
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic hydrogen evolution by co-catalyst-free TiO2/C bulk heterostructures synthesized under mild conditions†
Claudio Imparato*a,
Giuseppina Iervolinob,
Marzia Fantauzzi c,
Can Korald,
Wojciech Macyke,
Marcin Kobielusze,
Gerardino D'Erricof,
Ilaria Reag,
Rocco Di Girolamof,
Luca De Stefanog,
Antonello Andreoned,
Vincenzo Vaianob,
Antonella Rossic and
Antonio Aronne*a
c,
Can Korald,
Wojciech Macyke,
Marcin Kobielusze,
Gerardino D'Erricof,
Ilaria Reag,
Rocco Di Girolamof,
Luca De Stefanog,
Antonello Andreoned,
Vincenzo Vaianob,
Antonella Rossic and
Antonio Aronne*a
aDepartment of Chemical, Materials and Production Engineering, University of Naples Federico II, P.le V. Tecchio 80, 80125 Napoli, Italy. E-mail: claudio.imparato@unina.it; antonio.aronne@unina.it
bDepartment of Industrial Engineering, University of Salerno, Via Giovanni Paolo II 132, 84084 Fisciano (Salerno), Italy
cDepartment of Chemical and Geological Sciences, University of Cagliari, S.S. 554 Bivio per Sestu, 09042 Monserrato, Cagliari, Italy
dDepartment of Physics, University of Naples Federico II, CNR-SPIN, UOS Napoli, Via Cinthia, 80126 Napoli, Italy
eFaculty of Chemistry, Jagiellonian University, ul. Gronostajowa 2, 30-387 Kraków, Poland
fDepartment of Chemical Sciences, University of Naples Federico II, Via Cinthia, 80126 Napoli, Italy
gInstitute for Microelectronics and Microsystems, National Research Council, Via P. Castellino 111, 80131 Napoli, Italy
First published on 27th March 2020
Abstract
Hydrogen production by photocatalytic water splitting is one of the most promising sustainable routes to store solar energy in the form of chemical bonds. To obtain significant H2 evolution rates (HERs) a variety of defective TiO2 catalysts were synthesized by means of procedures generally requiring highly energy-consuming treatments, e.g. hydrogenation. Even if a complete understanding of the relationship between defects, electronic structure and catalytic active sites is far from being achieved, the band gap narrowing and Ti3+-self-doping have been considered essential to date. In most reports a metal co-catalyst (commonly Pt) and a sacrificial electron donor (such as methanol) are used to improve HERs. Here we report the synthesis of TiO2/C bulk heterostructures, obtained from a hybrid TiO2-based gel by simple heat treatments at 400 °C under different atmospheres. The electronic structure and properties of the grey or black gel-derived powders are deeply inspected by a combination of classical and less conventional techniques, in order to identify the origin of their photoresponsivity. The defective sites of these heterostructures, namely oxygen vacancies, graphitic carbon and unpaired electrons localized on the C matrix, result in a remarkable visible light activity in spite of the lack of band gap narrowing or Ti3+-self doping. The materials provide HER values ranging from about 0.15 to 0.40 mmol h−1 gcat−1, under both UV- and visible-light irradiation, employing glycerol as sacrificial agent and without any co-catalyst.
Introduction
Titanium dioxide is one of the most attractive semiconductor materials thanks to its relevant photocatalytic applications, among them environmental remediation and hydrogen production. The need to enhance specific functional properties of the material, for instance to extend the photoresponsivity to visible light and to increase the photocatalytic efficiency, leads to different strategies for the modification of its electronic structure. Doping with metals or non-metals, dye sensitization, coupling with other semiconductors or carbon-based materials, and the introduction of structural defects by harsh treatments such as hydrogenation are the most investigated approaches.1 Depending on the energy level of the electronic state formed within the band gap, doped TiO2 materials with different colorations can be obtained. They exhibit an extended absorption in the visible light range and an enhanced photocatalytic activity in oxidation reactions of pollutants, but their performance in hydrogen evolution does not usually appear improved.2,3 Conversely, the hydrogen evolution seems to be strongly favored by a prolonged hydrogenation treatment at high temperature and H2 pressure, which produces a high level of Ti3+ self-doping and/or oxygen vacancies, giving a dark coloration (grey or black titania).3–5 Hydrogenated materials show a core–shell structure with a thin amorphous shell coating the crystalline core and their band gap narrowing is mainly due to the upward shift of the valence band and tailing of both valence and conducting bands.3,6,7 However, they may suffer from some shortcomings. Although various methods to synthesize defective TiO2−x were tested, they generally involve drastic treatments or complicated procedures.5,8,9 The formation mechanism of different types of defects and their relationship with the photoinduced properties is still not completely understood4,10 and some reports attest a limited increase of photocatalytic activity in the visible range.11–13 A poor thermal stability of surface defects, particularly in the case of Ti3+, was observed on some materials, which tend to be gradually reoxidized in contact with air.4,5,14 Moreover, the electronic states associated to Ti3+ can act as recombination centers reducing the charge carriers' lifetime as they are located just below the conducting band, while some of them are too deep in the band gap so they do not possess suitable reduction potential for water splitting.13In this photocatalytic process the use of a sacrificial agent as electron donor (an organic compound, preferably derived from biomass) and the addition of a co-catalyst (commonly Pt or another noble metal) enhance the H2 evolution rate (HER), which has reached a benchmark value of about 40 mmol h−1 gcat−1.15,16 Interesting HER values were recently reported for carbon modified TiO2, ranging from about 6 to 37 mmol h−1 gcat−1.17 In the case of carbon doping, that usually requires a multi-steps procedure, the improved photocatalytic activity was generally related to the generation of midgap states or band gap narrowing as well as to the formation and stabilization of oxygen vacancies and Ti3+ sites.1,14,18 For heterostructures with graphene, graphene oxide, C nanotubes, C layers coating and also with graphite-like carbon the increased charge carriers' availability and lifetime, based on the separation of electrons and holes at the interface between the conjugated materials, were also proposed to explain the improved photocatalytic properties.19–21 Recently several studies focused on co-catalyst-free H2 evolution reaction on TiO2. The best result to date (3.5 mmol h−1 gcat−1) was reported for P25 TiO2 treated in a strongly reducing medium,22 whereas most works involving defective TiO2 do not exceed HER around 0.2 mmol h−1 gcat−1.16
Here we report the synthesis of TiO2/C bulk heterostructures formed by defective anatase TiO2 nanocrystals embedded in a carbonaceous matrix composed by amorphous and crystalline carbon, obtained by suitable thermal treatments of a hybrid chemical gel in which titanium is involved in a charge transfer complexation equilibrium with an organic compound.23 Both the sol–gel procedure and the following treatments of the gel under mild conditions have synergistic effects in determining the characteristics of the final product. These heterostructures exhibit different defective sites, oxygen vacancies without Ti3+ self-doping, carbon radicals localized on the C matrix and graphitic carbon, that determine their significant photocatalytic efficiency, tested in hydrogen generation using glycerol as an electron donor. Our materials show impressive co-catalyst-free HER under both UV irradiation (about 0.4 mmol h−1 gcat−1) and visible light (about 0.2 mmol h−1 gcat−1). Moreover, it is worthwhile to note that most of the works concerning H2 evolution employed methanol as a sacrificial agent,16 whereas in this paper glycerol was used since it is the major by-product of the biodiesel production process,15 opening a new perspective towards the valorization of this by-product coming from a renewable source.
