
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic thermodynamics and microkinetics of the reduction mechanism of electrolyte additives to facilitate the formation of solid electrolyte interphases in lithium-ion batteries†
Xiao Liua,
Jianhua Zhoua,
Zhen Xu*a and
Yixuan Wang *b
*b
aSchool of Chemical and Pharmaceutical Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250010, PR China. E-mail: xuzhen123@126.com
bComputational Chemistry Laboratory, Department of Chemistry and Forensic Sciences, Albany State University, Albany, GA31705, USA. E-mail: yixuan.wang@asurams.edu
First published on 24th April 2020
Abstract
The formation of a solid electrolyte interphase (SEI) between the anode surface and the electrolyte of lithium-ion batteries (LIBs) has been considered to be the most important yet the least understood issue of LIBs. To further our understanding in this regard, the density functional theory (DFT) B3PW91/6-311++G(3df,3pd) together with the implicit solvent model and the transition state theory were used for the first time to comprehensively explore the electroreduction mechanism of a novel additive, 4-chloromethyl-1,3,2-dioxathiolane-2-oxide (CMDO), and a few other solvents and additives, such as ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), fluoroethylene carbonate (FEC), and even ethylene sulfite (ES), for comparison. The one-electron reduction potential of Li+-coordinated compounds Li+(X) for forming decomposition precursors [c-Li+(X˙−)] decreases in the following sequence: CMDO (1.9–2.2 V vs. Li+/Li) ∼ ES(1.9 V) > FEC (0.7 V) > EC (0.47 V) > PC (0.45 V) > DMC (0.38 V); this implies that CMDO is reduced prior to other solvents or additives in the mixture. Although the ring opening of [c-Li+(CMDO˙−)] is the least kinetically favorable, as reflected by the highest energy barrier (Ea), i.e., CMDO (18.8–22.9 kcal mol−1) ∼ ES (23.4) > FEC (16.2) > PC (12.5) > EC (11.2) > DMC (8.0), CMDO still shows the highest overall reaction rate constant (∼1053 s−1) for forming an open ring radical [o-Li+(CMDO˙)−]. In addition, the termination reaction of [o-Li+(CMDO˙)−] for forming LiCl is thermodynamically more favorable than that of Li2SO3 or organic disulfite (LiSO3)2-R, which supports the experimental observation that the halogen-containing LiF or LiCl additives are predominant over all other halogen-containing species in the SEI layer. Moreover, the hybrid model by including the second solvation shell of Li+ via a supercluster [(CMDO)Li+(PC)2](PC)9 and the implicit solvent model (SMD) can result in a reduction potential (∼1.7 V) that is in excellent agreement with the experimental reduction peak.
1 Introduction
It is accepted that the stability, safety, and lifetime of rechargeable lithium ion batteries (LIBs) are closely related to the characteristics of the solid electrolyte interphase (SEI) formed between the electrolyte and anode surface.1–3 The composition, formation and growth mechanism of the SEI layer has therefore been the most important issue for the relevant investigations in the field of LIBs.2,4–6 The SEI film is initiated by the reduction of the electrolyte components, which include either solvents, additives, salts, or their combination, during the first couple of cycles.2,4,7–10 The nature of the SEI layer and its formation and growth mechanism therefore depend primarily on the composition of the electrolyte of LIBs.Because of its low melting point (∼–49 °C) and relatively high dielectric constant (∼64), PC is an excellent solvent for LIBs at low temperatures and is usually paired with low-viscosity linear carbonates like DMC or DEC. However, the mixture of PC and DMC as a solvent rapidly destroys the graphite anode and significantly decreases the reversible capacity of the LIB,11,12 which is usually attributed to the failure of the formation of an efficient SEI. It is very intriguing that the addition of a small amount (2–5% volume) of additives to the PC-based electrolyte can considerably improve the SEI layer formation and the consequent performance of the LIB.
In the past decades, numerous studies have been experimentally and theoretically devoted to examining various additives for PC-based electrolytes used to improve the quality of the SEI layer on the anode surface. The most studied additive is the unsaturated cyclic carbonate, vinylene carbonate (VC).13–20 Halogen-substituted carbonates such as fluoroethylene carbonate (FEC) and difluoroethylene carbonate (DFEC) have also been found to considerably improve the formation of the SEI.21,22 It was reported by Aurbach et al. that for FEC additives, LiF is predominant over all other F-containing species in the SEI layer.23
Other widely explored types of additives for PC-based electrolytes are sulfur-containing compounds ranging from sulfites (ethylene sulfite (ES) and propylene sulfite),10,16,19,24,25 to sultones (1,3-propane sultone, propene sultone)15,19,26 and sulfones (methyl vinyl sulfone, ethyl vinyl sulfone).16 It has been shown that the resistance of the sulfur-derived SEI layer is small.27–30 In addition, sulfur- and chlorine-containing additives have been able to improve the performance of the LIB at low temperatures.27,28 Although there are many theoretical and experimental efforts in this regard, the SEI is still considered the least understood component in LIBs, yet it presents the most critical problems.31
The above mentioned additives usually have higher reduction potentials than conventional solvents like EC, PC, and DMC, and the products from their reduction can build up a better SEI film.32 To explore the role of the additives in facilitating the formation of the SEI, the electroreductions of the additives and solvents for PC(EC)-based electrolytes have been investigated with first-principles based theoretical calculations and analysis such as density functional theory (DFT) and ab initio molecular dynamics in a few studies.13,17–20,33 The higher reduction potentials of the electrolyte additives imply that the additives are thermodynamically favorable and can be reduced prior to the solvents; however, the formation of SEI intermediates from the reduced cyclic carbonates and sulfites is kinetically unfavorable, reflected by the energy barriers higher than those of the solvents.33,34 To resolve this complication, we proposed a comprehensive model to estimate the overall reaction rate by applying a steady state to the formation of a reductive precursor and classical transition state theory (TST) to the ring opening.34 The overall reaction rate constants for forming the SEI intermediates, i.e., the ring opening radical of the electrolyte additives, are usually greater than those of EC and PC.
Very recently, a new electrolyte additive, 4-chloromethyl-1,3,2-dioxathiolane-2-oxide (CMDO), modified from ES by adding a chloromethyl group, was prepared to improve the commonly used PC-based electrolyte,35 and its performance was investigated in different combinations of EC, FEC, and CMDO. The addition of CMDO was shown to produce a thinner SEI film than that with FEC and was able to decrease the irreversible capacity loss. The combination of the three additives (CMDO, FEC, and EC) with PC/DMC plays a critical role in the enhanced reversible capacity of the carbon type anode at lower or ambient temperatures by producing a thin and uniform SEI film.
To provide a good understanding at a molecular level about the functional mechanism for the above CMDO mixed solvents, a DFT-based theoretical calculation and analysis was extensively performed for the Li+(X) (X = EC, PC, DMC, FEC, ES, and CMDO) and supermolecular clusters [(CMDO)Li+(PC)2] and [(CMDO)Li+(PC)2](PC)9. Using systematic investigations, we aim to (1) comprehensively investigate the reduction mechanism of CMDO for the first time in terms of thermodynamics (reduction potential and Gibbs free energy for the formation of the intermediate radicals and products), kinetics (energy barriers and rate constants), and major products; (2) compare the thermodynamics and kinetics of CMDO reductive decomposition with other additives and solvents after a careful and consistent theoretical calculation; (3) extend the previously developed hybrid model (supercluster + explicit solvent + implicit solvent)34 to CMDO, which may be able to provide excellent agreement with experiments for potential energy.
2 Models and computational details
For Li+(X) (X = EC, PC, DMC, FEC, and ES), the first electron reduction was previously investigated with different theoretical methods,8,9,13,34,36–38 yet most of them were in a vacuum phase or just supplemented by the single point calculation with implicit continuum models, such as the polarized continuum model (PCM) or SMD, rather than performing the full optimization in bulk solvent. In the present study, to consistently discuss the reduction mechanisms, a full optimization including the implicit solvent effect with SMD39 was first carried out with B3PW91/6-311++G(d,p) (SMD-B3PW91) for Li+(X) and Li+(CMDO). In the electrolyte solution of LIB, the most probable lithium ion species is the Li+ that is approximately solvated by three solvent molecules in the first solvation shell, such as Li+(PC)3, Li+(EC)3, and (ES)Li+(PC)2.30,34,37 Therefore, the cluster (CMDO)Li+(PC)2 was then used to more accurately explore the reduction thermodynamics and kinetics of CMDO. All the stationary points along the path of (CMDO)Li+(PC)2 were also fully optimized with SMD-B3PW91//6-311++G(3df,3pd). To make zero-point energy (ZPE) corrections as well as confirm the transition states, frequency analyses were done at the same level as the full optimization. To predict the charges carried by the atoms, especially Li+, the molecular electrostatic potential fitting (CHELPG) method was used.40 The implicit solvent effect herein was estimated using the SMD model,39 a recommended choice for computing the ΔG of solvation in Gaussian 09 with the Pauling radii for all atoms. We adopted a dielectric constant of 31.6 for weighting the mixed solvent of PC and DMC in relation to the condition implemented in experiment.35 All the above calculations were performed using the Gaussian 09 package.41To better simulate the solvent effects, 9 PC solvent molecules were finally explicitly supplemented to cluster (CMDO)Li+(PC)2, resulting in a supermolecule [(CMDO)Li+(PC)2](PC)9, where two PC and CMDO in the first solvation shell of Li+ form weak hydrogen bonds (C–H⋯O) with three PC molecules. The full geometry optimization for the supermolecules was first accomplished with the B3PW91/6-31G(d,p) method in a vacuum. Then, the single-point energies were obtained at the SMD-B3PW91/6-311++G(d,p)//B3PW91/6-31G(d,p) level.
The reduction potentials were estimated with two models, solvent or additives Li+(X) reduced to the closed reduction precursors [c-Li+(X˙)−] and open ring radicals [o-Li+(X˙)−], by directly comparing the Gibbs free energies in solvent.
| Li+(X) + e− → [c-Li+(X˙)−] |
| ΔG = G ([c-Li+(X˙)−]) − G(Li+(X)) |
| φ = −ΔG/F − 1.39 |
The overall rate constant for the reduction of Li+(X) was estimated by the combination of the steady state approximation and the classical transition state theory, which is similar to a procedure developed previously.34,42
3 Results and discussion
3.1 The first electron reduction of Li+ coordinated solvent or additive Li+X (X = EC, PC, DMC, FEC, ES, and CMDO) in bulk solvent
The lowest unoccupied molecular orbital (LUMO) of carbonates Li+(PC), Li+(EC), and Li+(DMC) are rather high (low negative, −0.18, −0.21, and −0.24 eV). As compared with Li+(EC), the inclusion of fluorine in Li+(FEC) just slightly decreases its LUMO (−0.26 eV). The LUMO of Li+(ES) is significantly lower than those of the above linear, cyclic and fluoro-substituted carbonates (−1.28 eV). It is intriguing that a synergistic effect from sulfur and chlorine seems to occur for Li+(CMDO), reflected by the LUMO energy level (−1.46 eV) that is even lower than Li+(ES). Fig. 1 also shows that the LUMO delocalizes over the solvents and additives, including functional groups such as F in FEC and Cl in CMDO, which implies that the functional groups may participate in the reduction process of these additives.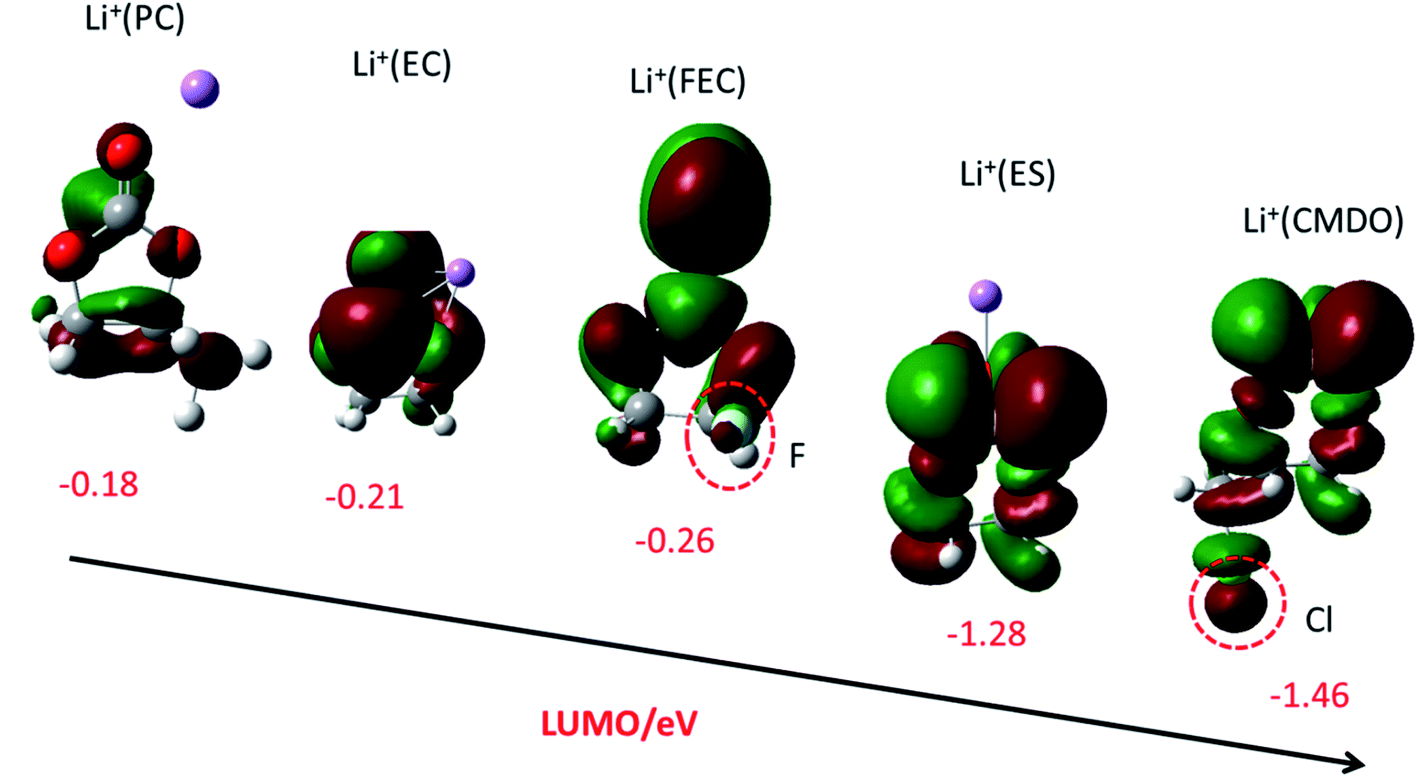 | ||
| Fig. 1 LUMO (isovalue = 0.02) and LUMO energy level (eV) of Li+(X) (from left to right, X = PC, EC, FEC, ES, and CMDO) with SMD-B3PW91 with the basis set of 6-311++G(d,p). | ||
Fig. 2 and 3 show the Gibbs free energy profiles with the selected geometries of the electroreduction path of Li+(EC), Li+(FEC), and Li+(CMDO). Generally, because of an electron affinity that is higher than Li+, an electron will preferentially go to the carbonyl carbon for carbonates (EC, PC, DMC and FEC) by approximately 0.8 eV (smd-B3PW91/6-311++(3df,3pd)) or the sulfur for S-containing additives (ES and CMDO) by around 2.1 eV, resulting in the dissociation precursors c-[Li+(X˙−)] (c: for the closed ring) for the open ring radicals, o-[Li+(X˙−)] (o: for the opened ring). The spin density distribution of c-[Li+(X˙)−] confirms that the location of the excess electron is mainly on the carbonyl carbon or sulfur (spin density ∼0.7–0.8e) but also can go to other functional groups, such as the F of FEC or Cl of CMDO. The excess electron in c-[Li+(X˙−)] (X = EC, PC, DMC, and FEC) slightly extends the C–O bonds for carbonates by approximately 0.1 Å but in sharp contrast, after Li+(ES) and Li+(CMDO) gain the first electron, the S1–O4 bond significantly increases to ∼2.7 Å to form a seven-membered ring.
