
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBonding properties of a superatom system with high-Z elements: insights from energy decomposition analysis
Xiaochen Wu†
,
Yang Gao†,
Weiyu Xie and
Zhigang Wang *
*
Institute of Atomic and Molecular Physics, Jilin University, Changchun 130012, China. E-mail: wangzg@jlu.edu.cn
First published on 9th April 2020
Abstract
Superatoms with high-Z elements have novel physicochemical properties, and a comprehensive and thorough view of their bonding properties plays a crucial role in the design of superatoms. Now, energy decomposition analysis shows increasingly prominent performance for understanding inter- and intra-molecular interactions, so the bonding properties of typical superatoms with high-Z elements, Th@Au14, have been investigated here. It is found that under different electron occupation types of the fragments, the electrostatic interaction energy, polarization, and exchange repulsion energy change significantly in their intramolecular interaction energy components, resulting in quantitative or even qualitative differences in their main interaction energy. This indicates that the bonding properties of fragments are related to their electronic structures, and even has extraordinary reference value for the future regulation and control of interactions in superatoms with high-Z elements, which has great significance for superatom development.
Introduction
Inter- and intra-molecular interactions carry considerable weight in the structure and properties of substances, as well as in chemical reaction processes.1–3 The research on them is of great significance to promote the development of many aspects, such as material synthesis, molecular assembly and medicinal design.4–8 Now, for superatoms,9,10 a distinctive material conformation of building blocks,11,12 their electron structures and chemical properties can be manipulated by changing the number, structure and composition of the atoms contained, bringing potential application value for many fields,13–15 so discussing the superatomic bonding properties plays an important role in the development of new materials. Especially, superatoms consisting of high-Z elements (f- and ds-elements) exhibit novel optical and magnetic properties.16–20 Hence, dissecting the physical origin of intra-molecular interactions in superatoms with high-Z elements is extremely interesting.Energy decomposition analysis (EDA) developed by Morokuma,21 Ziegler and Rauk22 is a powerful quantitative method to understand bonding properties based on the fundamental laws of quantum mechanics. It generally decomposes the instantaneous interaction energy between the fragments in a molecule into three terms with specific physicochemical meanings: the quasi-classical electrostatic interaction energy, Pauli repulsion energy, and orbital interaction energy.23 Through analyzing the results of the interaction energy components, the bonding properties can be explained intuitively and deeply. However, although EDA has made great developments in intramolecular and intermolecular interactions, the study about interactions in superatomic systems with high-Z elements is still in its infancy because the valence electrons of high-Z elements are relatively active24 and their interactions are also complex.
In this paper, due to the fundamental importance of gold-based clusters in many fields25–27 and the potential application value of superatom embedded actinide elements in optics, we use EDA to study the interaction energy in Th@Au14, a typical superatom with high-Z elements, formed by a thorium (Th) atom embedded into an Au14-cage structure. It is found that, under different electron occupation types, the main interaction energy of the Th atom and Au14-cage structure was significantly different. This provides a comprehensive interpretation about the relationship between the interaction energy and the electronic structures of fragments in superatoms with high-Z elements.
Methods
The energy decomposition analysis (EDA) method based on Morokuma and the ETS partitioning scheme of Ziegler and Rauk is widely used to analyze interactions quantitatively, which is limited to the RHF theoretical level.28 The interaction energy ΔE between the fragments in a molecular system is partitioned into several contributions, which can be identified as physically meaningful quantities. First, ΔE is separated into two major components, ΔEprep and ΔEint:| ΔE = ΔEprep + ΔEint. |
The EDA decomposes the instantaneous interaction energy ΔEint between the fragments in a molecule into three terms of specific physical and chemical meaning:
| ΔEint = ΔEelstat + ΔEPauli + ΔEorb |
The first term is the quasi-classical electrostatic interaction energy ΔEelstat between the charge densities of the fragments. The second is the exchange repulsion interaction ΔEPauli between fragments caused due to Pauli's principle. The last one is the orbital interaction energy ΔEorb owing to orbital mixing of the fragments. These items are defined by assigning the intermediate interaction during the bonding process of the fragments. These terms focus on the interpretability of the resulting energy terms in a physical and chemical sense. Meanwhile, the calculation makes use of the basic rules of quantum mechanics. Accordingly, EDA can connect well the relationship between the law of quantum mechanics and the chemical bond model.
Due to the strong electron correlation and relativity effect, it takes a large amount of computing resources to calculate systems with high-Z elements, and a long running time is required.
