
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro oxidations of low-density lipoprotein and RAW 264.7 cells with lipophilic O(3P)-precursors†
John T. Petroff IIa,
Ankita Isora,
Satyanarayana M. Chintala a,
Carolyn J. Albertb,
Jacob D. Frankeb,
David Weinsteina,
Sara M. Omlida,
Christopher K. Arnatta,
David A. Fordb and
Ryan D. McCulla*a
a,
Carolyn J. Albertb,
Jacob D. Frankeb,
David Weinsteina,
Sara M. Omlida,
Christopher K. Arnatta,
David A. Fordb and
Ryan D. McCulla*a
aDepartment of Chemistry, Saint Louis University, St. Louis, MO, USA. E-mail: ryan.mcculla@slu.edu
bDepartment of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, St. Louis, MO, USA
First published on 15th July 2020
Abstract
A beneficial property of photogenerated reactive oxygen species (ROS) is the capability of oxidant generation within a specific location or organelle inside a cell. Dibenzothiophene S-oxide (DBTO), which is known to undergo a photodeoxygenation reaction to generate ground state atomic oxygen [O(3P)] upon irradiation, was functionalized to afford localization within the plasma membrane of cells. The photochemistry, as it relates to oxidant generation, was studied and demonstrated that the functionalized DBTO derivatives generated O(3P). Irradiation of these lipophilic O(3P)-precursors in the presence of LDL and within RAW 264.7 cells afforded several oxidized lipid products (oxLP) in the form of aldehydes. The generation of a 2-hexadecenal (2-HDEA) was markedly increased in irradiations where O(3P) was putatively produced. The substantial generation of 2-HDEA is not known to accompany the production of other ROS. These cellular irradiation experiments demonstrate the potential of inducing oxidation with O(3P) in cells.
Introduction
Reactive oxygen species (ROS) are widely utilized and studied in their endogenous and exogenous forms through the lens of biology.1–5 These species come in the form of singlet oxygen (1O2), ozone (O3), hydroxy radicals (˙OH), superoxide (O2−), peroxides (ROOR), and others. Some ROS, such as O2− and ˙OH, are particularly biologically relevant since they are produced endogenously.6,7 However, others, especially 1O2, have been used in various applications to study the role of ROS in biology relating to oxidative stress.3,8–12 The role of oxidative stress in biology and medicine is of great importance as endogenous ROS are involved in normal cell signaling and diseases alike.10,13–17 With oxidative stress underpinning several of the global leading causes of death, expanding the capacity to generate ROS in cells to further elucidate the pathophysiology of oxidative stress is important.18Currently, there exists a variety of means to generate or introduce ROS and oxidative stress in cells, with photosensitizers and peroxides being leading methods.4,5 Photosensitizer upon irradiation typically excite oxygen to singlet oxygen or undergo electron transfer generating superoxide. One of the downsides to many of these methods is a lack of selectivity where an entire cell may be exposed to the exogenous ROS, and there exist cases where this ‘shotgun blast’ approach is not favorable.3,5,19 Ground state atomic oxygen [O(3P)] is an oxidant which has shown selectivity towards the oxidation of specific functional groups in biomolecules, and thus, O(3P) may be useful in biological oxidations.12,20,21 O(3P) is a frequent topic of research in atmospheric chemistry, but has only been studied in solution for the last few decades.22–25 An additional feature of photogeneration of O(3P) is the precursors carry the oxidant and are not reliant on endogenous molecular oxygen to create ROS, as is the case for photosensitizers. This freely diffusing oxidant is typically generated through irradiation of heterocycle oxides.26,27 For example, dibenzothiophene S-oxide (DBTO) generates O(3P) and the corresponding deoxygenated product dibenzothiophene (DBT) as shown in Fig. 1.23,25,26,28,29
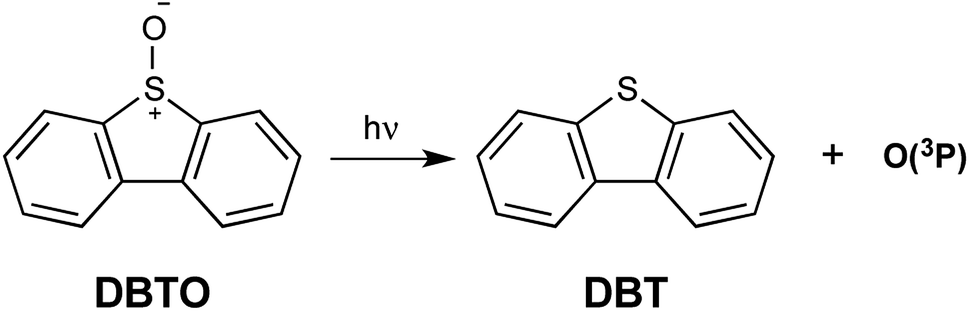 | ||
| Fig. 1 Generation of DBT and O(3P) through irradiation of DBTO. | ||
Recently, it has been confirmed through fluorescent microscopy that functionalizing dibenzothiophene S,S-oxides (DBTOOs) allows for non-toxic organelle-specific localization in HeLa cells.30,31 This was achieved with 4-(8-octyl-5,5-dioxidodibenzo[b,d]thiophen-2-yl)butanoic acid (1-SO2) whose analogous sulfoxide is 4-(8-octyl-5-oxidodibenzo[b,d]thiophen-2-yl)butanoic acid (1-SO) and with 4-(5,5-dioxidodibenzo[b,d]thiophen-2-yl)butanoic acid (2-SO2) whose sulfoxide analog is 4-(5-oxidodibenzo[b,d]thiophen-2-yl)butanoic acid (2-SO) as shown in Fig. 2. With confirmation that the DBT backbone can be localized in organelles, the plasma membrane microscopy dyes (1-SO2 and 2-SO2) were chosen as the model systems to mimic their sulfoxide analogs. The dyes, 1-SO2 and 2-SO2, have good overlap with known plasma membrane dyes.31 As the sulfone (1-SO2 and 2-SO2) and sulfoxide (1-SO and 2-SO) are structurally similar, it was posited 1-SO and 2-SO would localize within plasma membranes of the cell.
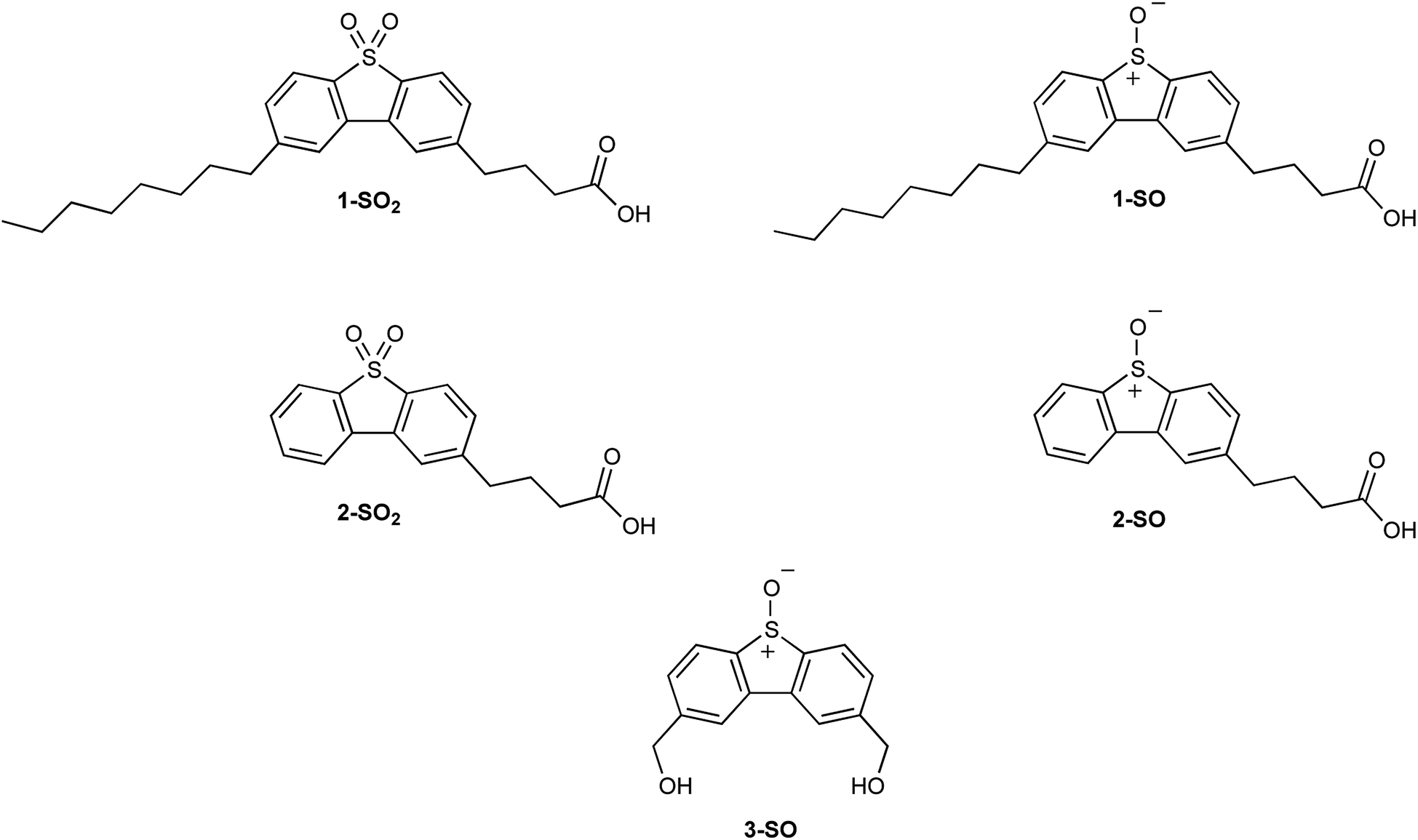 | ||
| Fig. 2 Compounds 1-SO2, 1-SO, 2-SO2, 2-SO, and 3-SO. | ||
Previous studies generating O(3P) with 2,8-bis(hydroxymethyl)dibenzo[b,d]thiophene 5-oxide (3-SO) in the presence of 1-O-hexadec-1′-enyl-sn-glycero-3-phosphocholine (pLPC), low-density lipoprotein (LDL), and RAW 264.7 cells produced evidence that O(3P) generates unique products unseen with other ROS.20 The formation of these unique oxidized lipid products (oxLP) was seen when pLPC and LDL, but not RAW 264.7 cells, were exposed to O(3P). In light of these results, it was hypothesized that O(3P)-precursors with increased lipophilicity would afford more oxLP in these systems. To test this hypothesis, sulfoxide analogs (1-SO & 2-SO) of the sulfone plasma membrane dyes were synthesized.