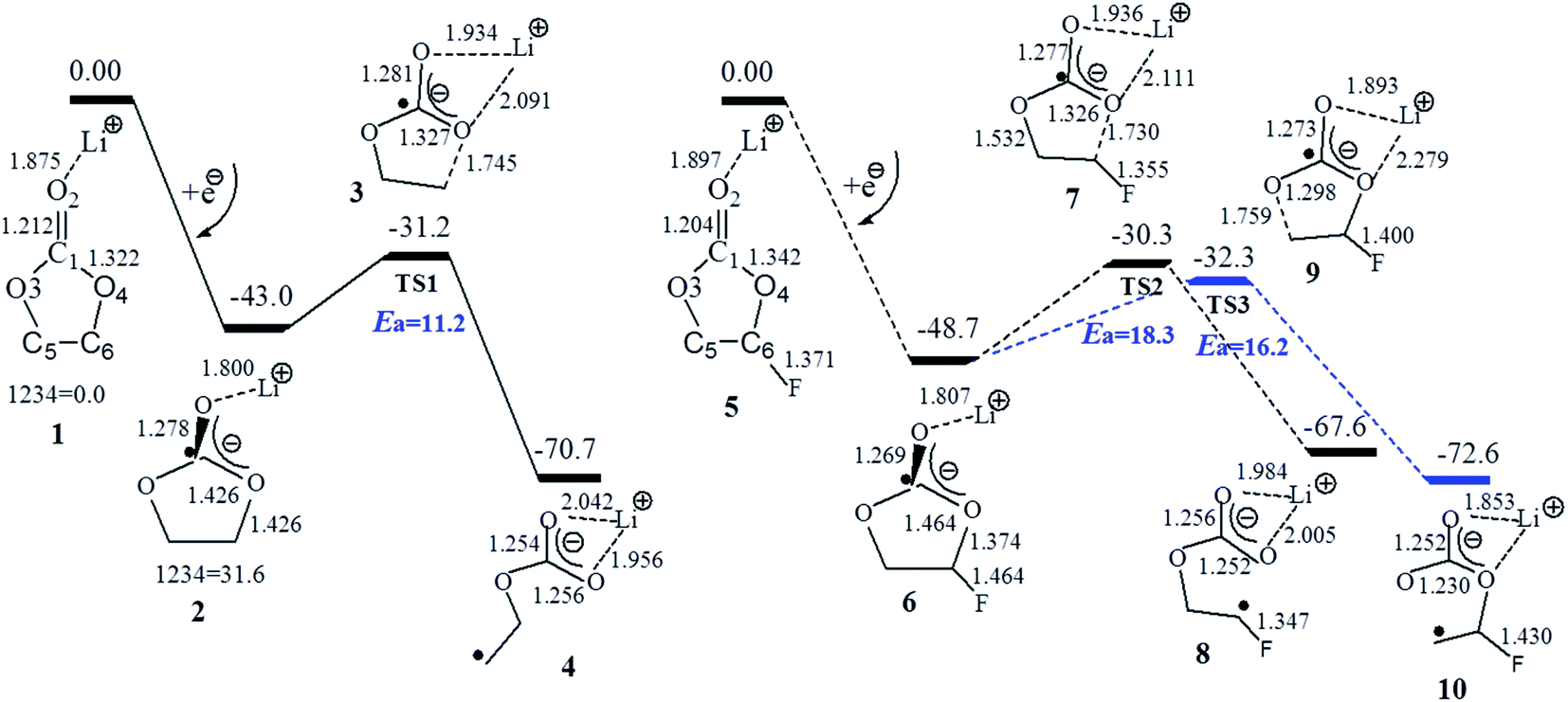 | ||
| Fig. 2 Gibbs free energy profiles at 298.2 K for the one-electron reduction mechanism of Li+(EC) (left) and Li+(FEC) (right) with SMD-B3PW91/6-311++G (3df,3pd). Ea stands for the energy barriers of the ring opening paths. | ||
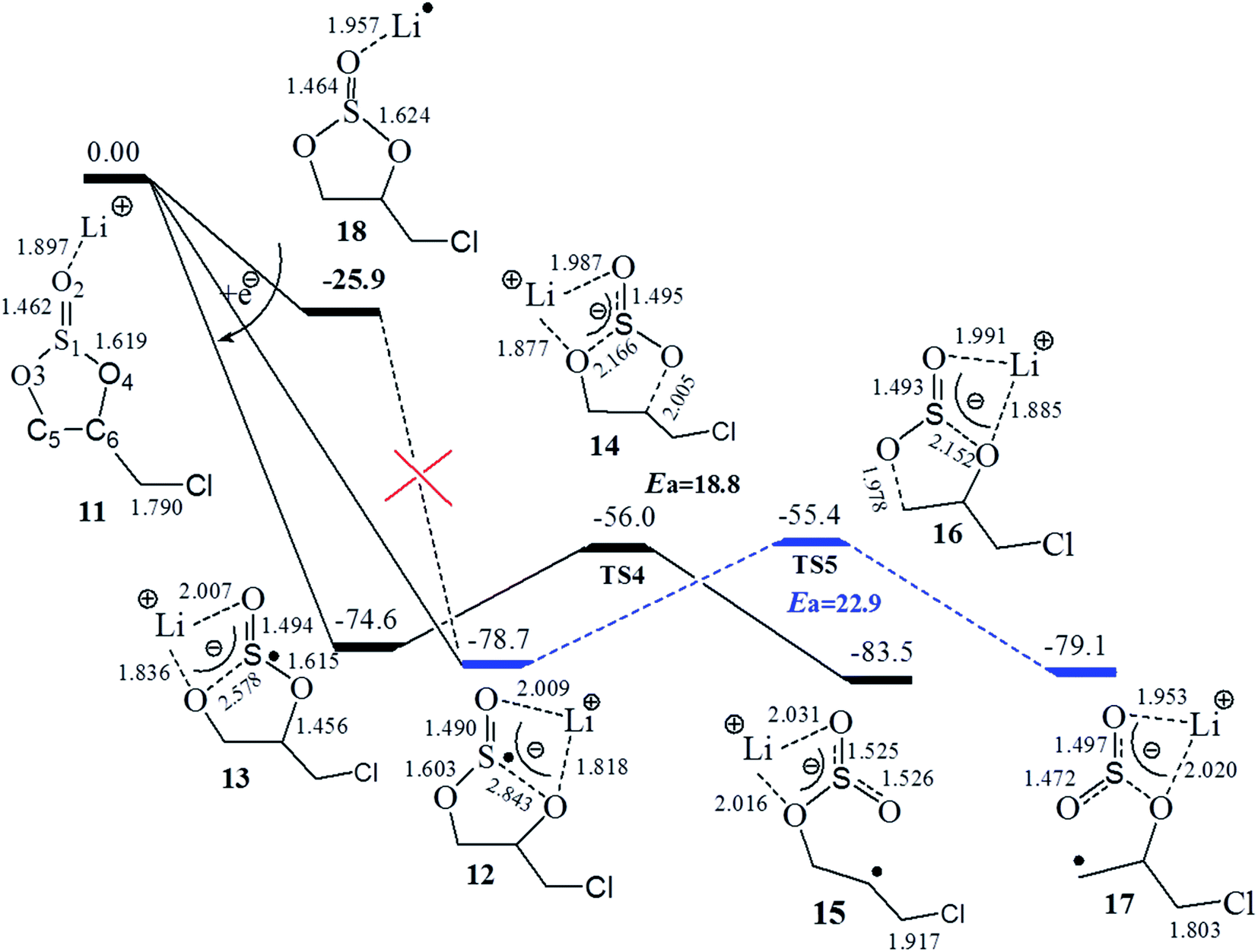 | ||
| Fig. 3 Gibbs free energy profile for the one-electron reduction path of Li+(CMDO) with SMD-B3PW91/6-311++G (3df, 3pd). Blue and black lines refer to the primary and secondary formation paths, respectively. | ||
PC, EC, and DMC have a higher tendency to bind to Li+ than the additives (FEC, ES and CMDO), as reflected by the higher negative binding energy (ΔEb) in Table 1, indicating that the additives do not preferentially solvate the lithium ion and consequently suppress the intercalation of Li+(PC).44 The binding energies as well as binding free energies of the investigated solvents and additives agree within 2 kcal mol−1. Such a small difference may not induce much difference in the solvation and desolvation, if any. Regarding the interface effect, our previous study shows that the supercluster of Li+, solvent and additive, e.g., Li+(ES)(PC), can be adsorbed well on a graphite surface by the interface (Li+-graphite = 2.48 Å),34 while due to the coordination competition of Li+ with PC and the interface, Li+(ES)(PC)2 is rather separated from the interface (Li+-graphite = 5.12 Å). Thus, the graphite interface may not change the clusters much.