Yet, density functional theory (DFT) can conveniently deal with the electron-correlation problems of heavy elements without obviously increasing the amount of computation.29–32 Therefore, we used the spin-polarized generalized gradient approximation (GGA) Becke–Perdew (BP86) exchange-correlation functional.33 Because the scalar and spin–orbit coupling relativity effect needs to be considered, the zeroth-order regular approximation method (ZORA) was also introduced.34–36 In addition, a triple-ζ with polarization functions (TZP) uncontracted Slater-type orbital (STO) basis set was used,37 with a [1s2–4f14] frozen core for the Th atom and a [1s2–4d10] frozen core for Au atoms. In the process of geometric optimization, no symmetric constraints were applied. Meanwhile, the vibration frequency of the obtained structure was analyzed under the same basis set and functional. There is no imaginary frequency, which ensures that the result is feasible. All the calculations were performed using the Amsterdam Density Functional package.38
Results and discussion
Th@Au14 is a typical superatom consisting of an f-element embedded in a coinage-metal structure (as shown in Fig. 1). The calculations of different initial structures show that its ground state is a singlet, and the superatom configuration can be summarized as 1S21P61D10, which is consistent with our previous report.19 Here, we are committed to using the EDA method to explore the interaction between the Th atom and Au14-cage structure with the electron configuration 1S21P61D6 because their interaction plays a major role in the bonding process of Th@Au14. | ||
| Fig. 1 A diagram showing the structures and properties of Th@Au14, including geometric structure, orbital energy levels and charge density difference, in which the possible orbitals of electron occupation have been listed, such as 6d, 37A1 and 25B1. The red solid lines and dotted lines represent the contribution of Frag. 1 and Frag. 2 to the Th@Au14 orbitals, respectively. In addition, in the lower left corner is the charge density difference of Th@Au14. Red and blue represent, respectively, losing and gaining electrons. | ||
It is known that the ground state electron structure of the Th atom is 6d27s2, and its valence electrons are excessively active. If the external environment changes, the 7s electrons of the Th atom may be excited to the 6d shell and create an excited state. Furthermore, during their bonding of the Th atom and Au14-cage structure, their charge distribution will undergo a strong charge rearrangement. These bring a variety of possible performances to their bond properties. However, in the studies of intramolecular interaction, the closed-shell singlet of fragments is generally considered, which is one-sided to the analysis of the interactions in systems with high-Z elements.
Thus, all the possible electron occupation types of the Th atom and Au14-cage structures are considered for a total of six types (Fig. 2). For convenience, the Th atom and Au14-cage structure are called Frag. 1 and Frag. 2, respectively. Meanwhile, the six calculation results of the EDA are listed in Table 1.
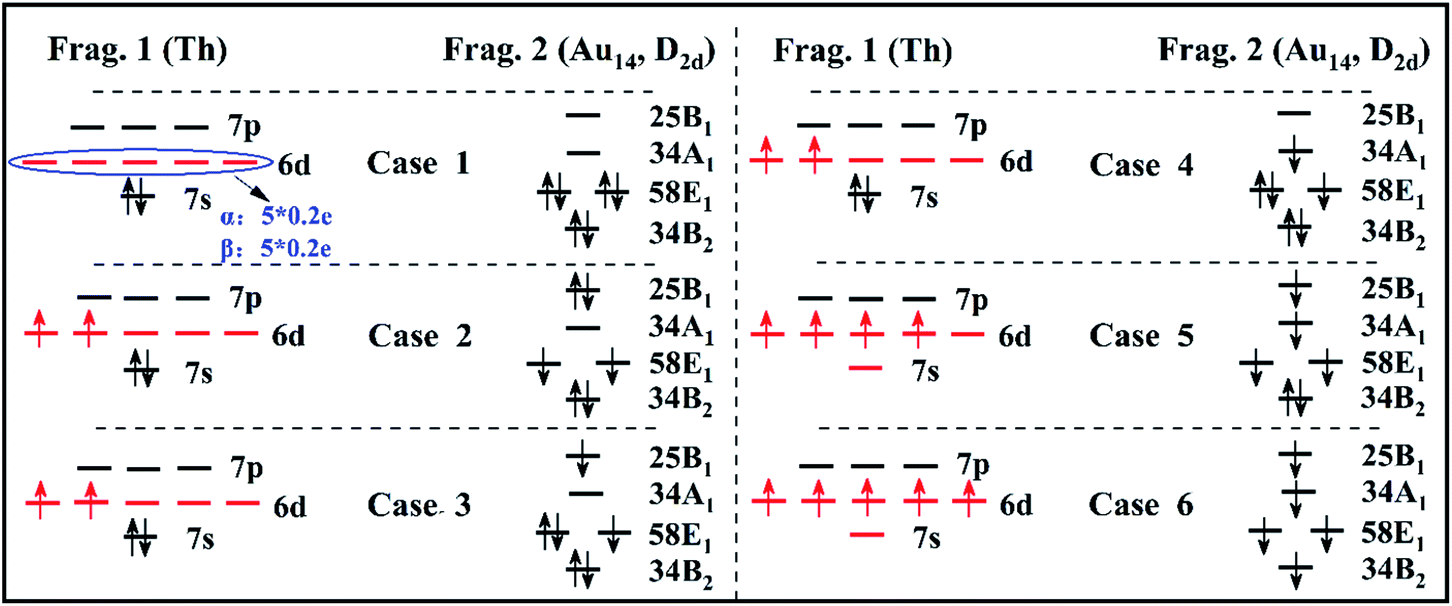 | ||