To complete the primary objective of testing the hypothesis stated above, the photochemistry and oxidation of LDL and RAW 264.7 cells by 1-SO, 2-SO, and 3-SO was investigated. Their oxidative photochemistry was vetted and their ability to generated O(3P) was verified. These O(3P) precursors were exposed to LDL and then, separately, RAW 264.7 cells in the presence of UVA light. The LDL and cells were subsequently analyzed by GC-MS to determine if unique oxidized lipid products could be found. Following analysis of the MS data, it was determined that irradiation of photolabile lipophilic O(3P) precursors in the presence of LDL or RAW 264.7 cells generate 2 hexadecenal (2-HDEA) in amounts significantly greater than control. This data suggests that unique, and previously unachievable, oxidation chemistry is achieved when O(3P) precursors are irradiated while inside of RAW 264.7 cell membranes.
Results and discussion
It has been shown, with fluorescent microscopy, that functionalization of dibenzothiophene sulfones (DBTOOs) with particular substituents will direct the small molecule to localize in specific cellular sites and organelles.30,31 The addition of a butanoic acid to the 2 position of a DBTOO with or without an octyl group at the 8 position (1-SO2 and 2-SO2) provided staining of the plasma membrane in cells.31 Extrapolating from the capacity for 1-SO2 and 2-SO2 to localize in the plasma membrane, it was posited their sulfoxide analogs 1-SO and 2-SO would be similarly lipophilic. Thus, 1-SO and 2-SO were synthesized to determine if increased lipophilicity of a O(3P)-precursor enhanced the formation of lipid oxidation products upon irradiation.The synthesis of 1-SO and 2-SO mirrors the previously reported synthesis of 1-SO2 and 2-SO2 until each molecule's respective last step.31 However, to produce 1-SO as shown in Scheme 1, a Kumada coupling was conducted with 2-bromodibenzothiophene (4) and n-octyl magnesium bromide in dry tetrahydrofuran (THF) with a nickel catalyst.31 Following workup and purification with normal phase chromatography, 2-octyldibenzo[b,d]thiophene (5) was isolated in a 64% yield.32 The 2-octyldibenzo[b,d]thiophene (5) was added to a solution of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloroethane (DCE)/nitrobenzene with succinic anhydride in the presence of aluminum trichloride (AlCl3) and the reaction was kept under an inert atmosphere. The addition of the oxobutanoic acid to form 4-(8-octyldibenzo[b,d]thiophen-2-yl)-4-oxobutanoic acid (6) was followed by reduction of the carbonyl to form 4-(8-octyldibenzo[b,d]thiophen-2-yl)butanoic acid (1-S). The deviation from the previously reported synthesis involves the formation of the sulfoxide, rather than a sulfone, which was performed with 1.1 equivalents of mCPBA at −30 °C which afforded 4-(8-octyl-5-oxidodibenzo[b,d]thiophen-2-yl)butanoic acid (1-SO) in a 69% yield.
:1 dichloroethane (DCE)/nitrobenzene with succinic anhydride in the presence of aluminum trichloride (AlCl3) and the reaction was kept under an inert atmosphere. The addition of the oxobutanoic acid to form 4-(8-octyldibenzo[b,d]thiophen-2-yl)-4-oxobutanoic acid (6) was followed by reduction of the carbonyl to form 4-(8-octyldibenzo[b,d]thiophen-2-yl)butanoic acid (1-S). The deviation from the previously reported synthesis involves the formation of the sulfoxide, rather than a sulfone, which was performed with 1.1 equivalents of mCPBA at −30 °C which afforded 4-(8-octyl-5-oxidodibenzo[b,d]thiophen-2-yl)butanoic acid (1-SO) in a 69% yield.
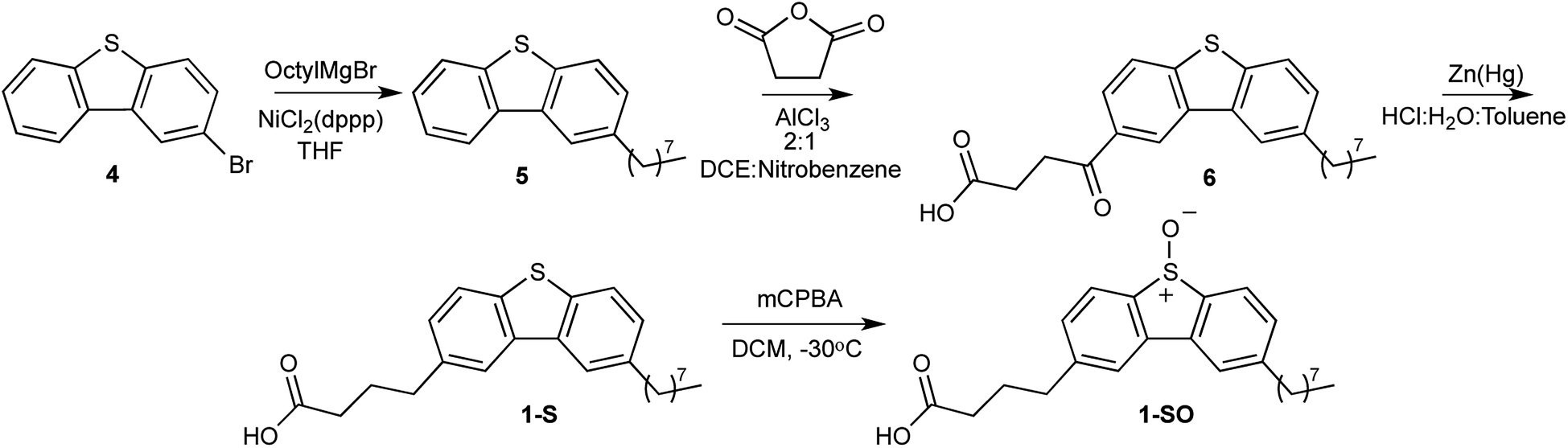 | ||
| Scheme 1 Synthesis of 1-SO. | ||
To synthesize 2-SO, a nearly identical approach was taken with 1-SO sans the use of 4 as a reactant. Dibenzothiophene (7) was treated with succinic anhydride in the presence of a catalyst to produce 4-(dibenzo[b,d]thiophen-2-yl)-4-oxobutanoic acid (8). The carbonyl of 8 is subsequently reduced using zinc amalgam in acidic conditions to form 4-(dibenzo[b,d]thiophen-2-yl)-butanoic acid (2-S). This sulfide is oxidized to the sulfoxide with mCPBA. This reaction produces 4-(5-oxidodibenzo[b,d]thiophen-2-yl)-butanoic acid (2-SO) in 52% yield (Scheme 2).
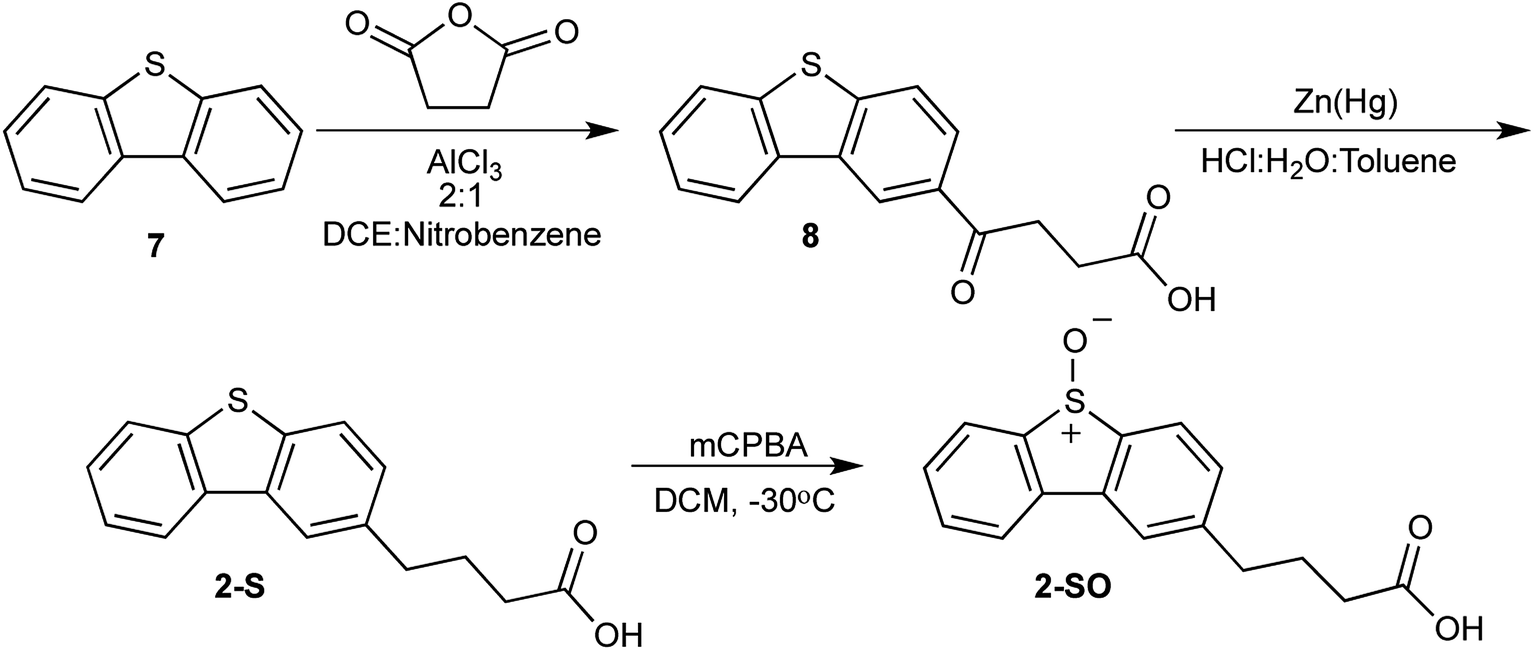 | ||
| Scheme 2 Synthesis of 2-SO. | ||
To examine the oxidation profiles of 1-SO and 2-SO, a solution of the respective sulfoxide in toluene was degassed by argon sparging and then irradiated with 14 broadly emitting LZC-UVA bulbs for four hours. In previous reports, the standard generator of O(3P), DBTO, produced benzaldehyde, benzyl alcohol, o-cresols, and m/p-cresols as shown in Fig. 3.26,33 However, the ratio of the oxidized products for toluene has been found to be very sensitive to degassing techniques and the particular sulfoxide undergoing deoxygenation.34 Incremental changes in residual molecular oxygen appear to affect benzylic oxidation in toluene as photo-generated O(3P) is speculated to react with residual O2 to form ozone that increases the formation of benzaldehyde and benzyl alcohol.34 For example, larger headspaces or longer irradiation periods led to increased benzaldehyde and benzyl alcohol even after argon sparging.34 Also, after performing freeze–pump–thaw (FPT) cycles,28 which is a superior degassing technique, irradiation of DBTO in toluene afforded no benzaldehyde. Functionalization or modification of the DBTO chromophore with aromatic substituents has been observed to decrease the yield of oxidized products.33,34 These observations with DBTO functionalization suggest that other additional processes, which do not involve O(3P), may produce benzaldehyde and enhance benzyl alcohol production in these experiments. Conversely, O(3P), when unadulterated by residual O2, favors cresols formation.26
