DOI:
10.1039/D0RA01335H
(Paper)
RSC Adv., 2020,
10, 10315-10321
Electrophilic alkylation of arenes with 5-bromopyrimidine en route to 4-aryl-5-alkynylpyrimidines†
Received
11th February 2020
, Accepted 5th March 2020
First published on 10th March 2020
Abstract
A new synthetic protocol for preparation of medicinally important 4-aryl-5-alkynylpyrimidines is described. The featured approach involves a sequence of chemo- and regioselective Brønsted acid-catalyzed electrophilic alkylation of arenes with 5-bromopyrimidine, followed by oxidative re-aromatization of the formed dihydropyrimidine ring. Finally, palladium-catalyzed Sonogashira cross-coupling reaction provided an end-game strategy.
Introduction
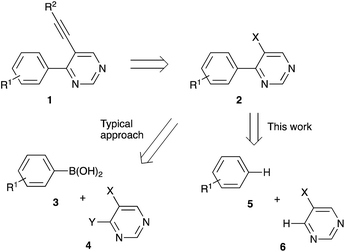
4-Arylpyrimidines 1 bearing acetylene substituents at C-5 (Scheme 1) were investigated as promising non-nucleoside-type adenosine kinase,1,2 tyrosine kinase,3,4 or lysine-specific demethylase5 inhibitors. These molecules were also tested as potential anti-proliferative agents6,7 or non-toxic selective herbicides,8 or used as a synthetic platform for further derivatization en route to medicinally relevant structures.9 Conveniently, structures of this type are accessible via Pd-catalyzed cross-coupling reaction involving terminal alkynes and hetaryl halides or sulfonates 2 (Scheme 1).10,11 Alternatively, a decarboxylative cross-coupling of alkynyl halides with hetarylcarboxylic acids can be employed.12 Although the synthetic route involving 2 as an electrophilic component in the Sonogashira reaction seems very straightforward, access to these starting materials typically relies on another transition metal-catalyzed cross-coupling step, usually chemoselective Suzuki reaction between arylboronic acid 3 and hetaryl dihalide 4 (Scheme 1).13–18 Since this conventional approach lacks any atom-economy appeal, we wondered, if an alternative synthetic strategy could be designed, utilizing a formal oxidative functionalization of two C–H bonds in arene 5 and pyrimidine 6 (Scheme 1). In perspective, this would allow for an easier assembly of 2 from more affordable precursors, while generating less chemical waste. Herein we wish to report on our synthetic studies towards this goal.
 |
| | Scheme 1 Synthetic approaches towards 4-arylpyrimidines. | |
Results and discussion
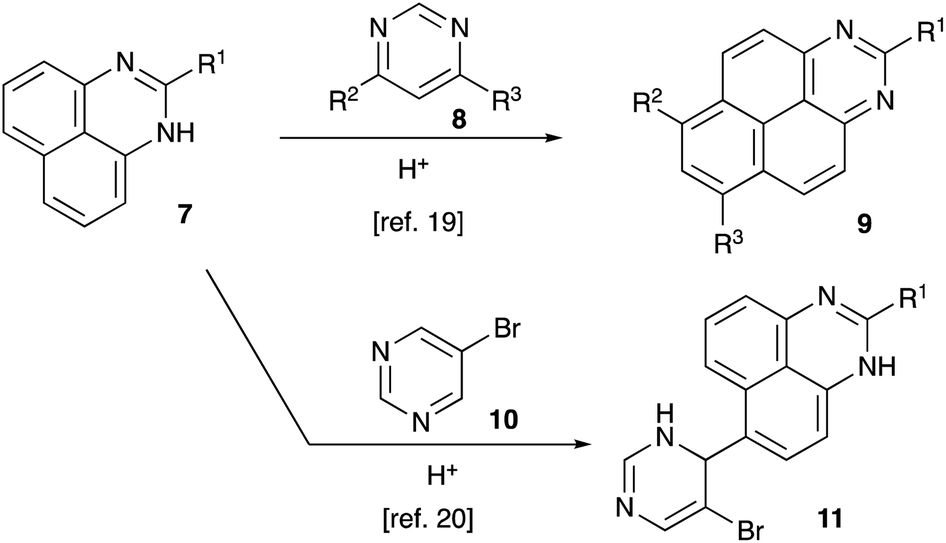
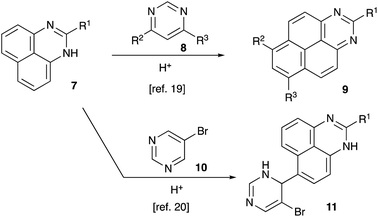
In recent years, our group was interested in designing novel acid-mediated multistep cascade transformations targeting material science and medicinal chemistry applications.19–23 In particular, an unusual annulation reaction was demonstrated, involving perimidines 7 and pyrimidines 8 and leading to the formation of 1,3-diazapyrenes 9 (Scheme 2).24 It was also shown that unlike most other pyrimidines, 5-bromopyrimidine 10 did not react according to this general scheme, forming mono-alkylation products 11 instead (Scheme 2).24,25 Apparently, this process is related to a Friedel–Crafts type SEAr alkylation reaction, in which pyrimidine in protonated form serves as an electrophile. We considered that an extension of the latter reaction to other nucleophilic arenes would help us to pursue our stated goal of installing an aryl substituent at C-4 of 5-bromopyrimidine ring.
 |
| | Scheme 2 Acid-assisted reactions of perimidines with pyrimidines. | |
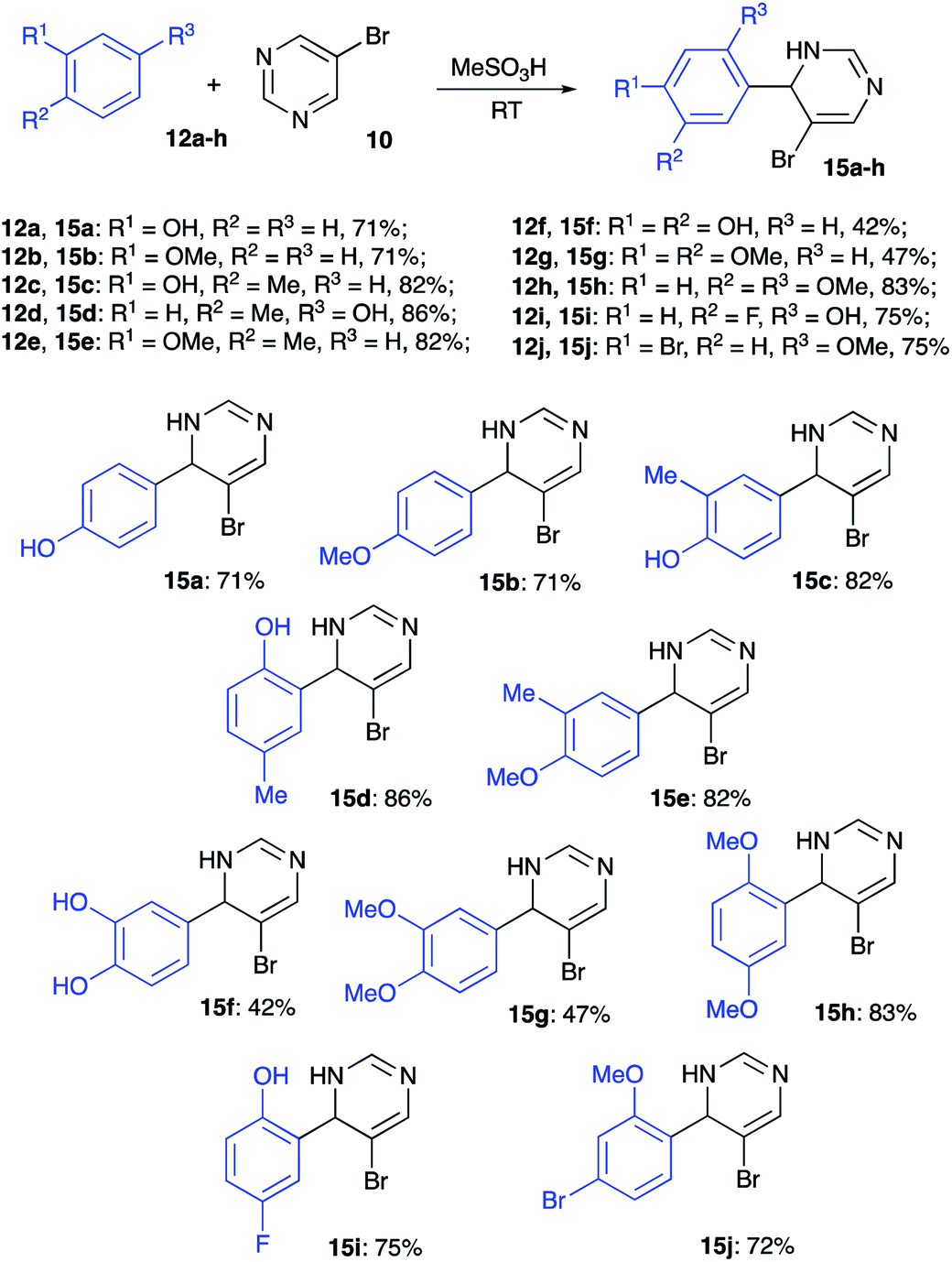
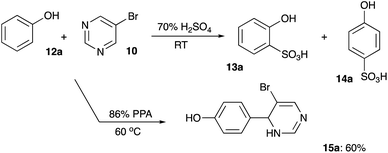
To test this idea, we carried out the reaction between phenol (12a) (as an example of electron rich nucleophilic arene) and 5-bromopyrimidine (10) in 70% aqueous sulfuric acid. Regrettably, this reaction led to the formation of a mixture of two sulfonic acids 13a and 14a, but the desired product 15a did not form under these conditions (Scheme 3). Next, we tried to carry out the same transformation in 86% polyphosphoric acid (PPA). We were pleased to find that in this case 15a was formed smoothly, and we managed to isolate it in 60% yield (Scheme 3). The same reaction carried out at room temperature in the presence of methanesulfonic acid afforded 15a in 71% yield (Scheme 4).
 |
| | Scheme 3 Reactions of phenol 12a with pyrimidine 15 in the presence of acids. | |
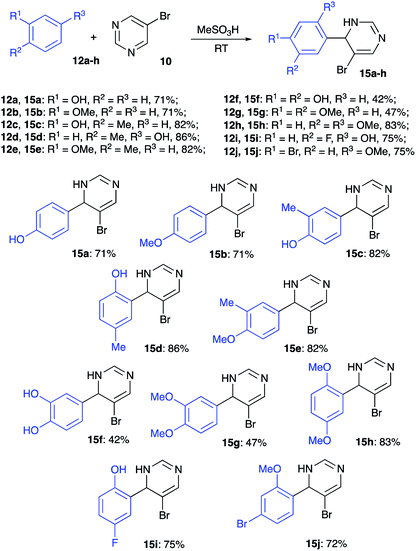 |
| | Scheme 4 Alkylation of different electron-rich benzenes with 5-bromopyrimidine. | |
Inspired by these results, we examined the reactivity of other electron rich arenes. To our delight, anisole (12b), o- and p-cresols (12c and 12d, respectively), and o-methylanisole (12e) reacted smoothly providing the corresponding alkylated products 15b–e in high yields (Scheme 4). Reactions of catechol (12f) and veratrole (12g) also proceeded smoothly, but isolation via extraction was accompanied with notable loss of products 15f and 15g, which proved to be quite water-soluble (Scheme 4). In contrast, reaction and isolation of 1,4-dimethoxybenzene (12h) proceeded uneventfully, leading to formation of 15h in high yield (Scheme 4). Next, we tested the reactivity of halogenated phenol derivatives. Remarkably, both p-fluorophenol (12i) and m-bromoanisole (12j) afforded the corresponding coupling products 15i, j very efficiently (Scheme 4).
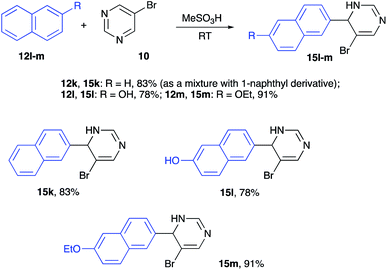
Reactions of naphthalenes with electrophilically activated 5-bromopyrimidine under the same conditions were also tested. Non-substituted naphthalene (12k) reacted very efficiently, but the regioselectivity was poor, and the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of inseparable 2-naphthyl- (15k) and 1-naphthyl-regioisomeric products was isolated in 83% combined yield (Scheme 5). Alkylation of 2-naphthol (12l) and 2-ethoxynaphthalene (12m) proceeded regioselectively and the corresponding structures 15l and 15m were obtained as sole products isolated in high yields (Scheme 5).
:1 mixture of inseparable 2-naphthyl- (15k) and 1-naphthyl-regioisomeric products was isolated in 83% combined yield (Scheme 5). Alkylation of 2-naphthol (12l) and 2-ethoxynaphthalene (12m) proceeded regioselectively and the corresponding structures 15l and 15m were obtained as sole products isolated in high yields (Scheme 5).