| Fig. 2 The electron occupations of the Th atom and Au14-cage structure. A total of six electron occupations (Case 1–6) are considered. 6d and 7s are the possible orbitals occupied by valence electrons in the Th atom. 25B1, 34A1, 58E1, 34B2, 23A1 are the possible orbitals occupied by valence electrons in the Au14-cage structure. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| ΔEint | −15.34 | −14.77 | −14.68 | −14.67 | −16.17 | −23.15 |
| ΔEelstat | −48.67 (46.21%) | −48.68 (59.04%) | −48.67 (58.69%) | −48.67 (58.72%) | −39.41 (64.08%) | −67.43 (76.97%) |
| ΔEorb | −56.66 (53.79%) | −33.77 (40.96%) | −34.26 (41.31%) | −34.22 (41.28%) | −22.09 (35.92%) | −20.17 (23.03%) |
| ΔEpauli | 89.99 | 67.68 | 68.25 | 68.22 | 45.33 | 64.45 |
The closed-shell singlet of the fragments is usually considered in an interaction analysis using EDA, so we first discuss the case that fragments 1 and 2 are both closed-shell singlet electron structures (see Fig. 2 Case 1). Yet the 6d shell of Frag. 1 has five degenerate orbitals which are occupied by two unpaired electrons. To conform that there be no spin electrons in the atom, the number of each 6d orbital electrons is defined as 0.4 e. The number of spin-up electrons is 0.2 e, and spin-down electrons is 0.2 e. At this point, the quantitative energy components were calculated by EDA. Herein, the Pauli repulsion energy is 89.99 eV. The absolute value of the orbital interaction energy is 56.66 eV, with a ratio in the total attraction term (orbital interaction energy and electrostatic interaction energy) of 53.79%. Moreover, the percentage of electrostatic interaction energy (−48.67 eV) to total attraction is 46.21%.
Because the valence electrons of the Th atom are 6d27s2 and the ground state is triplet, the next three electron occupation types (see Fig. 2 Case 2–Case 4) it is considered that the electronic structure of Frag. 1 is open-shell. In the 6d orbital, the spin direction of the two electrons is upward. Correspondingly, Frag. 2 needs to have two spin-down electrons to ensure that the ground state of Th@Au14 is a singlet. Since the symmetry of the Au14-cage structure is D2d, its orbitals are indicated by the irreducible representation (B2, E1, A1, B1) of D2d. In Case 2, there are two spin-down electrons occupying the 58E1 double degenerate orbitals of Frag. 2. Simultaneously, the interaction energy components change markedly. The Pauli repulsion energy becomes 67.68 eV. The absolute value of the orbital interaction energy reduces to 33.77 eV, and the ratio in the total attraction term becomes 40.96%. Their electrostatic interaction energy (−48.68 eV) has barely changed, but its percentage has changed to 59.04%.
Case 3 is to adjust the occupying position of the two spin-down electrons on the basis of Case 2. Here, there are three electrons in the 58E1 double degenerate orbital of Frag. 2 and one electron in the 25B1 orbital. The total number of unpaired electrons is two (spin directions are downward). Subsequently, the case of an unpaired electron occupying the 25B1 orbit adjusted to occupy the 34A1 orbit in Frag. 2 is regard as Case 4. Their energy components are also approximately the same as in Case 2. Their absolute value of orbital interaction energy reduces to 34.26 eV and 34.22 eV, and the ratio in the total attraction term becomes 41.31% and 41.28%, respectively. However, their electrostatic interaction energy (−48.67 eV) barely changes, but their percentage changes to 58.69% and 58.64%.