| ΔEb | ΔGb | ΔGred | φa | ΔGdiss | φb | Ea | k | φExp. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| SMD-B3PW91/6-311++G(d,p) | ||||||||||
| EC | −8.6 | −1.6 | −45.3 | 0.57 | −73.9 | 1.81 | 11.0 | 9.7 × 1037 | ||
| PC | −8.6 | −2.4 | −44.8 | 0.55 | −73.1 | 1.78 | 12.2 | 1.3 × 1036 | ||
| DMC | −8.3 | −2.5 | −41.9 | 0.43 | −70.3 | 1.66 | 8.0 | 1.1 × 1038 | ||
| FEC | −5.6 | 1.1 | −51.4 | 0.84 | −73.2 | 1.78 | 15.6 | 9.3 × 1037 | ||
| ES | −7.6 | −1.0 | −86.7 | 2.36 | −79.7 | 2.07 | 26.8 | 8.9 × 1055 | ||
| CMDO | −7.2 | −0.3 | −90.2 | 2.52 | −82.1 | 2.17 | 26.2 | 1.1 × 1059 | ||
| −86.0 | 2.34 | −84.9 | 2.29 | 22.1 | 3.6 × 1059 | |||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||
| SMD-B3PW91/6-311++G(3df,3pd) | ||||||||||
| EC | −9.3 | −2.9 | −43.0 | 0.47 | −70.7 | 1.68 | 11.2 | 2.8 × 1035 | 0.57 (ref. 43) | |
| PC | −9.5 | −3.2 | −42.4 | 0.45 | −70.0 | 1.65 | 12.5 | 2.0 × 1034 | 0.7–1.1 (ref. 35 and 44) | |
| DMC | −8.5 | −2.6 | −40.8 | 0.38 | −70.2 | 1.65 | 8.0 | 9.5 × 1035 | ||
| FEC | −8.4 | −1.9 | −48.7 | 0.72 | −72.6 | 1.76 | 16.2 | 2.3 × 1034 | 1.1–1.2 (ref. 45) | |
| ES | −7.4 | −1.0 | −75.9 | 1.90 | −78.5 | 2.01 | 23.4 | 1.3 × 1051 | 1.9–2.0 (ref. 24) | |
| CMDO | −7.0 | −1.3 | −78.7 | 2.02 | −79.1 | 2.04 | 22.9 | 1.1 × 1053 | 1.7–1.8 (ref. 35) | |
| −74.6 | 1.85 | −83.5 | 2.23 | 18.8 | 4.1 × 1053 | |||||
The Gibbs free energy change (ΔGred) for the formation of reductive decomposition precursors c-[Li+(X˙−)] in Table 1 shows that carbonates DMC, EC, and PC have the lowest potentials to initiate the reduction process, and their reduction potentials (φa) (0.38, 0.47, and 0.45 V vs. Li+/Li) are rather close to the reduction range of EC- and PC-based electrolyte solutions of LIBs (0.57 V for EC43 and 0.7–1.1 V for PC35,44). The reduction potential φa of FEC (∼0.84 V) is indeed higher than those of EC and PC by approximately 0.3 V. Another study shows that FEC reductively decomposes prior to EC.46 The reduction of FEC on a graphite electrode in PC-based electrolyte was reported to appear around 1.1–1.2 V.45 Because of the partial cleavage of SO after accepting an electron to the S-containing additives, Li+(ES) has a much higher reduction potential than the above carbonates (1.9–2.0 V), which is in an excellent agreement with the experimental value of 1.9–2.0 V.24 It is very interesting to note that CMDO has the highest reduction potential (φa: 1.85–2.02 V; φb: 2.04–2.23 V) among the solvents (EC, PC, and DMC) and the additives (FEC and ES). This reduction potential trend is in good agreement with the first cyclic voltammetry scan for the electrolyte consisting of PC, DMC, EC, CMDO, and FEC,35 where CMDO was reduced at ∼1.8 V prior to FEC by 0.6 V, and the consequent product during the first scan formed a good quality SEI layer to avoid the further reduction of the electrolyte. The calculated reduction potentials to form c-[Li+(X˙−)] are generally higher than the experimental ones. The difference is partially due to the solvent underestimation of model Li+(X). The explicit solvent effect in the computational model will decrease the calculated reduction potential,34 which will be further discussed later in this paper.
On the other hand, Table 1 shows that among the investigated solvents and additives, the cyclic and linear carbonates have the lowest energy barriers for the ring opening via the O4–C6 (ethylene carbon) homolytic cleavage of the reduction precursors (8.0, 11.0, and 12.0 kcal mol−1 for DMC, EC and PC). The additive FEC has two decomposition pathways, forming LiCO3CHFCH2· and LiCO3CH2CHF−˙ radicals with the respective energy barrier of 16.2 and 18.3 kcal mol−1, which are higher than EC by 4–6 kcal mol−1. In line with previous studies,34,36,37 Table 1 also shows that the energy barrier for the ring opening via O–C (ethylene carbon) for the reduction precursor of Li+(ES) (23.4 kcal mol−1) is much higher than those of cyclic and linear carbonates Li+(X) (X = EC, PC, and DMC) and even FEC.
The electron affinity of Li+ in Li+(CMDO), resulting in a radical Li·(CMDO) (18), is similar to that in Li+(EC) (−26.9 vs. −23.9 kcal mol−1 at SMD-B3PW91/6-311++(3df,3pd)), and the radical Li#x02d9;(X) is much less stable than c-[Li+(X˙−)]. According to Fig. 3, after Li+(CMDO) gains one electron, similar to the case of ES, the S1–O4 or S1–O3 bond of CMDO was considerably stretched to bring about two reductive decomposition precursors, 12 and 13, that vary with the location of Li+. In the gas phase, intermediates 12 and 13 can be generated from intermediate 18, where the excess electron goes to Li rather than S and the transition state was also located with an imaginary frequency of 500 cm−1. However, similar to the report from Liu et al. for Na+(ES),47 in solution as the electron partially transfers from Li to S, the much stronger solvation of Liσ+ than that of neutral Li can significantly decrease the energy of the compound. Thus, the transition state for the S–O bond cleavage does not exist in solution and the path from 18 to intermediates 12 and 13 may not be plausible.
Induced by the internal electron transfer process, intermediates 12 and 13 are advanced to primary and secondary radicals via the respective transition state 16 and 14. The black path (13–14–15) to the secondary radical (15) has a lower energy barrier than the blue path (12–16–17) to the primary radical (17), and the transition state of the former path is also slightly more stable than that of the latter. The secondary radical (15) has a lower energy than that of the primary one (17) by 3.5 kcal mol−1 (Table 2).
| ΔE0 | ΔG | q | sd | ω | ||
|---|---|---|---|---|---|---|
| S/C1 | C5 | |||||
| Li+(EC) | ||||||
| 1 | 0.00 | 0.00 | 0.85 | 0 | ||
| 2 | −42.5(−45.0) | −43.0(−45.3) | 0.78 | 0.85 | ||
| 3(TS1 2–4) | −31.2(−33.8) | −31.2(−34.4) | 0.91 | 0.55 | 0.47 | 994 |
| 4 | −69.5(−72.1) | −70.7(−74.0) | 0.78 | 1.07 | ||
|
||||||
| Li+(FEC) | ||||||
| 5 | 0.00 | 0.00 | 0.91 | 0 | ||
| 6 | −48.2(−50.6) | −48.7(−51.3) | 0.81 | 0.83 | ||
| 7 (TS2 6–8) | −30.0(−32.4) | −30.3(−33.1) | 0.77 | 0.51 | 0.38 | 955 |
| 8 | −66.1(−68.5) | −67.6(−70.4) | 0.69 | 0.97 | ||
| 9(TS3 6–10) | −32.0(−35.0) | −32.3(−35.7) | 0.83 | 0.51 | 0.52 | 1044 |
| 10 | −70.7(−73.8) | −72.6(−75.3) | 0.82 | 1.12 | ||
|
||||||
| Li+(CMDO) | ||||||
| 11 | 0.00 | 0.00 | 0.95 | |||
| 12 | −79.1(−89.4) | −78.7(−90.2) | 0.91 | 0.67 | ||
| 13 | −75.3(−85.6) | −74.6(−86.0) | 0.93 | 0.66 | ||
| 14(TS4 13–15) | −56.2(−63.1) | −55.4(−63.5) | 0.94 | 0.30 | 0.65 | 639 |
| 15 | −82.5(−84.0) | −83.5(−86.4) | 0.94 | 0.90 | ||
| 16(TS5 12–17) | −56.5(−63.6) | −55.4(−64.1) | 0.93 | 0.35 | 0.46 | 851 |
| 17 | −79.0(−80.5) | −83.5(−82.1) | 0.92 | 1.04 | ||
| 18 | −26.9 | −25.9 | ||||
3.2 Overall reaction rate vs. energy barrier and reduction potential
Similar to the case of additive ES,34 the above discussion reveals that additive CMDO also has a greater reduction potential than the carbonate solvents EC and PC; however, the energy barrier of the ring opening of CMDO is much greater than those of PC, EC, and FEC (22.1–26.2 vs. 11.0–16.0 kcal mol−1 (ref. 17)). Unlike the comparison of FEC with EC, the introduction of Cl in CMDO does not greatly change the energy barrier relative to ES. The comparison of the reduction potential and energy barrier between CMDO and solvents like EC and PC indicates that the reduction of CMDO to the closed ring precursor is also thermodynamically favorable, but the subsequent ring opening is kinetically unfavorable.17As shown in Table 1, relative to 6-311++G(d,p) the higher level basis set (3df,3pd) provides slightly higher or the same energy barriers for the carbonates, but lower energy barriers for the two sulfites, ES and CMDO by 3–4 kcal mol−1. The reduction potentials from (3df,3pd), smaller than those from 6-311++G(d,p), are rather close to the experimental ones for the S-additives (ES, CMDO).