 |
| | Scheme 5 Alkylation of different electron-rich naphthalenes with 5-bromopyrimidine. | |
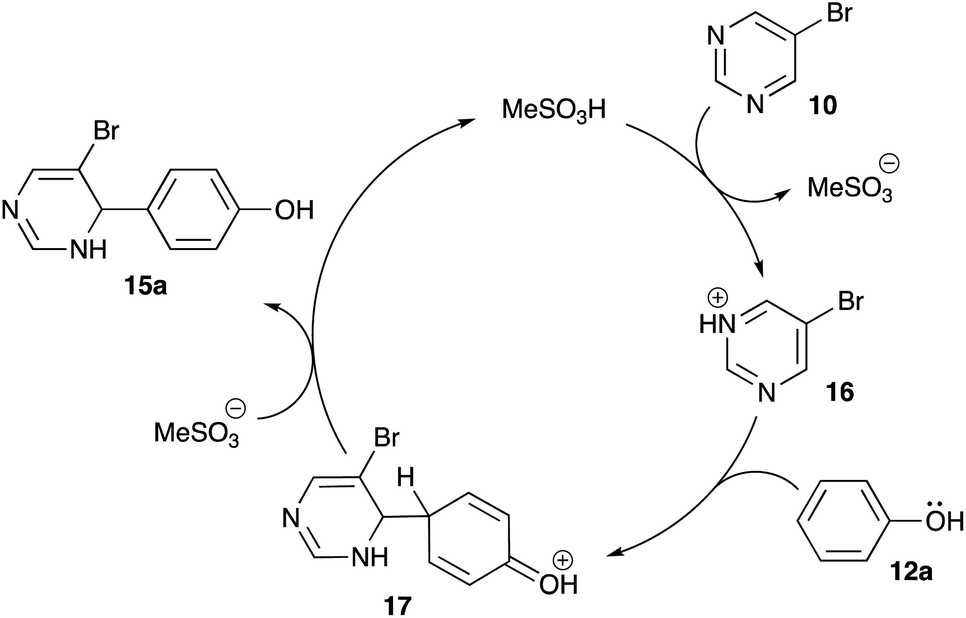
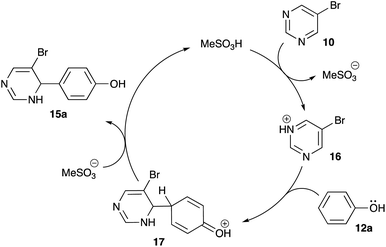
It is believed that this reaction proceeds via SEAr mechanism. Indeed, protonation of one of the nitrogen atoms in pyrimidine 10 in the presence of strong Brønsted acid should afford highly electrophilic pyrimidinium species 16, which could serve as an electrophile in Friedel–Crafts-like reaction involving electron-rich arenes (Scheme 6). To get an additional support to this mechanistic rationale we attempted to carry out this reaction employing electron deficient substrates: nitrobenzene, acetophenone, 2-hydroxyacetophenone, 2-acetonaphthone, 1-benzonaphthone, acetanilide, or 1,8-diaminonaphthalene (the latter upon protonation is converted in poorly nucleophilic ammonium form). Expectedly, the alkylation did not proceed with either of these substrates.
 |
| | Scheme 6 Mechanism of electrophilic alkylation. | |
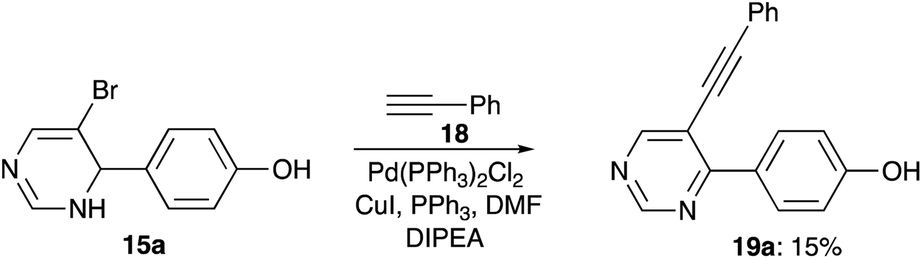
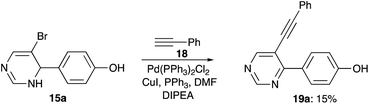
Next, we decided to investigate the possibility to use 5-bromo-3,4-dihydropyrimidin-4-yl arenes 15 in cross-coupling reaction with terminal alkynes. To this end, compound 15a was treated with phenyl acetylene 18 under standard conditions for Sonogashira reaction. Unfortunately, reaction produced a complex mixture of products, among which we only were able to isolate minute amounts of pyrimidine 19a, probably resulted from aerobic oxidation of the 3,4-dihydropyrimidine ring (Scheme 7).
 |
| | Scheme 7 Attempted Sonogashira coupling of dihydropyrimidine 15a. | |
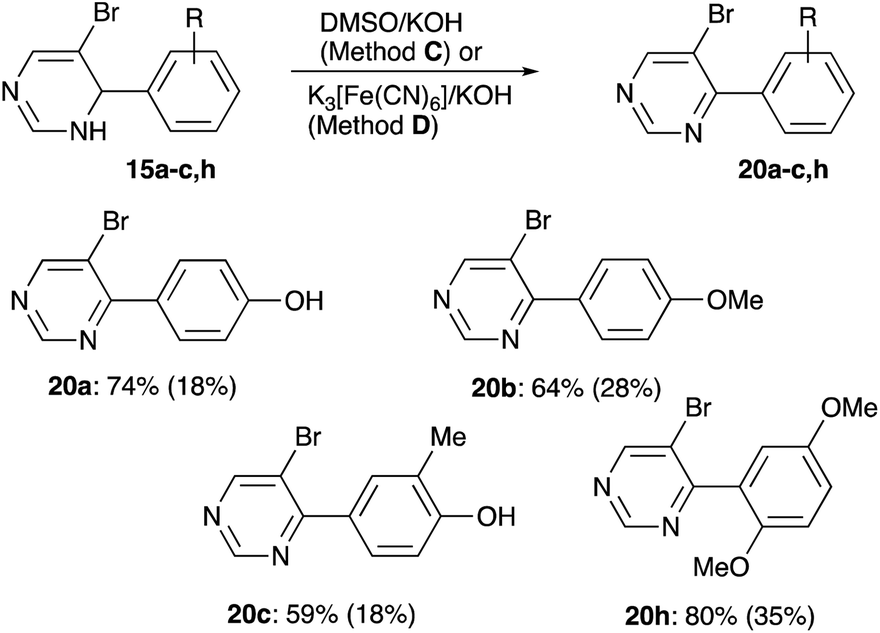
Since oxidation of dihydropyrimidine ring seemed to proceed spontaneously, it was decided to perform it first to achieve cleaner Sonogashira coupling at the next step. First, oxidation with DMSO was attempted (Method C), but this method was highly inefficient. Reaction of 19a with DDQ also proceeded inefficiently, affording a mixture of three products, none of which was identified as the desired product 20a. However, we managed to obtain practical yields of aromatized products 20 in reactions with aqueous potassium ferricyanide (Method D, Scheme 8). Structure of compound 20a was unambiguously confirmed by single crystal X-ray diffraction (CCDC #1983237).
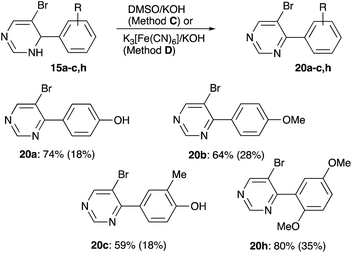 |
| | Scheme 8 Oxidative aromatization of dihydropyrimidines 15. Yields for Method D and for Method C (in parentheses) are shown. | |
Finally, acetylene substituent was installed into pyrimidine ring via Sonogashira coupling employing microwave irradiation. This allowed to obtain the target pyrimidylacetylenes 19 in good yields (Scheme 9).
