DOI:
10.1039/D0RA01366H
(Paper)
RSC Adv., 2020,
10, 14826-14836
Phase controlled synthesis of bifunctional TiO2 nanocrystallites via D-mannitol for dye-sensitized solar cells and heterogeneous catalysis†
Received
12th February 2020
, Accepted 27th March 2020
First published on 14th April 2020
Abstract
The crystal architecture of TiO2 was successfully tailored via a low-temperature (≤200 °C) hydrothermal process in the presence of D-mannitol for feasible applications in dye-sensitized solar cells (DSSCs) and heterogeneous catalysis. In the development of anatase-TiO2 (A-TiO2), D-mannitol does not merely acts as a complexing agent to manage the zigzag chains of octahedral TiO62− with dominant edge sharing but also performs as a capping agent by influencing the hydrolysis process during nucleation, as confirmed by Fourier-transform infrared spectroscopy and dynamic light scattering studies. After physical measurements, the as-synthesized nanocrystallites (NCs) of A-TiO2 were used in DSSCs, where a fascinating power conversion efficiency (PCE) of 6.0% was obtained, which showed excellent performance compared with commercial anatase-TiO2 (CA-TiO2: 5.7%) and rutile-TiO2 (R-TiO2) obtained without D-mannitol (3.7%). Moreover, a smart approach was developed via the A-TiO2 catalyst to synthesize pharmaceutically important C-3 alkylated 4-hydroxycoumarins through different activated secondary alcohols under solvent-free, and heat/visible light conditions. In addition, the catalytic activity of the so-produced A-TiO2 catalyst under solvent-free conditions exhibited remarkable recyclability with up to five consecutive runs with negligible reduction, which is superior to existing reports, and clearly reveals the novelty, and green, sustainable nature of the as-synthesized A-TiO2 catalyst. A plausible reaction mechanism of both coupling partners was activated through the interaction with the A-TiO2 catalyst to produce valuable C-3 alkylated 4-hydroxycoumarins with 95% yield and high selectivity.
Introduction
Titanium dioxide (TiO2) mainly exists in three polymorphs: anatase (tetragonal), i.e., A-TiO2; brookite (orthorhombic), and rutile (tetragonal), i.e., R-TiO2; these are regarded as different orientations of the shared edges/corners of the [TiO62−] octahedra.1 The TiO2 polymorphs play an important role due to their outstanding electrical, optical, and catalytic properties, as well as their chemical stability under both acidic and alkaline conditions, which have enabled their applications in energy storage, photovoltaic cells, and photocatalysis in the last few decades.2 For instance, A-TiO2 is largely preferred in photovoltaics and photocatalysis applications over the other two polymorphs, due to its abundance, environmental friendliness, low cost, excellent electron mobility, catalytic activity, and availability of several cleanroom-free chemical/physical synthesis methods. Generally, the higher performance of the A-TiO2 over R-TiO2 because of its maximum dye-loading capacity and superior incident photocurrent conversion efficiency. Moreover, the high refractive index and UV absorptivity of R-TiO2 make it a potential candidate in the application of optical communication devices.3 The crystal phase transition of TiO2 polymorphs using simple, cost-effective, and low-temperature solution-based methods with different morphologies has attracted considerable research interest for technologically important and practical applications. Normally, peptization, a modifier/surfactant/chelating agent, capping agent, cohesive agent, and annealing temperature play crucial roles during the phase transition pathways of these TiO2 polymorphs, in addition to intrinsic parameters for instance pore size, concentration of the Ti precursor, energy on the surface, aggregation rate, pH value of solution, and crystal growth dynamics.4,5 Many solution-based methods have been reported for the synthesis of different TiO2 polymorphs such as solvothermal, wet chemical, and sol–gel.6 Among these, hydrothermal synthesis is used to obtain phase homogeneity and morphological control at low temperatures.7 Zhou et al. reported the hydrothermal crystallization process for the synthesis of TiO2 via a dissolution–crystallization mechanism.8 At a specific temperature, hydrated Ti ions begin to dissolve to produce Ti4+ hydrated octahedral monomers, which, eventually, form polymers via a condensation reaction. Consequently, connection with octahedral monomers, the formation and growth of the TiO2 nuclei are initiated.9 Thus, the architecture of Ti4+ hydrated octahedral monomer and nucleation process under hydrothermal conditions are major components involved in the fabrication of TiO2 polymorphs. Recently, Oskam et al. reported the nucleation and growth mechanism of different TiO2 polymorphs by applying precursor chemistry, which was mainly dependent on the utilization of reactants.10 Joo et al. studied the crystal structures of TiO2 polymorphs that are controlled from rutile to anatase without and with D-sorbitol acting as a complexing agent.11 Yanqing et al. examined the impact of the TiCl4 concentration and pH of solution on nucleation processes of TiO2 polymorph.12 Until now, the formation of TiO2 polymorphs with a definite crystal structure, shape, surface orientation, and nucleation process under hydrothermal conditions remain uncharted; thus, the correlation between crystal structure formation, and the hydrolysis ratio needs to be investigated for various applications.13
In the present work, we successfully synthesized pure phase A-TiO2 NCs via a facile, low-temperature, and economical hydrothermal method in the presence of D-mannitol, which play role of both capping as well as complexing agent. The D-mannitol was selected due to its non-toxic biological origin, environmental friendliness, low-cost, capping ability, and capability to form an intermediate complex. The possible reaction mechanism to the formation of A-TiO2 is based on the different orientation of shared corners and edges of the TiO62− octahedral. The nucleation process of TiO2 has been derived by [Ti(OH)x(D-mannitol)yClz]n−, where, 4 ≤ x + y + z ≤ 6 during the hydrolysis process, and the relationship between the crystal structure formation and hydrolysis process has been explored. Furthermore, for comparison, we confirmed that the reactions that proceed without D-mannitol result in a phase of pure R-TiO2 rather than A-TiO2. The effect of the hydrothermal reaction conditions on the crystal architecture formation as well as the photovoltaic and photocatalytic properties was investigated, where A-TiO2, synthesized using D-mannitol, exhibited an enhanced DSSCs performance compared to that of the R-TiO2 prepared without it. The presented method for obtaining both A-TiO2 and R-TiO2 polymorphs is facile, environmentally friendly, exhibits long-term chemical stability, and involves a single-step. The TiO2 polymorphs synthesized through this method are used in various applications, including in DSSCs and heterogeneous catalysis.
Experimental details
Chemicals
AR grade chemicals were utilized without any refinement. Titanium(IV) chloride (TiCl4, 99.9%), D-mannitol (>98%), ethanol (96%), and α-terpineol (90%) were purchase from Sigma Aldrich company. Acetonitrile (99.8%), tert-butanol (99.5%), and ethyl cellulose were purchased from Alfa Aesar. The N-719 dye (Ruthenium-535, bis TBA) and iodide electrolyte (Iodolyte AN-50), were commercially available and used as received.
Hydrothermal synthesis of A- and R-TiO2
The NCs of A- and R-TiO2 polymorphs were produced by a simple and elegant one-step hydrothermal route. Typically, for the A-TiO2 NCs, 1 M TiCl4 and 2.5 g of D-mannitol were dissolved in DI-water water (DI, Milli-Q water; 18.2 MΩ cm). The reaction mixture was transferred to high pressure Teflon reactor and kept at constant temperature of 180 °C for 24 h. The as-procured yellowish-white precipitate was separated on centrifugation at 4000 rpm for 10 min, followed air annealing at 550 °C for 30 min to get white powder of A-TiO2NCs (Scheme 1). A similar experimental condition was applied for the synthesis of R-TiO2NCs, except there was no D-mannitol in the precursor solution. A 100% A-TiO2 paste of ∼20 nm sized NCs, named CA-TiO2, was purchased from ENB Korea and used without any modification as the reference.