To comprehensively discuss the overall rate constant (k) for the one-electron reduction consisting of the assumed electro-equilibrium (the formation of the reduction precursor, equilibrium K) and kinetic aspect (ring opening reaction, rate constant k′), on the basis of steady-state theory and the classical TST the overall rate constant was previously developed,34 which is roughly approximated as the product of (k = Kk′), an approximation from variation transition state.42
The calculated overall rate constants for the entire electroreduction of the clusters Li+(X) in bulk solvent are listed in Table 1. The plot of reduction potential (φ/V) against the energy barrier (Ea, kcal mol−1) with the inserted overall rate constant is given in Fig. 4. Although the energy barriers are relatively low (high k′), their reduction potentials for the investigated carbonates are also low (small K). The overall rate constants k for the linear and cyclic carbonates are in a range of 1034 to 1035 s−1 (2.0 × 1034, 9.5 × 1035, 2.8 × 1035, and 2.3 × 1034 for PC, DMC, EC, and FEC, respectively) at the SMD-B3PW91/6-311++G(3df,3pd) level. Although the equilibrium constant K for forming the reduction precursor of ES is rather high, in spite of the high energy barrier (low k′), the overall rate constant k of ES still reaches 1.3 × 1051 s−1. Similarly, for the reductive decomposition of CMDO, the high equilibrium constant comprimises the high energy barrier, which thus results in the highest overall rate constant (1053 s−1). The rate constant for the formation of the secondary radical (15) is roughly three times that of the primary radical (17) (4.1 × 1053 vs. 1.1 × 1053).
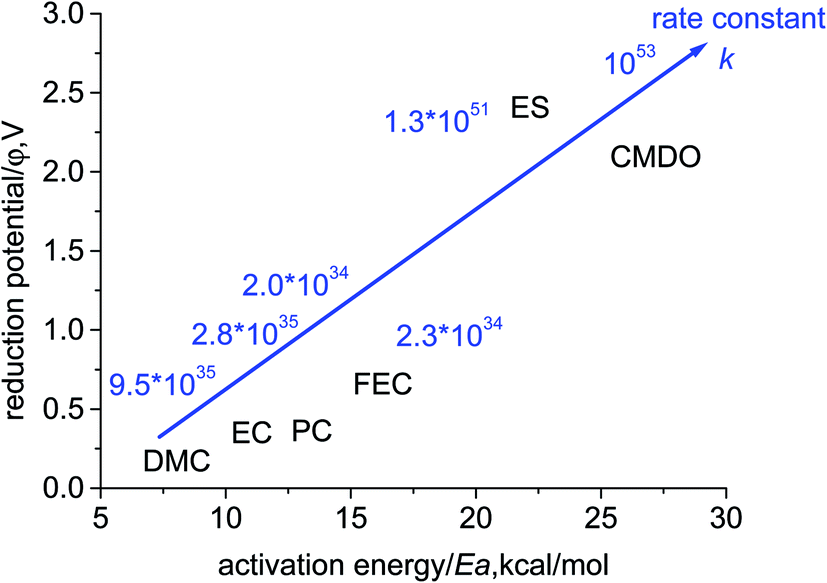 | ||
| Fig. 4 The plot of reduction potential (φ/V, vs. Li+/Li) against activation energy (Ea, kcal mol−1) for the electroreduction of Li+(X), with the inserted overall rate constant. | ||
3.3 Termination paths of radical 17
After the ring opening reaction of c-[Li+(CMDO)−˙], the termination of the radicals will occur via either dimerization, further reduction, or reaction with other species, forming the compositions of the SEI layer. Two major products were speculated for the reduction of EC, organic lithium dicarbonates and inorganic lithium carbonate.13 Two paths (A and B) for forming organic lithium disulfite were addressed via the barrierless polymerization of radical 17, resulting in lithium bisulfites LiSO3–CH2CH(CH2Cl)–SO3Li/CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2Cl and LiSO3–CH2CH(CH2Cl)–CH(CH2Cl)CH2–SO3Li, respectively. For further reduction by the second electron, four paths were investigated, where C and D involve Li+(CMDO), and E and F are engaged by Li+(PC). Paths C and E generate CMDO- or PC-solvated lithium sulfite (Li2SO3) while CH2CH–CH2Cl is eliminated. Another inorganic compound LiCl was produced along paths D and F. Fig. 5 indicates that the inorganic compound formation paths (C–F) are much more thermodynamically favorable than the organic compound formation paths (A and B). The termination paths for the ring opening radical of Li+(FEC) show a similar trend, which is summarized in Fig. S2.† In addition, the formation of LiCl from CMDO reduction (paths D and F) or that of LiF for FEC is thermodynamically more favorable than that of Li2SO3 or Li2CO3 by 17 kcal mol−1. This result supports the experimental observation that for the FEC additive, LiF is predominant over all other F-containing compounds in the SEI layer.23,35 Furthermore, the primary products for the novel additive are inorganic compounds LiCl, Li2SO3 and S-containing organic compounds. As compared with those from FEC, it was reported that the S-derived SEI has lower resistance.