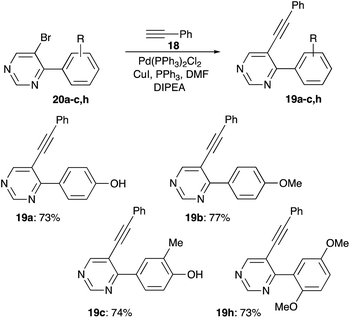 |
| | Scheme 9 Sonogashira cross-coupling reaction involving pyrimidines 20. | |
Conclusion
In conclusion, a new synthetic protocol was developed, allowing for a facile assembly of 4-aryl-5-alkynylpyrimidines involving chemo- and regioselective acid catalyzed electrophilic alkylation of arenes with 5-bromopyrimidine, followed by oxidative aromatization of the formed dihydropyrimidine moieties, and subsequent Sonogashira cross-coupling reaction.
Experimental part
General information
1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3, DMSO-d6, acetone-d6 using TMS as an internal standard. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart smp30 apparatus. All reactions were performed in oven-dried 5 mL round-bottomed flasks equipped with reflux condensers. The reaction progress and purity of isolated compounds were controlled by TLC on Silufol UV-254 plates, with EtOAc as eluent. Polyphosphoric acids (86%) were obtained by dissolving of precise amount of P2O5 in 85% orthophosphoric acid according to the published protocols.26,27 All reagents and solvents were purchased from commercial vendors and used as received.
Preparation of (5-bromo-3,4-dihydropyrimidin-4-yl)-benzenes (general procedure)
Method A (in methanesulfonic acid). A 10 mL round-bottomed flask equipped with magnetic stirring bar was charged with the corresponding substituted arene 4 (1.00 mmol), methanesulfonic acid (5 mL), and 5-bromopyridine 158 mg (1.00 mmol). The mixture was stirred at room temperature for 30 min (1 h for naphthalenes), then poured into cold water (50 mL), and aqueous ammonia was added dropwise to achieve a neutral reaction (pH 7–8). The formed precipitate was filtered, rinsed with small portions of cold water, and dried on air. This material was sufficiently pure for most application and did not require any additional purification.
Method A (in polyphosphoric acid). A 10 mL round-bottomed flask equipped with magnetic spin-bar was charged with the corresponding substituted arene 4 (1.00 mmol), 86% polyphosphoric acid (5 g), and 5-bromopyridine 158 mg (1.00 mmol). The mixture was stirred at 60 °C for 1 h. The subsequent quench, aqueous work up and isolation was identical to the one described for Method A. The formed precipitate was filtered, rinsed with small portions of cold water, and dried on air. This material was sufficiently pure for most application and did not require any additional purification.
4-(5-Bromo-3,4-dihydropyrimidin-4-yl)phenol (15a). Colorless solid, mp 183–185 °C. Yield 179 mg (0.71 mmol, 71% via Method A); 151 mg (0.60 mmol, 60% via Method B). 1H NMR (400 MHz, acetone-d6) δ 7.22 (d, J = 8.5 Hz, 2H, 2,6H), 7.12 (s, 1H, 2H, 4-pyr), 6.82 (d, J = 8.5 Hz, 2H, 3,5H), 6.64 (s, 1H, 6H, 4-pyr), 5.20 (s, 1H, 4H, 4-pyr). 13C NMR (101 MHz, DMSO-d6) δ 157.1, 143.1, 134.6, 128.4 (2C), 124.9, 115.0 (2C), 106.5, 59.4. FT IR (ZnSe, cm−1): 3244, 1601, 1575, 1241, 841, 650. HRMS (TOF-ES): calculated for C10H10BrN2O (M + H)+ 252.9971, found 252.9968.
5-Bromo-6-(4-methoxyphenyl)-1,6-dihydropyrimidine (15b). Colorless solid, mp 125–127 °C. Yield 189 mg (0.71 mmol, 71% via Method A). 1H NMR (400 MHz, acetone-d6) δ 7.31 (d, J = 8.7 Hz, 2H, 3,5H 4-methoxyphenyl), 7.13 (s, 1H, 2H), 6.92 (d, J = 8.7 Hz, 2H, 2,6H 4-methoxyphenyl), 6.64 (s, 1H, 4H), 5.23 (s, 1H, 6H), 3.79 (s, 3H, OMe). 13C NMR (101 MHz, CDCl3) δ 160.3, 145.6, 133.1, 129.1 (2C), 126.5, 114.5 (2C), 105.6, 60.4, 55.5. FT IR (ZnSe, cm−1): 2950, 1601, 1505, 1254, 1177, 1046, 761, 632. HRMS (TOF-ES): calculated for C11H12BrN2O (M + H)+ 267.0127, found 267.0130.
4-(5-Bromo-3,4-dihydropyrimidin-4-yl)-2-methylphenol (15c). Colorless solid, mp 155–157 °C. Yield 218 mg (0.82 mmol, 82% via Method A). 1H NMR (400 MHz, DMSO-d6) δ 7.04 (s, 1H, 2H 4-pyr), 7.00 (s, 1H, 3H), 6.94 (d, J = 8.1 Hz, 1H, 5H), 6.74 (d, J = 8.1 Hz, 1H, 6H), 6.59 (s, 1H, 6H 4-pyr), 5.05 (s, 1H, 4H 4-pyr), 2.11 (s, 3H, –Me). 13C NMR (101 MHz, DMSO) δ 155.2, 144.1, 134.4, 129.6, 127.4, 125.7, 123.7, 116.8, 114.4, 60.8, 16.1. FT IR (ZnSe, cm−1): 3422, 1623, 1575, 1450, 1379, 1278, 1250, 830, 625. HRMS (TOF-ES): calculated for C11H12BrN2O (M + H)+ 267.0127, found 267.0129.
2-(5-Bromo-3,4-dihydropyrimidin-4-yl)-4-methylphenol (15d). Colorless solid, mp 175–180 °C. Yield 229 mg (0.86 mmol, 86% via Method A); 1H NMR (400 MHz, acetone-d6) δ 7.16–7.10 (m, 2H, 3H, 2H 2-pyr), 6.94 (d, J = 8.0 Hz, 1H, 6H), 6.79–6.71 (m, 2H, 5H, 6H 2-pyr), 5.68 (s, 1H, 4H 2-pyr), 2.24 (s, 3H, –Me). 13C NMR (101 MHz, acetone-d6) δ 153.4, 145.2, 132.4, 130.0, 129.5, 129.3 (2C), 116.5, 102.1, 57.1, 20.6. FT IR (ZnSe, cm−1): 3310, 2500, 1873, 1745, 1590, 1267, 801. HRMS (TOF-ES): calculated for C11H12BrN2O (M + H)+ 267.0127, found 267.0126.