With a change of external environment, the 7s electrons may be excited to the 6d shell. So, different to the above four types, Case 5 is to consider its excited state (6d27s2 → 6d4) as Frag. 1. Then, for Frag. 2 there are four spin-down unpaired electrons, which occupy the 25B1, 34A1 and 58E1 double degeneracy orbitals. Simultaneously, the Pauli repulsion energy significantly decreased to 45.33 eV. The absolute value of the orbital interaction energy and electrostatic interaction energy also declined. However, the electrostatic interaction energy increases to 64.08% of the total attraction term.
To better elucidate the interaction between the two fragments, the charge density difference analysis of Th@Au14 was carried out (Fig. 1). It shows that Frag. 1 gains charges, and Frag. 2 loses charges. Through the previous studies on Th@Au14, the 6d, 7s of Frag. 1 are matched with 1D and 1S of Frag. 2, respectively. The composition of the 7s orbital only accounts for about 3.50% of the 1S superatomic orbital of Th@Au14. Accordingly, for Case 6 (see Fig. 2 Case 6) it can be judged that the two 7s electrons are internally excited to the 6d shell to form the 6d4 electronic state of Frag. 1. Then an electron is captured from Frag. 2 to form the 6d5 electronic state. Finally, Frag.1 turns into [Th]−, its electronic configuration is 6d57s0. Frag. 2 turns into [Au14]+. Owing to losing an electron, its superatom configuration becomes 1S21P61D5. In the resulting energy components, the absolute value of their electrostatic interaction energy (67.43 eV) increases significantly, accounting for 76.97% of the total attraction term.
Through the EDA results, it can be found that, in the same electron state, the interactions have obvious variances between the two fragments under different electron occupations. When the electronic structures of the two fragments are closed-shell singlets, their main interaction energy is the orbital interaction energy (53.79%), which is because the wave-function overlap dominates their interaction. Compared with the closed-shell singlet, if the electronic structures of the two fragments become open-shell, the main interaction energy will transform to electrostatic interaction energy (59.04%). Moreover, their Pauli repulsion interaction energy would be apparently decreased. Its decrement can be regarded as compensation for the orbital energy increase, which results in a slight change for the total interaction. The two fragments are considered to be combined in the form of an ion. This is consistent with interactions in some systems that have similar encapsulation structures and their fragments are in the open-shell.39,40 Yet the contribution of orbital interaction to the total interaction cannot be ignored.
Once the Th atom is in the excited state, the total interaction between the two fragments decreases slightly, indicating that the combination of the fragments is stronger. Furthermore, orbital anti-symmetrizing and renormalizing bring about a clear reduction of the Pauli repulsion energy resulting in the growth of their attractive effect. Herein, the proportion of electrostatic interaction energy increases distinctly. Although the sixth case is similar to the fifth, the total interaction energy becomes lower. Their interaction can be regarded as an electrostatic interaction energy (76.97%). Due to the charge transfer between Frag. 1 and Frag. 2, the attraction between the nucleus and the electrons is increased.
In predicting the structures and properties of superatoms directly using first principles, what is usually obtained is an intuitive understanding, such as the total system energy, bond energy, etc. Through decomposing them into physically meaningful items, EDA can help to predict the properties of difficult observations in experiments. However, high angular momentum electron orbital symmetries are complex; in theory, f-electrons can match electrons with s, p, d, and f orbital symmetry, which makes the electronic structure and bonding properties of systems with a high-Z element more complex. Here, using quantitative analysis of the interaction between fragments under different electron occupations using EDA, it can be found that the electronic structures of the fragments play a vital role in the bonding properties of superatoms with high-Z elements. When the electron occupation type of the fragments is a closed-shell singlet, their interaction is dominated by the overlap of wavefunction. Once the electron occupations of the fragments become open-shell, the electrostatic interaction is dominant. Thus, this provides a referenced route to regulate superatomic interactions of high-Z elements.
Conclusions
In conclusion, in the same electron state, the interaction between fragments with high-Z elements brings about obvious differences under different electron occupation types. During the bonding of the fragments, various electron occupation types can change the instantaneous interaction energy composition, such as electron relaxation, charge transfer and polarization. Through specific practical problems combined witn charge analysis to consider electron occupation types is of great significance for the interaction comprehensive analysis of f-element embedded in coinage-metal structure. Moreover, once all the different electron occupation types can be realized, then an important reference is generated for the regulation and control of the interactions in superatoms with high-Z elements.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (under grant numbers 11974136 and 11575071). Z. W. also acknowledges the High Performance Computing Center of Jilin University.Notes and references
- I. G. Kaplan, Intermolecular interactions: physical picture, computational methods and model potentials, John Wiley & Sons, 2006 Search PubMed.