Results and discussion
Synthesis of gel-derived defective TiO2 materials
Sol–gel is a versatile synthesis technique to produce different kinds of materials, starting from a colloidal solution of the precursors, which allows obtaining either physical (particulate) or chemical gels. TiO2 is frequently produced as bulk or nanoparticles from physical gels,24,25 while to attain chemical gels the stabilization of the sol against the uncontrolled hydrolysis of titanium alkoxides is required. It can be achieved using complexing agents of Ti4+, such as β-diketones, including acetylacetone (Hacac),23,26,27 forming heteroleptic alkoxide complexes able to give micelles by self-assembly.28 Chemical gels obtained in this way ensure a high homogeneity of the oxide structure and provide porous xerogels with macroscopic granulometry and specific features related to the organic complexing agent used. Concerning the materials derived from TiO2–acetylacetone systems, their photoresponsivity to visible light was mainly attributed to Ti3+ self-doping that was obtained by heating in air at 400 °C a hybrid dried gel with nominal Hacac/Ti molar ratio equal to 0.4 (ref. 23) or at 350 °C for 2 h a hybrid sol, containing variable amounts of Al(acac)3 and Hacac/Ti molar ratio equal to 1.27 In the latter case, the heavy Ti3+ self-doping in the surface layer of the sol-derived materials was interpreted as due to the catalytic role played by Al(acac)3 in the reduction of Ti4+.27 On the contrary, the yellow-colored hybrid chemical gel (hybrid sol–gel titania, HSGT) prepared by some of us exhibits both visible light absorption and a valuable percentage of Ti3+ self-doping (6%), which increases after calcination in air.23 Moreover, the most peculiar property of this material is the ability to generate and stabilize superoxide radical anions (O2˙−) on its surface for a very long time (years), as confirmed by EPR spectroscopy.23,26,29 The presence of the adsorbed radicals is responsible for the oxidative activity of HSGT without the need for light irradiation.23,26,29Superoxide radical can act as an oxidant, but also as a moderate reductant, through one-electron transfer, forming O2. We believe that adsorbed O2˙− plays a key role also in the self-reduction process occurring during thermal treatment of the hybrid gels, therefore the ability to control its formation at the surface of the material could be a new strategy for the preparation of defective TiO2 materials. Drying the TiO2-acac wet gel in air (HSGT) or under vacuum (HSGT-v) drastically influences the concentration of surface adsorbed superoxide radicals, which in turn affects the characteristics of the materials obtained by annealing. When the wet gel was dried at 30 °C in a vacuum oven, so in the absence of a prolonged contact with atmospheric O2, the formation of O2˙− was prevented, resulting in a hybrid xerogel showing no clear EPR signal.29 On the other hand, drying under a reducing atmosphere should favor the formation of a defective TiO2 structure in the subsequent annealing stage. At the same time the hybrid nature of the xerogels can generate carbon structures that should enhance both the photoresponsivity to visible light and the photocatalytic performance of TiO2.18,30 Consequently, to obtain a defective anatase TiO2 with a tailored distribution of defects the HSGT-v xerogels were annealed at 400 °C for 1 and 3 h under different atmospheres (see Table 1).
Morphological, structural and thermal characterization
The samples produced by annealing the HSGT-v xerogel are composed of particles with a broad size distribution in the micrometer range (from about 1 to 50 μm, as shown by SEM images, Fig. S1†), depending on the xerogel grinding and heat treatment. High-magnification SEM images, displayed in Fig. 1, reveal the presence of nanometric primary particles or grains with an average diameter of 10–20 nm. The analysis of TEM images (Fig. 1 and S1†) shows TiO2 nanocrystals embedded in a continuous carbonaceous matrix. A typical d-spacing value of 0.35 nm corresponding to the (101) lattice plane of anatase phase was detected for all annealed samples (Fig. 1). The aspect of HN1 and HN3 samples resemble that of N1 and N3 samples. Thermal treatments induce the partial crystallization of the amorphous xerogels in the anatase polymorph, as also attested by XRD profiles (Fig. S2†) and Raman spectra (Fig. 2a). The average size of the anatase nanocrystals, estimated to be about 10 nm by Scherrer's equation, is observed to lie in the 3–8 nm range, except for A3 which shows a larger value (8–12 nm). These results indicate that the adopted synthesis procedure generates a homogeneous hybrid gel from which a bulk TiO2/C heterostructure, formed by anatase nanocrystals uniformly dispersed in a carbon matrix, is obtained. The structure of HSGT-v heat-treated samples was analyzed by Raman spectroscopy (Fig. 2a and b). The six Raman active modes of anatase TiO2 (A1g + 2B1g + 3Eg) are clearly visible in the Raman spectra of all samples in the 100–800 cm1 range (Fig. 2a) and appear much more pronounced the A3 spectrum, in agreement with TEM analysis. Their peak positions well agree with the values predicted for the anatase polymorph31 indicating the lack of C-doping, in accordance with the XRD profiles, where no noticeable shifts of the main diffraction peaks are seen with respect to a reference anatase TiO2 (Fig. S2†), and with the XPS data, presented in the following. Except for A3, for which the prolonged annealing in air causes the combustion of carbon atoms belonging to the outer layers of the sample and the increase of the crystallization degree, all Raman spectra exhibit the characteristic G and D peaks of a defective graphitic material at about 1590 and 1360 cm−1 (Fig. 2b). They are related to the bond stretching of all pairs of sp2 carbon atoms of the graphitic network, and to breathing modes of sp2 atoms in rings, respectively.32 The D band, indicative of the defectivity of the graphitic network, is broad and exhibits few features suggesting the presence of additional components. To reveal them the curve fitting of these Raman signals was performed and the resulting parameters are reported in Table S1.† The curve fitting reveals the presence of three additional bands: D* (1150–1200 cm−1) due to the defectivity of graphitic lattice; D′′ (1500–1550 cm−1) due to the graphitic amorphous lattice; D′ (1600–1620 cm−1) due to the phonon density of states in the finite-size of graphite crystals.33 Therefore the carbonaceous matrix in which the anatase nanocrystals are embedded appears formed by sp2 carbon atoms arranged either in amorphous or in crystalline phases. For a graphitic material with a high structural disorder the graphitization degree can be better estimated by evaluating the integrated intensity ratio ID/(ID + IG) for the D and G bands obtained from curve fitting, rather than the ID/IG ratio.33,34 | ||
| Fig. 1 High-magnification SEM images (left) and TEM images (right, scale bar 2 nm) of some HSGT-v annealed samples. A d-spacing of 0.35 nm corresponding to the (101) lattice plane of TiO2 anatase phase is highlighted in all TEM images. | ||
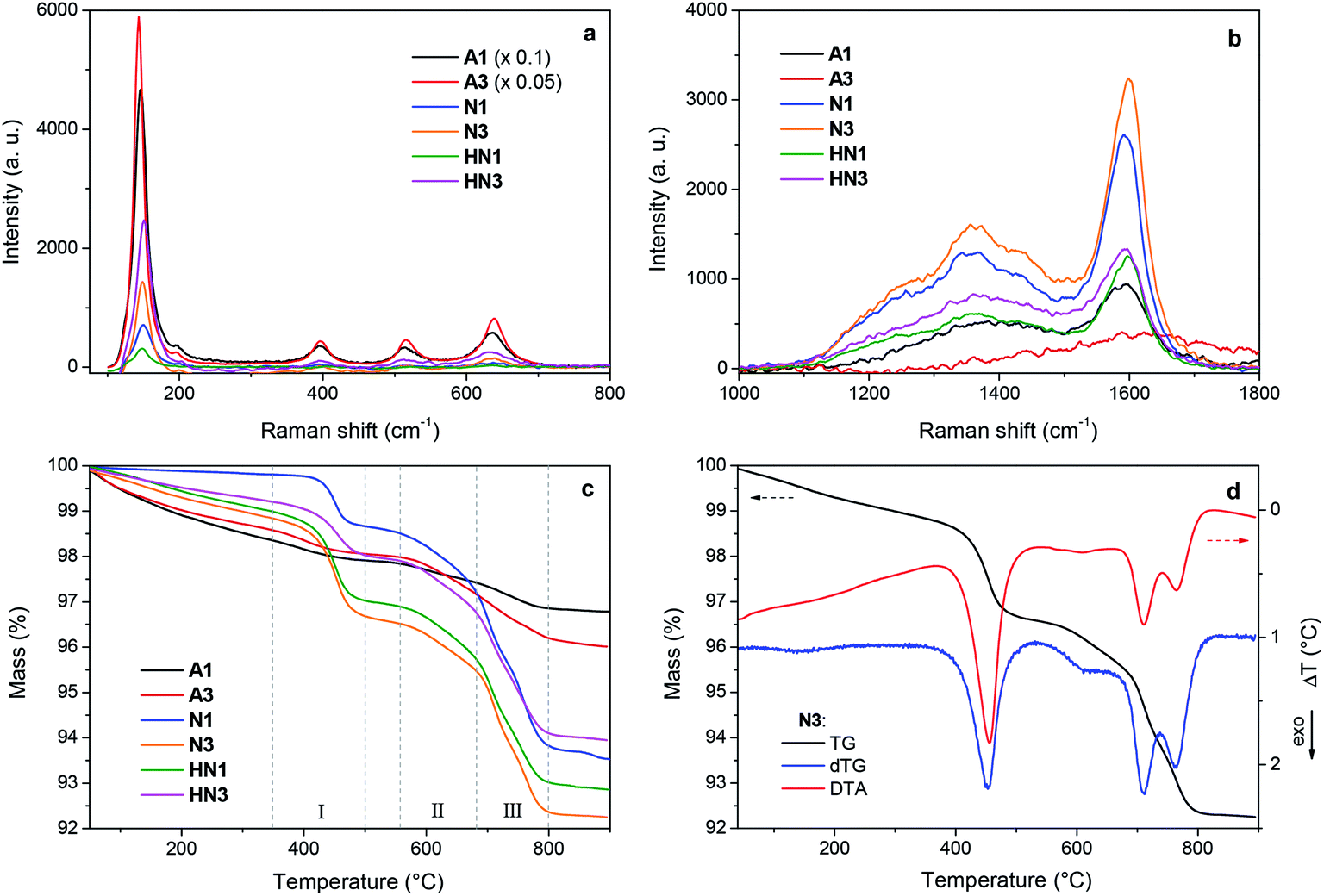 | ||
| Fig. 2 (a and b) Raman spectra and (c) thermogravimetric (TG) curves of HSGT-v annealed samples; (d) TG, DTG (differential TG) and DTA (differential thermal analysis) curves of N3 sample recorded in air at 10 °C min−1. | ||
A similar graphitization degree was estimated for all samples (Table 2) indicating that during the annealing in air the organic component creates a relative amount of graphitic carbon similar to that obtained under reducing atmospheres. Thermal analysis is a very useful tool to discern the different C species present in the HSGT-v annealed samples and to determine their whole amount (surface and bulk). TG curves of all samples, recorded in air at 10 °C min−1 heating rate, are displayed in Fig. 2c while in Fig. 2d the TG/DTA/DTG curves of N3 are reported as representative example of these measurements. Besides the gradual weight loss (about 1%) below 300 °C, mainly due to adsorbed water and surface –OH groups, three steep mass losses are clearly distinguished in the following ranges: 350–500 °C (I), 550–680 °C; (II), 680–800 °C (III).