5-Bromo-6-(4-methoxy-3-methylphenyl)-1,6-dihydropyrimidine (15e). Light-brown oil. Yield 230 mg (0.82 mmol, 82%). 1H NMR (400 MHz, acetone-d6) δ 7.56 (s, 1H, 2H), 7.24–7.19 (m, 2H, 2H, 5H 6-(4-methoxy-3-methylphenyl)), 6.92 (d, J = 8.9 Hz, 1H, 6H 6-(4-methoxy-3-methylphenyl)), 6.74 (s, 1H, 4H), 5.31 (s, 1H, 6H), 3.84 (s, 3H, –OMe), 2.18 (s, 3H, 2-Me). 13C NMR (101 MHz, acetone-d6) δ 158.8, 145.9, 135.1, 130.6, 128.7, 127.3, 127.2, 110.7, 104.8, 61.6, 55.7, 16.4. FT IR (ZnSe, cm−1): 2918, 1503, 1391, 1250, 1132, 1032, 776. HRMS (TOF-ES): calculated for C12H14BrN2O (M + H)+ 281.0283, found 281.0280.
4-(5-Bromo-3,4-dihydropyrimidin-4-yl)benzene-1,2-diol (15f). Light-grey solid, mp 175–177 °C. Yield 113 mg (0.42 mmol, 42%). 1H NMR (400 MHz, acetone-d6) δ 7.23 (s, 1H, 2H 4-pyr), 6.98 (s, 1H, 3H), 6.76 (d, J = 8.0 Hz, 1H, 6H), 6.67 (m, 2H, 5H, 6H 4-pyr), 5.18 (s, 1H, 4H 4-pyr). 13C NMR (101 MHz, acetone-d6) δ 146.8, 146.5, 145.7, 136.1, 132.1, 119.5, 115.4, 115.0, 105.7, 61.9; FT IR (ZnSe, cm−1): 3247, 2930, 1607, 1510, 1261, 1119, 827, 646. HRMS (TOF-ES): calculated for C10H10BrN2O2 (M + H)+ 268.9920, found 268.9924.
5-Bromo-6-(3,4-dimethoxyphenyl)-1,6-dihydropyrimidine (15g). Colorless solid, mp 94–96 °C. Yield 139 mg (0.47 mmol, 47%). 1H NMR (400 MHz, acetone-d6) δ 7.16 (brs, 1H, 2H 6-(3,4-dimethoxyphenyl)), 7.00 (s, 1H, 2H), 6.93 (m, 2H, 5H, 6H 6-(3,4-dimethoxyphenyl)), 6.66 (s, 1H, 4H), 5.24 (s, 1H, 6H), 3.80 (s, 6H, OMe). 13C NMR (101 MHz, acetone-d6) δ 150.4, 150.2, 144.5, 137.4, 131.3, 120.6, 112.5, 112.3, 103.6, 62.8, 56.2, 56.1. FT IR (ZnSe, cm−1): 2350, 1695, 1532, 1253, 1125, 1033, 763. HRMS (TOF-ES): calculated for C12H14BrN2O2 (M + H)+ 297.0233, found 297.0235.
5-Bromo-6-(2,5-dimethoxyphenyl)-1,6-dihydropyrimidine (15h). Colorless solid, mp 115–117 °C. Yield 246 mg (0.83 mmol, 83%). 1H NMR (400 MHz, acetone-d6) δ 7.06 (s, 1H, 2H), 6.98 (s, 1H, 6H 6-(2,5-dimethoxyphenyl)), 6.94 (d, J = 8.9 Hz, 1H, 3H 6-(2,5-dimethoxyphenyl)), 6.85 (d, J = 8.9 Hz, 1H, 4H 6-(2,5-dimethoxyphenyl)), 6.74 (s, 1H, 4H), 5.70 (s, 1H, 6H), 3.79 (s, 3H, OMe), 3.74 (s, 3H, OMe). 13C NMR (101 MHz, acetone-d6) δ 150.4, 150.2, 144.5, 137.4, 131.3, 120.6, 112.5, 112.3, 103.6, 62.8, 56.2, 56.1. FT IR (ZnSe, cm−1): 2350, 1695, 1532, 1253, 1125, 1033, 763. HRMS (TOF-ES): calculated for C12H14BrN2O2 (M + H)+ 297.0233, found 297.0233.
2-(5-Bromo-3,4-dihydropyrimidin-4-yl)-4-fluorophenol (15i). Light-yellow solid, mp 173–175 °C. Yield 203 mg (0.75 mmol, 75%). 1H NMR (400 MHz, acetone-d6) δ 7.20 (s, 1H, 2H 2-pyr), 7.04 (dd, J = 9.3, 3.0 Hz, 1H, 5H), 6.94–6.76 (m, 3H, 6H 2-pyr, 3H, 6H), 5.71 (s, 1H, 4H 2-pyr). 13C NMR (101 MHz, acetone-d6) δ 157.4 (d, J = 234.5 Hz), 151.9, 145.1, 131.7 (d), 131.2, 117.7 (d), 115.8 (d), 115.2 (d), 99.4, 57.3. FT IR (ZnSe, cm−1): 3287, 2400, 1635, 1576, 1447, 1356, 1254, 1179, 1024, 731. HRMS (TOF-ES): calculated for C10H9BrFN2O (M + H)+ 270.9877, found 270.9879.
5-Bromo-6-(4-bromo-2-methoxyphenyl)-1,6-dihydropyrimidine (15j). Colorless crystalline solid, mp 69–72 °C, Yield 248 mg (0.72 mmol, 72%). 1H NMR (400 MHz, acetone-d6) 7.48 (d, J = 8.6 Hz, 1H, 4H), 7.16–7.11 (m, 2H, 2H 4-pyr, 6H), 7.03 (dd, J = 8.6, 2.6 Hz, 1H, 3H), 6.74 (s, 1H 6H 2-pyr), 5.74 (s, 1H, 4H 2-pyr), 3.83 (s, 3H, –OMe). 13C NMR (101 MHz, acetone) δ 160.7, 144.1, 135.4, 131.8 (2C), 123.4, 118.3, 115.4, 101.3, 61.8, 56.0. FT IR (ZnSe, cm−1): 2820, 1589, 1485, 1225, 1029, 852. HRMS (TOF-ES): calculated for C11H11Br2N2O (M + H)+ 344.9233, found 344.9231.
5-Bromo-6-(naphthalene-1-yl)-1,6-dihydropyrimidine and 5-bromo-6-(naphthalene-2-yl)-1,6-dihydropyrimidine (15k). Pale-yellow solid, mp 110–115 °C. Yield 237 mg (0.83 mmol, 83%). 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 7.9 Hz, 1H), 8.02–7.76 (m, 4H), 7.54 (d, J = 7.5 Hz, 5.5H), 7.18 (s, 0.5H), 7.06 (s, 1H), 6.83 (s, 1H), 6.71 (s, 0.5H), 6.07 (s, 1H), 5.41 (s, 0.5H). 13C NMR (101 MHz, DMSO-d6) δ 144.5, 143.9, 141.5, 138.7, 134.1, 133.2, 133.1, 131.1, 129.0, 128.9, 128.6, 128.4, 128.0, 126.8, 126.7 (2C), 126.6, 126.3, 126.2 (2C), 126.1, 126.0, 124.4, 101.5, 99.7, 62.6, 59.6, 40.6. FT IR (ZnSe, cm−1): 1631, 1510, 1352, 1257, 1010, 771, 646. HRMS (TOF-ES): calculated for C14H12BrN2 (M + H)+ 287.0178, found 287.0181.
6-(5-Bromo-3,4-dihydropyrimidin-4-yl)naphthalen-2-ol (15l). Pale-yellow solid, mp 150–152 °C. Yield 237 mg (0.78 mmol, 78%). 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 8.7 Hz, 1H, 4H), 7.70 (d, J = 8.5 Hz, 1H,8H), 7.64 (s, 1H, 5H), 7.41 (d, J = 8.5 Hz, 1H, 7H), 7.18–7.05 (m, 3H, 1H, 3H, 2H 6-pyr), 6.68 (s, 1H, 6H 6-pyr), 5.31 (s, 1H, 4H 6-pyr). 13C NMR (101 MHz, acetone-d6) δ 156.5, 144.7, 139.1, 135.7, 131.3, 130.5, 128.9, 127.6, 126.8 (2C), 119.5, 109.7, 103,4, 63.4. FT IR (ZnSe, cm−1): 3150, 2354, 1610, 1350, 1247, 1150, 1010, 854, 673. HRMS (TOF-ES): calculated for C14H12BrN2O (M + H)+ 303.0128, found 303.0124.
5-Bromo-6-(6-ethoxynaphthalen-2-yl)-1,6-dihydropyrimidine (15m). Light-grey solid, mp 123–125 °C. Yield 303 mg (0.914 mmol, 91%). 1H NMR (400 MHz, acetone-d6) δ 7.82 (d, J = 9.4 Hz, 2H, 4H, 8H), 7.76 (s, 1H, 5H), 7.56 (d, J = 9.4 Hz, 1H, 7H), 7.29 (s, 2H, 1H, 2H 6-pyr), 7.16 (d, J = 8.9 Hz, 1H, 3H), 6.73 (s, 1H, 4H 6-pyr), 5.46 (s, 1H, 6H 6-pyr), 4.17 (q, J = 7.0 Hz, 2H, –Et), 1.43 (t, J = 7.0 Hz, 3H, –Et). 13C NMR (101 MHz, acetone-d6) δ 158.9, 144.9, 139.4, 135.5, 130.6, 130.4, 129.4, 128.2, 126.9 (2C), 120.1, 107.3, 103.4, 64.1, 63.3, 15.1. FT IR (ZnSe, cm−1): 2350, 1610, 1503, 1360, 1261, 1147, 1037, 803, 632. HRMS (TOF-ES): calculated for C16H16BrN2O (M + H)+ 331.0441, found 331.0445.
Preparation of (5-bromo-3,4-pyrimidin-4-yl)-benzenes (general procedure)
Method C. A 10 mL round bottomed flask equipped with magnetic stirring bar was charged with DMSO (5 mL), potassium hydroxyde (112 mg, 2.00 mmol), and the corresponding (5-bromo-3,4-dihydropyrimidine-4-yl)benzene (1.00 mmol). The mixture was stirred for 4 h at 60 °C, then poured into cold water (10 mL). Formed precipitate is collected, washed on a filter with small portions of water and dried on air. Thus obtained crude material was purified by column chromatography eluting with a mixture of petroleum ether and ethyl acetate (1:1).
Method D. 10 mL round bottomed flask equipped with magnetic stirring bar was charged with water (5 mL), potassium ferricyanide (658 mg, 2.00 mmol), and potassium hydroxyde (224 mg, 4.00 mmol). (5-Bromo-3,4-dihydropyrimidine-4-yl)benzene (1.00 mmol) was added to the formed solution, and the mixture was stirred at room temperature for 2 h. Aqueous work up, isolation and purification of the product was performed in the same way as described for Method C.
4-(5-Bromopyrimidin-4-yl)phenol (20a). Colorless solid, mp 120–121 °C. Rf 0.20 (EtOAc/hexanes, 1:1). Yield 44 mg (0.18 mmol, 18% via Method C); 138 mg (0.55 mmol, 55% via Method D). 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H, –OH), 9.13 (s, 1H, 2H 4-pyr), 9.02 (s, 1H, 6H 4-pyr), 7.73 (d, J = 8.6 Hz, 2H, 3,5H), 6.90 (d, J = 8.7 Hz, 2H, 2,6H). 13C NMR (101 MHz, DMSO-d6) δ 162.9, 160.1, 159.6, 156.8, 131.2 (2C), 127.1, 118.1, 115.0 (2C). FT IR (ZnSe, cm−1): 2923, 1701, 1558, 1436, 1390, 1150, 1028, 822. HRMS (TOF-ES): calculated for C10H8BrN2O (M + H)+ 250.9815, found 250.9816.
5-Bromo-4-(4-methoxyphenyl)pyrimidine28 (20b). Colorless solid, mp 99–101 °C. Rf 0.26 (EtOAc/hexanes, 1:1). Yield 73 mg (0.28 mmol, 28% via Method C); 169 mg (0.64 mmol, 64% via Method D). 1H NMR (400 MHz, CDCl3) δ 9.11 (s, 1H, 2H), 8.87 (s, 1H, 6H), 7.87 (d, J = 8.8 Hz, 2H, 2,6H 4-(4-methoxyphenyl)), 7.01 (d, J = 8.8 Hz, 2H, 3,5H 4-(4-methoxyphenyl)), 3.88 (s, 3H, OMe). 13C NMR (101 MHz, CDCl3) δ 163.7, 161.5, 160.3, 156.9, 131.3 (2C), 129.1, 118.8, 113.8 (2C), 55.6. FT IR (ZnSe, cm−1): 2955, 2325, 1895, 1726, 1607, 1554, 1431, 1259, 1154, 1023, 825, 776. HRMS (TOF-ES): calculated for C11H10BrN2O (M + H)+ 264.9971, found 264.9974
4-(5-Bromopyrimidin-4-yl)-2-methylphenol (20c). Colorless solid, mp 160–165 °C. Rf 0.22 (EtOAc/hexanes, 1:1). Yield 46 mg (0.18 mmol, 18% via Method C); 155 mg (0.59 mmol, 59% via Method D). 1H NMR (400 MHz, acetone-d6) δ 9.07 (s, 1H, 2H 4-pyr), 8.92 (s, 1H, 6H 4-pyr), 8.88 (s, 1H, –OH), 7.70 (s, 1H, 5H), 7.65 (d, J = 8.3 Hz, 1H, 3H), 6.96 (d, J = 8.3 Hz, 1H, 2H), 2.28 (s, 3H, –Me). 13C NMR (101 MHz, acetone-d6) δ 164.3, 160.9, 158.4, 157.6, 133.2, 129.6, 128.9, 119.1, 115.0, 16.2. FT IR (ZnSe, cm−1): 3049, 2358, 2217, 1565, 1485, 1401, 1279, 1116, 917, 788. HRMS (TOF-ES): calculated for C11H10BrN2O (M + H)+ 264.9971, found 264.9968.