- G. Zundel, Hydration and intermolecular interaction: infrared investigations with polyelectrolyte membranes, Elsevier, 2012 Search PubMed.
- J. W. Harbour, R. X. Luo, A. D. Santi, A. A. Postigo and D. C. Dean, Cell, 1999, 98, 859–869 CrossRef CAS PubMed.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- C. Yang, C. Wang, S. Zhang, J. Huang and P. Zhou, Mol. Simul., 2015, 41, 741–751 CrossRef CAS.
- Q. Wang, S. Pan, S. Lei, J. Jin, G. Deng, G. Wang, L. Zhao, M. Zhou and G. Frenking, Nat. Commun., 2019, 10, 3375 CrossRef PubMed.
- W. Xie, Y. Zhu, J. Wang, A. Cheng and Z. Wang, Chin. Phys. Lett., 2019, 36, 116401 CrossRef CAS.
- J. Wang, K. Gong, F. Lu, W. Xie, Y. Zhu and Z. Wang, Adv. Theory Simul., 2020, 1900226 CrossRef.
- Z. Luo and A. W. Castleman, Acc. Chem. Res., 2014, 47, 2931–2940 CrossRef CAS PubMed.
- P. Jena and Q. Sun, Chem. Rev., 2018, 118, 5755–5870 CrossRef CAS PubMed.
- A. C. Reber, S. N. Khanna and A. W. Castleman, J. Am. Chem. Soc., 2007, 129, 10189–10194 CrossRef CAS PubMed.
- J.-i. Nishigaki, K. Koyasu and T. Tsukuda, Chem. Rec., 2014, 14, 897–909 CrossRef CAS PubMed.
- S. A. Claridge, A. W. Castleman, S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 CrossRef CAS PubMed.
- T. Jayasekharan and T. K. Ghanty, J. Phys. Chem. C, 2010, 114, 8787–8793 CrossRef CAS.
- B. Yin, J. Li, H. Bai, Z. Wen, Z. Jiang and Y. Huang, Phys. Chem. Chem. Phys., 2012, 14, 1121–1130 RSC.
- X. Li, B. Kiran, L.-F. Cui and L.-S. Wang, Phys. Rev. Lett., 2005, 95, 253401 CrossRef PubMed.
- H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847–1859 RSC.
- Y. Gao, W. Jiang, L. Chen, J. Wang and Z. Wang, J. Mater. Chem. C, 2017, 5, 803–806 RSC.
- Y. Gao, B. Wang, Y. Lei, B. K. Teo and Z. Wang, Nano Res., 2016, 9, 622–632 CrossRef CAS.
- J. Wang, W. Xie, J. Wang, Y. Gao, J. Lei, R.-Q. Zhang and Z. Wang, Phys. Chem. Chem. Phys., 2018, 20, 27523–27527 RSC.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- M. v. Hopffgarten and G. Frenking, WIREs Comput. Mol. Sci., 2012, 2, 43–62 CrossRef.
- S. Cotton, Lanthanide and actinide chemistry, John Wiley & Sons, 2013 Search PubMed.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46–48 CrossRef CAS PubMed.
- V. Errico, G. Arrabito, S. R. Plant, P. G. Medaglia, R. E. Palmer and C. Falconi, Sci. Rep., 2015, 5, 12336 CrossRef PubMed.
- K. L. D. M. Weerawardene, H. Häkkinen and C. M. Aikens, Annu. Rev. Phys. Chem., 2018, 69, 205–229 CrossRef CAS PubMed.
- M. J. Phipps, T. Fox, C. S. Tautermann and C.-K. Skylaris, Chem. Soc. Rev., 2015, 44, 3177–3211 RSC.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- L. Andrews, B. Liang, J. Li and B. E. Bursten, J. Am. Chem. Soc., 2003, 125, 3126–3139 CrossRef CAS PubMed.
- J. Li, H.-S. Hu, J. T. Lyon and L. Andrews, Angew. Chem., Int. Ed., 2007, 46, 9045–9049 CrossRef CAS PubMed.
- J. T. Lyon, H.-S. Hu, L. Andrews and J. Li, Proc. Natl. Acad. Sci., 2007, 104, 18919 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- A. Marjolin, C. Gourlaouen, C. Clavaguéra, J.-P. Dognon and J.-P. Piquemal, Chem. Phys. Lett., 2013, 563, 25–29 CrossRef CAS.
- B. Vlaisavljevich, P. Miró, C. J. Cramer, L. Gagliardi, I. Infante and S. T. Liddle, Chem.–Eur. J., 2011, 17, 8424–8433 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |