most of the photocatalytic reactions can be excited by only about 3–5% ultraviolet (UV) light in the solar spectrum or expensive artificial UV sources. Thus, the demand for designing visible-light-driven photocatalysts with superior performance is very urgent. Bismuth oxyhalides (BiOX, X = I, Br, Cl)2 as promising ternary photocatalysts with special electronic structure and excellent photocatalytic performance have been widely used in several fields.3 Among them, BiOBr has a quite suitable band gap with a little visible-light absorption. The hybridization between O 2p and Bi 6s states induces the separation of photoexcited e−–h+ pairs.4 However, practical applications of BiOBr are still hampered by its insufficient light absorption and high recombination probability of photoexcited electron–hole pairs. Many approaches have been developed so as to overcome the drawback, such as the control of dominant exposed facets,5 doping metals and compound semiconductors,6,7 and construction of BiOBr-based heterojunction.8 Among those approaches, the heterojunction recombination of BiOBr with carbonaceous materials can effectively improve the separation of photoexcited e−–h+ pairs, thereby getting more absorption of light, such as BiOBr–C3N4,9 carbon nanofiber/BiOBr,10 g-C3N4/BiOBr11 and carbon nitride/BiOBr.12
The difficulty in separating and recovering the above photocatalysts from the treated liquid phase is another disadvantage, especially for powder catalysts. The introduction of magnetic materials, such as Fe3O4,13 MnO2,14 NiO,15 Fe2O3,16 CoFe2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 in composite systems, is a feasible way to overcome the limitations. Among them, Fe3O4 is one of the most widely used magnetic materials with low-cost, biocompatibility, outstanding photochemical stability and sufficient magnetism.18 Especially, it is interesting that the cubic phase Fe3O4 nanoparticle possesses 1.40–1.85 eV band gap.19,20 These features enable Fe3O4 to couple with other semiconductors for providing magnetism thereby promoting recovery in a magnetic field environment, and making the photocatalyst highly resistant to photocorrosion.21
17 in composite systems, is a feasible way to overcome the limitations. Among them, Fe3O4 is one of the most widely used magnetic materials with low-cost, biocompatibility, outstanding photochemical stability and sufficient magnetism.18 Especially, it is interesting that the cubic phase Fe3O4 nanoparticle possesses 1.40–1.85 eV band gap.19,20 These features enable Fe3O4 to couple with other semiconductors for providing magnetism thereby promoting recovery in a magnetic field environment, and making the photocatalyst highly resistant to photocorrosion.21
Graphene-based materials have emerged as promising candidates due to their large specific surface area, outstanding charge carrier mobility and high thermal/chemical stability. The presence of graphene can increase electric charges transfer rate, which prevents the recombination of the photogenerated electron–hole pairs and provides some active adsorption sites from oxygen-containing functional groups on the surface of the sheets. Therefore, graphene can promote the synergistic effect of adsorption-photocatalysis to accelerate the degradation of pollutants.22,23 Recently, many research efforts have been made to combine BiOBr with RGO to form BiOBr/RGO hybrid cocatalysts.24,25 For instance, Yu et al. successfully prepared a three-dimensional BiOBr/RGO heterostructure composites by a two steps hydrothermal method with GO as precursors, which exhibited an improved photocatalytic activity for degrading methyl orange.26 Tu et al. and Liu et al. have synthesized BiOBr/RGO photocatalyst for degradation of RhB under visible light, which illustrates that RGO sheets as an electronic transmitter in the photocatalytic reaction process expected to promote the synergistic effect of adsorption-photocatalysis to accelerate the degradation of pollutants.27,28 Unfortunately, these BiOBr/RGO composites are difficult to recycle after photodegradation reactions, increasing the cost of degradation. Nowadays, some studies have testified that the magnetic heterojunction system showed a favorable candidate to overcome the above problems.29,30
Recently, for the BiOBr/Fe3O4/RGO composite, Janani et al. ultrasonic dispersion method had been taken to obtain a photocatalyst, which is easy to magnetically separate and has a relatively low band gap energy for the need of visible-light absorption.31 Xu et al. prepared a Fe3O4/BiOBr/RGO composite photocatalyst by hydrothermal treatment, which has significantly improved photocatalytic activity for degrading RhB.32 Nevertheless, the effect of RGO or Fe3O4 on the interfacial properties of BiOBr has not been discussed in depth, so the photocatalytic mechanism of BiOBr/Fe3O4/RGO composites needs further investigation. In this work, we proposed we conducted a comprehensive and systematic study of the microscopic mechanisms of interface interactions and charge transfer in this system. The photocatalytic activity of BiOBr/Fe3O4/RGO on dyes under visible-light irradiation and the photocatalytic stability of magnetic recovery were systematically evaluated and discussed. In addition, the interface interaction and electron transfer mechanism of the composites were also verified by first-principles calculations.
Materials and methods
Chemicals and reagents
Bismuth nitrate (Bi(NO3)3·5H2O), potassium bromide (KBr), ethylene glycol (EG), natural flake graphite, sodium nitrate (NaNO3), iron(III) chloride hexahydrate (FeCl3·6H2O), ethylenediamine (EDA), sodium acetate trihydrate (CH3COONa·3H2O), hydrogen peroxide (H2O2), absolute ethyl ethanol, 2,2,6,6-tetramethylpiperidin-1-yloxy (TEMPO), 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), rhodamine B (RhB), methyl blue (MB), methyl orange (MO), acid red (AR), amino black (AB) were purchased from Sinopharm Chemical Reagent Co. Ltd., China. Other reagents used in this study, including concentrated sulfuric acid (H2SO4), potassium permanganate (KMnO4) and concentrate hydrochloric acid (HCl) were obtained from Xinyang Chemical Reagent Co. Ltd., China. All reagents were of analytical grade and used without further purification.
Preparation of GO and Fe3O4/RGO
Graphene oxide was prepared by a modified Hummers' method, and the detailed synthesis route was reported in our previous work.33 Fe3O4/RGO was prepared by a one-step hydrothermal process. Briefly, GO suspension (0.1 g, 1 wt%) was added to 40 mL of EG under magnetic stirring, recorded as liquid A. FeCl3·6H2O (0.3 g) was slowly added to 10 mL of EG solution under stirring, recorded as liquid B. Thereafter, the obtained liquid B, CH3COONa·3H2O (3 g) and EDA (10 mL) were slowly added to the liquid A and continuously stirred to form a homogeneous mixture. This homogeneous mixture was then hydrothermally heated in a Teflon stainless steel reactor at 200 °C for 8 h. Finally, the obtained precipitate was washed with ethanol, and dried under vacuum at 60 °C to obtain the Fe3O4/RGO powder. For comparison, the preparation of Fe3O4 is unchanged except that GO were involved.
Preparation of BiOBr/Fe3O4/RGO
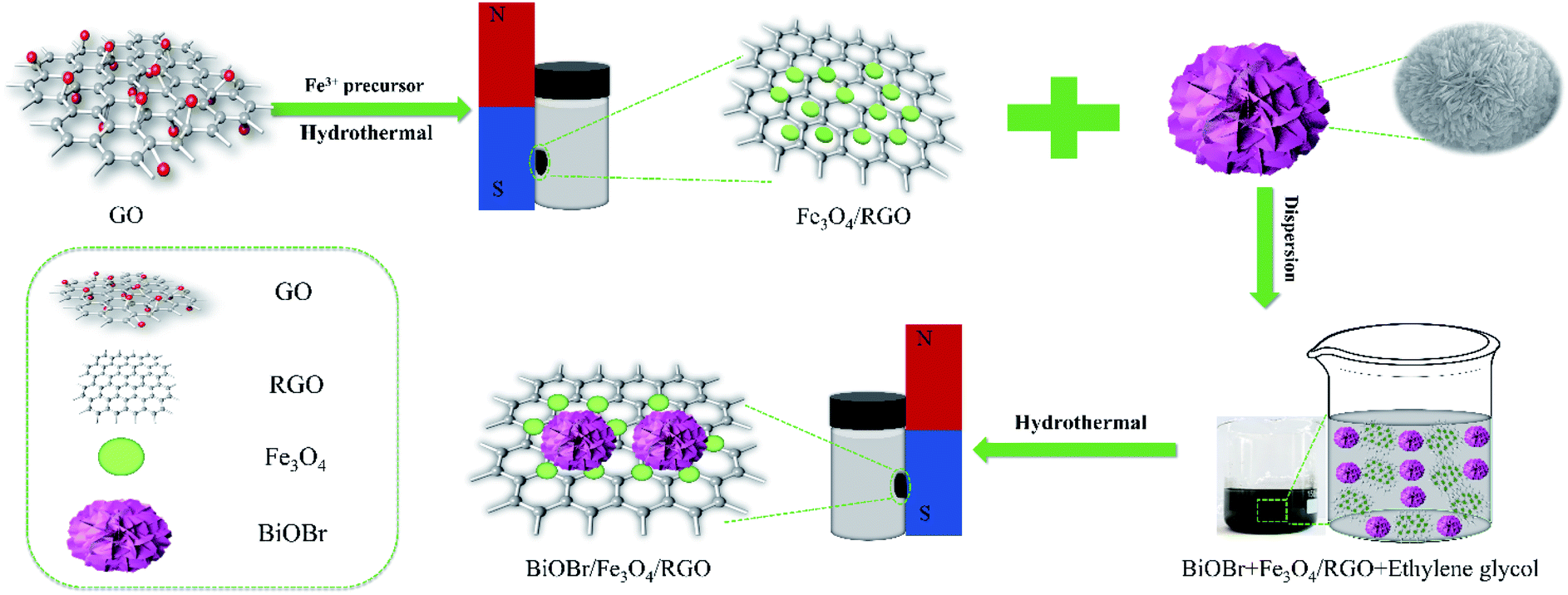
For the preparation of BiOBr, 0.7275 g Bi(NO3)3·5H2O and 0.1785 g KBr were added slowly into 16 mL EG solution and continuously stirred to form a homogeneous mixture. This homogeneous mixed solution was transferred into a 50 mL Teflon stainless steel reactor, and hydrothermal heated at 160 °C for 12 h to obtain BiOBr. The final product was washed with ethanol and dried in air at 60 °C to obtain a white BiOBr powder for further use. The BiOBr/Fe3O4/RGO composites were prepared by a simple solvothermal method. In brief, 10 mg of BiOBr was added to 75 mL of EG, respectively, and the solution was stirred to completely disperse for 1 h. 25, 12.5, 8.3 and 6.25 mg of Fe3O4/RGO (i.e., the mass fraction of Fe3O4/RGO in the precursor is 71%, 56%, 45%, and 38%) were slowly added to the above mixed solution and stirred for 4 h. The hydrothermal reaction was carried out at 160 °C for 12 h using a 100 mL Teflon stainless steel reactor lined with polytetrafluoroethylene (PTFE). Finally, the final product was washed with water and ethanol several times, and dried under vacuum for 12 h to obtain a powdery sample and labeled as BFG (Scheme 1). The BiOBr/Fe3O4/RGO samples with different mass percentage x% of Fe3O4/RGO were recorded as BFG-x%.
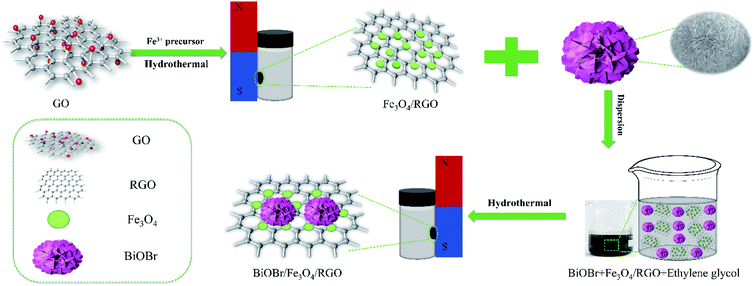 |
| | Scheme 1 The synthesis route of hydrothermal method for preparing BiOBr/Fe3O4/RGO composites. | |
Characterization
The X-ray diffraction (XRD) patterns were obtained on a D8-advance X-ray diffractometer (Bruker, Germany) equipped a Cu Kα radiation source (λ = 1.5418 nm). The microstructure and morphology of samples were investigated using a JSM-6390LV scanning electron microscope (SEM, JEOL, Japan) and a JEM-2100 transmission electron microscope (TEM, JEOL, Japan). The Raman spectrum was analyzed by a Lab RAM HR Evolution (Horiba Scientific, France) with an excitation wavelength of 633 nm. The surface functional groups of samples were determined by Fourier transform infrared (FT-IR) spectroscopy using a Nicolet Nexus 470 IR spectrometer ranging (Thermo Fisher Scientific, UK) from 4000 cm−1 to 400 cm−1. The analysis of X-ray photoelectron spectra (XPS) was performed at ambient temperature using an ESCALab 250Xi photoelectron spectrometer (Thermo Fisher Scientific, USA). UV-Vis spectra were recorded on a UV-2600 spectrometer using BaSO4 as a reference in the range of 200–800 nm (Shimadzu, Japan). Electrochemical impedance spectra (EIS) and transient photocurrent responses were characterized by an SP150 electrochemical system (Bio-logic, France). The magnetization measurements were carried out using a PPMS DynaCool vibrating sample magnetometer (VSM) (Quantum Design, USA). The activated species reacted in dyes were examined using visible light (λ > 420 nm) using an A300 type electron spin resonance (ESR) spectrometer (Bruker, Germany), in which DMPO and TEMPO were used as radical scavengers. The TOC-LCPH type analyzer (Shimadzu, Japan) was used to determine the total organic carbon (TOC) of the degraded solution.