5-Bromo-4-(2,5-dimethoxyphenyl)pyrimidine (20h). Light-brown oil. Rf 0.25 (EtOAc/hexanes, 1:1). Yield 102 mg (0.35 mmol, 35% via Method C); 236 mg (0.80 mmol, 80% via Method D). 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H, 2H), 8.89 (s, 1H, 6H), 7.01 (d, J = 9.0 Hz, 1H, 3H 4-(2,5-dimethoxyphenyl)), 6.94 (d, J = 9.0 Hz, 1H, 4H 4-(2,5-dimethoxyphenyl)), 6.86 (s, 1H, 6H 4-(2,5-dimethoxyphenyl)), 3.80 (s, 3H, 2-MeO–), 3.78 (s, 3H, 5-MeO–). 13C NMR (101 MHz, CDCl3) δ 166.8, 164.8, 158.7, 156.3, 153.7, 150.8, 126.9, 117.1, 115.1 (2C), 112.6, 56.2, 56.0. FT IR (ZnSe, cm−1): 2947, 2322, 1723, 1554, 1506, 1435, 1221, 1019, 810. HRMS (TOF-ES): calculated for C12H14BrN2O2 (M + H)+ 297.0233, found 297.0237.
Preparation of 4-(5-(phenylethynyl)pyrimidin-4-yl)benzenes via Sonogashira coupling (general procedure)
Pyrex microwave vessel was charged with phenylacetylene (102 mg, 1.00 mmol) and the corresponding substituted 4-(5-bromopyrimidin-4-yl)-2-benzene (1.00 mmol) in dry DMF (0.5 mL). Dry nitrogen was passed to the vessel for 50 min, then anhydrous DIPEA (1.5 mL, 1.11 g, 8.6 mmol), dry triphenylphosphine (23 mg, 0.087 mmol) were introduced, followed by bis(triphenylphosphine)palladium chloride (Pd(PPh3)Cl2, 15 mg, 0.021 mmol) and copper(I) iodide (CuI, 3.8 mg, 0.020 mmol). The vessel was sealed and microwaved at 115 °C for 25 min. Then the mixture was cooled down and extracted with ethyl acetate (2 × 50 mL). Combined organic extracts were consecutively washed with aqueous ammonium chloride solution (2 × 25 mL) and water (2 × 25 mL), and dried with sodium sulfate. Pure product was crystallized from concentrated solution obtained after evaporation of most solvent.
4-(5-(Phenylethynyl)pyrimidin-4-yl)phenol (19a). Pale-yellow solid, mp 194–195 °C. Rf 0.38 (EtOAc/hexanes, 1:1). Yield 199 mg (0.73 mmol, 73%). 1H NMR (400 MHz, acetone-d6) δ 9.09 (s, 1H, 2H 4-pyr), 8.92 (s, 1H, 6H 4-pyr), 8.27 (d, J = 8.8 Hz, 2H, 2,6H), 7.63–7.53 (m, 2H, 2,6H 4-(5-(phenylethynyl)pyr)), 7.46 (m, 3H, 3-5H 4-(5-(phenylethynyl)pyr)), 7.04 (d, J = 8.8 Hz, 2H, 3,5H). 13C NMR (101 MHz, acetone-d6) δ 164.7, 161.9, 161.0, 157.6, 132.3 (2C), 132.1 (2C), 130.2, 129.6 (2C), 129.1, 123.2, 116.1 (2C), 115.7, 97.9, 85.7. FT IR (ZnSe, cm−1): 3057, 2362, 2217, 1565, 1504, 1432, 1287, 1165, 845. HRMS (TOF-ES): calculated for C18H13N2O (M + H)+ 273.1022, found 273.1025.
4-(4-Methoxyphenyl)-5-(phenylethynyl)pyrimidine (19b). Light brown solid, mp 80–83 °C. Rf 0.47 (EtOAc/hexanes, 1:1). Yield 220 mg (0.77 mmol, 77%). 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H, 2H), 8.89 (s, 1H, 6H), 8.31 (d, J = 8.8 Hz, 2H, 3,5H 4-(4-methoxyphenyl)), 7.53–7.49 (m, 2H, 2,6H 5-(phenylethynyl)), 7.41–7.36 (m, 3H, 3-5H 5-(phenylethynyl)), 7.05 (d, J = 8.9 Hz, 2H, 2,6H 4-(4-methoxyphenyl)), 3.90 (s, 3H 4-MeO). 13C NMR (101 MHz, CDCl3) δ 164.9, 162.5, 159.7, 155.2, 131.7 (2C), 131.5 (2C), 129.5, 128.8, 128.7 (2C), 122.1, 115.8, 114.0 (2C), 98.5, 84.3, 55.6. FT IR (ZnSe, cm−1): 2929, 2221, 1726, 1607, 1509, 1435, 1259, 1173, 1027, 783. HRMS (TOF-ES): calculated for C19H15N2O (M + H)+ 287.1179, found 287.1180.
2-Methyl-4-(5-(phenylethynyl)pyrimidin-4-yl)phenol (19c). Light-brown solid, mp 168–170 °C. Rf 0.43 (EtOAc/hexanes, 1:1). Yield 210 mg (0.74 mmol, 74%). 1H NMR (400 MHz, CDCl3) δ 9.13 (s, 1H, 2H 4-pyr), 8.87 (s, 1H, 6H 4-pyr)), 8.12 (s, 1H, 5H), 8.07 (d, J = 8.4 Hz, 1H, 3H), 7.55–7.45 (m, 2H, 2,6H 4-(5-(phenylethynyl)pyr)), 7.41–7.35 (m, 3H, 3-5H 4-(5-(phenylethynyl)pyr), 6.94 (d, J = 8.4 Hz, 1H, 6H), 2.34 (s, 3H, –Me). 13C NMR (101 MHz, CDCl3) δ 164.7, 160.6, 157.2, 156.1, 132.6, 131.7 (2C), 129.4, 129.0, 128.7 (2C), 124.3, 122.4 (2C), 115.8, 114.9, 98.0, 84.7, 16.2. FT IR (ZnSe, cm−1): 3049, 2358, 2217, 1565, 1485, 1401, 1279, 1116, 917, 788. HRMS (TOF-ES): calculated for C19H15N2O (M + H)+ 287.1179, found 287.1175.