CH–CH2Cl and LiSO3–CH2CH(CH2Cl)–CH(CH2Cl)CH2–SO3Li, respectively. For further reduction by the second electron, four paths were investigated, where C and D involve Li+(CMDO), and E and F are engaged by Li+(PC). Paths C and E generate CMDO- or PC-solvated lithium sulfite (Li2SO3) while CH2CH–CH2Cl is eliminated. Another inorganic compound LiCl was produced along paths D and F. Fig. 5 indicates that the inorganic compound formation paths (C–F) are much more thermodynamically favorable than the organic compound formation paths (A and B). The termination paths for the ring opening radical of Li+(FEC) show a similar trend, which is summarized in Fig. S2.† In addition, the formation of LiCl from CMDO reduction (paths D and F) or that of LiF for FEC is thermodynamically more favorable than that of Li2SO3 or Li2CO3 by 17 kcal mol−1. This result supports the experimental observation that for the FEC additive, LiF is predominant over all other F-containing compounds in the SEI layer.23,35 Furthermore, the primary products for the novel additive are inorganic compounds LiCl, Li2SO3 and S-containing organic compounds. As compared with those from FEC, it was reported that the S-derived SEI has lower resistance.
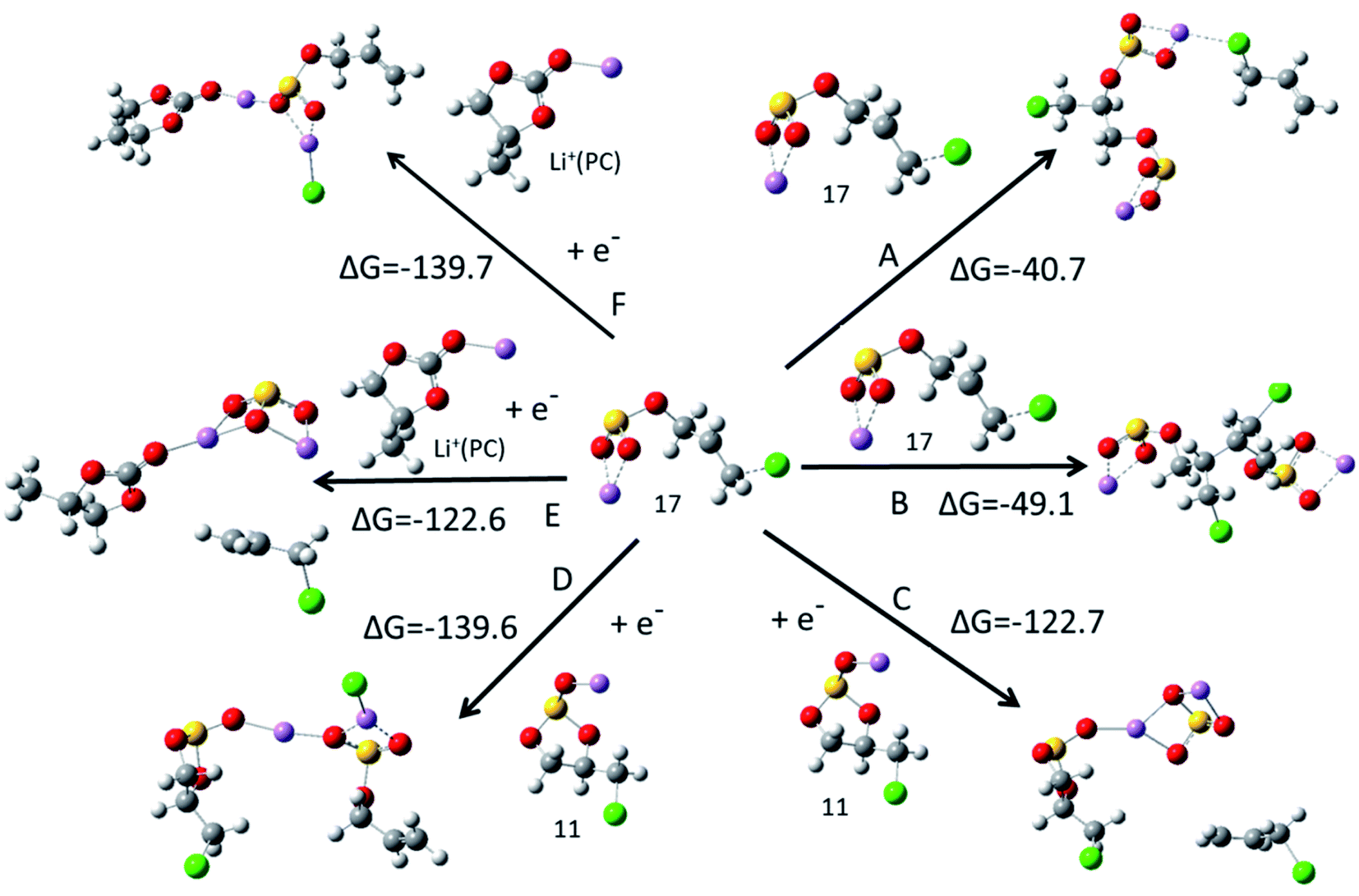 | ||
| Fig. 5 Termination paths of secondary radical 17 from SMD-B3PW91 with a basis set of 6-311++G(d,p). | ||
3.4 The electroreductive decompositions of CMDO in (CMDO)Li+(PC)2
In electrolyte solutions of LIBs, the most probable lithium ion species may be the Li+ solvated by three carbonate molecules in the first solvation shell, like Li+(PC)3, Li+(EC)3, and (ES)Li+(PC)2.30,37 To better account for the solvent effect, the Gibbs free energy path of the stepwise two-electron reductive pathway for (CMDO)Li+(PC)2 (19) was thus investigated and is shown in Fig. 6. The relevant energy data and main features are summarized in Table 3. The inclusion of two PC molecules in (CMDO)Li+(PC)2 decreases the reduction Gibbs free energy of CMDO for forming decomposition precursors 20 and 21 by 5.4 and 3.3 kcal mol−1, respectively, as compared with those of Li+(CMDO) from SMD-B3PW91 in Fig. 2. The estimated reduction potentials are reduced by 0.1–0.2 V to 2.11 and 2.37 V. Another density functional theory SMD-wB97XD results in rather similar reduction potentials to SMD-B3PW91/6-311++G(d,p), differing only by 0.8–1.6 kcal mol−1 (<0.1 V). Although transition states 1 and 2 were achieved and identified well with SMD-wB97XD, it failed to get them converged with SMD-B3PW91. Their energies at SMD-B3PW91 in Fig. 6 were estimated from partially optimized structures. TS6 for the formation of the secondary radical is again more stable than TS5 for the primary radical by 5.2 kcal mol−1 at B3PW91 and 6.9 kcal mol−1 at wB97XD.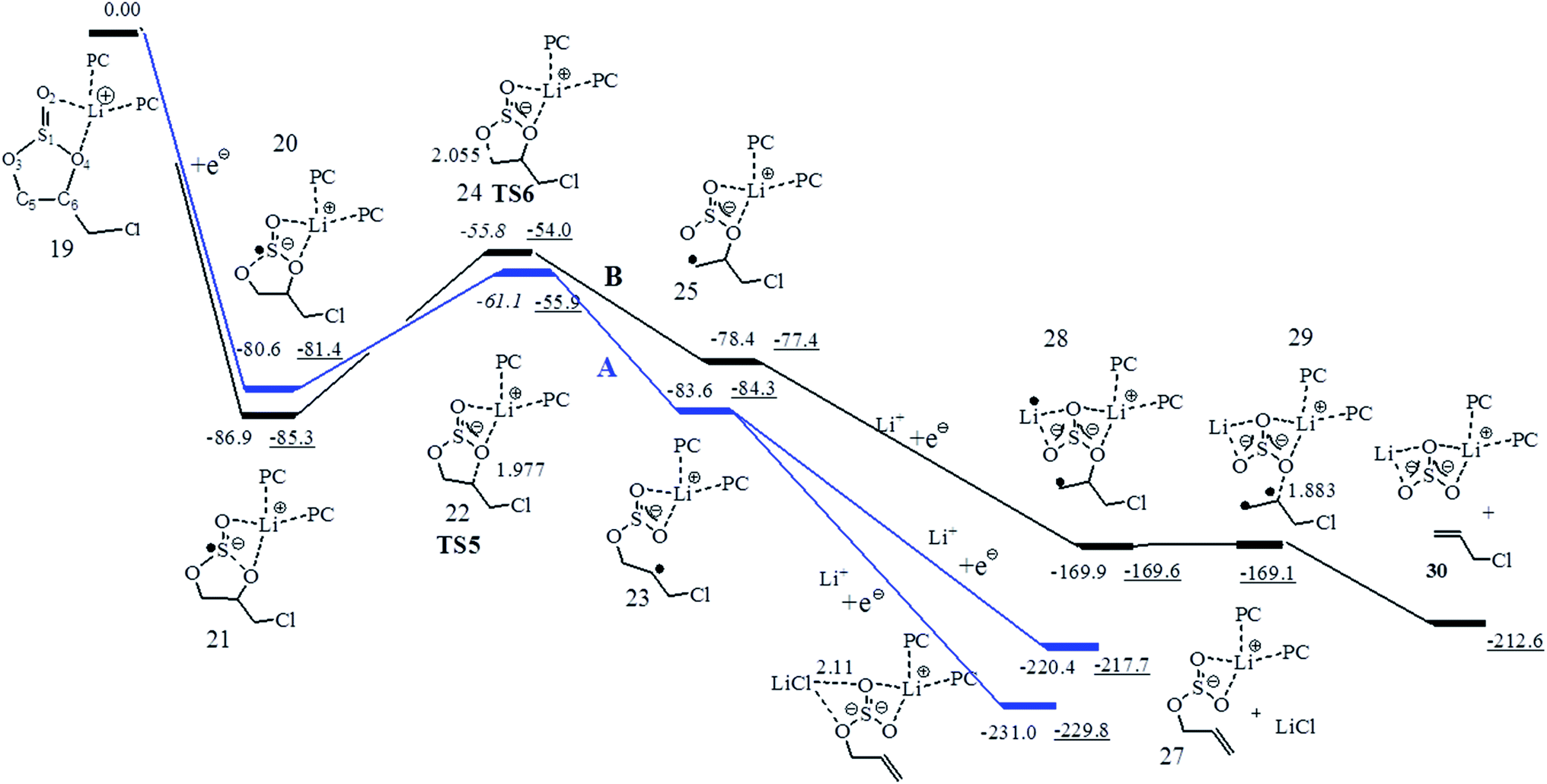 | ||
| Fig. 6 The Gibbs free energy (ΔG) profile for the reduction of CMDO of (PC)2Li+(CMDO) using SMD+B3PW91/6-311++G(d,p) (plain data) and SMD+wB97XD/6-311++G(d,p) (underline data). | ||
| Structures | ΔE0 | ΔG | sd | ω | |
|---|---|---|---|---|---|
| S | C | ||||
| 19 | 0 | 0 | |||
| 20 | −80.0/−80.1 | −80.6/−81.4 | 0.70/0.72 | ||
| 21 | −86.3/−85.2 | −86.9/−85.2 | 0.68/0.72 | ||
| 22 (TS5:19–22) | −62.4/−55.8 | −61.1/−55.9 | 0.32/0.33 | 0.52/0.51 | 710i/1005i |
| 23 | −80.7/−82.6 | −83.6/−84.3 | 0.95/0.87 | ||
| 24 (TS6:20–24) | −58.6/−53.8 | −55.8/−54.0 | 0.29/0.29 | 0.74/0.75 | 714i/973i |
| 25 | −75.8/−71.4 | −78.4/−77.4 | 1.16/1.09 | ||
|
|||||
| +Li+ | |||||
| 26 | −235.8/−236.3 | −231.0/−229.8 | |||
| 27 | −221.5/−222.3 | −220.4/−217.7 | |||
| 28 | −177.3/−178.4 | −169.9/−169.6 | |||
| 29 | /−177.4 | /−169.1 | /413i | ||
| 30 | /−216.7 | /−212.6 | |||
As compared with Li+(CMDO), the formation Gibbs energies of the ring opening radical from the reduction of (CMDO)Li+(PC)2 are also decreased (−83.6 vs. −90.2 kcal mol−1 for the secondary radical 23, and −78.4 vs. −86.0 kcal mol−1 for the primary one 25). The termination process for the radicals 23 and 25 through the further electron transfer was addressed. As shown in Fig. 6, the continuous electron reduction of the secondary radical with the addition of Li+ brings about the formation of an inorganic compound LiCl in 26 and 27 with a respective ΔG of −147.4 and −136.8 kcal mol−1 (relative to 23 and Li+ with SMD-B3PW91/6-311++G(d,p)). Complex 27, consisting of free LiCl and PC-solvated LiSO3CH2CHCH2, is less stable by approximately 10 kcal mol−1 than 26, in which LiCl is still coordinated to LiSO3R. The further electron reduction of the primary radical 25 results in a diradical intermediate (28) that has only a rather small barrier (∼1 kcal mol−1) through a transition state (29) to produce inorganic compound Li2SO3 by eliminating CH2CHCH2Cl. Similar to the case of Li+(CMDO), it is interesting that the formation of LiCl in 26 and 27 is thermodynamically more favorable than that of Li2SO3 by 5–17 kcal mol−1. This result further supports the experimental observation that for halogen-containing additives, LiF or LiCl are predominant over all other halogen-containing species in the SEI layer.23,35