| Sample | O/Ti4+ atomic ratio | ID/(ID + IG) | Ctotal graphitica (wt%) | Ctotala (wt%) | Celementalb (at%) | Spin density (1017 spin per g) | SSA (m2 g−1) | AQE (%) |
|---|---|---|---|---|---|---|---|---|
| a By TG/DTA analysis.b By XPS analysis. | ||||||||
| SA1 | 1.9 ± 0.1 | — | — | — | — | 3 ± 1 | — | — |
| SA3 | 2.1 ± 0.1 | — | — | — | — | 0.3 ± 0.1 | — | — |
| A1 | 1.8 ± 0.2 | 0.58 | 1.1 | 1.6 | 1.5% | 4 ± 1 | 56 | 0.97 |
| A3 | 1.8 ± 0.2 | — | 1.8 | 2.3 | 1.5–1.8% | 8 ± 1 | 45 | 0.51 |
| N1 | 1.6 ± 0.2 | 0.68 | 4.7 | 5.9 | 2.6% | 45 ± 5 | 11 | 0.66 |
| N3 | 1.8 ± 0.1 | 0.64 | 4.3 | 6.4 | 3.9–4.0% | 60 ± 5 | 4 | 1.2 |
| HN1 | 1.8 ± 0.1 | 0.68 | 4.0 | 6.0 | 3.4–4.0% | 45 ± 5 | 10 | 0.88 |
| HN3 | 1.8 ± 0.1 | 0.76 | 3.9 | 5.2 | 4.0–4.1% | 50 ± 5 | 7 | 1.3 |
Each event is associated with an exothermic DTA peak related to the combustion of different carbon species. According to the above Raman analysis the mass loss occurring in these ranges is related to the oxidation of amorphous sp2 carbon atoms (range I); carbon atoms in defective graphitic structures (range II); carbon atoms in graphitic network (range III).35 The temperatures of DTA and DTG peaks are very close indicating that the weight losses are strictly related to combustion processes. The first DTA exo-peak is generally sharp; the second is broad while the third one appears as two unresolved peaks as a consequence of the size distribution of the graphitic nanodomains.36 Similar results were obtained for all the samples. The total content of carbon (ranges I–III) and graphitic carbon (ranges II–III) is reported in Table 2.
Surface and electronic characterization
In a previous paper,23 the Ti 2p X-ray photoelectron spectra of the SA1 sample, obtained by drying and calcining in air the HSGT wet gel (Table 1), provided evidence of the presence of a considerable Ti3+ self-doping: two different components ascribed to Ti3+ and Ti sub-oxides were detected (at about 457.5 eV and at 455.8 eV respectively), together with the component due to Ti4+ (at about 459.3 eV). The percentage of reduced titanium was about 16% for Ti3+ and 10% for the Ti sub-oxides in SA1,23 and it strongly decreased to 3% for Ti3+ in the sample annealed in air for 3 h (SA3). The comparison of the Ti 2p spectra acquired on these samples is provided in Fig. S3a.† The O/Ti4+ ratio was 2.1 (±0.1) in the case of SA3, thus indicating the formation of stoichiometric titanium oxide (TiO2) within the experimental uncertainty. It is worth noting that no signal attributable to reduced titanium was revealed in an aged SA1 sample: the Ti 2p spectra of SA1 characterized again by XPS after about two years of storage show that only Ti4+ is present at the sample surface and that defective TiO2−x has turned into stoichiometric TiO2 (Fig. S3a†). On the other hand, a single component is observed in the Ti 2p spectra