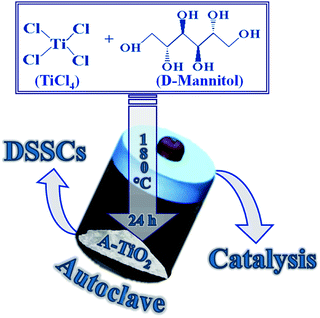 |
| | Scheme 1 The hydrothermal synthesis process of A-TiO2 in presence of D-mannitol for DSSCs and heterogeneous catalysis applications. | |
Fabrication of the DSSCs
A fluorine-tin-oxide substrate (FTO, TEC15, Pilkington, 15 ohm/sq.) was sonicated through detergent in DI-water, acetone, and isopropanol for 20 min, in sequence, and dried under nitrogen gas flow, followed by 15 min ultraviolet–ozone treatment to remove presence of any unwanted residues. Typically, the TiO2 paste was made by mixing of 1.0 g of TiO2 NCs, 2.0 g of α-terpineol, and 0.3 g of ethyl cellulose in 5.0 mL of ethanol with continuous stirring to obtain homogeneous paste for 6 h. Subsequently, the as-prepared NCs of A-, R-, and CA-TiO2 pastes were coated on the FTO substrate via a simple doctor-blade method using an adhesive tape on both side as a spacer. About eight-ten-micron thickness TiO2 films for each case were prepared and annealed at 550 °C in a muffle furnace under an air environment at a constant heating 5° min−1 rate for 30 min. For sensitization, these TiO2 photoanodes were heated to 60 °C and kept for adsorbing 0.3 mM N-719 dye solution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 tert-butanol:acetonitrile for 24 h at 27 °C in the dark. The Pt counter electrode having mirror finish, deposited by DC magnetron sputtering on the conducting FTO substrate, was used as the counter electrode. The two electrodes, i.e., TiO2 with N-719 dye and Pt were clipped together, and to prevent the leakage of the iodine electrolyte solution heat resistant sublimation tape was used as a sealant. The purpose of mask is to allow light to fall only onto a active area of all the devices, i.e. the area was 0.5 cm2 defined by the mask.
:1 tert-butanol:acetonitrile for 24 h at 27 °C in the dark. The Pt counter electrode having mirror finish, deposited by DC magnetron sputtering on the conducting FTO substrate, was used as the counter electrode. The two electrodes, i.e., TiO2 with N-719 dye and Pt were clipped together, and to prevent the leakage of the iodine electrolyte solution heat resistant sublimation tape was used as a sealant. The purpose of mask is to allow light to fall only onto a active area of all the devices, i.e. the area was 0.5 cm2 defined by the mask.
Catalyst activity
A combination of 4-hydroxycoumarin (1 mmol) and 4-methyl-phenylethanol (1 mmol) and A-TiO2 NCs powder (20 mg) was stirred at 70 °C for 5 h. After completion of the reaction (monitored by TLC) ethyl acetate was added to the reaction mixture and the catalyst was collected by filtration. The organic solvent was concentrated under reduced pressure. After purification by recrystallization with hot ethanol, C-3 alkylated 4-hydroxycoumarins were obtained. Moreover, to achieve the best reaction conditions, the effects of various amount of catalyst, reaction time, optimal temperature, and solvent were meticulously investigated. A model reaction between 4-hydroxycoumarin and activated alcohols was monitored to ascertain the optimal reaction parameters. After completing reaction, obtained products were confirm and characterized by 1H NMR spectral data.
Measurements and characterizations
The morphology observation of A-TiO2 and R-TiO2 were confirmed from the surface digital images by field emission scanning electron microscopy (FE-SEM, FEI Nova NanoSEM). High-resolution transmission electron microscopy and selected area electron diffraction (SAED) measurements analyses were conducted by (HR-TEM, FEI TECNAI G2 20 S-TWIN) by using a 200 kV accelerating voltage. The phases of the TiO2 NCs were validated by powder X-ray diffraction patterns (Shimadzu, XRD-6000, Japan) obtained using λ = 0.1542 nm. The phase was confirmed by Raman microscopy (Renishaw, inVia Raman microscope, UK) also. The laser beam (λ = 532 nm) was focused using a lens to produce a spot on the TiO2 film surfaces. The Fourier-transform infrared (FTIR) spectroscopy spectra were obtained to confirm intermediate reaction state from 500 to 4000 cm−1 using Nicolet, iS10, Smart MIRacle, Thermo Scientific. X-ray photoelectron spectroscopy (XPS) spectra were acquired using a PHI 5000 VersaProbe (Ulvac-PHI) to confirm the surface elements and the chemical states. The binding energies were used for internal calibration of C 1s peak (284.6 eV). The specific surface areas of A-TiO2 and R-TiO2 powders were measured through Brunauer–Emmett–Teller spectra on nitrogen physisorption instrument (BET, Belsorp II, BEL Japan INC). The amount of dye adsorbed over A-, R-, and CA-TiO2 photoelectrode was confirmed by desorbing in a 0.1 M NaOH water–ethanol (v/v = 1:1) mixed solution, followed by UV-Vis spectrophotometer (Varian Cary 5000 spectrophotometer) measurement by UV-Vis spectrophotometer (Shimadzu UV-2450) at 540 nm. Simulated sunlight provided by a solar simulator (Sun 2000 ABET 5 Technologies, USA) fitted with an A.M. 1.5G filter was used, and the intensity of one sun (100 mW cm−2) was calibrated on standard silicon solar cell. A Keithley 2400 source meter was used to estimate the current density–applied voltage (J–V) spectra of the A-, R-, and CA-TiO2 photoanodes. The computer-controlled electrochemical impedance spectroscopy (EIS) measurement spectra were recorded on IviumStat Technologies, Netherland at a bias voltage of 0.70 V in a frequency range of 150 kHz to 0.1 Hz under dark condition.
NMR spectral data
(1) 4-(1-p-Tolylethoxy)-2H-chromen-2-one (3a). 1H (400 MHz; CdCl3-d6): δ 1.59–1.63 (d, 3H, J = 16 Hz), 2.33 (s, 3H), 4.67–4.71 (q, 1H, J = 16 Hz), 6.04 (S, 1H), 7.22–7.63 (m, 8H).
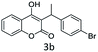
(2) 3-(1-(4-Bromophenyl)ethyl)-4-hydroxy-2H-chromen-2-one (3b). 1H (400 MHz; CdCl3-d6): δ 1.653–1.671 (d, 3H, J = 0.), 4.696–4.750 (q, 1H, J = 7.2 Hz, 14.4 Hz), 6.33 (s, 1H), 7.198–7.676 (m, 8H).
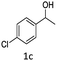
(3) 3-(1-(4-Chlorophenyl)ethyl)-4-hydroxy-2H-chromen-2-one (3c). 1H (400 MHz; CdCl3-d6): δ 1.65–1.69 (d, 3H, J = 16 Hz), 4.65–4.75 (q, 1H, J = 12 Hz and J = 28 Hz), 6.33 (bs, 1H), 7.22–7.72 (m, 8H).
(4) 4-(1-(2,4-Dichlorophenyl)ethoxy)-2H-chromen-2-one (3d). 1H (400 MHz; CdCl3-d6): δ 1.65–1.69 (d, 3H, J = 16 Hz), 4.65–4.72 (q, 1H, J = 28 Hz), 7.11–7.69 (m, 9H), 6.33 (s, 1H).
Results and discussion
Reaction mechanism
The TiO62− octahedral with different orientations of shared edges and corners are responsible for affording A- and R-TiO2. Basically, R-TiO2 is connected by ten octahedra, in which eight are corner-shared and two are edge-shared; the crystal nuclei for rutile TiO2 is made up of linear chains of TiO62− octahedra connected by corner sharing.14 In contrast, A-TiO2 is connected by eight octahedra, in which four are corner-shared and four are edge-shared, making zigzag chains of TiO62− octahedra that are linked to each other through edge-sharing bonding.15 The density of A-TiO2 (3.84 g cm−3) less than R-TiO2 (4.26 cm−3) due to more edge-shared, and larger interstitial spaces between the octahedra.16 Notably, the crystal structure of TiO2 can be agglomerated through different edge-shared and corner-shared TiO62− octahedra orientations during the condensation processes. Generally, when Ti precursor reacts with water molecules, the coordination number of Ti4+ increases from four to six, owing to the acceptance of oxygen lone pairs from nucleophilic ligands through its vacant d-orbitals. These six-fold octahedra structural units can be combined into the final crystal structure formation through condensation.17 Scheme 2 has illustrated the formation of A-TiO2 reaction mechanism. It was found that D-mannitol anions can be substituted for Cl anions during the hydrolysis process to form [Ti(OH)x(D-mannitol)yClz]n− intermediate complexes during crystallization. Similarly, Chen et al. demonstrated that BiOCl surface selectively adsorb molecules of D-mannitol to from hydrogen bonds with exposed oxygen atoms to decrease the surface energy during the crystal growth formation.18 Effect of D-mannitol on the crystal structure formation and nucleation process of A-TiO2 is worth for future investigations. In addition, the hydrochloric acid present in the TiCl4 aqueous solution could act as a catalyst not only facilitating the formation of crystal growth via condensing the intermediate complex but also nucleation process and related species.19
 |
| | Scheme 2 Plausible reaction mechanism to the formation of A-TiO2 NCs. | |
Intermediate complexes
To confirm the formation of intermediate complexes, the FTIR spectrum was investigated. On completing hydrothermal reaction between TiCl4 and D-mannitol, the white precipitate of intermediate complex was transferred into a centrifuge tube at 8000 rpm for 10 min, and washed many times with DI-water, and subsequently dried at room temperature before FTIR measurement. Fig. S1a in ESI† shows the FTIR spectrum of standard D-mannitol consisting of strong –OH and –C–O stretching vibration peaks at 3366, 1080, and 1015 cm−1, as well as the peaks indicative of the C–H stretching vibration at 2983, 2947, and 2901 cm−1. The peak indicative of –CH2 scissor vibrations is noted at 1417 cm−1.20 Fig. S1b† shows the FTIR spectrum of the intermediate complex between Ti4+ and D-mannitol ions, where the molecular interaction of D-mannitol ions with Ti ions, in addition to overlapping, is shifted positively and negatively, thereby validating the construction of a complex structure. The FTIR spectrum of the intermediate complex showed a broad peak at 3167 cm−1, which coincides to –OH stretching vibrations. A broad peak of the –OH stretching vibrations validated the presence of hydroxyl groups over different sites. The high-intensity FTIR peak near 600 cm−1 could be assinged to TiO2.21
Hydrolysis rate estimation
The effect of D-mannitol (0.1 M) on the hydrolysis process of TiCl4 (1 M) was measured through dynamic light scattering technique. As shown in Fig. 1a, in the presence of D-mannitol, TiCl4 in aqueous medium under goes hydrolysis to develop slow particle agglomeration with a small size distribution of ∼18 nm. TiCl4 in aqueous solution, in the absence of D-mannitol undergoes hydrolysis to form fast particles agglomeration with ∼70 nm size distribution (Fig. 1a), confirming the interaction of D-mannitol with TiCl4 that prevents the rapid hydrolysis (Fig. 1b). The coordination of D-mannitol anions with TiCl4 inhibit the crystal growth resulting the formation of small-sized A-TiO2 NCs. For an improved understanding of the hydrolysis reaction, we maintained both samples for more than thirty days (TiCl4 and TiCl4-0.1 M D-mannitol) at room temperature (Fig. 1c). After two days, aqueous medium of TiCl4 solution changed to white coloured precipitate with sediments. This white coloured precipitate subsequently agglomerated, and settle down slowly on the surface of bottom. However, the TiCl4-0.1 M D-mannitol solution remained transparent without precipitated even keep at one month, which is also supported by the dynamic light scattering method, where to preventing the fast hydrolysis process Ti cations could bond with D-mannitol anion at room temperature. We assumed that the slow hydrolysis process taking place between TiCl4-0.1 M D-mannitol solutions plays important role in the formation of small particles size, which ultimately supports the phase transformation process, from rutile to anatase. Though, to initiate the nucleation process during the hydrolysis of A-TiO2NCs from the D-mannitol comprising solution, suitable temperature is required. Xiong et al. reported, in their BiOCl study, that D-mannitol not only behaved as a cohesive agent to prompt the assembly of the structural morphology but also acted as a capping agent to prevent the crystal anisotropic growth.22 To validate the comparison, the acidity (pH = 0.6) of both solutions were kept the same for the dynamic light scattering measurement.
 |
| | Fig. 1 DLS spectra of; (a) 1 M TiCl4, (b) TiCl4-0.1 M D-mannitol in aqueous solution, and (c) diagram showing 30 days stability at a room-temperature where hydrolysis takes place in aqueous solution of TiCl4 without D-mannitol (R-TiO2) and TiCl4-0.1 M D-mannitol (A-TiO2). | |
Structural elucidation
XRD patterns were used to characterize the crystallite structures, phases, and average sizes of the as-obtained A- and R-TiO2. The XRD trends of the as-synthesized TiO2 NCs with and without D-mannitol are respectively shown in Fig. 2a and S2a.† The prior XRD pattern confirmed peaks at 25.3°, 37.8°, 48.0°, 53.8°, 55.0°, 62.6°, and 68.8°, which are ascribed to the 101, 004, 200, 105, 211, 204, and 116 reflection planes of A-TiO2 (JCPDS no. 21-1272),23 respectively, whereas in later XRD pattern, the peaks at 27.4°, 36.0°, 39.1°, 41.2°, 44.0°, 54.3°, 56.6°, 62.7°, 64.0°, 69.7°, and 72.4° in association with (110), (101), (200), (111), (210), (211), (220), (002), (310), (112), and (311) for R-TiO2 (JCPDS data file no. 21-1276) are respectively evidenced.24 There were no difficulties in separating the A and R phases of TiO2; as for anatase, the first two-theta peak was at 25.3° for d101, and for rutile, it was at 27.4° for d110. From the XRD patterns it was clear that, though both A- and R-TiO2 NCs were synthesized by the same chemical method, different phases were obtained just by adding D-mannitol. The average crystal size was calculated using the Debye–Scherrer equation. For the as-synthesized A-, CA-, and R-TiO2, the average crystallite sizes were 12.0, 16.2, and 19.5 nm, respectively. Raman spectroscopy is an effective tool for identifying the molecules in A-TiO2 NCs obtained from D-mannitol. In Fig. 2b, the major Raman shift peaks at 144, 199, 400, 519, and 635 cm−1 were attributed to the Eg, Eg, B1g (B1g/A1g), and Eg symmetries of A-TiO2.25 In the Raman spectrum, no additional peaks, except distinctive vibrations, were found, which confirms the formation of pure phase A-TiO2. The Raman spectrum demonstrated at 142, 235, 443, and 610 cm−1 are attributed to the B1g, two-phonon scattering, Eg, and A1g modes of the rutile phaseof TiO2 NCs, respectively (Fig. S2b†). The spin-orbital splitting of Ti 2p1/2 and Ti 2p3/2 photoelectrons corresponds to a doublet situated at binding energies of 463.9 and 458.1 eV, respectively, as shown in Fig. 2c. The peak separation of Ti 2p1/2 and Ti 2p3/2 was 5.8 eV, confirming the Ti4+ chemical state which is in magnificent consensus with the literature data for A-TiO2,.26 The O1S singlet of oxygen at 529.3 eV is shown in Fig. 2d. The Lorentzian–Gaussian mix function value was employed in the curve resolution of the individual O1S peaks in the two spectra. The curve resolved O1S signal revealed small sized secondary peak that is positioned at a binding energy of 531.86 eV for –OH species present on the surface.27
 |
| | Fig. 2 (a) X-ray diffraction, (b) Raman spectrum, (c) XPS analysis of Ti2p, and (d) O1S of A-TiO2 NCs with D-mannitol. | |
Surface morphology evolution
The surface morphology evolution of A-TiO2 NCs is shown in Fig. 3a. The FE-SEM images of A-TiO2 confirmed the formation of small-sized spheres, where all the grains are well-interlinked to one another, which would offer various facile transporting channels for electrons (or charge carriers) through the TiO2 NCs. The FE-SEM images demonstrate the well-interconnected ∼60 nm average diameter of nanocrystallites, when D-mannitol was absent in the precursor solution as shown in Fig. S2c.† The elemental stoichiometry determined by EDX mapping (Fig. S3†) has evidenced 30.8 and 69.1% atomic proportions for Ti and O, respectively, suggesting the formation of TiO2. The HR-TEM results indicate the high crystallinity of TiO2 interconnected NCs of about 15 nm in sizes (Fig. 3b). The 0.36 nm interplanar distance in compliance with the (101) plane of A-TiO2 NCs (Fig. 3c), and 0.32 nm interplanar distance in compliance with the (110) plane of R-TiO2 NCs (Fig. S2d†) were approved.28 The (101) facet of the A-TiO2 NCs has a relatively low surface energy; thus, it has better stability than those of other facets, and the HR-TEM image also has evidenced clear and sharp rings in reference to the (101), (004), (200), (105), and (211) lattice planes of A-TiO2 in the SAED pattern (Fig. 3d).
 |
| | Fig. 3 (a) FE-SEM, (b) EDX mapping for Ti and O. (c) HR-TEM, and (d) SAED pattern of A-TiO2 NCs. | |
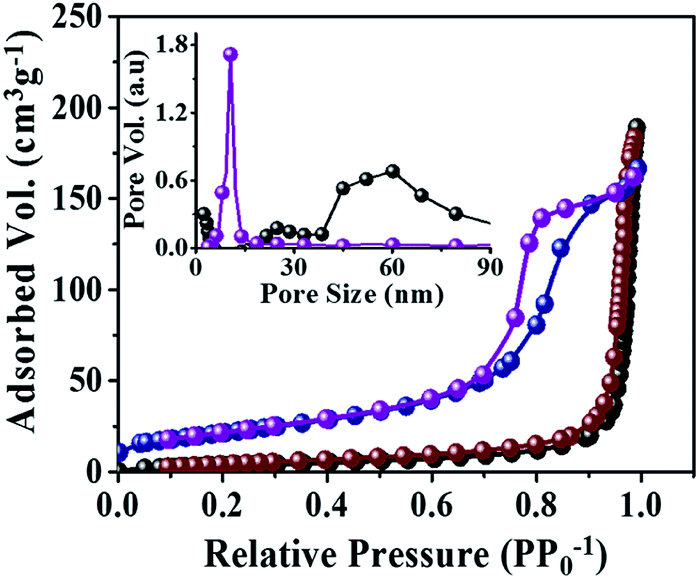
Specific surface area and pore-size distribution measurements
The specific surface area and porosity of both R-TiO2 and A-TiO2 NCs were investigated from the BET and BJH spectra. A type-IV isotherm (Brunauer–Deming–Deming–Teller classification) was due to their mesoporous nature. The hysteresis loop of type H1 suggested that the porosity of A-TiO2 is derived from interconnected NCs.29 Fig. 4 indicates 77.8 m2 g−1 as the specific surface area of A-TiO2 NCs. Furthermore, its narrow pore-size distribution centered at 13.0 nm was ascribed to the presence of relatively small inner pores between NCs and a large pore constructed by an independent structure, which agrees with the FE-SEM and HR-TEM analyses as well. Additionally, the standard multi-point BET technique was employed to calculated the specific surface area of 14.2 m2 g−1 for R-TiO2, which was only one-fifth that of A-TiO2. The narrow pore-size distribution exhibited in the as-prepared R-TiO2 was centered at 60.2 nm. Basically, both DSSCs and heterogeneous catalyst applications are porosity dependent due to the increase in the dye adsorption and active centers.
 |
| | Fig. 4 N2 adsorption desorption isotherms with inset depicts the pore size distribution of R-TiO2 (black and brown dot), and A-TiO2 (blue and purple dot), NCs in the presence of D-mannitol. The black and blue dot denoted adsorption and brown and purple dot denoted the desorption of R- and A-TiO2 NCs. | |
DSSC performance
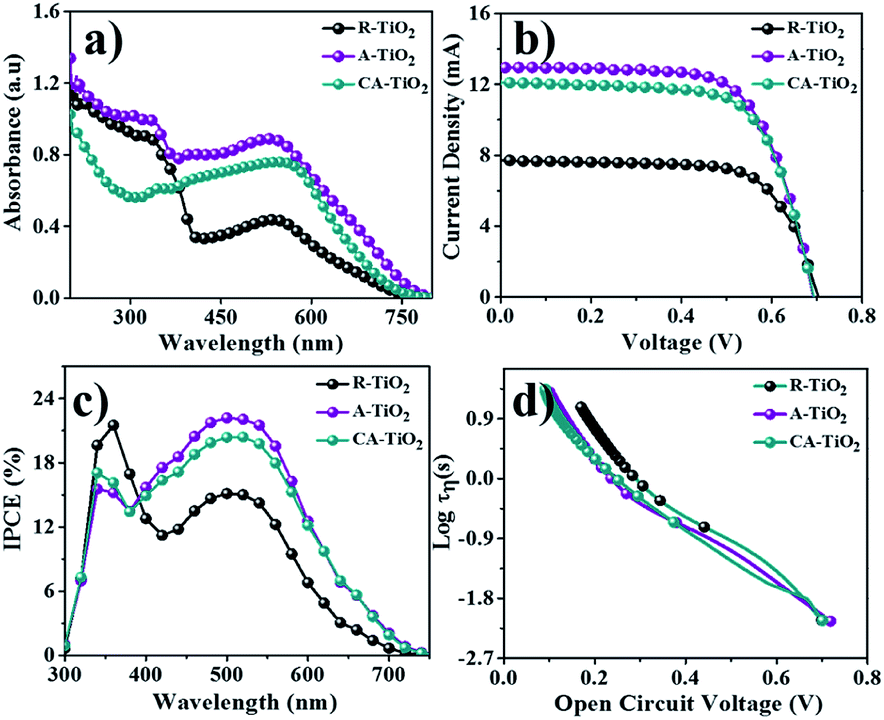
The DSSC performance of CA-TiO2 (∼20 nm particle-size, 100% anatase, ENB Korea) NCs was also measured as a reference. The performances of A- and CA-TiO2 NCs based DSSCs are given in Fig. 5. The UV-Vis absorption spectra of both photoanodes, with N-719 dye, were obtained in a wide visible wavelength range. The characteristic peak hump appeared at 530 nm was due to the N-719 dye (Fig. 5a). The N-719 dye adsorption over A-TiO2NCs (0.86 × 10−7 mol cm−2) was higher than those over CA-TiO2 (0.81 × 10−7 mol cm−2) and R-TiO2 NCs (0.42 × 10−7 mol cm−2), which possibly as a result of the higher surface area and narrow particle size of A-TiO2. To quantify the dye-loading amounts over two photoanodes, these sensitized photoanodes were immersed into 0.1 M sodium hydroxide solution to desorb the adsorbed dye molecules, followed by measuring the UV-Vis absorbance spectra. Using the Beer–Lambert law, the amounts of dye absorbed over these electrodes were determined and are listed in Table 1. The current density–voltage curves of both DSSCs, i.e., CA-TiO2 and, A-TiO2 NCs photoanodes, under one sun illumination were measured and are plotted in Fig. 5b. Table 1 presents a summary of the DSSCs outcome based on various parameters for both photoanodes, where, in both cases, the open circuit voltage (VOC) of 0.70 V was nearly same because VOC mainly is determined by the difference between the oxidation potential of the electrolyte and the Fermi level of the photoelectrode.30 Although, the as-obtained short-circuit current densities (JSC) of both DSSCs confirmed higher performance in A-TiO2 NCs photoanode compared to that in CA-TiO2, could be assigned to a narrow particle size as A-TiO2 demonstrates more dye molecules than that of CA-TiO2.31