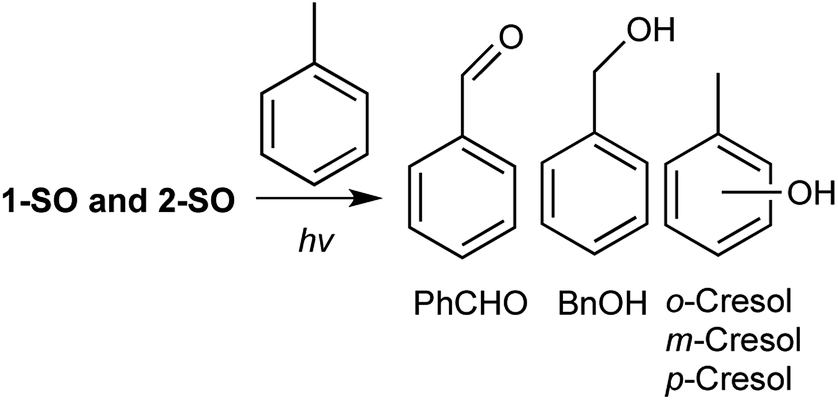 | ||
| Fig. 3 Toluene common intermediate experiment with the irradiation of 1-SO and 2-SO. | ||
The results of common intermediate experiments of 1-SO and 2-SO with toluene are listed in Table 1. Since benzylic oxidation is an unreliable indicator in this common intermediate experiment due to its sensitivity to dissolved molecular oxygen, the ratios of o-cresol and m/p-cresol formation were compared. Both 1-SO and 2-SO generated more o-cresol than m/p-cresols in a nearly 2:1 ratio. Likewise, DBTO generated more o-cresol; however, the ratio was closer to 1:1. While there may be other processes involved for 1-SO and 2-SO that increased the observed ratio, cresol formation in and of itself can indicate O(3P) is being produced during photo-deoxygenation.33 Additionally, the total yield of the oxidized products for 1-SO was half of that observed for DBTO. For 2-SO, nearly identical yields of cresols were observed compared to 1-SO. However, the yields of benzaldehyde and benzyl alcohol increased to 14.7% and 21.4%, respectively. The difference in yields among 1-SO, 2-SO, and DBTO, despite the similarity of the chromophores, underscores the sensitivity of this reaction to the reaction conditions. However, the formation of cresols in a similar ratio for 1-SO and 2-SO indicate that their photodeoxygenation yields some O(3P) even if other background processes cannot be ruled out.
| Compound | Toluene oxidation product yieldsa | ||||
|---|---|---|---|---|---|
| Benzaldehyde | Benzyl alcohol | o-Cresol | m/p-Cresolb | ϕsulfidec | |
| a Yields of toluene oxidation products were calculated relative to the corresponding sulfide produced during photodeoxygenation. Error was determined as a 95% confidence interval.b Measured as single peak.c Deoxygenation quantum yields of sulfide formation in acetonitrile.d Data from Satyanarayana et al.35e Data from Gregory et al.28f Data from ref Rockafellow et al.27g Data from this work. | |||||
| DBTOd | 5 ± 3 | 6 ± 3 | 17.6 ± 0.9 | 13 ± 1 | 0.0026 ± 0.0004e |
| DBTOf | 17 ± 3 | 13 ± 4 | 26 ± 5 | 22 ± 5 | 0.0046 ± 0.0007f |
| DBTOg | 1.8 ± 1.2 | 2.0 ± 0.1 | 11 ± 2 | 8.6 ± 3.5 | — |
| 1-SOg | 2.3 ± 1.0 | 3.1 ± 0.1 | 7.8 ± 0.5 | 4.1 ± 0.1 | 0.0012 ± 0.0002g |
| 2-SOg | 14.7 ± 4.1 | 21.4 ± 1.2 | 7.5 ± 0.7 | 4.8 ± 1.1 | 0.0020 ± 0.0003g |
To examine the efficiency of photodeoxygenation for 1-SO and 2-SO, the quantum yield of sulfide formation (ϕsulfide) for 1-S and 2-S, respectively, was determined. The ϕsulfide was determined by irradiating a degassed sample of a sulfoxide in an inert solvent, such as acetonitrile, at a given wavelength, typically 320 nm.26,27 The quantum yield of deoxygenation for DBTO was approximately 0.003.28 Saturated solutions of 1-SO and 2-SO were prepared in acetonitrile, argon sparged, and irradiated at 320 nm. The irradiations were halted prior to exceeding 10% sulfoxide conversation to ensure that the sulfoxide absorbed the majority of the light. After the corresponding sulfide formation was determined and flux was measured by chemical actinometry, the ϕsulfide for 1-SO and 2-SO were found to be 0.0012 and 0.0020, respectively. These values are lower than that of DBTO, with 1-SO preforming at nearly half the efficiency of 2-SO.
Low-density lipoprotein (LDL) is comprised of surface-containing phospholipids, which are known to react with O(3P), and a central core of other lipids including glycerides and cholesterol esters.20 A substantial portion of the esterified fatty acids of LDL are known to be polyunsaturated, providing a population of likely targets for O(3P), which has an affinity for unsaturated hydrocarbons.36,37 Previously, 3-SO has been used to generate O(3P) in solution with LDL and isolated phospholipids.20 In this previous work, UV-irradiation of LDL in the presence of 3-SO resulted in a substantial increase of four aldehyde products compared to UV-irradiation alone. These four aldehydes were: tetradecanal (TDA), pentadecanal (PDA), hexadecenal (HDA), and 2-hexadecenal (2-HDEA). In this work, the formation of these four aldehydes plus octadecanal (ODA) as shown in Fig. 4, were monitored as evidence for lipid oxidation induced by photodeoxygenation of 1-SO, 2-SO, and 3-SO.
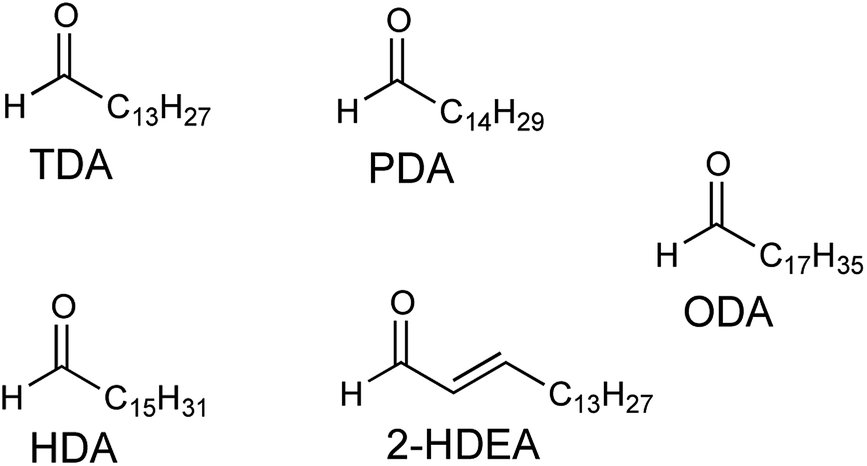 | ||
| Fig. 4 Potential lipid oxidation products arising from photodeoxygenation. | ||
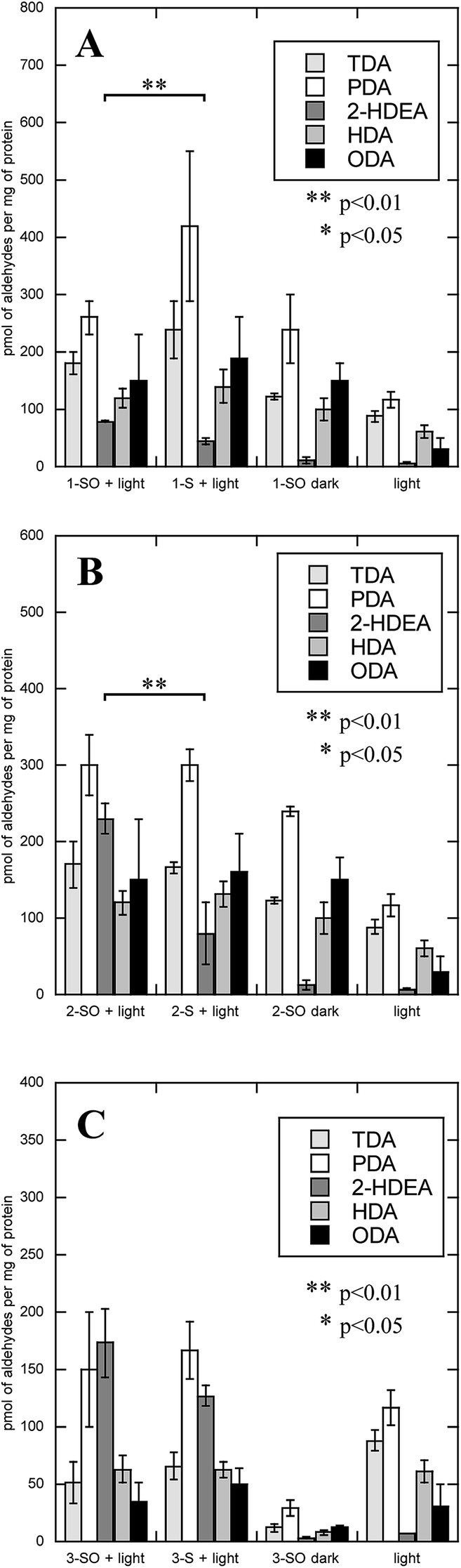
LDL also serves as a more complex system than an isolated phospholipid in solution, allowing one to determine if the lipophilicity of the O(3P)-precursor enhances oxLP. To measure oxLPs, 1 mL solutions of either 200 mM 1-SO, 2-SO, 3-SO, 1-S, 2-S, and dibenzo[b,d]thiophene-2,8-diyldimethanol (3-S) with 2 mg mL−1 of LDL were irradiated with broadly emitting UVA bulbs for 2 hours. Additional control experiments included 1-SO, 2-SO, 3-SO, 1-S, 2-S, and 3-S being incubated in the dark with LDL and irradiating LDL alone. After irradiation, the samples were subject to Bligh–Dyer extractions and derivatized with O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine hydrochloride (PFB) and then analyzed on GC-MS, where the molecular ions for PFB-derivatized TDA, PDA, HDA, 2-HDEA, and ODA were monitored. The results of these experiments are shown in Fig. 5.