recorded on the corresponding annealed samples derived from HSGT-v (Fig. 3a) thus indicating that no Ti3+ self-doping is detected. The binding energy of Ti 2p3/2 ranges between 458.9 and 459.2 eV for all the samples, a value typical for Ti in nanostructured nanocrystalline TiO2 (Table S2†),23,37,38 and no components at about 457 eV are found, which might be assigned to reduced titanium.23 The O/Ti4+ ratios for all the samples were calculated starting from curve fitting of O 1s XP-spectra. O 1s signals are multicomponent, with the most intense peak at 530.3 eV due to oxygen in TiO2, and the less intense components at 531.5 and 532.6 eV due to –OH and/or defective oxygen and to adsorbed water, respectively.23,38,39 Only the component at about 530 eV was considered to calculate O/Ti4+ ratio, and it was found that the O/Ti4+ ratios in A1 and A3 samples are 1.8 (±0.2). The absence of reduced Ti was also noticed when HSGT-v was annealed in nitrogen or in hydrogen/nitrogen mixtures for different times (Fig. 3a). For these samples the O/Ti4+ ratios were found to be lower than the expected stoichiometry, especially for N1 (Table 2).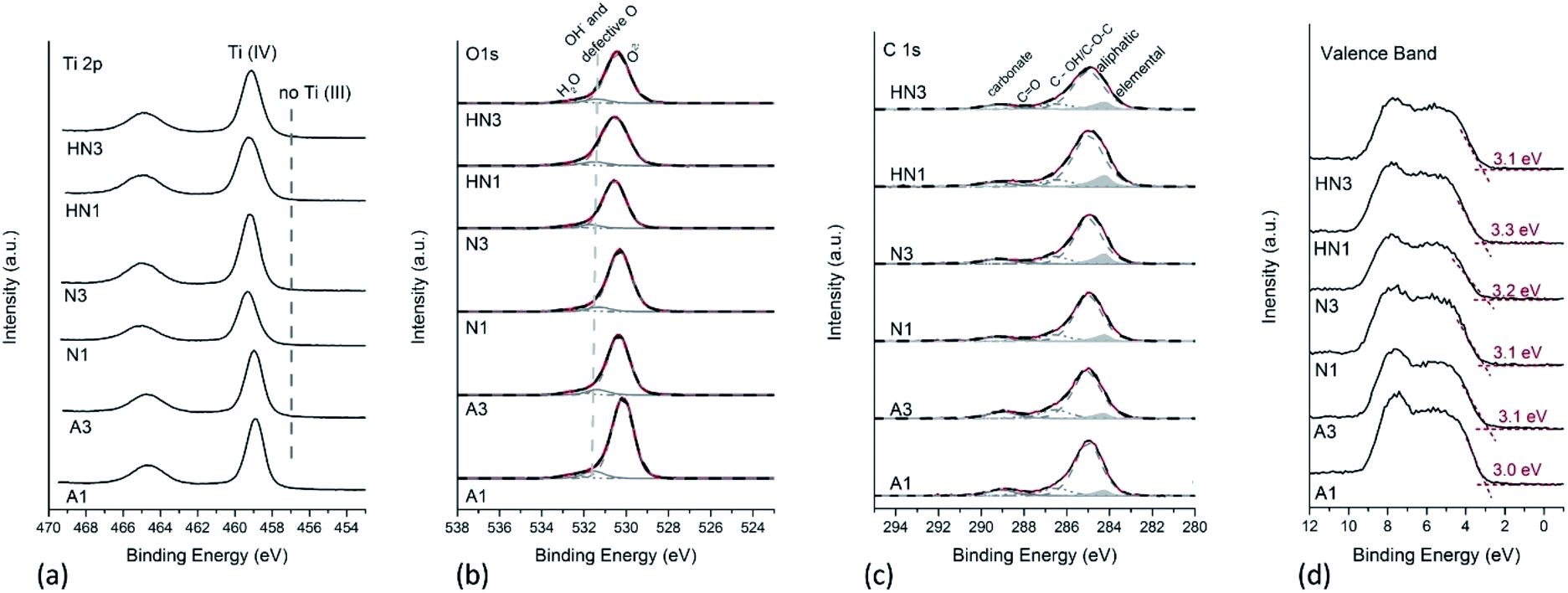 | ||
| Fig. 3 (a) Ti 2p, (b) O 1s, (c) C 1s and (d) valence band X-ray photoelectron spectra of annealed HSGT-v samples. The binding energy of the valence band edge, determined as the intersection of the horizontal offset line with the line representing the linear portion of the step, is reported. | ||
The presence of reduced titanium cannot be thus invoked to justify the photocatalytic properties of these materials (vide infra), but the O/Ti ratio lower than 2 and the component at 531.5 eV in O 1s peaks support the presence of oxygen defects influencing the catalytic properties. Although the formation of oxygen vacancies during annealing of TiO2 in inert or reducing atmosphere is often accompanied by a partial reduction of Ti4+, the lack of Ti3+ signal was also observed in some reduced TiO2−x samples.40 On the other hand, it was recently shown that in reduced ZrO2−x a distribution of neutral and charged oxygen vacancies not associated to the presence of Zr3+ species can be obtained,41 and a similar behavior might occur in TiO2.
To assess the surface carbon species, the curve fitting of C 1s signals has been also performed, adding a component due to elemental carbon, according to the curve-fitting strategy proposed in literature for resolving the different contributions to C 1s signals in PM10 samples.42 The results are provided in Fig. 3c. A component due to elemental carbon is observed at 284.2 eV, whose relative intensity with respect to the total carbon content increases from 7% for the air-annealed samples to 11–12% for the samples heated under H2/N2 mixture. Such component was not detected in the case of the C 1s signals acquired on SA1 and SA3 samples (Fig. S3b†).
It can be observed that the higher is the elemental C content, the higher the Ti 2p binding energy (Table S2†). This result might be ascribed to the presence of C–TiO2 heterostructures, in agreement with Liu et al.43 They reported also a small shift towards higher BE values in the most intense component at about 530 eV of O 1s peak on C–TiO2 heterostructures compared to TiO2. In this paper we observe the same trend (Fig. 3b). However, the absence of carbon and oxygen components at 282.7 eV and 527.6 eV respectively, ascribed by those authors to Ti–O–C bonds in C-doped TiO2, suggests us that carbon does not behave as a dopant. To further clarify if the elemental carbon presence is due to C-doping of TiO2, the XPS valence bands (VB) of the samples were also acquired and processed. The valence bands (Fig. 3d) show two prominent peaks at about 5.5 eV and 7.7 eV, due to π-nonbonding and σ-bonding O 2p electrons respectively; the shape of the VB is typical for anatase being the peak at 7.7 eV more intense than the one at 5.5 eV. The absence of additional electronic states above the VB edge, which are supposed to be present in the case of C-doped TiO2, leads us to exclude that carbon behaves as a dopant.44
Electron paramagnetic resonance spectroscopy points out further differences between the samples derived from HSGT and HSGT-v dried gels. All their spectra (Fig. 4a and b) consist in single peaks with g factor about 2.0024, close to the free electron value. Several literature reports on chemically modified or physically treated TiO2 show this kind of EPR signal, with g factors between 2.002 and 2.005, and most of them attribute it to single electrons trapped in oxygen vacancies.40,45–48 Even though a singlet with similar features has been found in TiO2 composites containing carbon-based materials and related to C-centered radicals,20,49–52 this eventuality is not considered by the majority of the authors.
 | ||
| Fig. 4 EPR spectra recorded at room temperature on: (a) HSGT samples annealed in static air at 400 °C for 1 (SA1) and 3 h (SA3) and HSGT-v samples annealed in air flow at 400 °C for 1 (A1) and 3 h (A3); (b) HSGT-v samples annealed in different conditions. The intensity scale is different in the two graphs. (c) Real part of conductivity σ′ as a function of frequency for A1, A3, SA1 and SA3 samples; (d) real part σ′ (full symbols) and imaginary part σ′′ (open symbols) of the complex conductivity for the annealed HSGT-v samples as a function of frequency. Blue (1 h) and red (3 h) colors correspond to the annealing duration. Squares (A1, A3), circles (N1, N3) and triangles (HN1, HN3) refer to air, N2, and H2/N2 mixture gas flow respectively. The dashed black curves show the fit obtained by applying the Drude–Smith model. | ||
Despite an apparent similarity, a more detailed lineshape analysis on the SA1 and A1 spectra reveals clear discrepancies. Indeed, the samples contain a comparable concentration of paramagnetic species (3 × 1017 and 4 × 1017 spin per g, respectively), but the SA1 signal has a somewhat larger peak-to-peak width (5.5 vs. 4.3 G) and a mixed Gaussian–Lorentzian lineshape (Table S3†), while the A1 signal is mostly Lorentzian. Notably, upon a longer (3 h) annealing in air an opposite variation of the signals is observed (Fig. 4a, Table S3†): in SA3 it becomes narrower and very weak, while in A3 the intensity doubles with respect to A1 (8 × 1017 spin per g). This opposite behavior suggests that the EPR signals of the two kinds of materials have a different origin. The spectrum of SA1 may be mainly due to defects such as oxygen vacancies, while that of A1 have a large contribution from a species more stable under oxidizing conditions (a longer heating in air). To further investigate the behavior of these EPR signals, microwave power saturation measurements were performed (Fig. S4†). For both samples the amplitude (proportional to the intensity) tends to reach a plateau at high microwave power, indicating partial saturation of the signal, but the two curves have a quite different shape. As suggested by a sharper slope change, SA1 apparently comprises two components with different relaxation rates, which are not observed for A1. The dependence on microwave power of the intensity of signals associated with oxygen vacancy defects in TiO2 was described either as reaching a plateau46 or decreasing after a maximum,53 indicating relatively long relaxation times for these radicals. The latter trend was also attributed to unpaired electrons in a point defect at the interface between particles.54 In the case of TiO2 coupled with carbon materials (e.g. graphene-like), peaks were found with g about 2.0035 and different linewidth and power saturation trends, according to the spatial distribution of the radical centers in the delocalized π systems.50 The partially Gaussian lineshape, the power saturation trend and the evolution with the annealing time suggest the SA1 spectrum to be a composite signal, originated by a heterogeneous radical distribution with a contribution from oxygen vacancies. Keeping in mind the close relationship often found between Ti3+ and vacancy sites,5 this interpretation would be in accordance with the high Ti3+ content in SA1.23 Considering the presence of different carbon species in the HSGT-v derived materials, excess electrons localized on such species probably represent the prevalent source of their EPR signal (Fig. 4b). The peaks of the xerogels treated under N2 and H2/N2 mixture, showing totally or mainly Lorentzian shape, are distinguished by a spin density one order of magnitude higher with respect to A1 and A3 (Table 2). The correlation between total carbon content and spin density supports the attribution of the EPR signal to C-centered unpaired electrons, as proposed by some authors.30 On the other hand, the contribution of electrons trapped in oxygen vacancies, whose presence is indicated by XPS and PL spectra (see below), cannot be excluded in these samples as well.
Terahertz spectroscopy characterization
The difference in behavior of annealed samples obtained from HSGT and HSGT-v is confirmed by Terahertz Time Domain Spectroscopy (THz-TDS). This technique allows to extract the complex optical properties of a material in a contactless way and to explore the electronic dynamics over nanometer length scales, without resorting to the Kramers–Kronig relations. Therefore, it directly probes the conducting state within nanodomains, in contrast with traditional dc conductivity measurements probing long-range transport phenomena only.55 It is also worth mentioning that THz is an ideal frequency range for the investigation of inorganic semiconductors, since scattering times are typically in the range 0.05–1 ps, and plasma frequencies also fall in the far-infrared region.The comparative analysis of the samples produced by annealing in air HSGT-v (A1 and A3) and HSGT (SA1 and SA3) evidences a clear difference of the transport properties in the THz spectrum. In Fig. 4c the data retrieved for A1 and A3 are compared with SA1 and SA3, in the range 0.3–1 THz. The real conductivity σ′ in HSGT-v-derived samples exceeds 103 S m−1 at 1 THz, which is much larger (by one order of magnitude or more) than in HSGT-derived ones, clearly indicating that there is an additional conduction mechanism. In HSGT-derived samples, moreover, conductivity is very sensitive to the annealing time, whereas A1 and A3 display only a slight difference in terms of their conducting properties even at the lowest frequencies.
For all HGST-v derived samples, it is important to stress that both real (Re[![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10d.gif) (ω)]) and imaginary (Im[(ω)]) components of the measured conductivity are found to be much larger than previous results reported in literature. In fact, THz spectroscopy studies carried out on pure pellets56 or on nanopowders with different dopants57 show that conductivity usually does not exceed some tens of S m−1 at 1 THz. On the other hand, I–V measurements and potential-dependent capacity characterization of carbon-doped and/or carbon-modified anatase TiO2 nanopowders present lower resistance and in turn higher conductivity values with respect to pure samples by several orders of magnitude.58
(ω)]) and imaginary (Im[(ω)]) components of the measured conductivity are found to be much larger than previous results reported in literature. In fact, THz spectroscopy studies carried out on pure pellets56 or on nanopowders with different dopants57 show that conductivity usually does not exceed some tens of S m−1 at 1 THz. On the other hand, I–V measurements and potential-dependent capacity characterization of carbon-doped and/or carbon-modified anatase TiO2 nanopowders present lower resistance and in turn higher conductivity values with respect to pure samples by several orders of magnitude.58
Re[(ω)] and Im[(ω)] are then simultaneously fit to the Drude-Smith (DS) model, keeping background permittivity (ε∞), relaxation time (τ), plasma frequency (ωp) and c1 as free parameters (see eqn (1), Experimental). The complex conductivity comparison of the samples prepared under various atmospheres and annealing temperatures is shown in Fig. 4d. The background permittivity extracted from the DS fit yields an average value of 5.0 ± 0.7, with less than 15% distribution among the samples. Moreover, from the fitting procedure we find c1 = −1 for all samples, which is consistent with the observed frequency behavior for the complex conductivity, with the real part approaching zero at dc whereas the imaginary part being negative at low frequencies (below 1 THz). This is interpreted in the DS model as conductivity dominated by carriers backscattering,59 therefore outlining a picture where transport in all HSGT-v samples derives, regardless of the annealing conditions, from electrons localized either on oxygen vacancies or on carbon radicals located on the carbonaceous matrix, and not from excess charge released into the conduction band because of the formation of Ti3+. The observed increase in conductivity absolute values can therefore be explained with the high density of structural defects induced by the adopted synthesis conditions.
The extracted plasma frequencies (ωP ≈ 12 ± 1 THz) and relaxation times (τ ≈ 76 ± 8 fs) are found to merge within the deviation of the 5 spot measurements, confirming in a direct way that all HGST-v samples display the same complex THz response. While scattering values are in line with previous results,56,57 the retrieved plasma frequency differs quite significantly from what has been reported on both anatase/rutile mixed nanopowders56 and nanostructured anatase TiO2 samples.60 Reported works on the intrinsic transport properties of anatase have proven that the metallic transition occurs for charge carrier concentrations ≥1019.61 In our samples, given a plasma frequency ωp ∼ 10 THz and even assuming a polaron effective mass m* ∼ 10m0 (electron mass) as in rutile,61 we estimate a charge carrier density N = ωp2ε0m*/e2 in the order of 1016 to 1017 cm−3, which is far smaller than the metallic transition condition. This behavior proves that the enhanced conductivity for the samples derived from HSGT-v shows non-degenerate electrical transport properties.61 It is worth to mention that the recent works on carbon-doped TiO2 materials attribute the conductivity increase to the narrowing effect of doping on the band gap by introducing tails in both valence and conduction bands.62 On the contrary, for the HSGT-v-derived samples the XPS valence band spectra do not show any side tails. In this context, the observed reduction in plasma frequency is consistently explained as a change in the electron-phonon interaction induced by the presence of carbon species, that leads to a larger polaronic effective mass, similarly to what has been observed in anatase films doped with Ni.63
Moreover, from the knowledge of the bulk conductivity (σ), the surface impedance (ZS) can be easily determined for an ideal system using the eqn (2) (see Experimental). We evaluated the real part (the surface resistance Z′) and the imaginary part (the surface reactance Z′′) of the impedance for the N3 sample. Results are plotted in Fig. S5,† showing that the surface impedance varies in the range of tens of Ohms and is relatively reactive (inductive), since Z′′ > Z′. Comparatively, in the near-THz frequency the surface resistance and the surface reactance of bulk copper present values of the order of tenths of Ohms.