 |
| | Fig. 5 (a) UV-Vis, (b) J–V, (c) IPCE, and (d) τ (vs. Voc) measurements of R-, A- and CA-TiO2 photoanodes. | |
Table 1 Summary of DSSCs average efficiency were obtained using six devices under AM 1.5G illumination (100 mW cm−2)
| Photoanodes |
JSC (mA cm−2) |
VOC (V) |
ff (%) |
PCE (%) |
Dye adsorption mol cm−2 |
Crystal size (nm) |
| R-TiO2 |
7.7 |
0.71 |
69 |
3.7 |
0.42 × 10−7 |
19.5 |
| Std. dev. |
0.089 |
0.023 |
0.018 |
0.019 |
| A-TiO2 |
12.9 |
0.70 |
67 |
6.0 |
0.86 × 10−7 |
12.0 |
| Std. dev. |
0.082 |
0.020 |
0.011 |
0.012 |
| CA-TiO2 |
12.0 |
0.70 |
65 |
5.8 |
0.81 × 10−7 |
16.2 |
| Std. dev. |
0.088 |
0.022 |
0.016 |
0.017 |
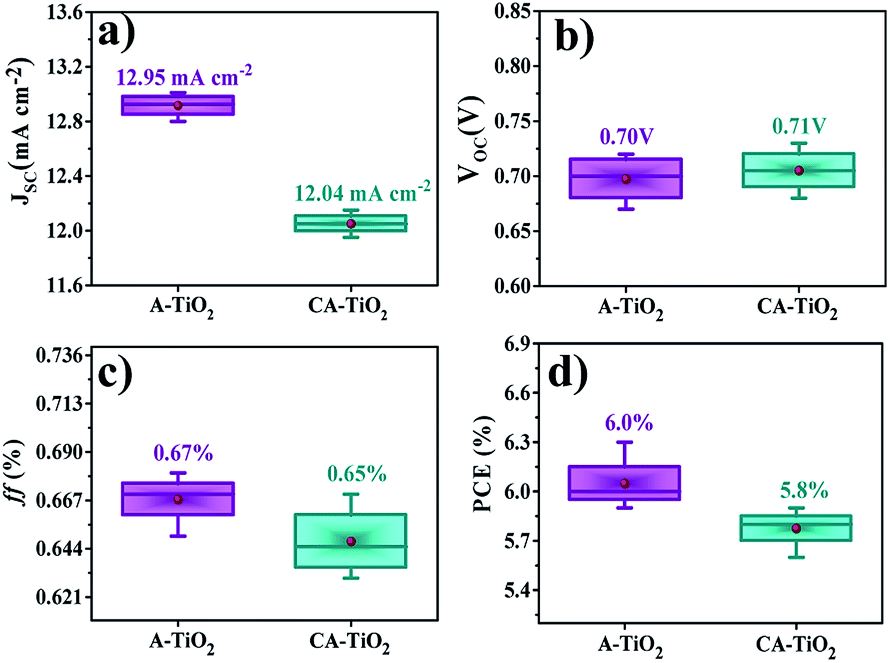
The presence of an interconnected architecture of NCs in A-TiO2 electrode could provide a long-range charge-transfer channels, resulting in 12.9 mA cm−2 JSC, 0.70 V VOC, 67% ff, and 6.0% PEC, which were considerably higher than those of CA-TiO2 (12.0 mA cm−2 JSC, 0.70 V VOC, 65% ff, and 5.8% PEC) and R-TiO2 electrodes (7.7 mA cm−2 JSC, 0.71 V VOC, 69% ff, and 3.7% PEC), Fig. 5. The significant enhancement in the DSSCs performance of A-TiO2NCs photoanode could be due to its unique morphology, high electrical conductivity (2.96 × 10−7 Ω cm), relatively high specific surface area (77.8 m2 g−1), and optimum electron mobility (0.124 cm2 V−1 s−1). Fig. 5c reveals the incident photon-to-electron conversion efficiency (IPCE) spectra of R-TiO2, A-TiO2, and CA-TiO2 photoanodes, where two peaks, one at around 350 nm (TiO2) and the other at around 520 nm, indicative of the absorbed N-719 dye, were observed.32 The as-synthesized A-TiO2 photoanode confirmed a maximum IPCE of 22.08%, which was 20.31% in CA-TiO2, and 15.12% in R-TiO2 photoanodes, revealing a higher N-719 dye adoption of A-TiO2 over CA-TiO2 and R-TiO2. The open-circuit voltage decay plots of both electrodes measured under applying open circuit voltage conditions with respect to time are shown in Fig. 5d. At zero current, there was an exponential decrease in VOC, which supports the recombination of electrons.33 In the graph, the A-TiO2 photoanode exhibited a slow decrement in VOC, which is due to the electron recombination mitigation. The calculated lifetime of the electron, i.e., the time taken by an excess minority carrier to recombine, of A-TiO2, determined by the VOC decay method, was higher than those of the others, suggesting that the electrons are more likely to reach the working electrode than CA-TiO2 and R-TiO2 photoanodes.34
Fig. 6a–d summarizes the average performances of CA-TiO2 and A-TiO2 photovoltaic devices. The as-synthesized A-TiO2 photoanode exhibited a high reproducibility of JSC, VOC, ff, and PEC with a comparatively small standard deviation of 1.72%, which is substantially higher than those for the CA-TiO2 photoanode-based DSSCs devices (1.20%), suggesting the possibility of its reusability compared to the case with others.
 |
| | Fig. 6 Summary of average performance of six devices: (a) Jsc, (b) Voc, (c) ff, and (d) PCE of CA- and A-TiO2-based. The maximum/minimum and average values are denoted by — and ●, respectively. | |
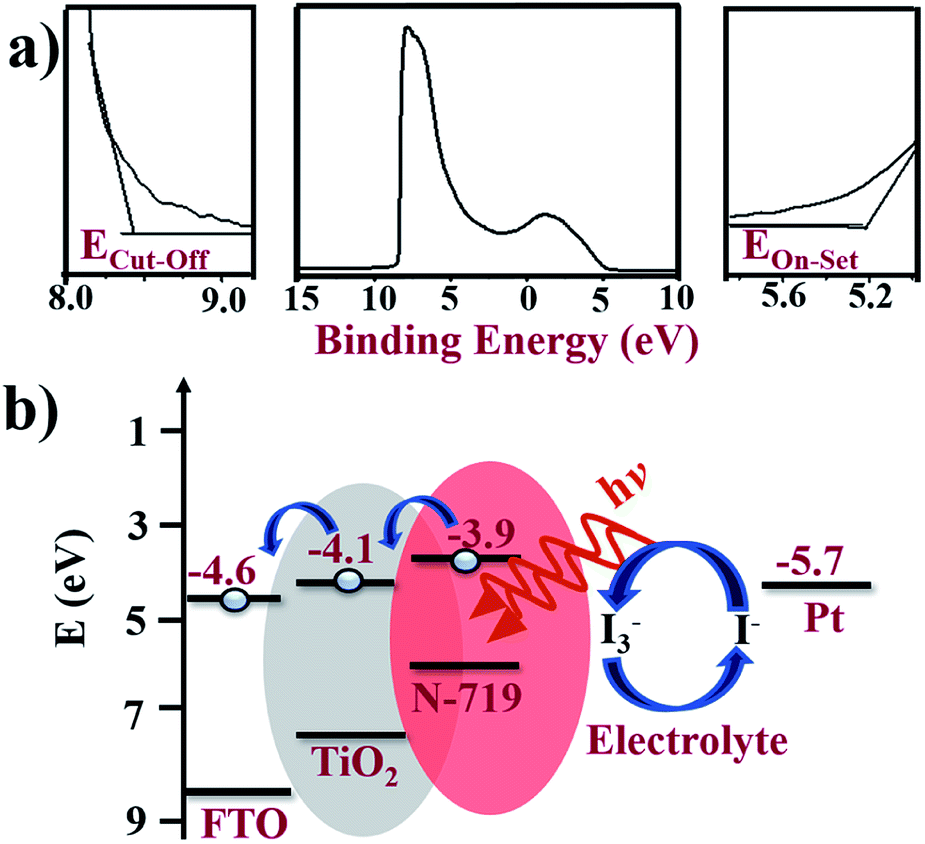
Energy levels
To confirm the energy alignment of dye and TiO2, UPS measurement was performed, which is shown in Fig. 7a, where the various energy edge ranges such as the cut-off region, survey binding energy spectra, and onset region for A-TiO2 NCs are clearly indicated. A schematic energy band diagram of the DSSCs based on the A-TiO2 NCs photoanode charge transfer process is shown in Fig. 7b. The high binding energy cut-off (Ecut-off) of the A-TiO2 NCs photoanode was 8.56 eV. The binding energy onset (Eonset) was −5.30 eV, relative to the Fermi level of Au (at 0 eV). The valence band minimum (VBM) calculated using VBM = hν − (Ecut-off − Eonset) equation, whereas, ν is the frequency of vibration of light and h is the Planck's constant estimates VBM of −7.35 eV for A-TiO2 NCs. The band gap energy denoted by (Eg) of the A-TiO2 NCs electrode was estimated using the following equation:where, α is the absorption coefficient, and A is the proportionality constant. Fig. S4† presents a UV-Vis spectrum of the A-TiO2 photoanode, where 3.2 eV Eg was obtained for A-TiO2NCs film, which closely matches to the reported value.35 Using this band gap energy, the conduction band minimum (CBM) of A-TiO2 was calculated using the following equation:
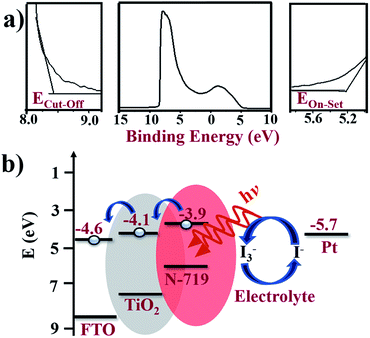 |
| | Fig. 7 (a) UPS spectra shown three different energy edge ranges: cut-off region, survey binding energy spectra, and onset region of A-TiO2. (b) The schematic energy bandgap diagram of FTO/A-TiO2/N-719 dye/electrolyte/Pt photoanode device calculated from UPS analyses. | |
The calculated CBM of the A-TiO2 NCs electrode was −4.1 eV. To understand the electron transfer pathways of this photoanode, the energy band diagram of the FTO/A-TiO2/N-719 dye/electrolyte/Pt DSSCs was also designed. In DSSCs, on capturing photons over the N-719 dye layer, the electron–hole pairs are created, and the excited electrons are injected into the CBM of the A-TiO2NCs, which are then collected at the FTO substrate of the −4.6 eV work function more easily.36
Electrochemical charge transfer process
The photogenerated charge transfer and recombination processes are crucial in DSSCs performance, which can be explained with EIS measurement operated bias potential 0.70 V under dark condition. Fig. 8 shows the Nyquist curve of the A-TiO2 NCs photoanode which demonstrates one semicircle at high-frequency region (>1 kHz), and another semicircle at a low-frequency region (100–0.1 Hz). A relatively small semicircle at high-frequency region corresponds to the charge-transfer resistance (R1), and the capacitance (CPE1) at the counter electrode (platinum)/redox electrolyte interface. The second semicircle, at the low-frequency range, corresponds to the charge-transfer interactions occurring at the photoanode-dye/electrolyte interface (R2), and the chemical capacitance (CPE2) at the TiO2/dye/redox electrolyte interface.37 The equivalent circuit and the corresponding values obtained by the Z view software are shown as an inset of Fig. 8, and also in Table 2.
 |
| | Fig. 8 EIS plot of the R-, A- and CA-TiO2 NCs photoanodes. | |
Table 2 The R-TiO2 and A-TiO2 NCs photoanodes fitted electronic parameters of Nyquist plots
| |
RS |
R1 |
R2 |
CPE1-T |
CPE1-P |
CPE2-T |
CPE2-P |
τ (ms) |
| R-TiO2 |
5.2 |
4.4 |
31.0 |
5.32 ×10−3 |
0.59 |
7.90 ×10−3 |
0.91 |
1.58 × 10−4 |
| A-TiO2 |
5.9 |
2.4 |
19.7 |
1.06 ×10−3 |
0.73 |
21.0 ×10−3 |
0.87 |
2.24 × 10−4 |
Effects of the catalyst amount, temperature, time, and solvent system
For the synthesis of C-3 alkylated 4-hydroxycoumarins, we successfully analyzed the reaction conditions, for instance catalyst amount, temperature, and required time for the completion of the reaction (Scheme 3, Table 3). In first, of A-TiO2 catalyst amount was tested; among them a model amount of 20 mg exhibited a notable yield. Although, when the catalyst amount was increased to 30 mg, no obvious change in the yield was observed, this may be due to the exhaustion of the catalytic sites or the maximum conversion of the catalyst efficiency.
 |
| | Scheme 3 Synthesis of the substituted C-3 alkylated 4-hydroxycoumarins using the A-TiO2 catalyst. | |
Table 3 The optimization of reaction conditions determines
| No |
Amount of catalyst (mg) |
Temperature (°C) |
Solvent |
Time (h) |
Yieldb (%) |
| Reaction conditions: 4-hydroxycoumarin (1 mmol), 4-methylphenylethanol (1 mmol), catalyst (20 mg), solvent free. Isolated yields. |
| 1 |
0 |
Reflux |
MeCN |
10 |
— |
| 2 |
5 |
Reflux |
MeCN |
1 |
0 |
| 3 |
10 |
50 °C |
MeCN |
1 |
0 |
| 4 |
15 |
50 °C |
MeOH |
1 |
0 |
| 5 |
20 |
50 °C |
EtOH |
1 |
0 |
| 6 |
20 |
50 °C |
THF |
1 |
0 |
| 7 |
20 |
50 °C |
DCM |
1 |
0 |
| 8 |
20 |
50 °C |
IPA |
1 |
0 |
| 9 |
20 |
50 °C |
Toluene |
1 |
0 |
| 10 |
20 |
50 °C |
Solvent free |
5 |
75 |
| 11a |
20 |
70 °C |
Solvent free |
5 |
95 |
| 12 |
30 |
70 °C |
Solvent free |
5 |
95 |
| 13 |
20 |
Visible light |
Solvent free |
5 |
95 |
Under the optimized experimental conditions, we then performed the reactions with different solvents and varying reaction conditions (reflux, 50 °C, 70 °C, and under visible light irradiation) for different time periods (1–10 h). In the presence of 20 mg of A-TiO2 catalyst, at 70 °C for a reaction time of 4.5 h, 95% yield was obtained, which is one of the maximum values, so far, reported in the literature.38–47 It was observed that after extended reaction time and high temperature, the reaction did not proceed in the forward direction. In our study, 20 mg of A-TiO2 catalyst under heat/visible light and solvent free conditions demonstrated a notable product yield of 95% with stipulated time period of 4.5 h. Similarly, the nature of the solvent can interrupt the reaction environment by affecting the catalytic stability, solubility, and rate of the reaction. After careful examination of all the results, a solvent-free condition was preferred for the synthesis of further derivatives. Thereafter, this solvent-free specific condition was used for all the reactions. Table 3 summarizes the percentage yield of C-3 alkylated 4-hydroxycoumarins under distinct reaction condition i.e. catalyst amount, reaction temperature, various solvents, and time.
Synthesis of substituted C-3 alkylated 4-hydroxycoumarins using the A-TiO2 catalyst
Under the optimal re substituted C-3 alkylated 4-hydroxycoumarins using the A-TiO2 catalyst (Table 4).
Table 4 C-alkylation of 4-hydroxycoumarin with various alcohols
| No |
Alcohol |
Product |
Time (h) |
Yield % |
| 1 |
 |
 |
4.5 |
97 |
| 2 |
 |
 |
5 |
95 |
| 3 |
 |
 |
5 |
94 |
| 4 |
 |
 |
5 |
90 |
The plausible reaction mechanism
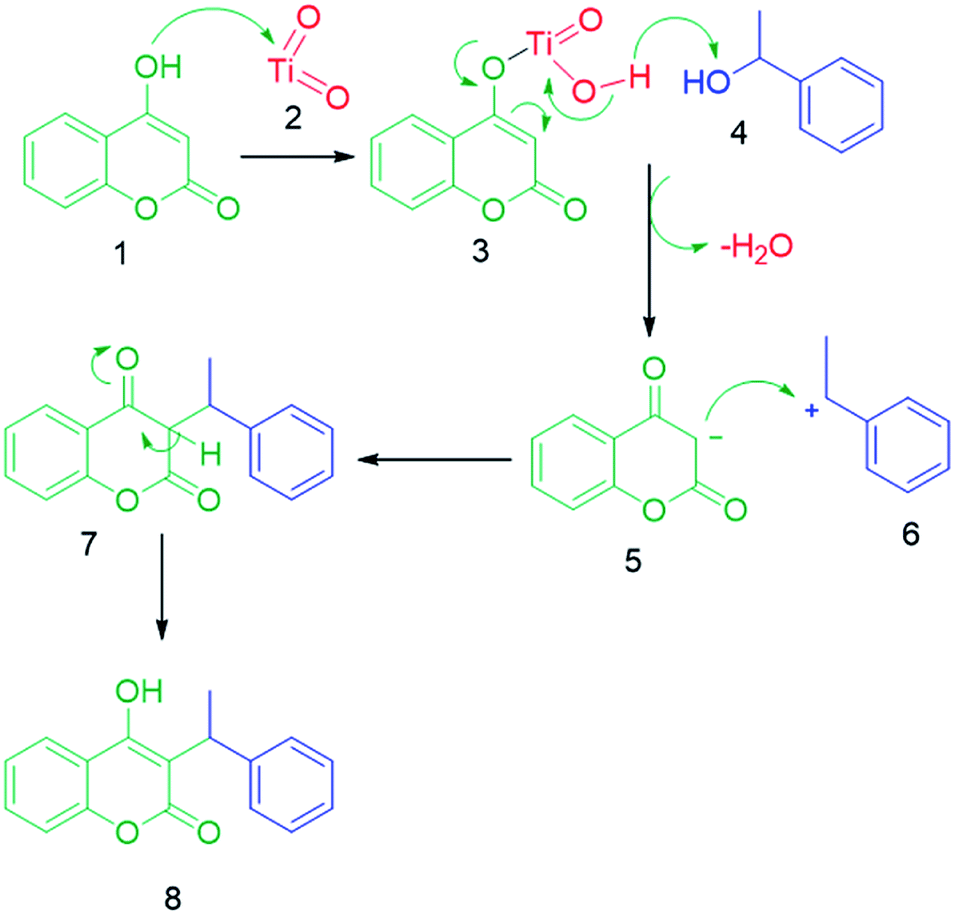
A-TiO2-catalyzed synthesis of C-3 alkylated 4-hydroxycoumarins occurred by removal of a water molecule, which results in the formation of a stable carbocation, as alkylating species that were derived from alcohol on the Lewis acidic sites of A-TiO2 (C-3 alkylation). The enolic hydroxy group of 4-hydroxycoumarin was activated by Ti metal via Lewis acid catalysis to increase the nucleophilicity of the 3-position of 4-hydroxycoumarin. The final product C-3 alkylated 4-hydroxycoumarins was produced through an electrophilic attack activated secondary benzyl alcohols on the nucleophilic enolic hydroxy group of 4-hydroxycoumarin (Scheme 4).
 |
| | Scheme 4 Plausible reaction mechanism for the synthesis of C-3 alkylated 4-hydroxycoumarins. | |
Recyclability of the A-TiO2 nanocatalyst
After evaluating the activity and flexibility of the A-TiO2 catalyst for different kinds of reactions, the recyclability of the catalyst was observed using the reaction between 4-hydroxycoumarin and 4-methyl-phenylethanol as a model reaction under the optimal conditions. Typically, after completion of reaction, the A-TiO2 catalyst was separated from the reaction mixture through a simple filtration method. The reusability of the catalyst was assessed after activating the catalyst at 200 °C for 1 h; the catalyst was employed for five runs to test the consistency of its activity. After five runs, the product yields obtained over the A-TiO2 catalyst exhibited no noticeable decrease, suggesting a high consistency of the activity of the as-synthesized A-TiO2 catalyst. This reusability study reveals the high stability and turnover of A-TiO2 nanocatalysts under optimal operating conditions. The reusability of the catalysts is one and significant advantage that makes them beneficial for marketable applications.
Conclusions
In summary, A-TiO2 was successfully prepared via a simple, elegant and cost-effective hydrothermal method in the presence of D-mannitol. In this synthesis, D-mannitol plays a crucial role in the fabrication of A-TiO2 NCs, as a; (a) complexing agent that regulates the zigzag chains of the TiO62− octahedral by dominant edge sharing, and (b) capping agent that influences the nucleation for NCs during the hydrolysis process. The FTIR spectrum impeccably suggests the formation of an intermediate complex structure between Ti4+ and D-mannitol ions. A PCE of 6.0% is achieved from A-TiO2 NCs photoanode, which is about 1.6 times higher than R-TiO2 (3.7%); this may be due to the difference in the structure, surface area, dye intake capacity, and charge transfer resistance. In addition, A-TiO2 catalyst was used for synthesizing of pharmaceutically significant C-3 alkylated 4-hydroxycoumarins under solvent-free conditions. An excellent yield of 95% at very less reaction time of 4.5 h is obtained. The all synthesized compounds are confirmed by 1H NMR spectroscopy. The catalytic activity of produced A-TiO2 has remarkable recyclability up to five consecutive runs with slight change in formed product yields, which clearly demonstrates the novel, efficient, recyclable, and economical nature of the as synthesized A-TiO2 catalyst. Therefore, the A-TiO2 catalyst can be a potential candidate, over other expensive and rare-earth catalysts, during organic transformation processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to extend their sincere appreciation to the Researchers supporting project number (RSP-2019/55), King Saud University, Riyadh, Saudi Arabia for funding this research.
References
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- H. Zhang and J. F. Banfield, J. Mater. Chem., 1998, 8, 2073–2076 RSC.
- R. Boppella, A. Mohammadpour, S. Illa, S. Farsinezhad, P. Basak, K. Shankar and S. V Manorama, Acta Mater., 2016, 119, 92–103 CrossRef CAS.
- J. Zhang, F. Huang and Z. Lin, Nanoscale, 2010, 2, 18–34 RSC.
- J. Yang, S. Mei and J. M. F. Ferreira, J. Am. Ceram. Soc., 2000, 83, 1361–1368 CrossRef CAS.
- M. Wu, G. Lin, D. Chen, G. Wang, D. He, S. Feng and R. Xu, Chem. Mater., 2002, 14, 1974–1980 CrossRef CAS.
- A. A. Chaudhry, S. Haque, S. Kellici, P. Boldrin, I. Rehman, F. A. Khalid and J. A. Darr, Chem. Commun., 2006, 2286–2288 RSC.
- J. Xie, X. Wang and Y. Zhou, J. Mater. Sci. Technol., 2012, 28, 488–494 CrossRef CAS.
- J. Zhang, P. Sun, P. Jiang, Z. Guo, W. Liu, Q. Lu and W. Cao, J. Mater. Chem. C, 2019, 7, 5764–5771 RSC.
- D. Reyes-Coronado, G. Rodŕiguez-Gattorno, M. E. Espinosa-Pesqueira, C. Cab, R. de Coss and G. Oskam, Nanotechnology, 2008, 19, 145605 CrossRef CAS PubMed.
- S. F. Shaikh, R. S. Mane, B. K. Min, Y. J. Hwang and O. Joo, Sci. Rep., 2016, 6, 20103 CrossRef CAS PubMed.
- Z. Yanqing, S. Erwei, C. Zhizhan, L. Wenjun and H. Xingfang, J. Mater. Chem., 2001, 11, 1547–1551 RSC.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- Y. Zhang, Z. Jiang, J. Huang, L. Y. Lim, W. Li, J. Deng, D. Gong, Y. Tang, Y. Lai and Z. Chen, RSC Adv., 2015, 5, 79479–79510 RSC.
- P. M. Oliver, G. W. Watson, E. Toby Kelsey and S. C. Parker, J. Mater. Chem., 1997, 7, 563–568 RSC.
- A. Tricoli, M. Righettoni and S. E. Pratsinis, Nanotechnology, 2009, 20, 315502 CrossRef PubMed.
- S. G. Kumar and K. S. R. K. Rao, Nanoscale, 2014, 6, 11574–11632 RSC.
- F. Tian, J. Xiong, H. Zhao, Y. Liu, S. Xiao and R. Chen, CrystEngComm, 2014, 16, 4298–4305 RSC.
- Z. Wang, D. Xia, G. Chen, T. Yang and Y. Chen, Mater. Chem. Phys., 2008, 111, 313–316 CrossRef CAS.
- S. Pfeffer-Hennig, P. Piechon, M. Bellus, C. Goldbronn and E. Tedesco, J. Therm. Anal. Calorim., 2004, 77, 663–679 CrossRef CAS.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- J. Xiong, G. Cheng, G. Li, F. Qin and R. Chen, RSC Adv., 2011, 1, 1542–1553 RSC.
- Y.-G. Guo, Y.-S. Hu and J. Maier, Chem. Commun., 2006, 2783–2785 RSC.
- B. Pal, M. Sharon and G. Nogami, Mater. Chem. Phys., 1999, 59, 254–261 CrossRef CAS.
- G. C. Vásquez, M. A. Peche-Herrero, D. Maestre, B. Alemán, J. Ramírez-Castellanos, A. Cremades, J. M. González-Calbet and J. Piqueras, J. Mater. Chem. C, 2014, 2, 10377–10385 RSC.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492–3495 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- B. Wang, J. Liu, Q. Sun, B. Xiao, R. Li, T.-K. Sham and X. Sun, Adv. Mater. Interfaces, 2016, 3, 1600369 CrossRef.
- D. Chen, F. Huang, Y.-B. Cheng and R. A. Caruso, Adv. Mater., 2009, 21, 2206–2210 CrossRef CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- J. H. Yang, C. W. Bark, K. H. Kim and H. W. Choi, Materials, 2014, 7, 3522–3532 CrossRef CAS PubMed.
- S. Mayumi, Y. Ikeguchi, D. Nakane, Y. Ishikawa, Y. Uraoka and M. Ikeguchi, Nanoscale Res. Lett., 2017, 12, 513 CrossRef PubMed.
- S. F. Shaikh, H.-C. Kwon, W. Yang, H. Hwang, H. Lee, E. Lee, S. Ma and J. Moon, J. Mater. Chem. A, 2016, 4, 15478–15485 RSC.
- E. Tea and C. Hin, Phys. Chem. Chem. Phys., 2016, 18, 22706–22711 RSC.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS PubMed.
- D. Han, C. Wu, Y. Zhao, Y. Chen, L. Xiao and Z. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 42029–42034 CrossRef CAS PubMed.
- J. Yu, J. Fan and B. Cheng, J. Power Sources, 2011, 196, 7891–7898 CrossRef CAS.
- D.-U. Chen, P.-Y. Kuo and D.-Y. Yang, Bioorg. Med. Chem. Lett., 2005, 15, 2665–2668 CrossRef CAS PubMed.
- J.-W. Xie, L. Yue, W. Chen, W. Du, J. Zhu, J.-G. Deng and Y.-C. Chen, Org. Lett., 2007, 9, 413–415 CrossRef CAS PubMed.
- M. I. Naumov, S. A. Sutirin, A. S. Shavyrin, O. G. Ganina, I. P. Beletskaya, V. Bourgarel-Rey, S. Combes, J.-P. Finet and A. Y. Fedorov, J. Org. Chem., 2007, 72, 3293–3301 CrossRef CAS PubMed.
- J. Kischel, K. Mertins, D. Michalik, A. Zapf and M. Beller, Adv. Synth. Catal., 2007, 349, 865–870 CrossRef CAS.
- C. R. Reddy, B. Srikanth, N. N. Rao and D.-S. Shin, Tetrahedron, 2008, 64, 11666–11672 CrossRef CAS.
- X. Lin, X. Dai, Z. Mao and Y. Wang, Tetrahedron, 2009, 65, 9233–9237 CrossRef CAS.
- P. Thirupathi and S. S. Kim, Tetrahedron, 2010, 66, 2995–3003 CrossRef CAS.
- P. Theerthagiri and A. Lalitha, Tetrahedron Lett., 2010, 51, 5454–5458 CrossRef CAS.
- G. Aridoss and K. K. Laali, Tetrahedron Lett., 2011, 52, 6859–6864 CrossRef CAS.
- M. A. Gonzalez, S. Takkellapati, K. Tadele, T. Li and R. S. Varma, ACS Sustain. Chem. Eng., 2019, 7, 6744–6757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, X-ray diffraction pattern, Raman analysis, FE-SEM images, HR-TEM, UV-Vis, JV, IPCE, electrochemical impedance spectroscopy, BET analysis NMR spectral data etc. NMR spectral data etc. See DOI: 10.1039/d0ra01366h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
T. A. J. Siddiquib,
Shoyebmohamad F. Shaikh
a,
T. A. J. Siddiquib,
Shoyebmohamad F. Shaikh