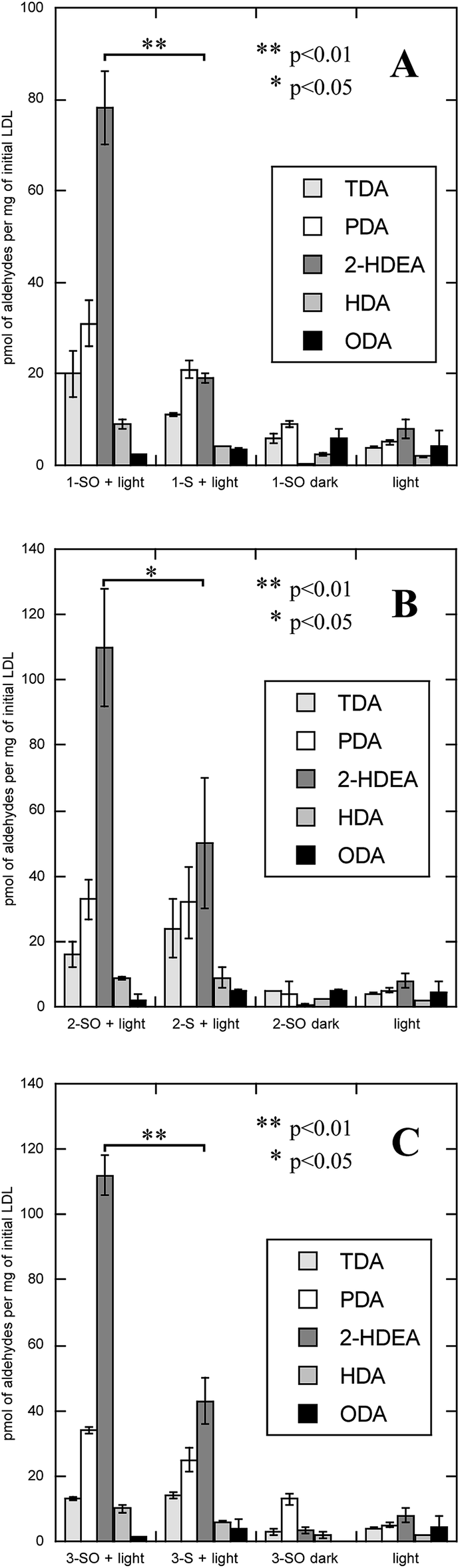 | ||
| Fig. 5 Amount of oxidized lipid product arising from LDL after irradiation of O(3P)-precursors and additional control experiments. (A) LDL oxidized by the photodeoxygenation of 1-SO (1-SO + light) and controls, which were the corresponding sulfide in light (1S + light), 1-SO without irradiation (1-SO dark), and irradiation of cells without addition of compounds (light). (B) LDL oxidized by the photodeoxygenation of 2-SO (2-SO + light) and controls similar to (A) with 2-SO or 2-S. (C) LDL oxidized by the photodeoxygenation of 3-SO (3-SO + light) and controls similar to (A) with 3-SO or 3-S. | ||
The oxidation of LDL by all three sulfoxides (1-SO, 2-SO, and 3-SO) upon UV irradiation gave similar results (Fig. 5A: 1-SO + light, Fig. 5B: 2-SO + light, Fig. 5C: 3-SO + light). For all three sulfoxides, the amount of 2-HDEA had the largest increase compared to control experiments, and the total amounts of lipid aldehydes detected generally increased. In the control experiments with only irradiation and no compounds (Fig. 5, light) or no light (Fig. 5A: 1-SO + dark, Fig. 5B: 2-SO + dark, Fig. 5C: 3-SO + dark), less than 10 pmol per mg LDL for any one aldehyde was detected. To verify that photodeoxygenation of 1-SO, 2-SO, and 3-SO was the sole cause of the observed increase of aldehydes, the corresponding sulfides 1-S, 2-S, and 3-S were tested (Fig. 5A: 1-S + light, Fig. 5B: 2-S + dark, Fig. 5C: 3-S + light). For all three sulfides, a significant increase in TDA, PDA, and 2-HDEA compared to the other control experiments was observed. Compared to their corresponding sulfoxides, the sulfides yielded approximately the same, if not more, TDA and PDA. Additionally, 2-HDEA was the dominant aldehyde product for both 2-S and 3-S. However, the three sulfides all produced at least 60 pmol less 2-HDEA per mg of LDL than their corresponding sulfoxides. Compared to all three controls, only the increase in 2-HDEA for 1-SO, 2-SO, and 3-SO and light had p-values of less than 0.05 indicating significance. These results led to the conclusion that photodeoxygenation has the most substantial effect on the formation of 2-HDEA.
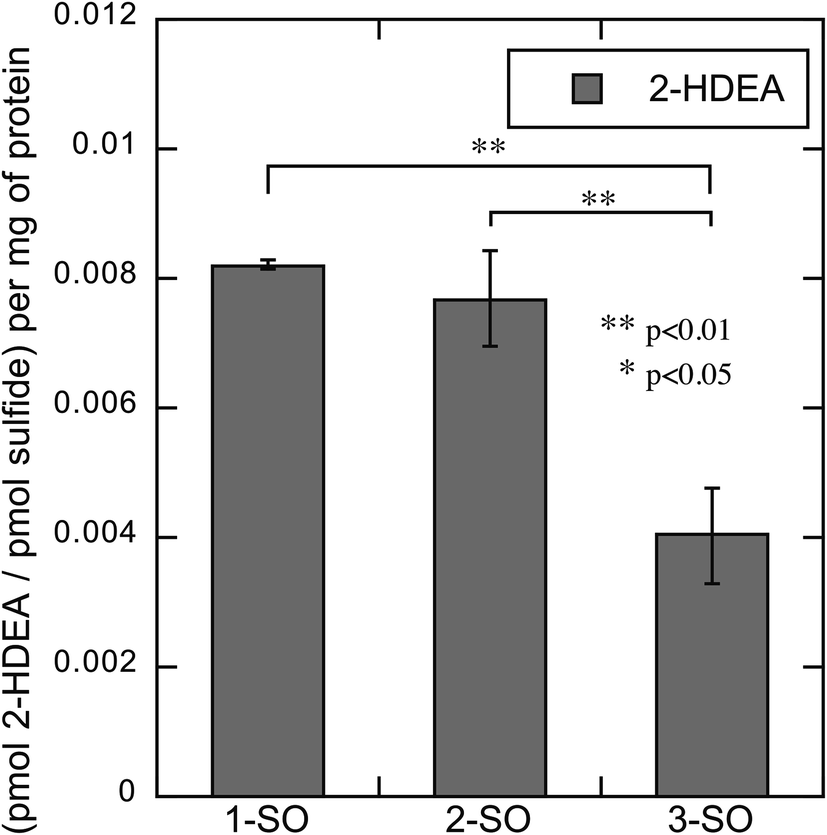
The rate of photodeoxygenation for 1-SO, 2-SO, and 3-SO was not the same as seen in Table 1 and previous work.38 This discrepancy stems from quantum yield measurements being done under anaerobic conditions in organic solvent, which is the standard approach.27,34 However, these irradiations reported here occurred in aqueous media under aerobic conditions, which is known to influence quantum ϕsulfide.38 Thus, to quantify the extent to which photodeoxygenation was affecting 2-HDEA formation, the amount of 2-HDEA formed compared to sulfide formation (i.e. 1-S, 2-S, and 3-S) was determined. To determine the extent of photodeoxygenation, 1-SO, 2-SO, and 3-SO were irradiated in the same conditions as were used to oxidize LDL except without LDL present. The results of these experiments are used to accurately account for 2-HDEA formation and are shown in Fig. 6. Both 2-SO and 3-SO produced similar amounts of 2-HDEA relative to the amount of deoxygenation. This was consistent with the similar quantum yields for 2-SO and 3-SO, 0.0012 and 0.002,38 respectively, and the nearly identical amounts of 2-HDEA formed as shown in Fig. 5. The most lipophilic sulfoxide, 1-SO, produced over double the amount of 2-HDEA per sulfide generated compared to 2-SO and 3-SO. Thus, the smaller quantum yield for 1-SO compared to 2-SO and 3-SO is likely the reason for the decreased amount of 2-HDEA for 1-SO in Fig. 5. The logP values of 1-SO, 2-SO, and 3-SO were calculated or measured at 6.26, 2.86, and 1.88, respectively.21 Overall, these results indicate the increased lipophilicity of 1-SO makes it more efficient at producing 2-HDEA in LDL.