Photocatalytic experiment
Photodegradation of the dye aqueous solution was carried out using a 300 W Xenon lamp (PLS-SEX300D, Beijing Perfect Light Technology Co. Ltd., China) with a 420 nm cut filter as a light source. In degradation experiment, 10 mg BiOBr, 35 mg BFG-71%, 22.5 mg BFG-56%, 18.3 mg BFG-45%, and 16.25 mg BFG-38% were added to 50 mL of 20 mg L−1 RhB, placed in a 100 mL glass jacketed reactor with the bilayer cooled by circulating water to keep the temperature constant for ensure the same experimental conditions, where the amount of photocatalysts is designed to ensure that each composite includes 10 mg of BiOBr. The suspension was vigorously stirred in the dark for 30 min before irradiation to reach an adsorption–desorption equilibrium, and then irradiated for 60 min under visible light, which the light source was placed 15 cm above the solution. At the certain time interval, 5 mL of the suspension was taken out from the reactor and centrifuged, and the degradation was monitored with a SP-2500 series UV-Vis spectrophotometer (Shanghai Spectrum Instruments Co. Ltd., China). The absorbance was then measured as a concentration, and a dye removal efficiency were calculated using Ct/C0, where Ct (mg L−1) and C0 (mg L−1) were the concentrations of dyes at time t (min) and at original, respectively. In a cycle experiment, the catalyst recovered from the solution by an external magnet, washed with deionized water and ethanol, and then dispersed in another solution of the dye solution (50 mL, 20 mg L−1).
Photoelectrochemical test
For electrochemical testing, measurements were made using a traditional three-electrode system. A fluorine-doped tin oxide conductive glass (FTO) coated with a sample, a standard calomel electrode and a Pt plate were selected from the working electrode, the reference electrode and the counter electrode, respectively. In this experiment, 10 mg sample was added to a mixture of 200 μL of 5% Nafion solution and 600 μL of absolute ethanol, and the mixture was uniformly coated on a 1.0 cm × 1.0 cm FTO glasses to form a uniform film. Further, a 0.1 mol L−1 potassium ferricyanide solution and a 300 W Xe lamp with a 420 nm cutoff filter was used as the light source.
Theoretical calculations
Complete the theoretical work was done using projector augmented wave (PAW) pseudopotentials, in which exchange and correlation in Perdew–Burke–Ernzerhof (PBE) formalism of density functional theory (DFT) as implemented in CASTEP.34 The valence atomic configurations are 6s26p3 for Bi, 4s24p5 for Br, 2s22p4 for O, and 2s22s2 for C, respectively. The energy cutoff for planewave function set is 400 eV, and a Monkhorst–Pack grid of 2 × 10 × 1 are used. In geometric optimization, the self-consistent convergence criterion and the maximal force between atoms are 1 × 10−5 eV per atom and 0.05 eV Å−1, respectively. The maximum displacement is 5 × 10−4 Å, and the stress is less than 0.02 GPa. Here, the hybrid semi-empirical solutions of Tkatchenko and Scheffler (TS) schemes are given to represent vdW interactions, and damped atom paired dispersion correction in the form of C6R−6 in the DFT formalism.35 The dispersion-corrected total energy Etot is expressed as| | |
Etotal = EKS-DFT + EvdW
| (1) |
where EKS-DFT is the conventional Kohn–Sham DFT energy and EvdW is the dispersion correction. The semi-empirical method provides the best compromise between the first-principles assessment of the dispersion terms and the need to improve the non-bond interactions in the standard DFT description.
Results and discussion
Structural characterization
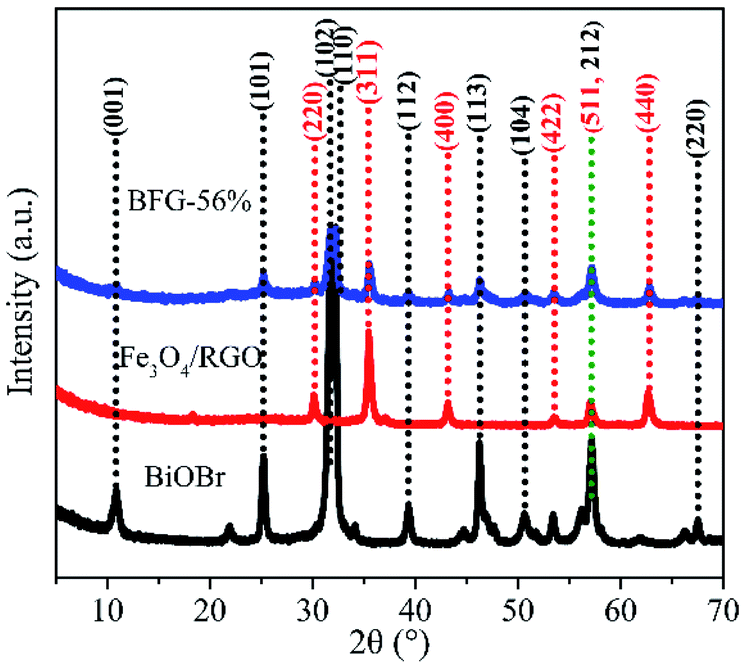
The X-ray diffraction (XRD) patterns of BiOBr microspheres, Fe3O4/RGO and BFG-56% were recorded to confirm their crystallinity and phase purity in Fig. 1. It can be observed that the nine diffraction peaks of BiOBr powers could be indexed to crystal planes of tetragonal BiOBr phase, which match perfectly with the standard powder diffraction pattern (JCPDS#09-0393). The Fe3O4/RGO sample exhibits six diffraction peaks that were also consistent with XRD patterns of cubic Fe3O4 nanoparticles (JCPDS#65-3170).36 However, the obvious diffraction peak of C is not detected, which is probably because of the low RGO content and weak intensity of RGO. As for the BFG-56%, composite has a similar XRD spectrum to the combination of pure BiOBr spectrum and Fe3O4/RGO spectrum, which reveal that the structure of Fe3O4 and BiOBr do not decompose or convert to other crystals after a series of functionalization processes. In addition, the degree of defect in the composite and its carbon structural changes are further explored by Raman techniques in Fig. S1.† The extent of defects/disorder can be evaluated through the intensity ratio of D-band and G-band, ID/IG.37 The ID/IG values for Fe3O4/RGO (1.204) and BFG-56% (1.167) are higher than that for GO (0.919), which is ascribed to the reduction of GO to RGO after high-temperature hydrothermal synthesis,38 accompanied by the surface of the RGO layer filled with Fe3O4 and BiOBr, indirectly suggesting the effective synthesis of the ternary composite.
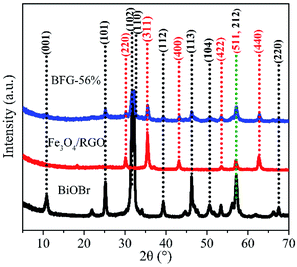 |
| | Fig. 1 XRD patterns of BiOBr, Fe3O4/RGO and BFG-56%. | |
The microstructures of BiOBr, Fe3O4/RGO and BFG-56% has been examined by SEM and TEM. A plurality of BiOBr microspheres with average diameter of 1–2 μm are observed in Fig. 2(a). The enlarged image of BiOBr (Fig. 2(b)) clearly shows a 3D flower-like microsphere structure self-assembled from nanoleaflets with tens of nanometers thick, which allows it to obtain more active sites and makes it more beneficial for light absorption. Fig. 2(c) shows the TEM image of Fe3O4/RGO, revealing that Fe3O4 nanoparticles with an average diameter of 30 to 35 nm are uniformly supported on the transparent RGO sheets, where an average spacing of 0.251 nm in the HRTEM (Fig. 2(d)) corresponding to the (311) lattice plane of Fe3O4. While in Fig. 2(e and f), BiOBr and Fe3O4/RGO sheets can be clearly seen in the BFG-56% composites. Here, Fe3O4/RGO sheets only cover a part of the surface of BiOBr, which hardly affects the capture of photons by the BiOBr. Further, the adjacent connection between Fe3O4/RGO sheets and BiOBr enables the transfer of charge carriers during the photocatalytic process, enhancing the catalytic activity of composites. The TEM, elemental mappings and EDS spectrum (Fig. 2(g–i) and S2†) further demonstrate the formation of BFG-56% photocatalysts comprising BiOBr, Fe3O4 and RGO, which is consistent with the XRD and Raman results.
 |
| | Fig. 2 SEM images of the synthesized samples: BiOBr (a and b) and BFG-56% (e and f); TEM images (c and g) of Fe3O4/RGO and BFG-56%; HRTEM images (d and h) of Fe3O4/RGO and BFG-56% and EDS mapping (i) of BFG-56%. | |
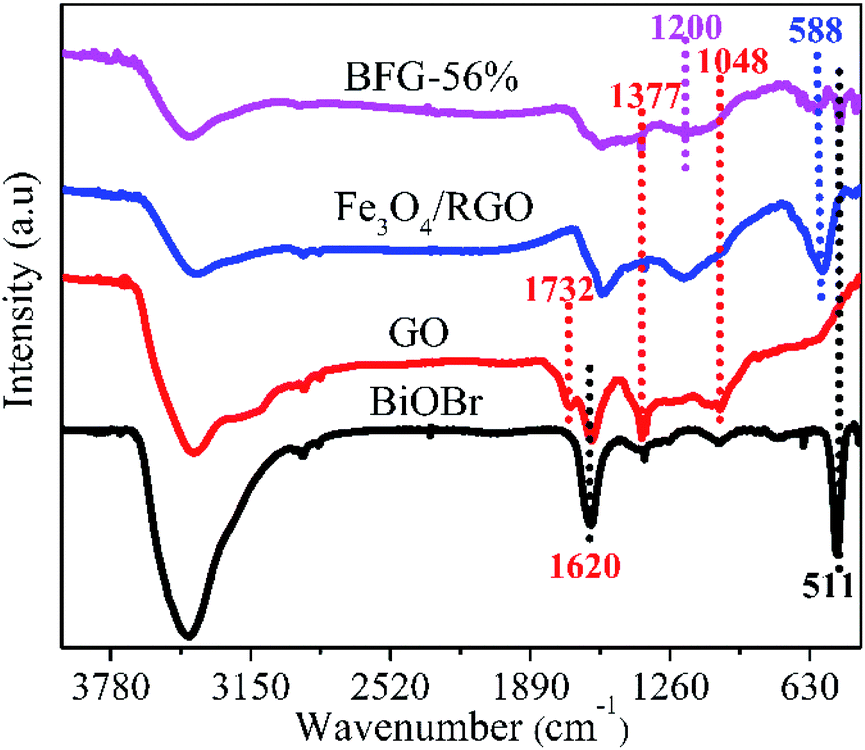
FT-IR analysis as shown in Fig. 3 is used to investigate the chemical bonding attached on the surface of as-incorporated composites. The spectrum of GO depicts a strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak at 1620 cm−1 and the other functional groups of containing C–O peak at 1048 cm−1, C–O–H peak at 1377 cm−1 and CO peak at 1732 cm−1 which are clearly visible.39 After modification by Fe3O4 nanoparticles, a new characteristic peak appears at 588 cm−1, mainly due to the vibration of Fe–O, proving the existence of Fe3O4 nanoparticles.40 Based on this, another new characteristic peak at 511 cm−1 appears in the BFG-56% spectrum, which corresponds to the Bi–O bond in the BiOBr spectrum. However, the characteristic peak of Bi–O appearing in the BFG-56% spectrum is significantly weakened, due to the influence of the introduction of Fe3O4 and RGO in composites. Furthermore, the disappearance of some of the oxygen-containing functional groups in the Fe3O4/RGO and BFG-56% composites or the weakening of their characteristic peaks also confirmed that GO was reduced to RGO after hydrothermal treatment, which is consistent with the Raman spectrum. It is worth noting that the BFG-56% exhibits a new broad absorption at 1200 cm−1 and its peak intensity is significantly reduced, which may be attributed to Bi–C vibration, indicating the possible presence of chemical bonding between BiOBr and RGO.41
C peak at 1620 cm−1 and the other functional groups of containing C–O peak at 1048 cm−1, C–O–H peak at 1377 cm−1 and CO peak at 1732 cm−1 which are clearly visible.39 After modification by Fe3O4 nanoparticles, a new characteristic peak appears at 588 cm−1, mainly due to the vibration of Fe–O, proving the existence of Fe3O4 nanoparticles.40 Based on this, another new characteristic peak at 511 cm−1 appears in the BFG-56% spectrum, which corresponds to the Bi–O bond in the BiOBr spectrum. However, the characteristic peak of Bi–O appearing in the BFG-56% spectrum is significantly weakened, due to the influence of the introduction of Fe3O4 and RGO in composites. Furthermore, the disappearance of some of the oxygen-containing functional groups in the Fe3O4/RGO and BFG-56% composites or the weakening of their characteristic peaks also confirmed that GO was reduced to RGO after hydrothermal treatment, which is consistent with the Raman spectrum. It is worth noting that the BFG-56% exhibits a new broad absorption at 1200 cm−1 and its peak intensity is significantly reduced, which may be attributed to Bi–C vibration, indicating the possible presence of chemical bonding between BiOBr and RGO.41
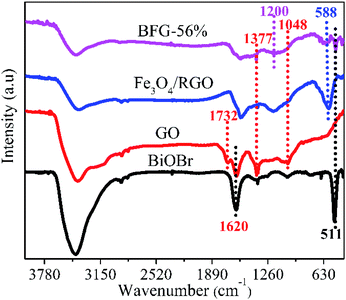 |
| | Fig. 3 FT-IR spectra of GO, BiOBr, Fe3O4/RGO and BFG-56%. | |
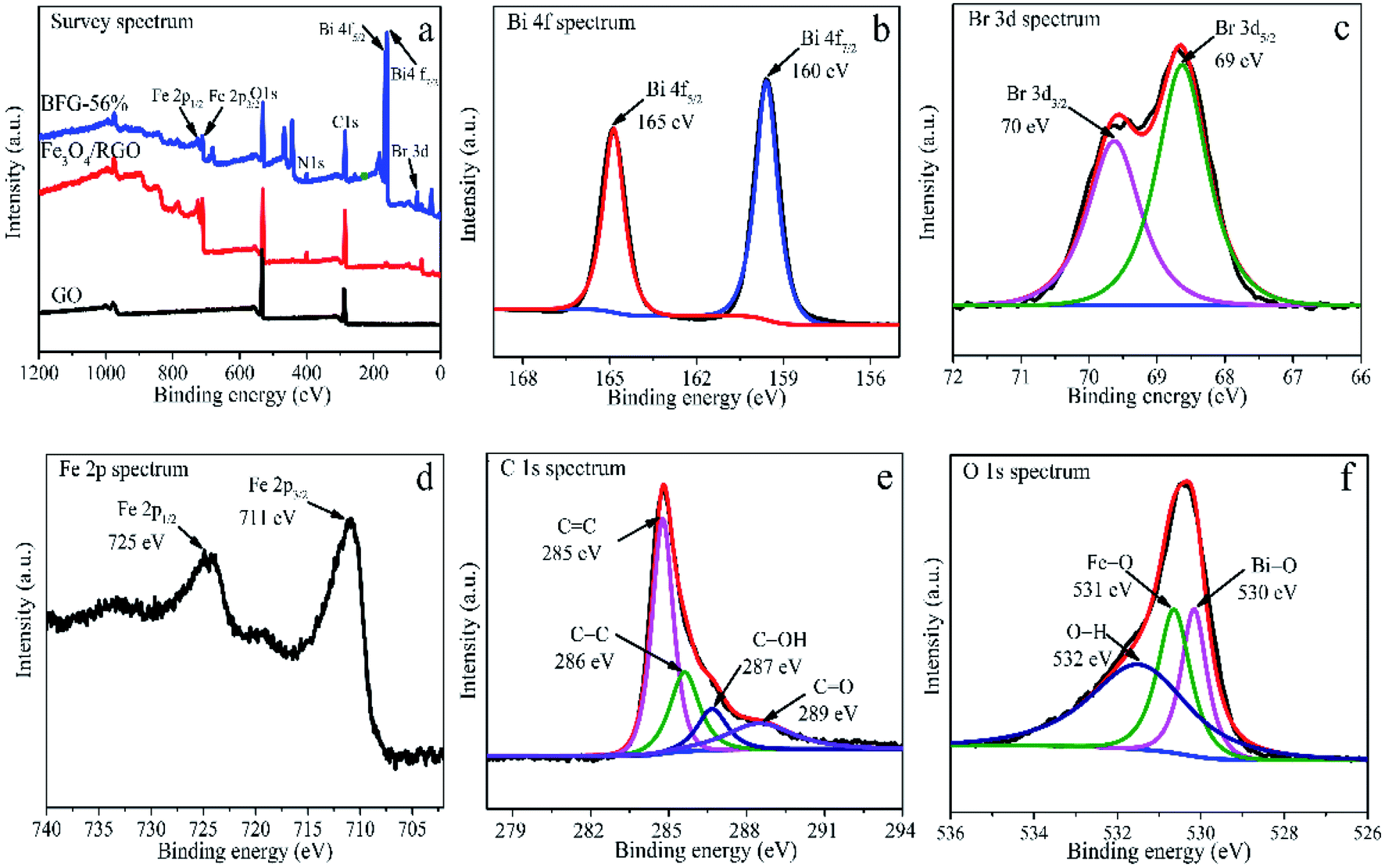
The XPS were analyzed to further determine the chemical bond in BFG-56% composite. As shown in Fig. 4(a), the survey XPS spectrum confirmed the presence of, Bi 4f, Br 3d, Fe 2p, C 1s and O 1s in BFG-56% with corresponding chemical binding energies of ∼163, ∼70, ∼718, ∼287, ∼531 eV. In the high resolution For Bi 4f spectrum (Fig. 4(b)), two peaks at 165 and 160 eV correspond to Bi 4f5/2 and Bi 4f7/2, respectively, and the chemical energy difference between the two peaks is 5 eV, illustrating the presence of Bi3+.42 Compared with the XPS spectrum of BiOBr in the literature,43 the above two characteristic peaks in Bi 4f have a slight shift to lower binding energy, indicating that a strong chemical bond exists between BiOBr and Fe3O4/RGO.44 As depicted in Fig. 4(c), the similar shifts are also noticed in Br 3d spectrum of BFG-56%, where the binding energies of 70 and 69 eV correspond to Br 3d3/2 and Br 3d5/2 due to the presence of Br−.45 As for Fe 2p spectrum of BFG-56%, Fe 2p1/2 and Fe 2p3/2 correspond to chemical energies of 725 and 711 eV, as shown in Fig. 4(d), respectively. Fig. 4(e) shows that the deconvolution of C 1s consists of sp2 CC (285 eV), C–C (286 eV), C–OH carbon atom (287 eV) and carbonyl CO (289 eV), which is almost consistent with the FT-IR result,46 further explaining that GO has been reduced successfully. Fig. 4(f) shows that O 1s can be divided into three peaks, corresponding Fe–O bond with the peak of 531 eV, O–H bond with the peak of 532 eV and Bi–O bond with the peak of 530 eV. According to the above analysis, it is concluded that Fe3O4, RGO and BiOBr coexist in the composite.
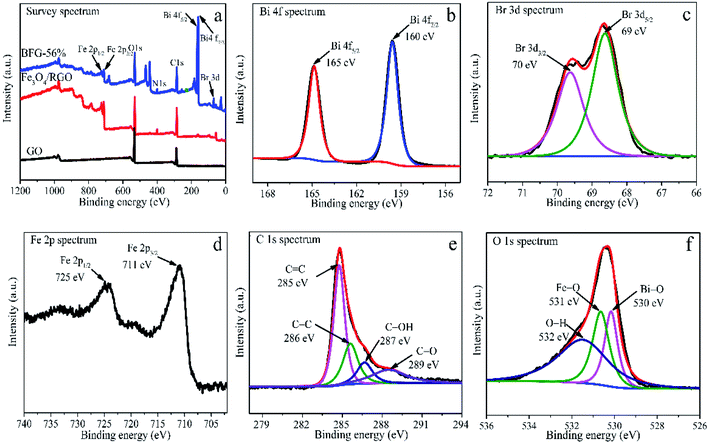 |
| | Fig. 4 (a) XPS of GO, Fe3O4/RGO and BFG-56%; BFG-56% spectra curve fitting: Bi 4f (b), Br 3d (c), Fe 2p (d), C 1s (e), O 1s (f). | |
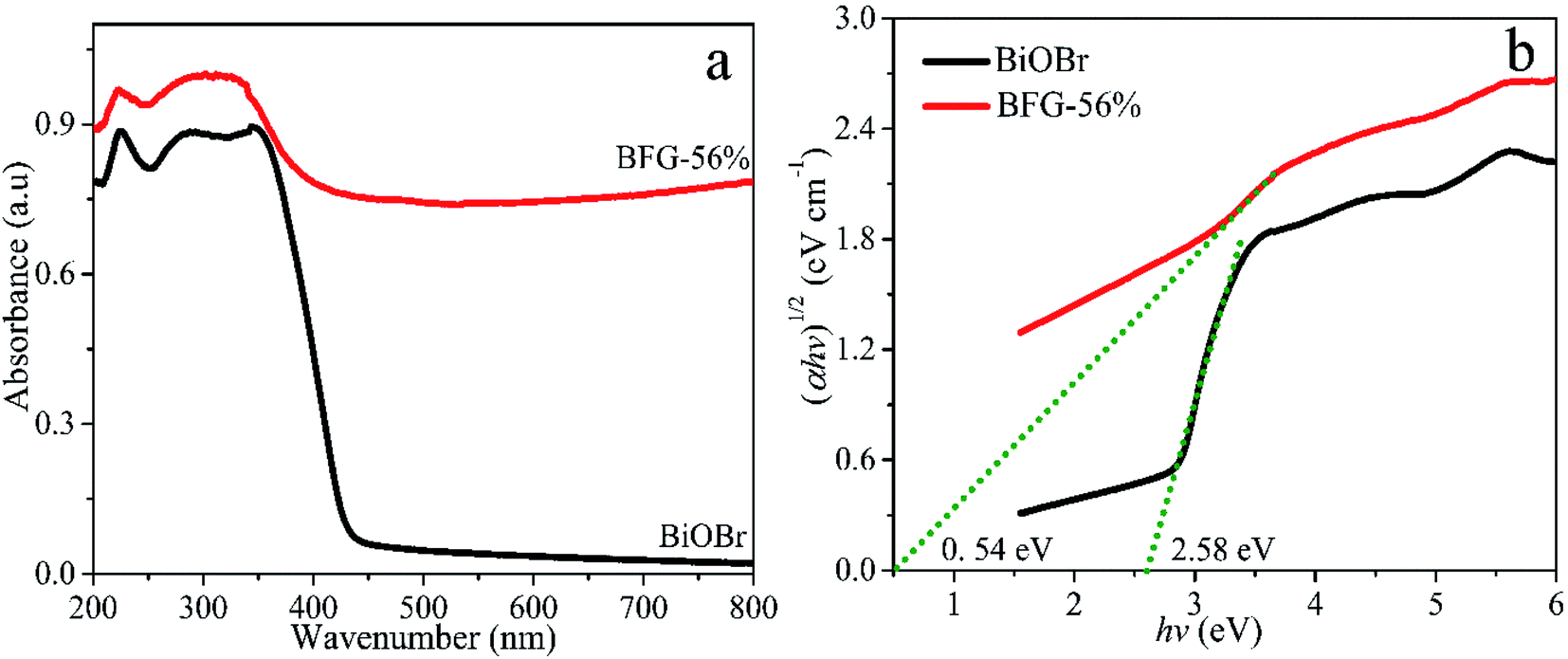
The photocatalytic properties of the catalyst are primarily related to the light absorbing ability. UV-vis diffuse reflectance spectra (DRS) curve fitting was used to characterize the optical properties of BiOBr and BFG-56%, as shown in Fig. 5(a). The optical energy bandgap was calculated using the Tauc equation47 and the relationship between (αhν)1/2 and photon energy (hν) is plotted in Fig. 5(b). Each band gap Eg is obtained by making a tangent to the plotted relationship curve. The band gap energy of BFG-56% is 0.54 eV, which is much lower than that of pure BiOBr (2.58 eV). Thereby, it is calculated that the absorption edges of BFG-56% and BiOBr are 2296 and 481 nm, respectively. This clearly shows that Fe3O4/RGO can effectively narrow the bandgap of BiOBr, and enhance the visible light absorption of the photocatalyst.
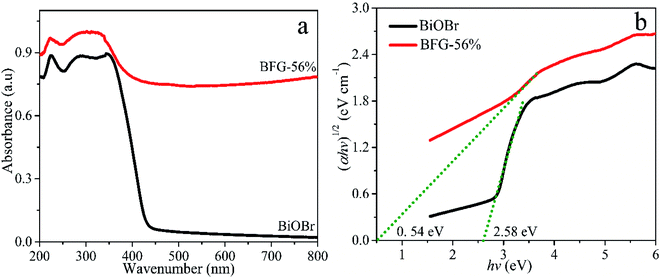 |
| | Fig. 5 (a) UV-vis DRS of BiOBr and BFG-56%. (b) The band gap energies (Eg) of BiOBr and BFG-56% are estimated by the relationship between (αhν)1/2 and photon energy (hν). | |
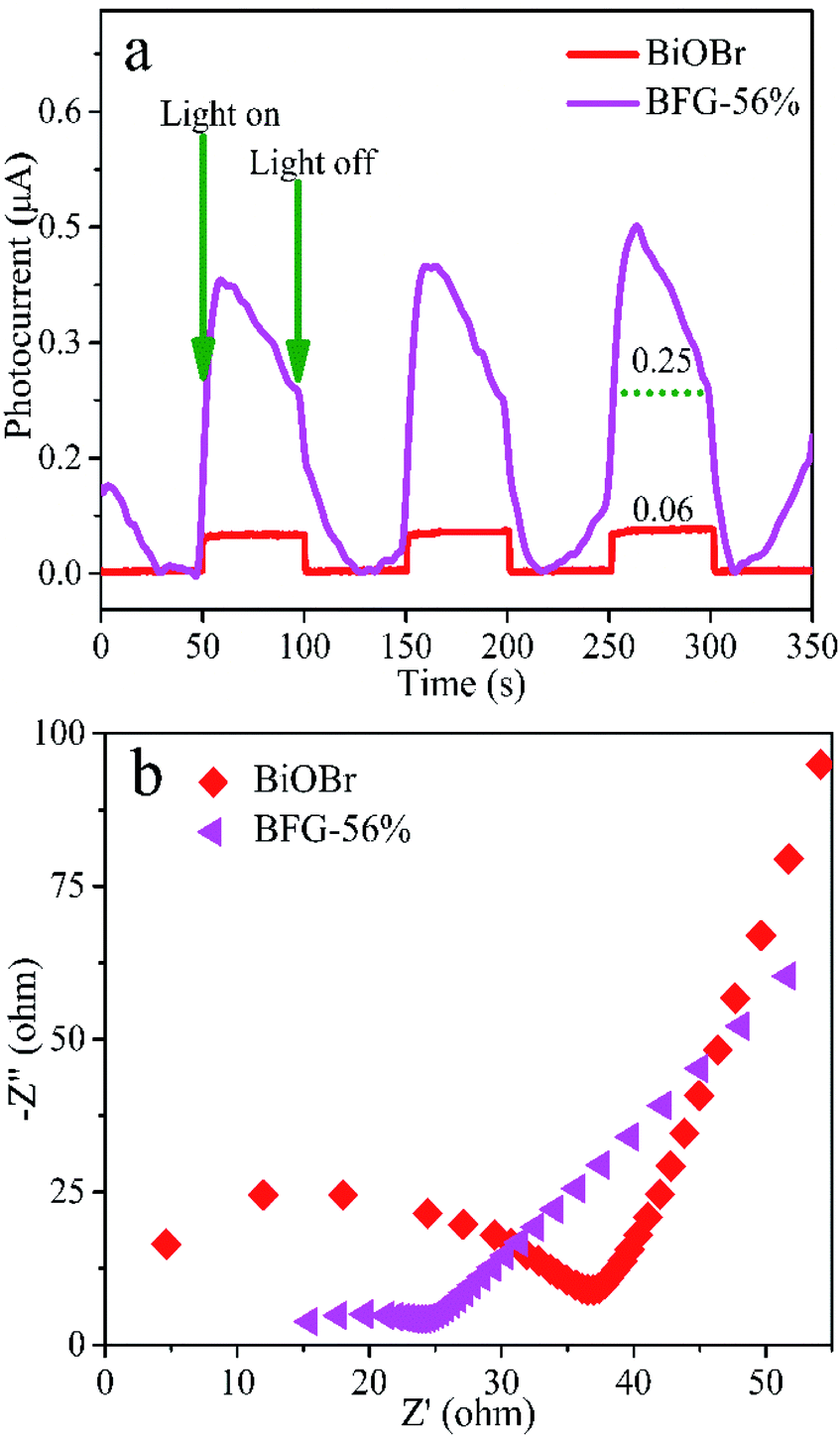
The fluorescence lifetime of photo-induced electrons can be obtained by transient photocurrent measurements. Fig. 6(a) shows that the transient photocurrent responses of the three switch intermittent illumination cycles for BiOBr and BFG-56% under visible-light irradiation. Here, the transient photocurrent densities of BiOBr and BFG-56% are 0.06 μA and 0.25 μA, respectively. The photocurrent response of BFG-56% is 4 times that of BiOBr, which is more effective for the separation of photoexcited electrons and holes. The results indicate that the introduction of Fe3O4/RGO can effectively enhance the interface charge migration. The electrochemical properties of composites were investigated by EIS, which is also a method to evaluate the separation efficiency of e−–h+ pairs of photocatalysts. It is well known that the higher the e−–h+ separation efficiency, the smaller the semicircle, and thus the higher the photocatalytic activity.48 The semicircular radius of EIS Nyquist curve for BFG-56% is significantly smaller than those of other samples in Fig. 6(b). It is confirmed that the introduction of Fe3O4/RGO not only reduces the charge transfer resistance, but also promotes charge transfer and inhibits charge recombination.
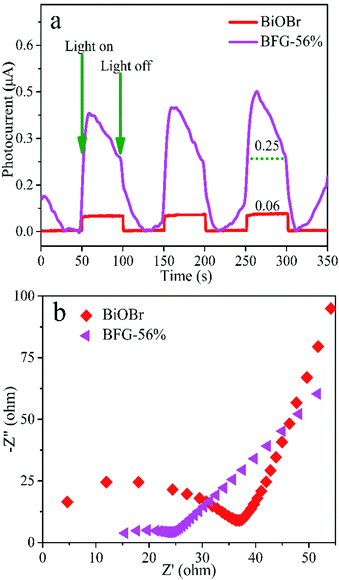 |
| | Fig. 6 (a) Photocurrent response curves and (b) EIS Nyquist plots of BiOBr and BFG-56%. | |
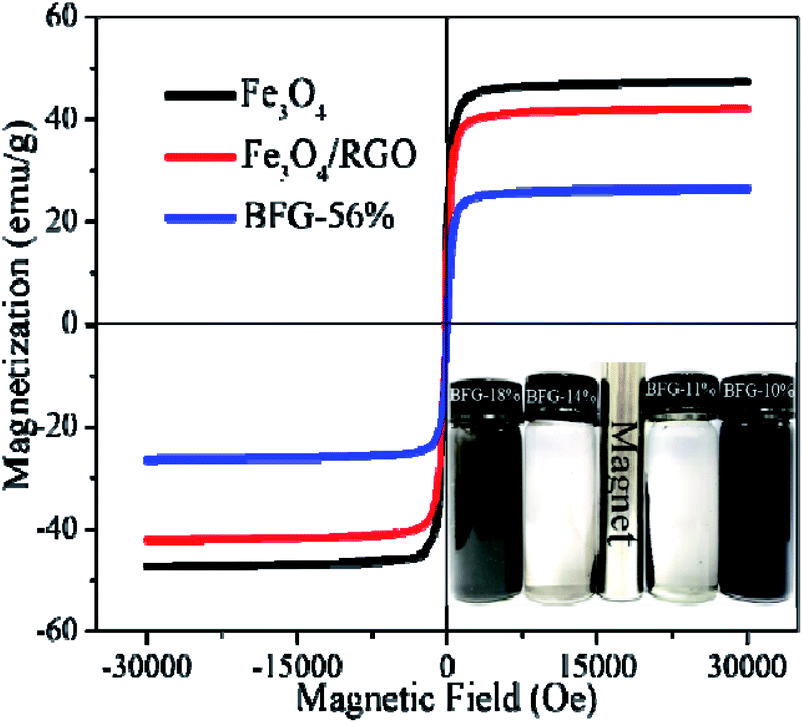
A vibrating sample magnetometer (VSM) is used to characterize the magnetic properties of composites at room temperature. The Fe3O4, Fe3O4/RGO and BFG-56% samples have typical S-type hysteresis loops, and the residual magnetization tends to zero, demonstrating that three materials are superparamagnetic, as shown in Fig. 7. The saturation magnetizations (Ms) of pure Fe3O4, Fe3O4/RGO and BFG-56% are 47.78, 42.39 and 26.49 emu g−1, respectively. It is obvious that Fe3O4 is the most magnetic. Compared with pure Fe3O4, Fe3O4/RGO causes a slight decrease (5.39 emu g−1) in Ms value due to the introduction of RGO. The magnetic field strength Ms of BFG-56% is 15.90 emu g−1 lower than that of Fe3O4/RGO, which is ascribed to the influence of nonmagnetic BiOBr on BiOBr/Fe3O4/RGO.22 In addition, the magnetic separation of BFG-56% was also investigated, as shown in the inset of Fig. 7. Fe3O4/RGO and BFG-56% are well dispersed in water. However, the suspension can be changed from dark gray to transparent within 22 s when an external magnetic field approaches the bottle. All the composites are separated from water under an external magnetic field, which verifies that BiOBr/Fe3O4/RGO composite has the excellent magnetic response. Thus, the work provides a convenient and efficient magnetic separation method for recovery and recycling of photocatalyst.
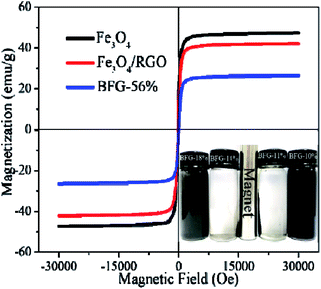 |
| | Fig. 7 Magnetization curves of Fe3O4, Fe3O4/RGO and BFG-56%. | |
Adsorption and photocatalytic performances
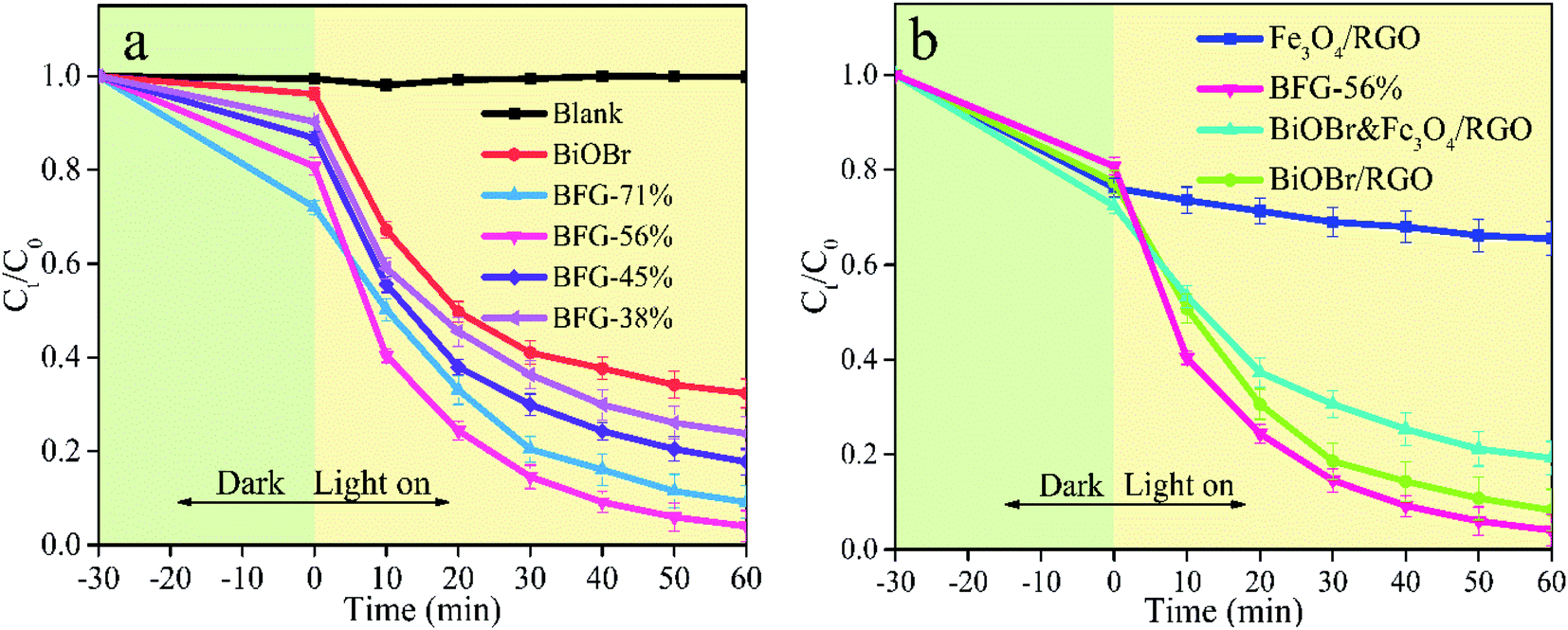
In order to evaluate the photocatalytic performance of BiOBr/Fe3O4/RGO photocatalysts, the abilities of photocatalysts with different ratios of Fe3O4/RGO (BFG-71%, BFG-56%, BFG-45% and BFG-38%) to remove RhB in aqueous solution under dark and visible light (λ > 420 nm) at the temperature of 30 °C are investigated to determine the optimal ratio of BiOBr in composites. Fig. 8(a) shows that all BiOBr/Fe3O4/RGO composites moderately reached the adsorption–desorption equilibrium after dark treatment in the RhB aqueous solution for 30 min. The overall BiOBr/Fe3O4/RGO composites with various doping amounts of Fe3O4/RGO have higher adsorption capacity and photocatalytic activity than pure BiOBr. With the reduction of Fe3O4/RGO doping ratio, the adsorption capacities of BiOBr/Fe3O4/RGO composites decrease gradually (28% for BFG-71% > 19% for BFG-56% > 13% for BFG-45% > 10% for BFG-38%). It can be reasonably believed that the more active sites of RGO, the greater affecting the capture for RhB molecules. Upon visible light irradiation, the concentration of all RhB solutions continued to decreased, which is mainly caused by the photocatalytic degradation of BiOBr in composites. It is worth noting that the degradation rate of RhB for BFG-56% increased from 19% to 96% within 60 minutes with a range of increase of 77%, which is significantly higher than 63% for BFG-71%, 69% for BFG-45% and 66% for BFG-38%, indicating that BiOBr/Fe3O4/RGO composite with 56% Fe3O4/RGO loading has the topgallant photocatalytic degradation of RhB in all composite photocatalysts, which is believed that excess Fe3O4/RGO does not effectively improve the photocatalytic performance of composites.
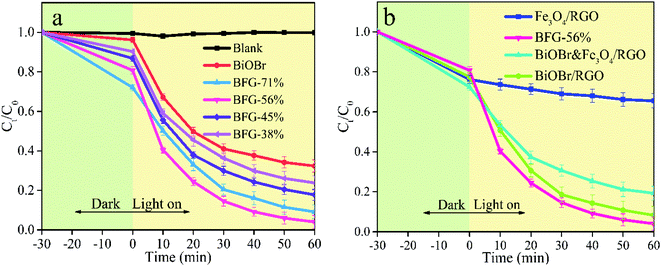 |
| | Fig. 8 Effect of reaction time (a) performance test of pure BiOBr and BiOBr/Fe3O4/RGO composites in different masses of Fe3O4/RGO under dark and visible light and (b) performance testing of Fe3O4/RGO, BFG-56%, BiOBr&Fe3O4/RGO and BiOBr/RGO in dark and visible light. | |
It is expected that Fe3O4/RGO has a synergistic effect on BiOBr during photocatalysis. In order to determine whether RGO or Fe3O4 in composites improve the photocatalytic activity, we synthesized BiOBr/RGO composites in which the ratio of BiOBr to RGO is consistent with that of BFG-56%. The detailed preparation procedures and structure characterization of BiOBr/RGO composites could be obtained from the ESI (Fig. S3 and S4†). To prove this hypothesis, we tested the removal capacities of 12.5 mg Fe3O4/RGO, their physical mixtures (BiOBr&Fe3O4/RGO) and 13.1 mg BiOBr/RGO for RhB under the same conditions. As shown in Fig. 8(b), the overall removal capacity of BiOBr&Fe3O4/RGO for RhB is approximately equal the sum of pure BiOBr and Fe3O4/RGO, indicating that the physical mixing of Fe3O4/RGO with BiOBr does not effectively improve the photocatalytic ability. In comparison, the prepared BFG-56% improved the overall removal capacity by 15%, which indicates that a chemical combination between BiOBr and Fe3O4/RGO is in favor of their photocatalytic performance. It is worth noting that the removal effect of BiOBr/RGO is close to that of BFG-56%, which shows that RGO in BFG-56% plays a major role in promoting photocatalysis, and Fe3O4 in BFG-56% mainly plays a magnetic recovery role.
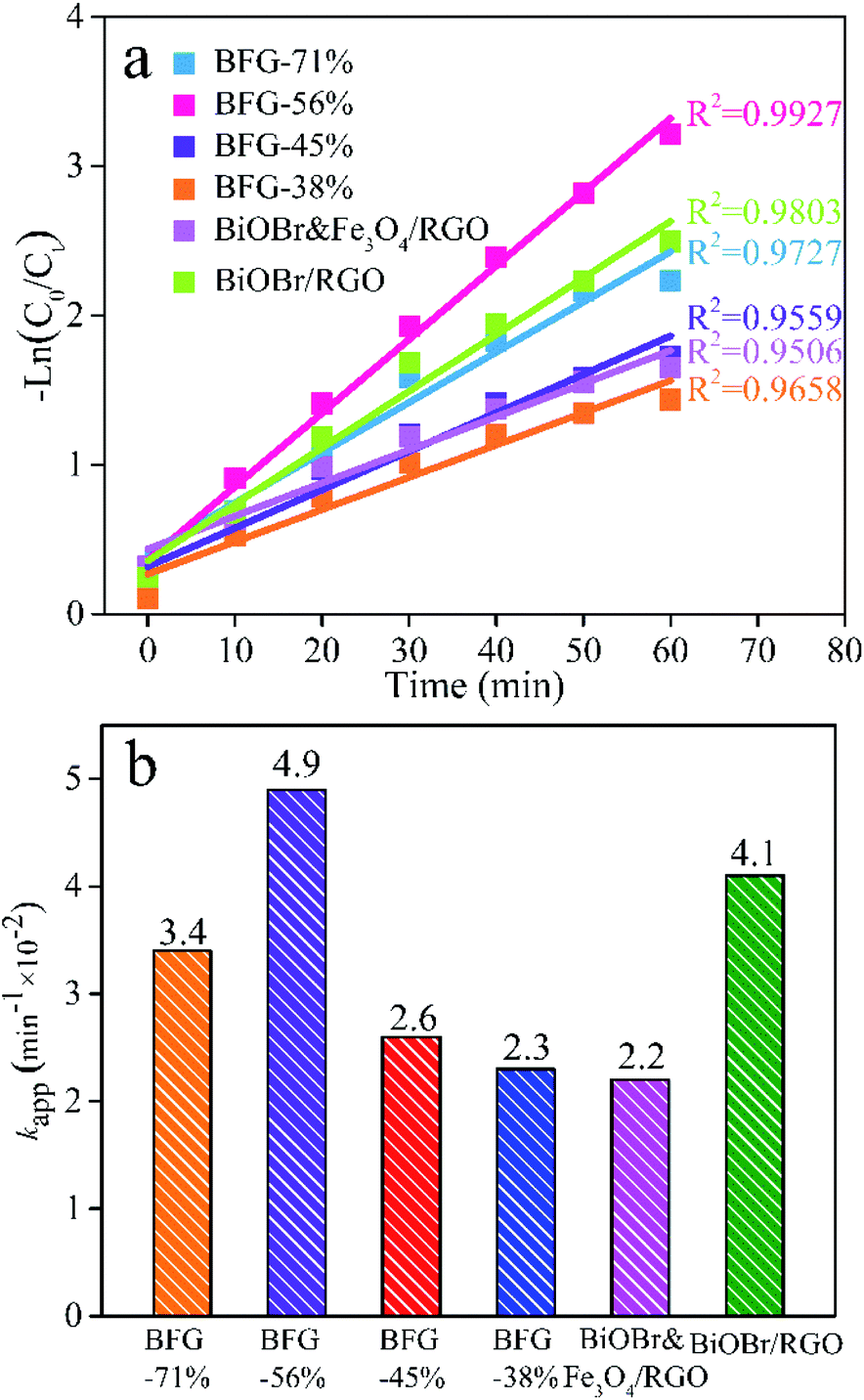
To further illustrate the enhancement, the photocatalytic activity of BFG-71%, BFG-56%, BFG-45%, BFG-38%, BiOBr&Fe3O4/RGO and BiOBr/RGO from 0 to 60 min is compared using a pseudo-first-order kinetic equation with a Langmuir–Hinshelwood (L–H) model:
| |
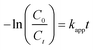 | (2) |
where
kapp is the pseudo-first-order rate constant, which can be obtained from the slope of the −ln(
C0/
Ct)
versus t plot.
Fig. 9(a) shows that the L–H model matches the photocatalytic phase of the above five samples, and the comparison of
kapp values of samples is shown in
Fig. 9(b). It can be seen intuitively that the order of
kapp from large to small is: BFG-56%, BiOBr/RGO, BFG-71%, BFG-45%, BFG-38% and BiOBr&Fe
3O
4/RGO, further demonstrating that the chemical bonding of RGO can delay the recombination of e
−–h
+ pairs, thereby enhancing the photocatalytic activity of BiOBr. For further demonstration of the excellent photocatalysis performances of the optimized BFG-56% for removal on RhB aqueous solution, the
kapp values of BiOBr-based composite photocatalysts degradation for RhB under visible light irradiation reported in the literature are summarized in Table S1.
†
 |
| | Fig. 9 Photocatalytic degradation of RhB in aqueous solution: (a) Plots for corresponding −ln(C0/Ct) versus photoirradiation time; (b) histogram of kapp value of BFG-71%, BFG-56%; BFG-45%, BFG-38%, BiOBr&Fe3O4/RGO and BiOBr/RGO. | |
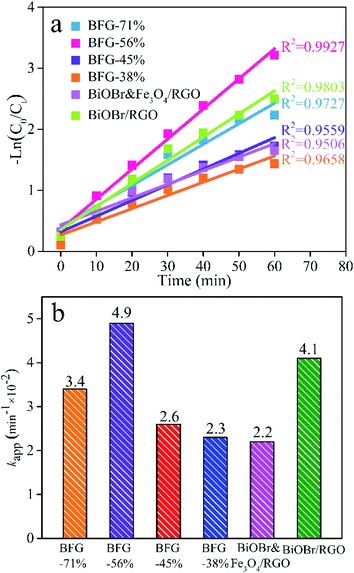
Besides the activity and removal capacity of photocatalysts, evaluation on the stability and reusability of photocatalyst is indispensable in practical applications. The removal ability of BFG-56% for five dyes (RhB, MB, MO, AB and AR) has also been explored. Fig. 10(a) shows that the total removal efficiency of BFG-56% for the cationic dyes RhB and MB are 96% and 86%, respectively. In comparison, the total removal efficiency of the anionic dyes MO, AB and AR are reduced to 75%, 45% and 26%, respectively. This mainly illustrates the high response of BiOBr/Fe3O4/RGO to cationic dyes during the adsorption process and visible-light degradation. Besides the activity and removal capacity of photocatalysts, the evaluation on the stability and reusability is indispensable in practical applications, from the economic and environmental points of view. The applied BFG-56% was quickly separated from the degraded RhB solution by an external magnetic field, washed with ethanol and water, and vacuum dried to obtain the sample. Fig. 10(b) shows that BFG-56% retains more than 90% removal efficiency even after four cycles in a short contact time, demonstrating its great stability and reusability for practical application. As shown in Fig. S5,† despite 4 cycles of recycling, the crystal form of BFG-56% sample did not change significantly, indicating that the prepared sample with good stability.
 |
| | Fig. 10 (a) The removal ability of BFG-56% for five dyes (RhB, MB, MO, AB, AR); (b) the cycle test of BFG-56% for photocatalytic degradation of RhB. | |
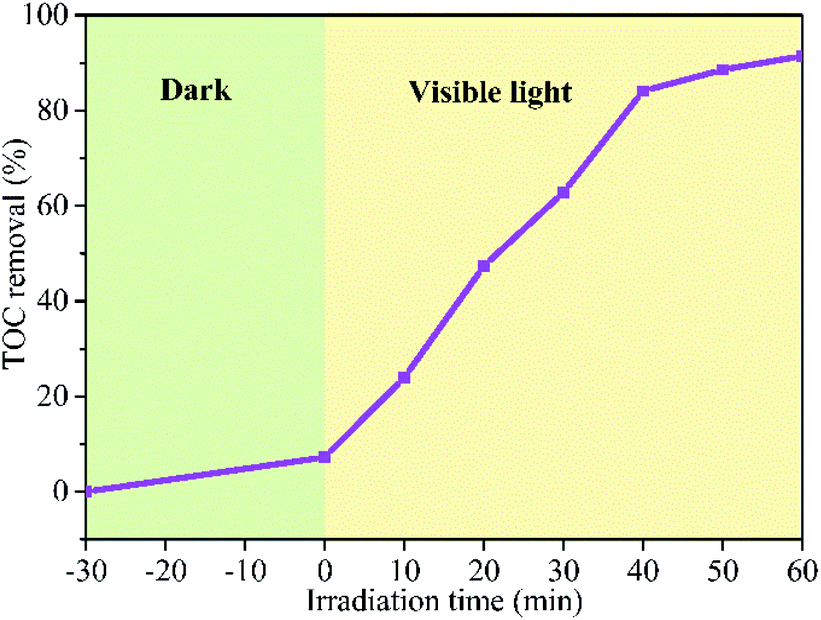
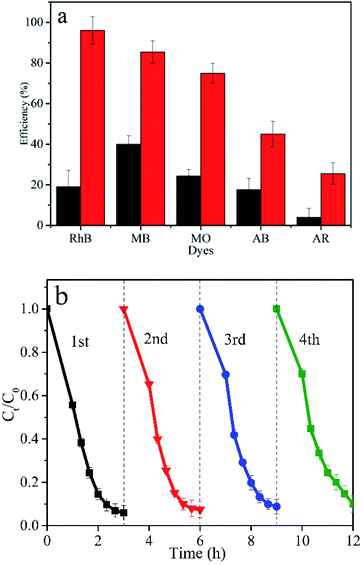
The degree of mineralization of the pollutant is very important as the intermediate product might possess a more virulent form. Therefore, it is necessary to investigate the TOC removal efficiency during the photocatalytic degradation process. The TOC removal ratio of RhB over BFG-56% is shown in Fig. 11 which increase with the irradiation time. About 91.45% of RhB is removed and mineralized to CO2 and H2O under visible light irradiation for 60 min. The mineralization process indicates that the BFG-56% photocatalyst has very strong mineralization ability.
 |
| | Fig. 11 TOC removal efficiency of RhB over BFG-56% under visible light irradiation. | |
Discuss degradation mechanisms
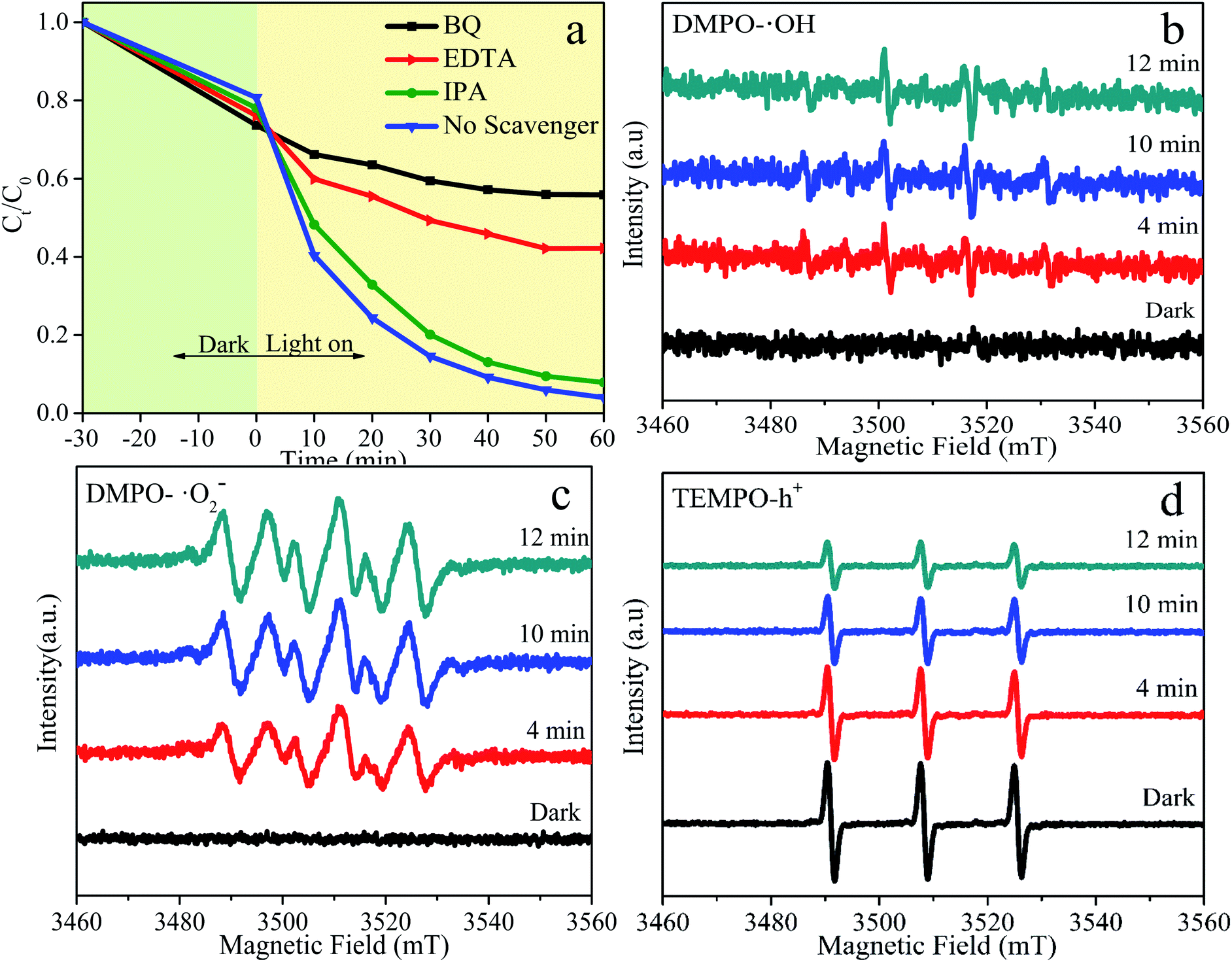
In order to systematically investigate the photocatalytic degradation mechanism of BFG-56%, free radical trapping experiments are performed to explore activated species that play a vital part in the reaction system, as shown in Fig. 12. Generally, the ordinary active species is hydroxyl radicals (˙OH), superoxide radicals (˙O2−) and photo-excited holes (h+) in photocatalytic degradation. Isopropanol (IPA), benzoquinone (BQ) and ethylenediaminetetraacetic acid (EDTA) are used to remove ˙OH, ˙O2− and h+, respectively. Fig. 12(a) illustrates that the effects of the addition of different capture agents on the degradation of RhB. When the IPA inhibitor is added to the solution, degradation efficiency of RhB is almost unchanged, proving that ˙OH has almost no effect on the photocatalytic reaction. However, when BQ and EDTA are added as scavengers for ˙O2− and h+, the degradation efficiency of RhB decreases sharply, confirming that ˙O2− and h+ are the primary activated species. To further verify the main activated species of BFG-56% in the photocatalytic process, ESR spin-trap of DMPO and TEMPO techniques is carried out under visible-light irradiation. The signal of DMPO-˙OH is relatively weak, and there is no signal in the darkness, which indicates that ˙OH has negligible influence in Fig. 12(b). The characteristic peak of DMPO-˙O2− has no signal in the dark, but gradually increases with time under visible-light irradiation, indicating that the ˙O2− plays a major role in the overall photocatalytic reaction, as shown in Fig. 12(c). The characteristic peaks in Fig. 12(d) of TEMPO-h+ have a strong signal in the dark, and its reduction under visible light is conspicuous, which means that h+ is also significant. The influence order of the activated species in the process of RhB photodegradation is ˙O2− > h+ > ˙OH. In summary, ˙O2− and h+ are the significant activated species in the photocatalytic reaction.
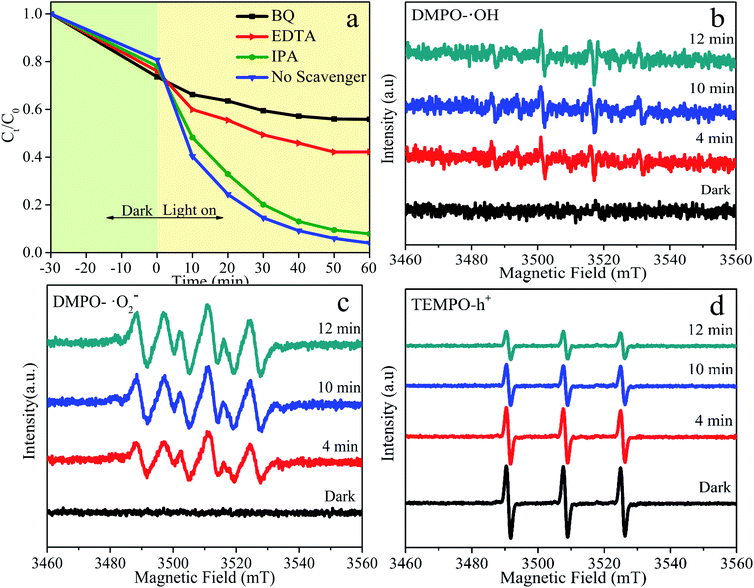 |
| | Fig. 12 Photocatalytic degradation of RhB using BFG-56% catalyst with different capture agents under visible-light irradiation (λ > 420 nm) (a); ESR BFG-56% spectra curve fitting of (b) TEMPO-h+ (c) DMPO-˙OH and (d) DMPO-˙O2− adducts in BFG-56% samples in dark and visible light. | |
In order to explore the reaction mechanism of degradation of RhB by BiOBr/Fe3O4/RGO composites, the entire process of efficient generation, separation and transfer of photoexcited e−–h+ pairs should be considered. It is well known that the potentials of the VB and CB of BiOBr determine the separation and transfer of e−–h+ pairs at the interface. The VB potential (EVB) and CB potential (ECB) of the semiconductor can be obtained from the absolute electronegativity of the atoms and the band gap as follows:12
where
χ and
Eg are the average absolute electronegativity and band gap of the semiconductor, respectively.
Ee is 4.5 eV
vs. the normal hydrogen electrode (NHE),
49 and the
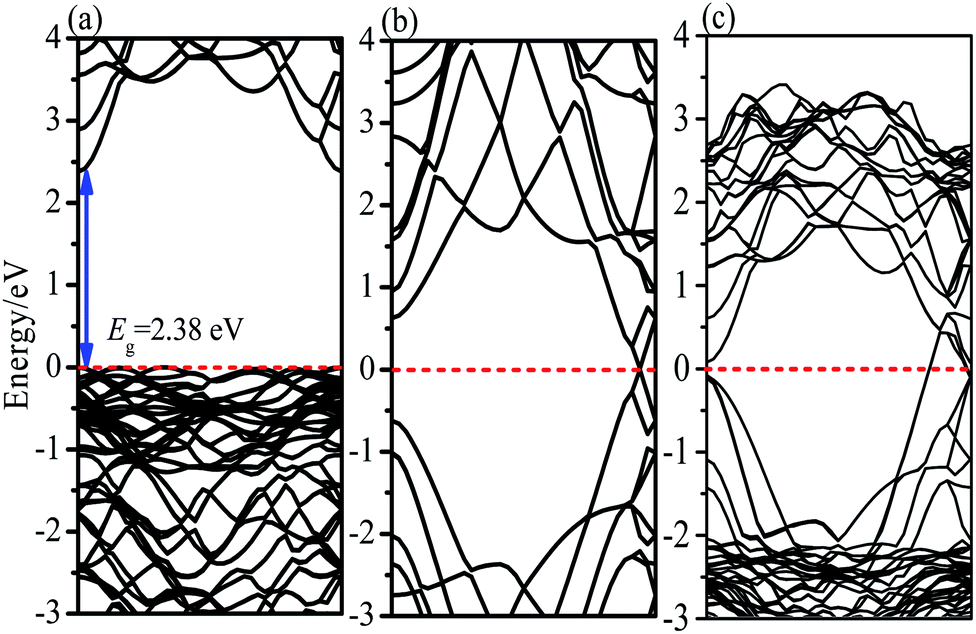
χ values for BiOBr are 6.45 eV. According to the calculated band gap result of BiOBr (
Fig. 13), the obtained
ECB and
EVB of BiOBr were 0.76 and 3.14 eV
vs. NHE with a band gap of 2.38 eV.
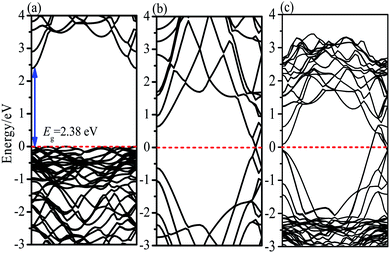 |
| | Fig. 13 Calculated energy band structures of (a) BiOBr (b) RGO (c) BiOBr/RGO heterostructure. | |
With the formation of BiOBr/Fe3O4/RGO composites, the relative positions of the CB and VB of BiOBr changed due to the tendency of an equalized Fermi level after intimate contact. It is well known that the inherent reference for band alignment of materials is the work function. Here, work function is defined as follows:
where
Evac is the energy of a stationary electron in the vacuum near the surface.
EF is the Fermi level. The geometry structures of BiOBr/Fe
3O
4/RGO heterostructure were constructed and described in
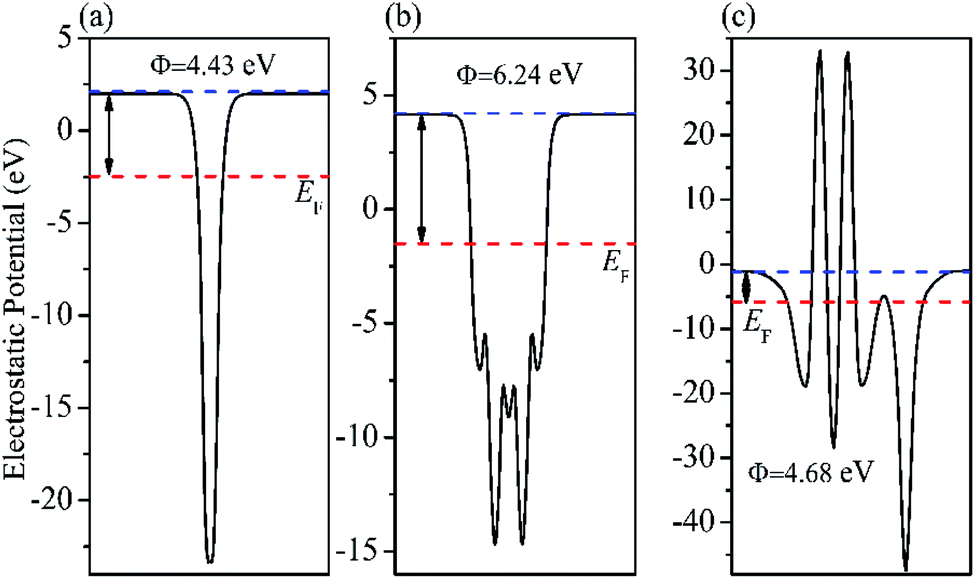
Fig. 14. The
Fig. 14(a) and (b) indicate that the calculated electrostatic potentials for RGO sheets, and BiOBr are 4.43, and 6.24 eV (
vs. vacuum), respectively. The work function of RGO is lower than that of BiOBr, which caused electrons to move from RGO to BiOBr until the Fermi energy of the two semiconductors were aligned. The work function of the BiOBr/RGO heterostructure is 4.68 eV, as shown in
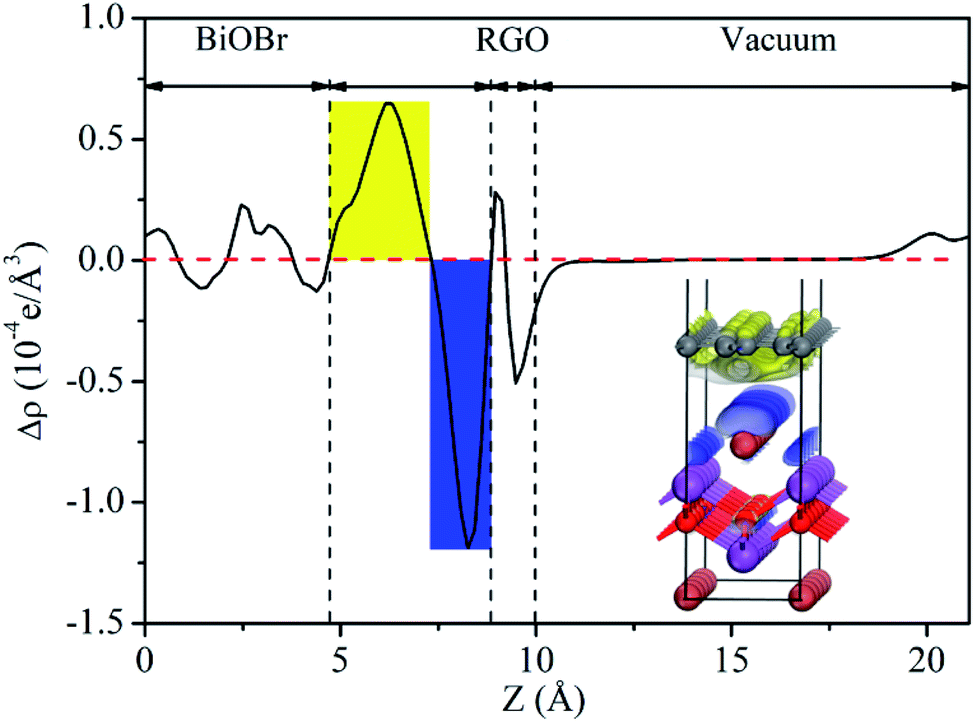
Fig. 14(c), which between the work function of RGO and BiOBr. As a result, a lot of negative charges accumulate on the surface of BiOBr, while RGO gathers positive charges. The net positive and negative charges induced an internal electric field directed from RGO to BiOBr, which is consistent with the result of charge density difference (
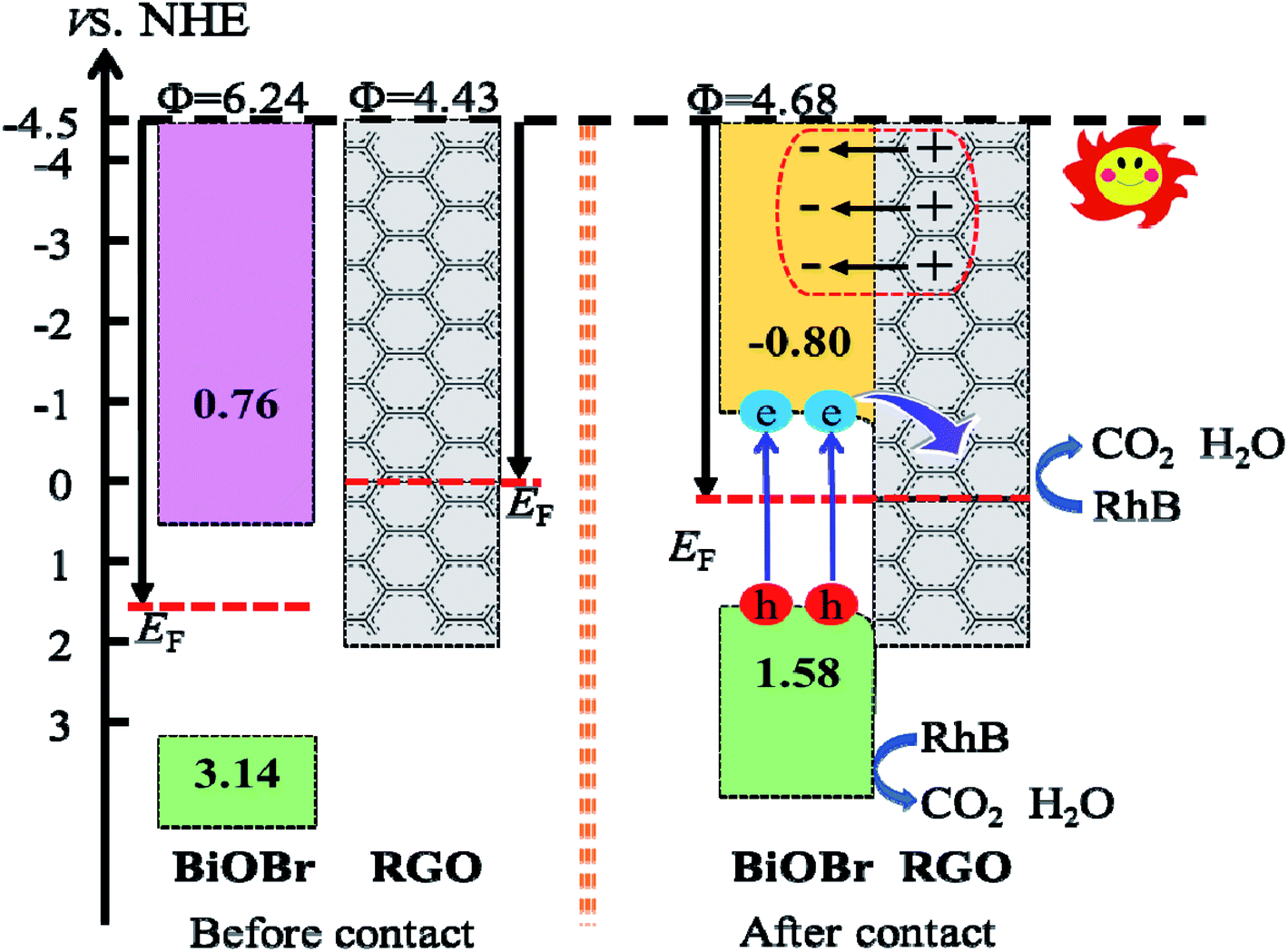
Fig. 15). With the changes of Fermi level, the VB and CB of BiOBr shift downward 1.56 eV. Thus, the CB and VB edge of BiOBr change to −0.80 and 1.58 eV
vs. NHE, respectively, which eventually builds up an n-type Schottky barrier (−0.80 eV,
Fig. 16). Here, according to the Schottky–Mott model at the metal/semiconductor interface,
50 the n-type Schottky barrier height is expressed as the energy difference between the minimum value (CBM) of the conduction band and the Fermi level. It is noteworthy that due to the energy difference between CBM and Fermi level of BiOBr is less than the energy difference between VBM (the maximum value of valence band) and Fermi energy, RGO doped with Fe
3O
4 and BiOBr form an n-type Schottky contact.
51
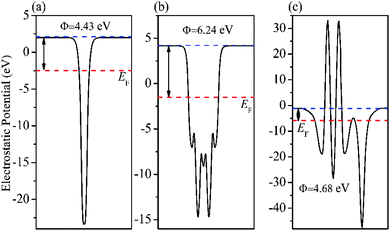 |
| | Fig. 14 Calculated electrostatic potentials of (a) RGO (b) BiOBr (001) and (c) BiOBr/RGO heterojunction. | |
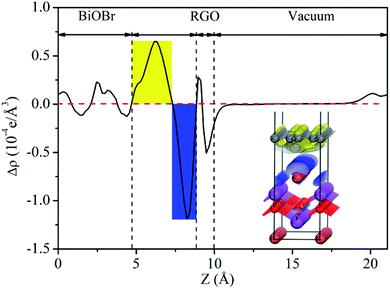 |
| | Fig. 15 Planar-averaged electron density difference Δρ(z) for BiOBr/RGO heterostructure. The blue and yellow areas indicate electron depletion and accumulation, respectively. | |
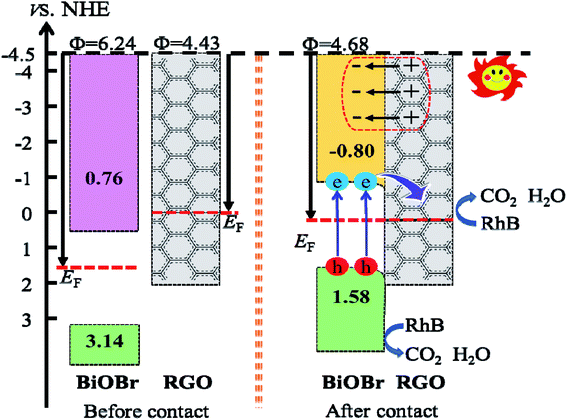 |
| | Fig. 16 Diagram of the band edge positions before and after contact of BiOBr and RGO. | |
The composition of the heterojunction interface plays a significant role in charge transfer and separation. As mentioned above, the Fermi level moves towards the negative direction of BiOBr and the positive direction of RGO until they are equal, when the BiOBr/RGO (001) interface is equilibration. At this moment of the CB and VB of BiOBr are pulled in the negative direction a little, and then form a slightly downward band bending close to the interface between BiOBr and RGO. When the BiOBr/Fe3O4/RGO heterojunction absorbs visible light, the BiOBr layer receives photon energy, causing electrons to be transmitted from VB to CB, and then continuously flow into RGO through the permeation mechanism,52 while it is no way for electrons in RGO to migrate back to BiOBr, and thus are trapped in RGO. Therefore, this n-type Schottky junction promotes the flow of electrons from BiOBr to RGO and leaves holes in BiOBr, which promotes the separation of photo-generated carriers.53 In effect, it is easily for the adsorbed O2 molecules to capture electrons on the surface of RGO sheets to generating ˙O2−, due to the standard redox potential of O2/˙O2− (−0.28 V vs. NHE) is more negative than the CB potential of RGO in the interface.54 In this case, the standard redox potential of ˙OH/OH− (1.99 V vs. NHE) is more positive than the VB potential (1.58 V vs. NHE) of the BiOBr layer, indicating that OH− cannot be oxidized ˙OH free radicals with a very strong oxidation capacity by the photogenerated holes in the VB of the BiOBr layer. Certainly, the VB edge is more positive than the redox potential of RhB (1.43 V).55 It is possible to oxidize holes directly through photoexcitation to generate energy in this heterojunction, thereby degrading RhB.56,57 Thus, the responsible causes of RhB degradation is the superoxide radical anions and photogenerated holes.
Conclusion
In summary, we were constructed recyclable magnetic BiOBr/Fe3O4/RGO composites with a notable catalytic activity by a simple solvothermal strategy. The prepared BiOBr/Fe3O4/RGO composite with 56 wt% content of Fe3O4/RGO used as photocatalysts for the removal of dyes in aqueous solution exhibit an excellent adsorption–photocatalysis synergistic effect, and its reaction rate is about 2.23 times that of pure BiOBr. The heterojunctions interface properties of the composite for photoexcited charge transfer are proposed by experiments and theoretical calculations. RGO doped with Fe3O4 regulates the BiOBr interface to form a n-type Schottky junction, which promotes the separation of photo-excited electrons. More importantly, the developed photocatalysts could be separated easily from the solution through an external magnetic field and demonstrated excellent reusability over multiple cycles. Therefore, the BiOBr/Fe3O4/RGO composites have a remarkable prospect in the treatment of industrial wastewater without secondary pollution.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (51472081), Cooperative Foundation of China-UK Research and Innovation Bridges (2016YFE0124300), Leading Plan of Green Industry (YXQN2016005), the Development Funds of Hubei Collaborative Innovation Center (HBSKFMS2015004, HBSDB201704). The authors would like to thank Chaofeng He from Shiyanjia Lab (https://www.shiyanjia.com) for materials characterizations.
References
- J. Xu, W. Meng, Y. Zhang, L. Li and C. Guo, Appl. Catal. B Environ., 2011, 107, 355–362 CrossRef CAS.
- B.-G. Aleksandra, P. Wilczewska and A. Malankowska, Sep. Purif. Technol., 2019, 217, 164–173 CrossRef.
- H. Zhang, H. Zheng, Y. Wang, R. Yan, D. Luo and W. Jiang, Ind. Eng. Chem. Res., 2019, 58, 1875–1887 CrossRef CAS.
- Y. Chen, M. Wen and Q. Wu, CrystEngComm, 2011, 13, 3035–3039 RSC.
- J. Chen, M. Guan, W. Cai, J. Guo, C. Xiao and G. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 20909–20914 RSC.
- G. Jiang, X. Wang, Z. Wei, X. Li, X. Xi, R. Hu, B. Tang, R. Wang, S. Wang and T. Wang, J. Mater. Chem. A, 2013, 1, 2406–2410 RSC.
- J. Guo, X. Liaoa, M.-H. Lee, G. Hyett and C.-C. Huang, Appl. Catal. B Environ., 2019, 243, 502–512 CrossRef CAS.
- F. Qiu, W. Li, F. Wang, H. Li, X. Liu and J. Sun, J. Colloid Interface Sci., 2017, 493, 1–9 CrossRef CAS PubMed.
- J. Fu, Y. Tian, B. Chang and F. Xi, J. Mater. Chem., 2012, 22, 21159–21166 RSC.
- A. Geng, L. Meng, J. Han, Q. Zhong and M. Li, Cellulose, 2018, 25, 4133–4144 CrossRef CAS.
- J. Xia, J. Di, S. Yin, H. Li, H. Xu, L. Xu, H. Shu and M. He, Mater. Sci. Semicond. Process., 2014, 24, 96–103 CrossRef CAS.
- P. Yan, D. Jiang, Y. Tian, L. Xu, J. Qian, H. Li, J. Xia and H. Li, Biosens. Bioelectron., 2018, 111, 74–81 CrossRef CAS PubMed.
- J. Li, F. Yang, Q. Zhou, R. Ren, L. Wu and Y. Lv, J. Colloid Interface Sci., 2019, 546, 139–151 CrossRef CAS PubMed.
- X. Luo, C. Wang, S. Luo, R. Dong, X. Tu and G. Zeng, Chem. Eng. J., 2012, 187, 45–52 CrossRef CAS.
- L. Ding, H. Tao, C. Zhang, J. Li, W. Zhang and J. Wang, Mater. Sci. Semicond. Process., 2020, 107, 104798–104804 CrossRef CAS.
- Z. Wu, J. Pei, X. Song, N. Liu, J. Li, M. Zhang, J. Zhang, D. Zhang and F. Zhao, New J. Chem., 2020, 1, 2–17 Search PubMed.
- S. Huang, Y. Xu, M. Xie, H. Xu, M. He, J. Xia, L. Huang and H. Li, Colloid. Surface. Physicochem. Eng. Aspect., 2015, 478, 71–80 CrossRef CAS.
- C. Guo, S. Gao, J. Lv, S. Hou, Y. Zhang and J. Xu, Appl. Catal. B Environ., 2017, 205, 68–77 CrossRef CAS.
- P.-K. Boruah, S. Szunerits, R. Boukherroub and M.-R. Das, Chemosphere, 2018, 191, 503–513 CrossRef CAS PubMed.
- E. Mohammadiyan, H. Ghafuri and A. Kakanejadifard, Optik, 2018, 166, 39–48 CrossRef CAS.
- N. Guo, H. Li, X. Xu and H. Yu, Appl. Surf. Sci., 2016, 389, 227–239 CrossRef CAS.
- A.-K. Agegnehu, C.-J. Pan, M.-C. Tsai, J. Rick, W.-N. Su, J.-F. Lee and B.-J. Hwang, Int. J. Hydrogen Energy, 2016, 41, 6752–6762 CrossRef CAS.
- X. Chen, Y. Dai, J. Guo, T. Liu and X. Wang, Ind. Eng. Chem. Res., 2016, 55, 568–578 CrossRef CAS.
- X. Li, C. Dong, K. L. Wu, S.-H. Xia, Y. Hu, M. Ling and K. Liu, J. Mater. Sci. Lett., 2016, 164, 502–504 CrossRef CAS.
- B. Sun, X. Yu, L. Wang, L. Feng and C. Li, J. Fuel Chem. Technol., 2016, 44, 1074–1081 CrossRef CAS.
- X. Yu, J. Shi, L. Feng, C. Li and L. Wang, Appl. Surf. Sci., 2017, 369, 1775–1782 CrossRef.
- X.-M. Tu, S.-L. Luo, G.-X. Chen and J.-H. Li, Chem.–Eur. J., 2012, 18, 14359–14366 CrossRef CAS PubMed.
- W. Liu, J. Cai and Z. Li, ACS Sustain. Chem. Eng., 2015, 3, 277–282 CrossRef CAS.
- P.-K. Boruah and M.-R. Das, J. Hazard. Mater., 2020, 385, 121516–121579 CrossRef PubMed.
- G.-U. Rehman, M. Tahi, P.-S. Goh, A.-F. Ismail and I.-U. Khan, Powder Technol., 2019, 356, 547–558 CrossRef CAS.
- S. Janani, S. Rani, P. Ellappan and L.-R. Miranda, J. Environ. Chem. Eng., 2016, 4, 534–541 CrossRef.
- H. Xu, Z. Xu, J. Zhou, G. Yan, X. W. Li and S. P. Zhuo, Ceram. Int., 2019, 45, 15458–15465 CrossRef CAS.
- Y. Lv, B. Xing, M. Zheng, G. Yi, G. Huang, C. Zhang, R. Yuan, Z. Chen and Y. Cao, Nanomaterials, 2018, 8, 670–687 CrossRef PubMed.
- J.-P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. Benjwal, M. Kumar, P. Chamoli and K.-K. Kar, RSC Adv., 2015, 5, 73249–73260 RSC.
- J. Wang, S. A. Kondrat, Y. Wang, G. L. Brett, C. Giles, J. K. Bartley, L. Li, L. Qian, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 3575–3587 CrossRef CAS.
- X. Cai, X. Shen, L. Ma, Z. Ji, C. Xu and A. Yuan, Chem. Eng. J., 2015, 268, 251–259 CrossRef CAS.
- R. Madhuvilakku, S. Alagar, R. Mariappan and S. Piraman, Sens. Actuators, B, 2017, 253, 879–892 CrossRef CAS.
- F. Gholamian, M. Shabanian and M. Shahrokh, J. Cluster Sci., 2013, 24, 177–188 CrossRef CAS.
- X. Tu, S. Luo, G. Chen and J. Li, Chem. - Eur. J., 2012, 18, 14359–14366 CrossRef CAS PubMed.
- X. Dong, W. Zhang, Y. Sun, J. Li, W. Cen, Z. Cui, H. Huang and F. Dong, J. Catal., 2018, 357, 41–50 CrossRef.
- J. Xia, S. Yin, H. Li, H. Xu, L. Xu and Y. Xu, Dalton Trans., 2011, 40, 5249–5258 RSC.
- X. Zeng, Z. Wang, N. Meng, D.-T. McCarthy, A. Deletic, J.-H. Pan and X. Zhang, Appl. Catal. B Environ., 2017, 202, 33–41 CrossRef CAS.
- W. S. Wang, H. Du, R. X. Wang, T. Wen and A. W. Xu, Nanoscale, 2013, 5, 3315–3321 RSC.
- L. Zhou, H. Deng, J. Wan, J. Shi and S. U. Tong, Appl. Surf. Sci., 2013, 283, 1024–1031 CrossRef CAS.
- C. Guo, Y. He, P. Du, X. Zhao, J. Lv, W. Meng, Y. Zhang and J. Xu, Appl. Surf. Sci., 2014, 320, 383–390 CrossRef CAS.
- C. Sun, Y. Guo, X. Xu, Q. Du, H. Duan, Y. Chen, H. Li and H. Liu, Composites, Part A, 2017, 92, 33–41 CrossRef CAS.
- J. Hou, C. Yang, Z. Wang, S. Jiao and H. Zhu, Appl. Catal. B Environ., 2013, 129, 333–341 CrossRef CAS.
- J. Bardeen, Phys. Rev. A, 1947, 71, 717–727 CrossRef.
- J. Hu, W. Duan, H. He, H. Lv, C. Huang and X. G. Ma, J. Mater. Chem. C, 2019, 7, 7798–7805 RSC.
- X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- T. Berger, D. Monllor-Satoca, M. Jankulovska, T. Lana-Villarreal and R. Gomez, ChemPhysChem, 2012, 13, 2824–2875 CrossRef CAS PubMed.
- T. Shen, Z.-G. Zhao, Q. Yu and H.-J. Xu, J. Photochem. Photobiol., A, 1989, 47, 203–212 CrossRef CAS.
- S.-B. Gawande and S.-R. Thakare, Int. Nano Lett., 2012, 2, 11–17 CrossRef.
- Y. Li, Z. Sun, S. Zhu, Y. Liao, Z. Chen and D. Zhang, Carbon, 2015, 94, 599–606 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01668c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Jisong Hua,
Xinxin Zhang*b,
Di Li
*a,
Jisong Hua,
Xinxin Zhang*b,
Di Li