3.5 The reduction profile of CMDO by explicitly including solvent molecules in a supercluster [(CMDO)Li+(PC)2](PC)9 and implicit solvent model
To more accurately account for the solvent effect on the CMDO electroreduction, nine PC were explicitly supplemented to cluster (CMDO)Li+(PC)2 in the second solvation shell of Li+, forming supermolecule [(CMDO)Li+(PC)2](PC)9. The Gibbs free energy profile for the reduction of the supermolecule is shown in Fig. 7 together with [(CMDO)Li+(PC)2] for comparison. The energetic data including relative energy, Gibbs free energy and reduction potential are collected in Table 4. The H⋯O distances of weak hydrogen bonds (C–H⋯O) between PC in the second solvation shell and PC of (CMDO)Li+(PC)2 are around 2.3–2.7 Å.48 Following the trend that the inclusion of explicit PC molecules in the first solvation shell of Li+ decreases the reduction potentials of [(CMDO)Li+], the reduction potential of CMDO trends down as nine PC molecules are added to [(CMDO)Li+(PC)2] in the second solvation shell (2.43 and 2.81 V for [(CMDO)Li+(PC)2](PC)9 vs. 3.08 and 3.28 V for [(CMDO)Li+(PC)2] in vacuum).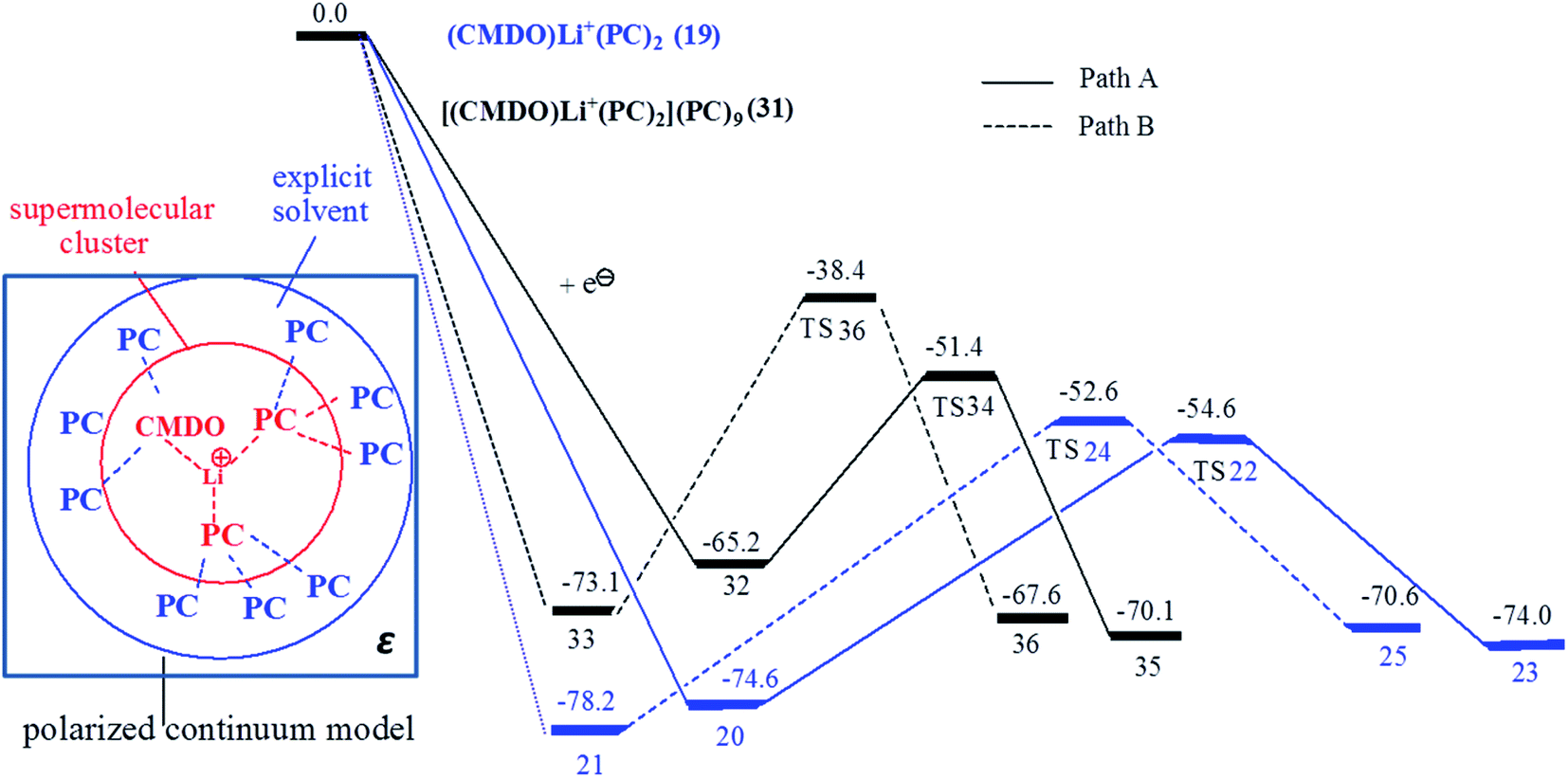 | ||
| Fig. 7 The Gibbs free energy surface of paths A (solid line for forming the secondary radical) and B (dashed line for forming the primary radical) of the superclusters (CMDO)Li+(PC)2 (blue line) and [(CMDO)Li+(PC)2](PC)9 (black line) with SMD-B3PW91/6-311++G(d,p)//B3PW91/6-31G(d,p). | ||
| Compounds | ΔE0a | ΔGb | φ0c | ν/cm−1 |
|---|---|---|---|---|
| a The data in the parentheses are in a vacuum and estimated at a B3PW91/6-311++G(d,p) single point calculation from the geometries at the B3PW91/6-31G(d,p) level.b G = E[SMD-B3PW91/6-311++G(d)] + thermal correction at 298.2 K to ΔG at B3PW91/6-31G(d,p).c Estimated with a thermodynamic cycle (the standard reduction potential φ0 vs. Li+/Li).34 | ||||
| (CMDO)Li+(PC)2 | ||||
| 19 | 0 | 0 | ||
| 20 | −77.5 (−105.9) | −74.6 (−103.1) | 1.9 (3.1) | |
| 21 | −81.4 (−110.9) | −78.2 (−107.7) | 2.0 (3.3) | |
| 22 (TS5 19–22) | −57.5(-86.5) | −54.6 (−83.6) | 626i | |
| 23 | −75.9(-102.3) | −74.0 (−100.4) | 1.8 (3.0) | |
| 24 (TS6 20–24) | −55.5(-84.0) | −52.6 (−81.0) | 629i | |
| 25 | −72.3(−101.2) | −70.6 (−99.5) | 1.7 (2.9) | |
|
||||
| [(CMDO)Li+(PC)2](PC)9 | ||||
| 31 | 0 | 0 | ||
| 32 | −66.8 (−89.8) | −65.2 (−88.2) | 1.4(2.4) | |
| 33 | −74.9 (−98.7) | −73.1 (−96.9) | 1.8(2.8) | |
| 34 (TS 31–34) | −53.9 (−78.8) | −51.4 (−76.3) | 660i | |
| 35 | −72.9 (−99.0) | −70.1 (−96.2) | 1.7(2.8) | |
| 36(TS 32–36) | −43.9(-69.1) | −38.4 (−63.7) | 792i | |
| 37 | −69.1 (−93.9) | −67.6 (−92.4) | 1.5(2.6) | |
|
||||
| Exp.35 | 1.7–1.8 | |||
The SMD model for bulk solvent was also supplemented to supermolecule [(CMDO)Li+(PC)2](PC)9. As shown in Fig. 7, a hybrid model was proposed to account for the solvent effects by jointly using explicit solvent molecules in the second solvation shell of Li+ and implicit solvent models such as SMD and PCM. The energetic data of SMD-[(CMDO)Li+(PC)2](PC)9 also qualitatively confirms the conclusions from models SMD-Li+(CMDO) and SMD-[(CMDO)Li+(PC)2]. For instance, the path for forming the secondary radical has a lower energy barrier than that of the primary radical. As compared with those in the gas phase, the inclusion of SMD further decreases the reduction Gibbs free energies of [(CMDO)Li+(PC)2](PC)9 by roughly 23 kcal mol−1, resulting in 65.2 and 73.1 kcal mol−1 (88.2 and 96.9 kcal mol−1 in the gas phase). For (CMDO)Li+(PC)2, the SMD reduces ΔG by ∼30 kcal mol−1, which implies that the supplemented explicit solvent molecules in [(CMDO)Li+(PC)2](PC)9 partially (∼7 kcal mol−1) screen the solvent effects from SMD. The reduction potentials (1.7–1.8 V) of CMDO from SMD-[(CMDO)Li+(PC)2](PC)9 agree much better with the experimental peak (1.7–1.8 V (ref. 35)) than those from the SMD-[(CMDO)Li+(PC)2] (1.42–1.58 V). The explicit supplement of the second solvation shell of Li+ through a supermolecule [(CMDO)Li+(PC)2](PC)9 is able to further compromise the solvent effects arising from the SMD model.
4 Conclusions
Density functional theory B3PW91 together with the implicit solvent model (SMD) were used to systematically investigate the electroreduction mechanism for a novel electrolyte additive CMDO and other additives and solvents such as EC, PC, DMC, FEC, and ES. The one-electron reduction potential of Li+-coordinated compound Li+(CMDO) to form decomposition precursor [c-Li+(CMDO˙−)] is the highest (1.9–2.0 V vs. Li+/Li) at SMD-B3PW91/6-311++G(3df,3pd), followed by ES (1.9 V), FEC (0.72 V), EC (0.47 V), PC (0.45 V), and DMC (0.38 V). The results imply that CMDO is reduced prior to other solvents or additives in the electrolyte mixture of CMDO, FEC, EC and PC/DMC. On the other hand, the ring opening of [c-Li+(CMDO˙−)] is the least kinetically favorable, reflected by the highest energy barrier (Ea): CMDO (18.8–22.9 kcal mol−1) ∼ ES (23.4) > FEC (16.2) > PC (12.5) > EC (11.2) > DMC (8.0). However, with a steady state approximation and classical transition state theory, CMDO still shows the highest overall reaction rate constant (∼1053 s−1) for forming the open ring radical [o-Li+(CMDO˙)−]. In addition, the termination reaction of [o-Li+(CMDO˙)−] for forming LiCl is thermodynamically more favorable than that of Li2SO3 by ∼20 kcal mol−1, and much more favorible than the paths of organic disulfite (LiSO3)2-R, supporting the experiment observation that for halogen-containing additives, LiF or LiCL is predominant over all other halogen-containing species in the SEI layer. The above results were further confirmed by a more realistic model (CMDO)Li+(PC)2. Moreover, even with small basis sets in the hybrid model, the supercluster [(CMDO)Li+(PC)2](PC)9 supplemented by SMD for bulk solvent provides the reduction potentials (1.7–1.8 V) for CMDO that agree well with the experimental reduction peak, suggesting that the explicit second solvation shell of Li+ via a supermolecule [(CMDO)Li+(PC)2](PC)n can further comprimises solvent effects arising from the SMD model.Conflicts of interest
There are no conflicts to declare.References
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824–832 CrossRef CAS.
- D. J. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- J.-M. Trarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood III, Carbon, 2016, 105, 52–76 CrossRef CAS.
- M. R. Busche, T. Drossel, T. Leichtweiss, D. A. Weber, M. Falk, M. Schneider, M.-L. Reich, H. Sommer, P. Adelhelm and J. Janek, Nat. Chem., 2016, 8, 426 CrossRef CAS PubMed.
- K. Schroder, J. Alvarado, T. A. Yersak, J. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531 CrossRef CAS.
- M. D. Bhatt and C. O'Dwyer, Phys. Chem. Chem. Phys., 2015, 17, 4799–4844 RSC.
- J. M. M. d. l. Hoz and P. B. Balbuena, Phys. Chem. Chem. Phys., 2014, 16, 17019–17098 Search PubMed.
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- H. H. Zheng, G. Liu and V. Battaglia, J. Phys. Chem. C, 2010, 114, 6182–6189 CrossRef CAS.
- W. Märkle, J. F. Colin, D. Goers, M. E. Spahr and P. Novák, Electrochim. Acta, 2010, 55, 4964–4969 CrossRef.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708–11718 CrossRef CAS PubMed.
- Y. Ein-Eli, S. R. Thomas and V. R. Koch, J. Electrochem. Soc., 1997, 144, 1159–1165 CrossRef CAS.
- H. M. Jung, S.-H. Park, J. Jeon, Y. Choi, S. Yoon, J.-J. Cho, S. Oh, S. Kang, Y.-K. Han and H. Lee, J. Mater. Chem. A, 2013, 1, 11975–11981 RSC.
- R. Wagner, S. Brox, J. Kasnatscheew and D. R. Gallus, Electrochem. Commun., 2014, 40, 80–83 CrossRef CAS.
- Y. X. Wang and P. Balbuena, J. Phys. Chem. B, 2002, 106, 4486–4495 CrossRef CAS.
- U. Keisuke, S. Keitaro, O. Yukihiro and T. Yoshitaka, J. Am. Chem. Soc., 2013, 135, 11967–11974 CrossRef PubMed.
- E. G. Leggesse and J.-C. Jiang, RSC Adv., 2012, 2, 5439–5446 RSC.
- U. Purushotham, N. Takenaka and M. Nagaoka, RSC Adv., 2016, 6, 65232 RSC.
- U. Purushotham, N. Takenaka and M. Nagaoka, RSC Adv., 2016, 6, 65232–65242 RSC.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CrossRef CAS.
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Letter, 2017, 2, 1337 CrossRef CAS.
- G. H. Wrodnigg, J. O. Besenhard and M. Winter, J. Electrochem. Soc., 1999, 146, 470–472 CrossRef CAS.
- W. Yao, Z. Zhang, G. Jun, J. Li, J. Xu, Z. Wang and Y. Yang, Energy Environ. Sci., 2009, 2, 1102–1108 RSC.
- R. Petibon, L. Madec, L. M. Rotermund and J. R. Dahn, J. Power Sources, 2016, 313, 152–163 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, J. Electrochem. Soc., 2000, 147, 1245–1250 CrossRef CAS.
- S. Li, W. Zhao, Z. Zhou, X. Cui, Z. Shang, H. Liu and D. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4920–4926 CrossRef CAS PubMed.
- S. K. Jurng, H. Kim, J. G. Lee, J. H. Ryu and S. M. Oh, J. Electrochem. Soc., 2016, 163, A223–A228 CrossRef CAS.
- M. D. Bhatt and C. O’Dwyer, J. Electrochem. Soc., 2014, 161, A14115–A14121 Search PubMed.
- Y. Li, K. Leung and Y. Qi, ACC. Chem. Res., 2016, 49, 2363–2370 CrossRef CAS PubMed.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- Y. Wang, S. Nakamura, K. Tasaki and P. B. Balbuena, J. Am. Chem. Soc., 2002, 124, 4408–4421 CrossRef CAS PubMed.
- Y. Sun and Y. Wang, Phys. Chem. Chem. Phys., 2017, 19, 6861–6870 RSC.
- A. S. Wotango, W.-N. Su, A. M. Haregewoin, H.-M. Chen, J.-H. Cheng, M.-H. Lin, C.-H. Wang and B. J. Hwang, ACS Appl. Mater. Interfaces, 2018, 10, 25252–25262 CrossRef CAS PubMed.
- L. Xing, W. Li, M. Xua, T. Li and L. Zhou, J. Power Sources, 2011, 196, 7044–7047 CrossRef CAS.
- E. G. Leggesse and J.-C. Jiang, J. Phys. Chem. A, 2012, 116, 11025–11033 CrossRef CAS PubMed.
- F. Ren, W. Zuo, X. Yang, M. Lin, L. Xu, W. Zhao, S. Zheng and Y. Yang, J. Phys. Chem. C, 2019, 123, 5871–5880 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, et. al., Gaussian09 D01., Gaussian Inc., Pittsburgh, PA, 2009 Search PubMed.
- Z.-F. Xu and Y. Wang, J.Phys.Chem.C, 2011, 115, 20565–20571 CrossRef CAS PubMed.
- H. Ota, Y. Sakata, X. Wang, J. Sasahara and E. Yasukawa, J. Electrochem. Soc., 2004, 151, A437–A446 CrossRef CAS.
- M. Inaba, Z. Siroma, Y. Kawatate, A. Funabiki and Z. Ogumi, J. Power Sources, 1997, 68, 221 CrossRef CAS.
- S.-K. Jeong, M. Inaba, R. Mogi, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2001, 17, 8281 CrossRef CAS.
- N.-S. Y. Choi, K. H., K. Y. Lee, M. Sung, H. Kim and S.-S. Kim, J. Power Sources, 2006, 161, 1254–1259 CrossRef CAS.
- Q. Liu, D. Mu, B. Wu, L. Wang, L. Gai and F. Wu, ChemSusChem, 2017, 10, 786 CrossRef CAS PubMed.
- Y. X. Wang and P. P. Balbuena, J. Phys. Chem. A, 2001, 105, 9972–9982 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01412e |
| This journal is © The Royal Society of Chemistry 2020 |