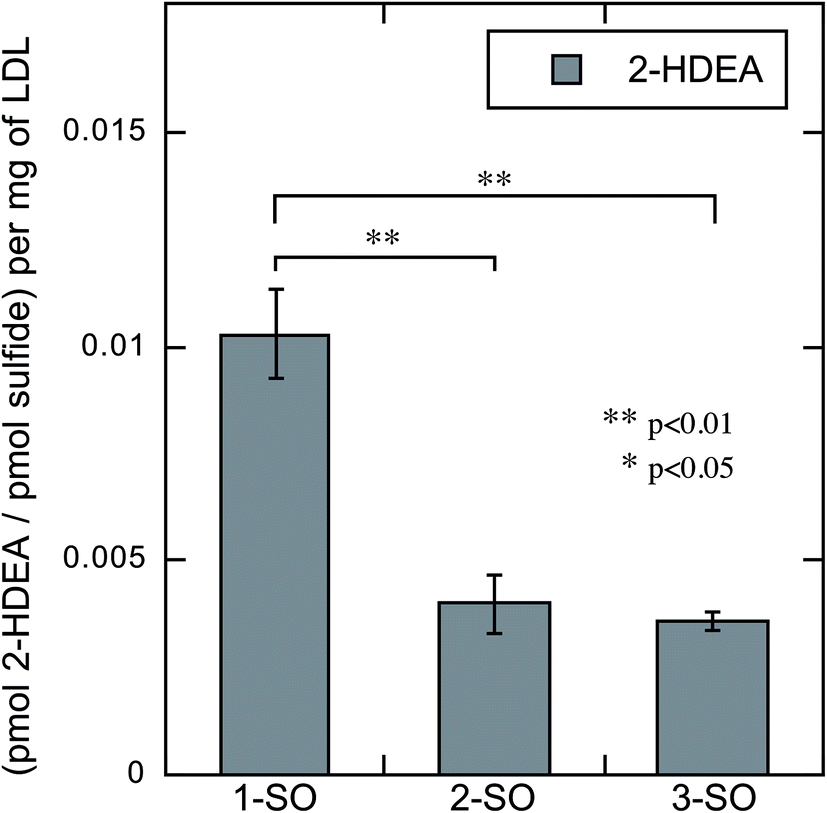 | ||
| Fig. 6 Amount of 2-HDEA produced in LDL relative to the extent of photodeoxygenation of 1-SO, 2-SO, and 3-SO upon irradiation. | ||
In previous oxidations of LDL with 3-SO, 2-HDEA was suspected of arising from oxidation of the vinyl ether group of plasmalogens, which is known to react with various ROS.20,39,40 While 2-HDEA was the major product when isolated plasmalogens and LDL were exposed to O(3P), 2-HDEA has not been observed to form with other ROS. Singlet oxygen (1O2) formed long-chain aldehydes when generated in Chinese hamster ovarian cells.41 Hydroxyl radical (˙OH) formed α-hydroxyaldehydes when reacted with LDL, and ozone (O3) forms long-chain saturated aldehydes when generated in the presence of plasmalogens.20,40–42 This indicates the 2-HDEA was produced from a reaction between O(3P) and plasmalogens.
While several other ROS have been generated in cells, O(3P) has not.4,5,20,43–45 The previously reported efficacy of 1-SO2 and 2-SO2 as microscopy dyes suggested that 1-SO and 2-SO would also incorporate into cells.31 To examine the capacity of 1-SO, 2-SO, and 3-SO to produce oxLP, RAW 264.7 cells were chosen as the target cell line, since plasmenylethanolamine comprises approximately 36% of the ethanolamine glycerolipid pool.46 An analog of plasmenylethanolamines in the form of pLPC has previously been shown to react readily with O(3P) in solution.20
As with many cell lines, RAW 264.7 cells are sensitive to UV light, and previous work had found that 5 minutes of irradiation was enough to induce cell death.47 Thus, cell viability was examined to determine the amount of cell death under the irradiation conditions used in this study. RAW 264.7 cells were grown in RPMI supplemented with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin (Thermo Fisher 10000 U mL−1), and 1% GlutaMAX. Cells were split and then seeded into 96-well plates with 10000 cells per well. The plate was then incubated for 24 hours. Vehicle control was added to the wells to reach a final concentration of 0.2% DMSO in DPBS. The plates were then incubated for 10 minutes and transferred to the photoreactor where they were irradiated with 14 broadly emitting UVA bulbs. Cell viability was then determined using a MTS assay. While the cells tolerated 5 minutes UV exposure, cell viability decreased to 23% and −3% at 1 and 2 hours, respectively, as shown in Fig. 7.
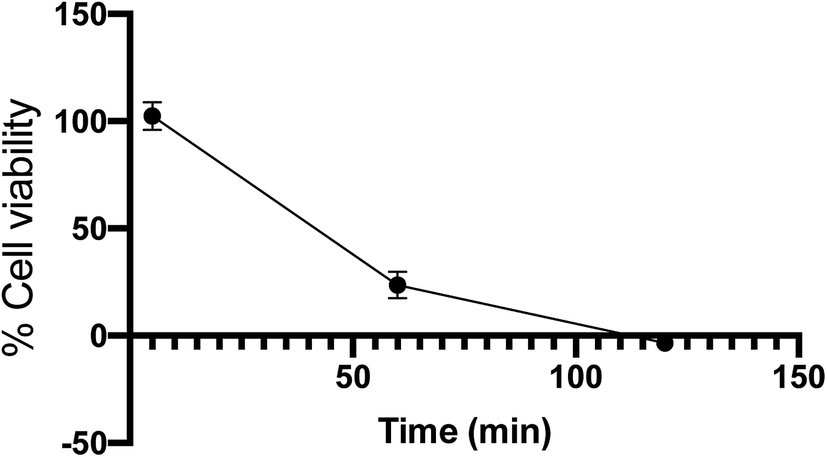 | ||
| Fig. 7 Cell viability of RAW 264.7 cells determined by MTS assay after irradiation with UVA bulbs over time. Error bars are 95% confidence intervals. | ||
To examine the morphology of the cells at 2 hours, the cells were plated on a cell culture dish (10 × 200 mm) with 88000 cells per mL density. The cell culture dish was incubated for 24 hours and then placed in a photoreactor with 14 broadly emitting UV-A bulbs. The cell culture dish was then examined on Olympus BX60 microscope (Fig. S2†). As expected, there morphological changes were consistent with cell death; however, the cell membrane remained largely intact. Thus, while cell viability was significantly decreased, the ability more lipophilic O(3P)-precursors to generate lipid oxidation products in cells could still be examined.
As described above, RAW 264.7 cells were grown in media as described above before being split and seeded into 6-well plates with 1 × 106 cells per well. The plate was incubated for 24 hours, allowing the cells to multiply to confluency. After the incubation media was removed, the given compound (1-SO, 2-SO, 3-SO, 1-S, 2-S, or 3-S) was added to the well in a final concentration of 200 μM in 1 mL of PBS. The 6-well plate was then incubated for an additional 10 minutes. After incubation, the plate was transferred to the photoreactor where it was irradiated with broadly emitting UVA bulbs for 2 hours. Once irradiation was complete, Bligh–Dyer extractions were performed. Once Bligh–Dyer extractions were complete, the sample was purified over a SupelCo SupelClean LC-Si column using chloroform as the eluant. The isolated eluent was evaporated, reconstituted, and PFB derivatized. The crude derivatization solution was then purified by liquid–liquid extraction. The extract was then prepared for analysis by GCMS using the same method as was used with LDL.
The irradiation of RAW 264.7 cells with (1-SO, 2-SO, 3-SO, 1-S, 2-S, or 3-S), the associated dark controls, and the untreated control afforded oxLPs in the form of TDA, PDA, HDA, 2-HDEA, and ODA for all conditions. These results are shown in Fig. 7. Unlike LDL, the control experiments of irradiation without compounds (Fig. 8, light) and no light (Fig. 8A, 1-SO dark, Fig. 8B, 2-SO dark) demonstrated an overall increase in oxLP, with the exception of 3-SO in the dark. In fact, 3-SO in the dark gave nearly identical amounts of the OLPs as untreated cell as shown in the ESI (Fig. S3†). Compared to 3-SO dark, the exposure of RAW 264.7 cells to UV irradiation resulted in significant increases of TDA, PDA, HDA, and ODA. This was not surprising since UV exposure is known to generate aldehydes in cells.48,49 Additionally, substantial increases in TDA, PDA, HDA, and ODA were observed for 1-SO and 2-SO compared to 3-SO when incubated in the dark. This is consistent with the previous studies that have shown destabilization of the cellular plasma membrane make it more susceptible to oxidation.50,51 Thus, the addition of lipophilic small molecules and UV-irradiation appears to increase the rate of oxidation leading to TDA, PDA, HDA, and ODA; however, these processes have no effect on 2-HDEA formation.
 | ||
| Fig. 8 Amount of oxidized lipid product arising from RAW 264.7 cells after irradiation of O(3P)-precursors and additional control experiments. (A) RAW 264.7 cells oxidized by the photodeoxygenation of 1-SO (1-SO + light) and controls, which were the corresponding sulfide in light (1S + light), 1-SO without irradiation (1-SO dark), and irradiation of cells without additional compounds (light). (B) RAW 264.7 cells oxidized by the photodeoxygenation of 2-SO (2-SO + light) and controls similar to (A) with different compounds. (C) RAW 264.7 cells oxidized by the photodeoxygenation of 3-SO (3-SO + light) and controls similar to (A) with different compounds. | ||
Since small lipophilic molecules and UV-irradiation naturally resulted in an increase in TDA, PDA, HDA, and ODA, it was not surprising that the irradiation of RAW 264.7 cells with 1-SO, 2-SO, 3-SO, 1-S, 2-S, or 3-S resulted in increase of these four aldehydes. However, unlike the controls which formed little 2-HDEA, irradiation of RAW 264.7 cells with 1-SO, 2-SO, 3-SO, 1-S, 2-S, or 3-S all showed an increase in 2-HDEA. All the sulfoxides yield more 2-HDEA than the corresponding sulfide, which was consistent with the results observed for LDL. For 1-SO and 2-SO, they produced approximately double the amount of 2-HDEA compared to 1-S and 2-S, respectively. This increase in 2-HDEA for 1-SO and 2-SO compared to their corresponding sulfides was found to correspond to p-values of less than 0.01 indicating significance. Interestingly, 3-SO only produced 30% more 2-HDEA than 3-S, which could not be used to support the hypothesis that photodeoxygenation of 3-SO had any effect on 2-HDEA formation. This was consistent with previous reports where no noticeable quantity of oxLP could be measured compared to the controls for 3-SO.20 These results indicated that increased lipophilicity of the O(3P)-precursors 1-SO and 2-SO assists in producing more 2-HDEA upon irradiation and concomitant photodeoxygenation.
The increase in TDA, PDA, HDA, and ODA in the dark when RAW 264.7 cells were exposed to the sulfoxides suggested that 1-SO, 2-SO, and 3-SO may have some toxicity. The cell viability of RAW 264.7 cells upon incubation of 1-SO and 1-S without the subsequent irradiation was examined. While 1-S showed no dark toxicy, a decrease of cell viability to 24% was observed at 200 μM for 1-SO (Fig. S1†). Lower concentrations had no significant effect on cell viability.
As with LDL, the amount of deoxygenation for 1-SO, 2-SO, and 3-SO was not expected to be the same. Thus, the amount of 2-HDEA formed compared to sulfide formation was determined to quantify the effect of photodeoxygenation on 2-HDEA formation. The sulfide formation was quantified in the same manner as how the LDL sulfide formation was measured, except the samples were housed in a 6-well plate. The results are shown in Fig. 9. When the extent of photodeoxygenation is considered, 1-SO and 2-SO had nearly the same yield of 2-HDEA and nearly double of what was observed for 3-SO. This was unlike LDL, where only 1-SO had a higher yield of 2-HDEA. A potential reason for this difference would be if 1-SO and 2-SO both incorporated into the plasma membrane of the RAW 264.7 cells to a similar extent. This supposition is supported by the similar amounts of oxLP observed for 1-SO and 2-SO when incubated with RAW 264.7 cells in the dark (Fig. 8A 1-SO dark, Fig. 8B 2-SO dark).
 | ||
| Fig. 9 Amount of 2-HDEA produced in RAW 264.7 cells relative to the extent of photodeoxygenation of 1-SO, 2-SO, and 3-SO upon irradiation. | ||
Conclusion
Two lipophilic DBTO derivatives (1-SO and 2-SO) were synthesized, and these molecules were concluded to generate O(3P) by common intermediate experiments with toluene, where they both generated cresols upon photodeoxygenation. The O(3P) precursors, 1-SO and 2-SO, have ϕsulfide of 0.0012 and 0.002, respectively. These sulfoxides, as well as 3-SO and the control experiments, generated OLPs in the form of TDA, PDA, HDA, 2-HDEA, and ODA when irradiated in the presence of LDL. The principal product for the LDL irradiations was 2-HDEA which agrees with previous reports and has only been attributed to O(3P) formation. When controlling for total sulfide formation for the respective sulfoxide and LDL irradiations, it becomes apparent that the greater the lipophilicity of the molecule, as measured by logP, the more oxLP formed. RAW 264.7 cells were treated with 1-SO, 2-SO, 3-SO and associated controls then irradiated. The sulfoxides and sulfides afforded meaningful amounts oxLP in the form of TDA, PDA, HDA, and ODA; however, 2-HDEA, the oxLP attributed to O(3P), was only generated in appreciable amounts with the sulfoxides 1-SO, 2-SO, and 3-SO. When this data was corrected for sulfide formation it again became apparent that an increase of lipophilicity of the O(3P)-precursor led to increased oxLP formation. This work with RAW 264.7 cells is the first report of O(3P)-precursors being used to generate oxidized products in cells. This opens the possibility that O(3P)-precursors may be functionalized to direct their photocleaved oxidant in some specified manner in cells.
Experimental
Materials
2,8-Bis(hydroxymethyl)dibenzo[b,d]thiophene 5-oxide (3-SO) and the corresponding sulfide (3-S) were prepared from dibenzothiophene as previously reported.38 2-[15,15,16,16,16-d5]-hexadecenal (2-[d5]-hexadecenal) was also prepared by a previously reported method as well.52 Low-density lipoprotein was purchased from Lee Biosolutions. Compounds 1-S, 2-S, 4, 5, 6, 7, and 8 were all prepared following previously reported procedures.31 All other chemicals were purchased from Alfa Aesar, Oakwood Chemical, Ark Pharm, Thermo Fisher Scientific, and Sigma-Aldrich and were used without further purification. Flash chromatography was performed using a Biotage Horizon with silica gel (SiliaFlash P60) purchased from SiliCycle. 1H and 13C NMR spectra were gathered on a Bruker DRX-400 in either DMSO-d6 or CDCl3. logP calculations were completed using ChemDraw Ultra 12.0. For gas chromatograph analysis, a Shimadzu GC-2010 Plus with an AOC-201 Auto Injector on a Restek Rxi-5ms column was used. An Agilent 1200 Series HPLC fitted with a quaternary pump and diode array detector was used for HPLC chromatographs run on an Agilent Eclipse XDB-C18 column (5 mm, 150 × 4.6 mm). High-resolution mass spectra were measured using a Thermo Scientific Q Exactive Orbitrap equipped with a Nano ESI ionization source. Absorption spectra were recorded using a Shimadzu UV-1800 UV-Vis spectrophotometer using samples dissolved in acetonitrile contained in a 10 × 10 mm quartz cell at a concentration of 0.025 and 0.035 mM for 2-SO and 1-SO, respectively.
:1 EtOAc:hexanes with 0.1% acetic acid. The silica associated with the product band was washed with 95% EtOAc and 5% MeOH. The filtrate was evaporated under reduced pressure to yield an off-white solid (101.3 mg, 69%). 1H NMR (chloroform-d,400 MHz): δ (ppm) 7.89 (t, J = 7.6 Hz, 2H), 7.62 (d, J = 6.8 Hz, 2H), 7.30 (d, J = 7.8 Hz, 2H), 2.80 (t, J = 7.7 Hz, 2H), 2.72 (t, J = 7.7 Hz, 2H), 2.42 (t, J = 7.3 Hz, 2H), 1.97–2.08 (m, 2H), 1.68 (quin, J = 7.3 Hz, 2H), 1.21–1.41 (m, 10H), 0.82–0.94 (m, 3H) 13C NMR (chloroform-d, 101 MHz): δ (ppm) 177.3, 148.4, 146.6, 143.2, 143.2, 142.6, 142.6, 137.7, 137.3, 129.9, 129.7, 127.6, 127.4, 36.2, 35.2, 32.9, 31.8, 31.3, 29.4, 29.2, 26.1, 22.6, 14.1, 14.1, 26.0 MS (ESI) calculated for C24H29O3S+ m/z: 399.1988, found m/z: 399.1982.:1 EtOAc:hexanes with 0.1% acetic acid. The silica associated with the product band was washed with 95% EtOAc and 5% MeOH. The filtrate was evaporated under reduced pressure to yield an off white solid (81.5 mg, 52%). 1H NMR (chloroform-d,400 MHz): δ (ppm) 7.98 (d, J = 7.5 Hz, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.79 (d, J = 7.5 Hz, 1H), 7.55–7.65 (m, 2H), 7.46–7.53 (m, 1H), 7.31 (d, J = 7.9 Hz, 1H), 2.72–2.84 (m, 2H), 2.34–2.44 (m, 2H), 1.96–2.05 (m, 2H) 13C NMR (DMSO-d6,101 MHz): δ (ppm) 174.3, 147.3, 145.2, 142.4, 136.8, 136.5, 132.7, 129.9, 129.7, 127.5, 127.4, 122.6, 122.4, 34.5, 33.4, 26.1 MS (ESI) calculated for C16H15O3S+ m/z: 287.0742, found m/z: 287.0729.Common intermediate experiments with toluene
2.0–3.5 mM solutions of the sulfoxides (DBTO, 1-SO and 2-SO) were prepared in toluene with 0.125 mM or 1 mM dodecane, which was used as an internal standard. The undissolved sulfoxide was filtered off, and 4.0 mL of the filtered solution was transferred into a 5 mL quartz cuvette. The solution was degassed by argon sparging for 15–30 min and the solution was irradiated using 14 Luzchem UVA bulbs centered at 350 nm (LZC-UVA). The concentrations of toluene oxidation products were obtained by GC-FID analysis, and the increase in the concentration of sulfide was obtained through HPLC analysis.Quantum yield of deoxygenation as monitored by sulfide formation
2.7–3.5 mM solutions of the sulfoxides (2-SO and 1-SO) were prepared in acetonitrile. The undissolved sulfoxide was filtered, and 4.0 mL of the filtered solution was transferred into a quartz cuvette. The solution, which had an absorption of 2 or greater at 320 nm, was degassed by purging it with argon gas for 10 minutes. The solution was then irradiated with 320 ± 6 nm light using a 75 W xenon arc lamp focused on a monochromator. The reactions were carried out for less than 10% conversion. The concentration increase of the sulfide was obtained using HPLC analysis. Photolysis of azoxybenzene to yield the rearranged product, o-hydroxyazobenzene, was used as a chemical actinometer.34,53Irradiation of LDL with O(3P) precursors, their respective sulfides, and other conditions
2 mg (2 μL) of LDL was added to a 40 mm (length) quartz test tube. To the test tube was added 974 μL of Milli-Q treated water, along with 22.2 μL of 0.009 M sulfoxide (1-SO, 2-SO, or 3-SO) or sulfides (1-S, 2-S, and 3-S) in 10% DMSO PBS solution. For untreated controls, 22.2 μL of a 10% DMSO PBS solution was added in lieu of the compound solution. The test tube was sealed with a SubaSeal septa fitting 11 mm internal diameter. Samples which were to not be exposed to light were then wrapped in aluminum foil. These samples were then placed in a rotating carousel designed to suspend test tubes and the carousel with the samples was then placed into Luzchem LZC-4X with 14 Hitachi FL8BL-B broadly emitting fluorescent bulbs centered at 352 nm (UVA) then irradiated for 2 hours. Immediately following irradiation samples were placed on dry ice prior to analysis.Cell line culture
RAW 264.7 cell line was purchased from ATCC (Manassas, VA). RAW 264.7 cells were incubated at 37 °C in 95% relative humidity with 5% CO2. The cells were cultured in RPMI media containing 10% FBS, 1% glutamine, and 1% penicillin and streptomycin.RAW 264.7 cell lipid oxidation studies
RAW 264.7 cells (1 × 106) were seeded on a six-well cell culture plate in 2 mL of RPMI supplemented with 10% FBS, 1% penicillin–streptomycin (Thermo Fisher 10000 U mL−1), and 1% GlutaMAX. The cells were then incubated for 24 hours at 37 °C in a humidified atmosphere of 5% CO2. Prior to experimental conditions, the cell culture media was removed and 978 μL of PBS was added. To the well was added 22.2 μL of 0.009 M sulfoxide (1-SO, 2-SO, or 3-SO) or sulfides (1-S, 2-S, or 3-S) in 10% DMSO PBS solution. For untreated controls, 22.2 μL of a 10% DMSO PBS solution was added in lieu of the compound solution. The plate was incubated for 10 minutes in the aforementioned conditions. Following the incubation, the plates were set on a rotating flat carousel inside a Luzchem LZC-4C with 14 Hitachi FL8BL-B broadly emitting fluorescent bulbs centered at 352 nm (UVA) and irradiated for 2 hours.