4-(2,5-Dimethoxyphenyl)-5-(phenylethynyl)pyrimidine (19h). Light-brown oil. Rf 0.36 (EtOAc/hexanes, 1:1). Yield 220 mg (0.73 mmol, 73%). 1H NMR (400 MHz, CDCl3) δ 9.21 (s, 1H, 2H), 8.90 (s, 1H, 6H), 7.34–7.29 (m, 5H), 7.08–7.01 (m, 2H, 3,6H 4-(2,5-dimethoxyphenyl)), 6.97 (d, J = 8.6 Hz, 1H, 4H 4-(2,5-dimethoxyphenyl)), 3.82 (s, 3H, 2-MeO), 3.78 (s, 3H 4-MeO). 13C NMR (101 MHz, CDCl3) δ 166.3, 158.2, 154.1, 153.6, 151.7, 131.8 (2C), 129.6, 128.7 (2C), 125.6, 122.0, 120.3, 118.4, 115.6, 112.7, 98.8, 83.1, 56.3, 56.1. FT IR (ZnSe, cm−1): 2943, 2831, 2217, 1726, 1498, 1431, 1397, 1274, 1221, 1034, 810. HRMS (TOF-ES): calculated for C20H17N2 (M + H)+ 317.1285, found 317.1286.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant #18-33-00849 mol_a) and by the Ministry of Education and Science of the Russian Federation (grant #0795-2020).
Notes and references
- M. A. Matulenko, E. S. Paight, R. R. Frey, A. Gomtsyan, S. DiDomenico, M. Jiang, C.-H. Lee, A. O. Stewart, H. Yu, K. L. Kohlhaas, K. M. Alexander, S. McGaraughty, J. Mikusa, K. C. Marsh, S. W. Muchmore, C. L. Jakob, E. A. Kowaluk, M. F. Jarvis and S. S. Bhagwat, Bioorg. Med. Chem., 2007, 15, 1586–1605 CrossRef PubMed.
- S. Schneider, D. Kessler, L. Van der Veen and T. Wunberg, WO2011092198A1, 2011.
- M. Takasaki, T. Tsujino, S. Tanabe, M. Ookubo, K. Sato, A. Hirai, D. Terada, S. Inuki and S. Mizumoto, WO2013157540A1, 2013.
- Y. K. Chen, T. Kanouni, S. W. Kaldor, J. A. Stafford and J. M. Veal, WO2015089192A1, 2015.
- Y. K. Chen, T. Kanouni, J. A. Stafford and J. M. Veal, WO2016037005A1, 2016.
- T. Wunberg, R. Brueckner, D. Kessler, O. Kraemer, D. McConnell, S. Schneider and L. Van der Veen, WO2010012740A1, 2010.
- T. Wunberg, L. Van der Veen and O. Kraemer, WO2012101184A1, 2012.
- T. Kuragano and H. Ikeda, JP2004115431A, 2004.
- M. Beesu, A. C. D. Salyer, K. L. Trautman, J. K. Hill and S. A. David, J. Med. Chem., 2016, 59, 8082–8093 CrossRef PubMed.
- T. Lechel, S. Moehl and H.-U. Reissig, Synlett, 2009, 1059–1062 Search PubMed.
- T. Lechel and H.-U. Reissig, Eur. J. Org. Chem., 2010, 2555–2564 CrossRef.
- N. S. Le Pham, J. Lee, H. Shin and J.-H. Sohn, Tetrahedron, 2018, 74, 3843–3851 CrossRef.
- J. Liu, A. E. Fitzgerald and N. S. Mani, J. Org. Chem., 2008, 73, 2951–2954 CrossRef PubMed.
- A. Mollard, S. L. Warner, L. T. Call, M. L. Wade, J. J. Bearss, A. Verma, S. Sharma, H. Vankayalapati and D. J. Bearss, ACS Med. Chem. Lett., 2011, 2, 907–912 CrossRef PubMed.
- A. Steffen, M. I. Sladek, T. Braun, B. Neumann and H.-G. Stammler, Organometallics, 2005, 24, 4057–4064 CrossRef.
- L. M. Toledo-Sherman, M. E. Prime, L. Mrzljak, M. G. Beconi, A. Beresford, F. A. Brookfield, C. J. Brown, I. Cardaun, S. M. Courtney, U. Dijkman, E. Hamelin-Flegg, P. D. Johnson, V. Kempf, K. Lyons, K. Matthews, W. L. Mitchell, C. O'Connell, P. Pena, K. Powell, A. Rassoulpour, L. Reed, W. Reindl, S. Selvaratnam, W. W. Friley, D. A. Weddell, N. E. Went, P. Wheelan, C. Winkler, D. Winkler, J. Wityak, C. J. Yarnold, D. Yates, I. Munoz-Sanjuan and C. Dominguez, J. Med. Chem., 2015, 58, 1159–1183 CrossRef PubMed.
- A. D. William, A. C. H. Lee, S. Blanchard, A. Poulsen, E. L. Teo, H. Nagaraj, E. Tan, D. Chen, M. Williams, E. T. Sun, K. C. Goh, W. C. Ong, S. K. Goh, S. Hart, R. Jayaraman, M. K. Pasha, K. Ethirajulu, J. M. Wood and B. W. Dymock, J. Med. Chem., 2011, 54, 4638–4658 CrossRef PubMed.
- J. Xiao, J. J. Marugan, W. Zheng, S. Titus, N. Southall, J. J. Cherry, M. Evans, E. J. Androphy and C. P. Austin, J. Med. Chem., 2011, 54, 6215–6233 CrossRef PubMed.
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, L. V. Frolova, A. Kornienko, I. V. Magedov and M. Rubin, Chem. Commun., 2013, 49, 9305–9307 RSC.
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, J. P. Matheny and M. Rubin, RSC Adv., 2015, 5, 8647–8656 RSC.
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 RSC.
- A. V. Aksenov, A. N. Smirnov, I. V. Magedov, M. R. Reisenauer, N. A. Aksenov, I. V. Aksenova, A. L. Pendleton, G. Nguyen, R. K. Johnston, M. Rubin, A. De Carvalho, R. Kiss, V. Mathieu, F. Lefranc, J. Correa, D. A. Cavazos, A. J. Brenner, B. A. Bryan, S. Rogelj, A. Kornienko and L. V. Frolova, J. Med. Chem., 2015, 58, 2206–2220 CrossRef PubMed.
- N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC.
- A. V. Aksenov, S. V. Shcherbakov, I. V. Lobach, I. V. Aksenova and M. Rubin, Eur. J. Org. Chem., 2017, 2017, 1666–1673 CrossRef.
- A. V. Aksenov, S. V. Shcherbakov, I. V. Lobach, L. G. Voskressensky and M. Rubin, Eur. J. Org. Chem., 2018, 2018, 4121–4127 CrossRef.
- F. Uhlig, Angew. Chem., 1954, 66, 435–436 CrossRef.
- A. L. Huhti and P. A. Gartaganis, Can. J. Chem., 1956, 34, 785–797 CrossRef.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem.–Eur. J., 2014, 20, 2960–2965 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data. CCDC 1983237. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01335h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Dmitriy A. Domenyukb,
Vladimir A. Zelenskyb and
Michael Rubin
a,
Dmitriy A. Domenyukb,
Vladimir A. Zelenskyb and
Michael Rubin