Lipid extraction for LDL and RAW 264.7 cells
The 1 mL volume from the either the LDL test tubes or the RAW 264.7 plates was collected for analysis. Cells were scraped prior to removing the PBS which they were in. To the respective vessel, 1 mL of 154 mM saline solution was added. The LDL test tubes were vortexed and the RAW 264.7 plates were again scrapped. The saline was then combined with the original collection. From the RAW 264.7 combined suspension, 200 μL was removed for protein quantification. The cells and the LDL samples were extracted using the Bligh and Dyer lipid extraction technique in the presence of 10 pmol of 2-[d5]-hexadecenal.54 Extraction was conducted twice per sample. After lipid extraction, samples underwent silica extraction on LC-Si (Supelco) columns using chloroform to separate plasmalogens from free fatty aldehydes.55 The samples were then prepared for GC-MS analysis by derivatization with pentafluorobenzyl (PFB) hydroxylamine. Derivatization with PFB hydroxylamine was performed by resuspending the reaction products in 300 μL of ethanol and 300 μL of PFB hydroxylamine (6 mg mL−1) in water. The ethanol–water mixture was vortexed for 5 min at room temperature, with further incubation at room temperature for 25 min. Following the incubation, 1.2 mL of water was added to the reaction products, and the reaction products were extracted with 4:1 v:v cyclohexane/diethyl ether followed by resuspension in petroleum ether prior to GC-MS analysis.
Aldehyde quantification for LDL and RAW-264.7 cells
After derivatization with PFB hydroxylamine, the samples were analyzed by capillary gas chromatography-mass spectrometry (GC-MS). GC-MS analysis was performed in the negative ion chemical ionization mode (NICI) with methane as the reagent gas using an Agilent J&W DB-1 column (12 m, 0.2 mm inner diameter, 0.33 μm film thickness) with a 6890 gas chromatograph-5973 mass spectrometer (Agilent). The source temperature was set at 150 °C. The electron energy was 193.3 eV and the emission current was 49.4 A. The injector and transfer lines were maintained at 250 °C and 280 °C, respectively. The GC oven was maintained at 150 °C for 3.5 min, increased at the rate of 25 °C min−1 to 310 °C and held at 310 °C for another 5 min. Products quantification was performed using selected ion monitoring (SIM) by comparing integrated area of the respective product to the integrated area produced by the internal standard 2-[d5]-hexadecenal at m/z = 418. The five products monitored were tetradecanal (m/z = 387), pentadecanal (m/z = 401), 2-hexadecenal (m/z = 413), hexadecanal (415) and octadecanal (m/z 443). All statistical comparisons between samples were made using a one-way ANOVA with post-hoc Tukey HSD test.Quantification of protein from RAW 264.7 cell studies
Utilizing the 200 μL aliquot from the RAW 264.7 cell studies, a Bradford assay was conducted using 25 μL of the 200 μL sample, in duplicate, following the standard procedure.56Quantification of sulfide formation in 6-well plates and quartz test tubes
To either the well of a 6-well plate (for RAW 264.7 cells) or a 40 mm quartz test tube (for LDL) 978 μL of PBS (RAW 264.7 cells) or Milli-Q treated water (LDL) was added. To the well or test tube, 22.2 μL of 0.009 M sulfoxide (1-SO, 2-SO, or 3-SO) in 10% DMSO PBS solution was also added. Each condition was produced in duplicate. These samples were then placed in (or on for 6-well plates) a rotating carousel which was placed into Luzchem LZC-4C with 14 Hitachi FL8BL-B broadly emitting fluorescent bulbs centered at 352 nm (UVA) and irradiated for 2 hours. Following irradiation, samples were immediately analyzed on HPLC to measure sulfide formation. The sulfide formation was subsequently quantified using standard curves.RAW 264.7 cell toxicity studies
RAW 264.7 cells were incubated at 37 °C in 95% relative humidity with 5% CO2. The cells were cultured in RPMI media containing 10% FBS, 1% glutamine, and 1% penicillin and streptomycin. The cells were split 1:20 on reaching ∼90% confluency. RAW 264.7 cells were plated at 10 000 cells per well on six 96-well plates (CellStar – Greiner bio-one clear) with a cell suspension volume of 100 μL per well. Three sets of two plates (UV and No-UV) were marked for 5 min UV-A irradiation, 1 hour UV-A irradiation, and 2 hour UV-A irradiation each. The plates were then incubated for 24 hours. After incubation, 50 μL of 0.6% DMSO in DPBS was introduced in the wells (final concentration – 0.2% DMSO in DPBS). The plates were then incubated for 10 minutes. Three plates were irradiated with UV-A light in the photoreactor for 5 min, 1 hour, and 2 hours, respectively. Correspondingly, thermal controls (No-UV) were placed next to the photoreactor wrapped in aluminum foil. After UV-A irradiation, the UV-A along with No-UV plates were placed back into the incubator. The plates were then analyzed the next day using a MTS assay.
To examine the toxicity of 1-SO and 1-S, RAW 264.7 cells were plated at 10000 cells per well on six 96-well plates (CellStar – Greiner bio-one clear) with a cell suspension volume of 100 μL per well. The plates were then incubated for 24 hours. 1 mM stock solutions of 1-SO and 1-S were prepared in 1% DMSO in DPBS. The solutions were vortexed and sonicated for ∼5 minutes. The stock solutions were then used to prepare 3× concentrations (600 μM, 60 μM, 6 μM, 600 nM, 60 nM, 6 nM) in DPBS. Additionally, VC of 0.6% DMSO in DPBS was prepared. After incubation, 50 μL of the 3× solutions (final concentrations – 200 μM, 20 μM, 2 μM, 200 nM, 20 nM, 2 nM) and VC (final concentrations 0.2% DMSO in PBS) were added to the wells and incubated for 10 minutes. The plates were wrapped in aluminum foil and placed outside of the incubator for 2 hours. The plates were then incubated for 24 hours after the experiment and MTS assay was performed the next day.
Cell viability was determined by an MTS assay. A fresh solution of 2 mg mL−1 MTS solution in DPBS and 0.92 mg mL−1 PMS solution in DPBS was prepared. 25 μL of MTS solution (100 μ;L of PMS solution for every 2 mL MTS solution) was added to each well. The plates were then placed back in the incubator for ∼1.5 h and analyzed using Flexstation 3 multimode plate reader and percent viability was calculated. Statistical analysis was performed ad-hoc t-tests were performed with Welch correction using GraphPad Prism. Cell viability for No-UV (control) plates were calculated using (Abssample − Absblank) × 100/(AbsVC − Absblank) and for UV plates were calculated using (Abssample − Absblank) × 100/(Abs(VC-NoUV) − Absblank(NoUV))
Visualization of RAW 264.7 cells pre- and post-UV treatment
Cells were plated on two 10 × 200 mm cell culture dishes (Cellstar by Greiner bio-one) in 5 mL media (cell density 88000 cells per mL) and incubated for 24 hours. One culture dish was irradiated in a Luzchem photoreactor with 14 UV-A LZC bulbs for 2 hours and a control dish was wrapped in foil and kept adjacent to the photoreactor. The dishes were then visualized using a Olympus BX60 Microscope and images were captured at 200× magnification.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under CHE-1900417. A portion of this study was supported by research funding from the National Institutes of Health R01 GM-115553 to D. A. F.References
- R. W. Redmond and J. N. Gamlin, A Compilation of Singlet Oxygen Yields from Biologically Relevant Molecules, Photochem. Photobiol., 1999, 70(4), 391–475 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Photosensitized singlet oxygen and its applications, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- B. V. Chernyak, D. S. Izyumov, K. G. Lyamzaev, A. A. Pashkovskaya, O. Y. Pletjushkina and Y. N. Antonenko, et al., Production of reactive oxygen species in mitochondria of HeLa cells under oxidative stress, Biochim. Biophys. Acta, Bioenerg., 2006, 1757(5), 525–534 CrossRef CAS PubMed.
- J. J. P. Gille and H. Joenje, Cell culture models for oxidative stress: superoxide and hydrogen peroxide versus normobaric hyperoxia, Mutat. Res., DNAging: Genet. Instab. Aging, 1992, 275(3), 405–414 CrossRef CAS.
- A. Pinto, Y. Mace, F. Drouet, E. Bony, R. Boidot and N. Draoui, et al., A new ER-specific photosensitizer unravels 1O2-driven protein oxidation and inhibition of deubiquitinases as a generic mechanism for cancer PDT, Oncogene, 2015, 35, 3976 CrossRef PubMed.
- R. J. Aitken and S. D. Roman, Antioxidant systems and oxidative stress in the testes, Oxid. Med. Cell. Longevity, 2008, 1(1), 15–24 CrossRef PubMed.
- M. P. Murphy, How mitochondria produce reactive oxygen species, Biochem. J., 2009, 417(1), 1–13 CrossRef CAS PubMed.
- I. Liguori, G. Russo, F. Curcio, G. Bulli, L. Aran and D. Della-Morte, et al., Oxidative stress, aging, and diseases, Clin. Interventions Aging, 2018, 13, 757–772 CrossRef CAS PubMed.
- M. Schieber and S. Chandel Navdeep, ROS Function in Redox Signaling and Oxidative Stress, Curr. Biol., 2014, 24(10), R453–R462 CrossRef CAS.
- Y. Posen, V. Kalchenko, R. Seger, A. Brandis, A. Scherz and Y. Salomon, Manipulation of redox signaling in mammalian cells enabled by controlled photogeneration of reactive oxygen species, J. Cell Sci., 2005, 118(9), 1957–1969 CrossRef CAS PubMed.
- J. Korang, I. Emahi, W. R. Grither, S. M. Baumann, D. A. Baum and R. D. McCulla, Photoinduced DNA cleavage by atomic oxygen precursors in aqueous solutions, RSC Adv., 2013, 3(30), 12390–12397 RSC.
- M. Zhang, G. E. Ravilious, L. M. Hicks, J. M. Jez and R. D. McCulla, Redox Switching of Adenosine-5′-phosphosulfate Kinase with Photoactivatable Atomic Oxygen Precursors, J. Am. Chem. Soc., 2012, 134(41), 16979–16982 CrossRef CAS PubMed.
- T. Finkel, Signal transduction by reactive oxygen species, J. Cell Biol., 2011, 194(1), 7–15 CrossRef CAS PubMed.
- K. Venkataraman, S. Khurana and T. C. Tai, Oxidative stress in aging--matters of the heart and mind, Int. J. Mol. Sci., 2013, 14(9), 17897–17925 CrossRef CAS PubMed.
- V. J. Thannickal and B. L. Fanburg, Reactive oxygen species in cell signaling, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2000, 279(6), L1005–L1028 CrossRef CAS PubMed.
- G. Testa, F. Cacciatore, G. Galizia, D. Della-Morte, F. Mazzella and A. Langellotto, et al., Waist Circumference but Not Body Mass Index Predicts Long-Term Mortality in Elderly Subjects with Chronic Heart Failure, J. Am. Geriatr. Soc., 2010, 58(8), 1433–1440 CrossRef PubMed.
- J. K. Paik, J. S. Chae, R. Kang, N. Kwon, S. H. Lee and J. H. Lee, Effect of age on atherogenicity of LDL and inflammatory markers in healthy women, Nutr., Metab. Cardiovasc. Dis., 2013, 23(10), 967–972 CrossRef CAS PubMed.
- N. J. Pagidipati and T. A. Gaziano, Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement, Circulation, 2013, 127(6), 749–756 CrossRef PubMed.
- X. Zhang, Y. Yuan, L. Jiang, J. Zhang, J. Gao and Z. Shen, et al., Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy, Autophagy, 2014, 10(10), 1801–1813 CrossRef CAS PubMed.
- M. T. Bourdillon, B. A. Ford, A. T. Knulty, C. N. Gray, M. Zhang and D. A. Ford, et al., Oxidation of Plasmalogen, Low-Density Lipoprotein and RAW 264.7 Cells by Photoactivatable Atomic Oxygen Precursors, Photochem. Photobiol., 2014, 90(2), 386–393 CrossRef CAS PubMed.
- J. Korang. Photochemical Generation Of Atomic Oxygen O(3P) in Aqueous Medium and Its Biological Application. Doctor of Philosphy thesis, Saint Louis University, Ann Arbor, MI, 2012.
- E. Lucien and A. Greer, Electrophilic Oxidant Produced in the Photodeoxygenation of 1,2-Benzodiphenylene Sulfoxide, J. Org. Chem., 2001, 66(13), 4576–4579 CrossRef CAS.
- K. B. Thomas and A. Greer, Gauging the Significance of Atomic Oxygen [O(3P)] in Sulfoxide Photochemistry. A Method for Hydrocarbon Oxidation, J. Org. Chem., 2003, 68(5), 1886–1891 CrossRef CAS PubMed.
- O. R. Wauchope, S. Shakya, N. Sawwan, J. F. Liebman and A. Greer, Photocleavage of plasmid DNA by dibenzothiophene S-oxide under anaerobic conditions, J. Sulfur Chem., 2007, 28(1), 7 Search PubMed.
- G. Bucher and J. C. Scaiano, Laser Flash Photolysis of Pyridine N-Oxide: Kinetic Studies of Atomic Oxygen [O(3P)] in Solution, J. Phys. Chem., 1994, 98(48), 12471–12473 CrossRef CAS.
- R. D. McCulla and W. S. Jenks, Deoxygenation and Other Photochemical Reactions of Aromatic Selenoxides1, J. Am. Chem. Soc., 2004, 126(49), 16058–16065 CrossRef CAS.
- E. M. Rockafellow, R. D. McCulla and W. S. Jenks, Deoxygenation of dibenzothiophene-S-oxide and dibenzoselenophene-Se-oxide: a comparison of direct and sensitized photolysis, J. Photochem. Photobiol., A, 2008, 198(1), 45–51 CrossRef CAS.
- D. D. Gregory, Z. Wan and W. S. Jenks, Photodeoxygenation of Dibenzothiophene Sulfoxide: Evidence for a Unimolecular S−O Cleavage Mechanism 1, J. Am. Chem. Soc., 1997, 119(1), 94–102 CrossRef CAS.
- S. M. Omlid, S. A. Dergunov, A. Isor, K. L. Sulkowski, J. T. Petroff, E. Pinkhassik and R. D. McCulla, Evidence for diffusing atomic oxygen uncovered by separating reactants with a semi-permeable nanocapsule barrier, Chem. Commun., 2019, 55(12), 1671–1846 RSC.
- J. T. Petroff II, K. N. Skubic, C. K. Arnatt and R. D. McCulla, Asymmetric Dibenzothiophene Sulfones as Fluorescent Nuclear Stains, J. Org. Chem., 2018, 83(22), 14063–14068 CrossRef PubMed.
- J. T. Petroff 2nd, S. Grady, C. K. Arnatt and R. D. McCulla, Dibenzothiophene Sulfone Derivatives as Plasma Membrane Dyes, Photochem. Photobiol., 2020, 96(1), 67–73 CrossRef PubMed.
- J. Liu, S. Hu, W. Zhao, Q. Zou, W. Luo and W. Yang, et al., Novel Spectrally Stable Saturated Blue-Light-Emitting Poly[(fluorene)-co-(dioctyldibenzothiophene-S,S-dioxide)]s, Macromol. Rapid Commun., 2010, 31(5), 496–501 CrossRef CAS PubMed.
- X. Zheng, S. M. Baumann, S. M. Chintala, K. D. Galloway, J. B. Slaughter and R. D. McCulla, Photodeoxygenation of dinaphthothiophene, benzophenanthrothiophene, and benzonaphthothiophene S-oxides, Photochem. Photobiol. Sci., 2016, 15(6), 791–800 RSC.
- J. T. Petroff II, S. M. Omlid, S. M. Chintala and R. D. McCulla, Wavelength dependent photochemistry of expanded chromophore and asymmetric dibenzothiophene S-oxide derivatives, J. Photochem. Photobiol., A, 2018, 358, 130–137 CrossRef.
- S. M. Chintala, J. T. Petroff Ii, A. Barnes and R. D. McCulla, Photodeoxygenation of phenanthro[4,5-bcd]thiophene S-oxide, triphenyleno[1,12-bcd]thiophene S-oxide and perylo[1,12-bcd]thiophene S-oxide, J. Sulfur Chem., 2019, 1–13 Search PubMed.
- Z. Wan and W. S. Jenks, Photochemistry and Photophysics of Aromatic Sulfoxides. 2. Oxenoid Reactivity Observed in the Photolysis of Certain Aromatic Sulfoxides, J. Am. Chem. Soc., 1995, 117(9), 2667–2668 CrossRef CAS.
- L. Viens, A. Athias, G. Lizard, G. Simard, S. Gueldry and S. Braschi, et al., Effect of lipid transfer activity and lipolysis on low density lipoprotein (LDL) oxidizability: evidence for lipolysis-generated non-esterified fatty acids as inhibitors of LDL oxidation, J. Lipid Res., 1996, 37(10), 2179–2192 CAS.
- J. Korang, W. R. Grither and R. D. McCulla, Photodeoxygenation of Dibenzothiophene S-oxide Derivatives in Aqueous Media, J. Am. Chem. Soc., 2010, 132(12), 4466–4476 CrossRef CAS PubMed.
- S. Stadelmann-Ingrand, R. Pontcharraud and B. Fauconneau, Evidence for the reactivity of fatty aldehydes released from oxidized plasmalogens with phosphatidylethanolamine to form Schiff base adducts in rat brain homogenates, Chem. Phys. Lipids, 2004, 131(1), 93–105 CrossRef CAS PubMed.
- K. M. Wynalda and R. C. Murphy, Low-concentration ozone reacts with plasmalogen glycerophosphoethanolamine lipids in lung surfactant, Chem. Res. Toxicol., 2010, 23(1), 108–117 Search PubMed.
- O. H. Morand, R. A. Zoeller and C. R. Raetz, Disappearance of plasmalogens from membranes of animal cells subjected to photosensitized oxidation, J. Biol. Chem., 1988, 263(23), 11597–11606 CAS.
- W. Jira and G. Spiteller, Plasmalogens and their oxidative degradation products in low and high density lipoprotein, Chem. Phys. Lipids, 1996, 79(2), 95–100 CrossRef CAS.
- W. M. Sharman, C. M. Allen and J. E. van Lier, Photodynamic therapeutics: basic principles and clinical applications, Drug Discovery Today, 1999, 4(11), 507–517 CrossRef CAS PubMed.
- R. L. Lipson, E. J. Baldes and M. J. Gray, Hematoporphyrin derivative for detection and management of cancer, Cancer, 1967, 20(12), 2255–2257 CrossRef CAS.
- T. Wei, C. Chen, J. Hou, W. Xin and A. Mori, Nitric oxide induces oxidative stress and apoptosis in neuronal cells, Biochim. Biophys. Acta, Mol. Cell Res., 2000, 1498(1), 72–79 CrossRef CAS.
- R. A. Zoeller, S. Rangaswamy, H. Herscovitz, W. B. Rizzo, A. K. Hajra and A. K. Das, et al., Mutants in a macrophage-like cell line are defective in plasmalogen biosynthesis, but contain functional peroxisomes, J. Biol. Chem., 1992, 267(12), 8299–8306 CAS.
- T. Ikehara, M. Nakahashi, S. Zehong, M. Akutagawa, K. Tsuchiya and M. Kitamura, et al., Effects of UV-A LED light irradiation on growth of cultured RAW 264.7 cells, Toxicol. Environ. Chem., 2015, 97(2), 243–255 CrossRef CAS.
- S. A. Marchitti, Y. Chen, D. C. Thompson and V. Vasiliou, Ultraviolet radiation: cellular antioxidant response and the role of ocular aldehyde dehydrogenase enzymes, Eye Contact Lens, 2011, 37(4), 206–213 CrossRef PubMed.
- M. N. Jones, Surfactants in membrane solubilisation, Int. J. Pharm., 1999, 177(2), 137–159 CrossRef CAS PubMed.
- K. Parsi, Interaction of detergent sclerosants with cell membranes, Phlebology, 2015, 30(5), 306–315 CrossRef PubMed.
- M. Nazari, M. Kurdi and H. Heerklotz, Classifying surfactants with respect to their effect on lipid membrane order, Biophys. J., 2012, 102(3), 498–506 CrossRef CAS PubMed.
- V. V. Brahmbhatt, F.-F. Hsu, J. L. F. Kao, E. C. Frank and D. A. Ford, Novel carbonyl and nitrile products from reactive chlorinating species attack of lysosphingolipid, Chem. Phys. Lipids, 2007, 145(2), 72–84 CrossRef CAS PubMed.
- N. J. Bunce, J. Lamarre and S. P. Vaish, Photorearrangement of azoxybenzene to 2-hydroxyazobenzene: a convenient chemical actinometer*, Photochem. Photobiol., 1984, 39(4), 531–533 CrossRef CAS.
- E. G. Bligh and W. J. Dyer, A rapid method of total lipid extraction and purification, Can. J. Biochem. Physiol., 1959, 37(8), 911–917 CrossRef CAS PubMed.
- V. V. Brahmbhatt, C. Nold, C. J. Albert and D. A. Ford, Quantification of Pentafluorobenzyl Oxime Derivatives of Long Chain Aldehydes by GC-MS Analysis, Lipids, 2008, 43(3), 275–280 CrossRef CAS PubMed.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72(1), 248–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01517b |
| This journal is © The Royal Society of Chemistry 2020 |