Optical, spectro- and photo-electrochemical characterization
UV-vis DRS spectra reveal that all HSGT-v samples exhibit a visible light absorption reaching the NIR range, besides the typical absorption of TiO2 in the UV range (Fig. 5a). For the A1 and A3 dark-grey colored samples this absorption appears as an extended tail, while for the N1, N3, HN1 and HN3 black colored samples it is more pronounced. It should be emphasized that similar visible-light photoresponsivity was previously obtained for “black” titania nanomaterials prepared by hydrogenation and treatments in harsh conditions,3,4,11 and was tightly connected to the band gap narrowing and band tailing originated by the formation of an amorphous hydrogenated outer layer and to the midgap states associated with Ti3+-self doping.4 Conversely, in this study such photoresponsivity was achieved by much milder treatments. The band gap value (Eg) of all HSGT-v annealed materials was estimated to be about 3.1 eV by photoluminescence (PL) (see below). For A1 and A3 samples it was also estimated by Tauc plot (Fig. S6†) using a procedure proposed by some of us.64 The same value was found within the experimental error (±0.1 eV) for both samples, thus confirming the reliability of the Eg estimation. The lack of either band gap narrowing or Ti3+-self doping indicates that the extended visible light absorption of our materials is related to the synergistic effect of carbon species and oxygen vacancies, in agreement with the PL data.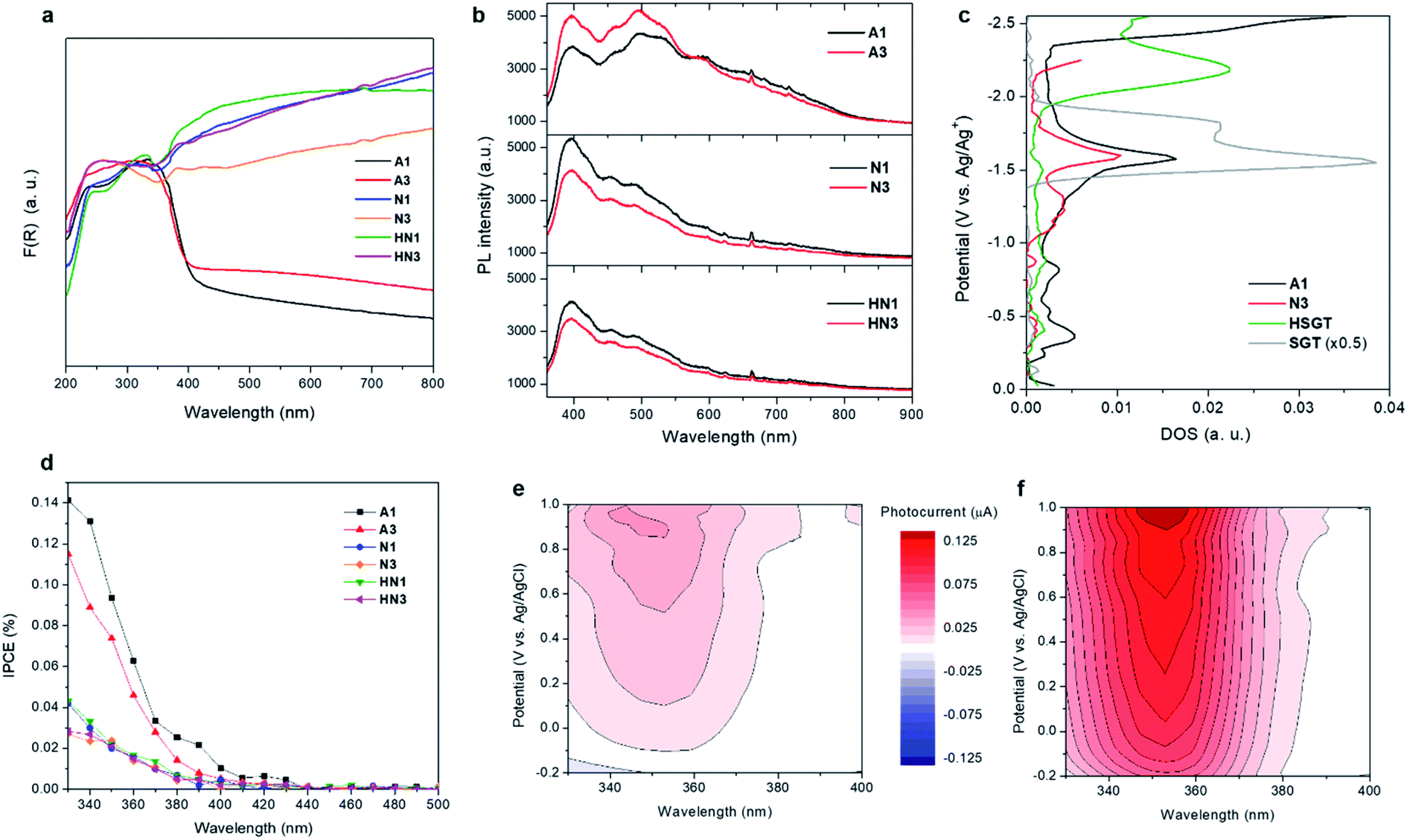 | ||
| Fig. 5 (a) UV-visible diffuse reflectance (UV-vis DRS) spectra of the annealed HSGT-v samples, expressed as Kubelka–Munk function, F(R). (b) Steady-state photoluminescence (PL) spectra of the annealed HSGT-v samples (excitation wavelength 325 nm). (c) Density of electronic states (DOS) patterns for samples A1, N3, HSGT dried gel and a reference TiO2 prepared by sol–gel (SGT), measured in 0.1 mol L−1 LiClO4 acetonitrile electrolyte. The measurements were carried out for each working electrode under the same conditions, in inert atmosphere. (d) Incident-photon-to-current conversion efficiency (IPCE) measured at applied potential 0.9 V vs. Ag/AgCl (electrolyte: aqueous 0.1 mol L−1 KNO3 solution, saturated with Ar); (e) photocurrent as a function of potential (vs. Ag/AgCl) and incident light wavelength, recorded on PET/ITO electrode covered with A1 sample, under regular conditions, and (f) after the addition of 5 wt% glycerol to the electrolyte (aqueous 0.1 mol L−1 KNO3 solution, saturated with Ar). | ||
The analysis of the PL emission spectra gives a further evidence of the presence of oxygen vacancy defects. A pronounced emission band at 400 nm plus other contributions in the visible region of the wavelength spectrum are seen in all PL spectra (Fig. 5b). The emission centered at 400 nm (3.1 eV) is attributed to the excitonic recombination, in particular the radiative recombination of self-trapped excitons. The self-trapped excitons are generated by the self-localization of photoinduced charges due to the strong electron–phonon coupling. The PL emission seen in the visible region between 440 and 650 nm is due to interband transitions. The green contributions centered at about 455 and 500 nm as well as the red one at about 600 nm are related to the radiative recombination of free electrons and holes trapped into oxygen vacancy sites (F+-centers) and between free holes and electrons that relax from either conduction band or shallow electron states associated with subsurface oxygen vacancies, respectively.65 These transitions appear to be enhanced in A1 and A3 with respect to the other samples indicating the presence of a larger amount of oxygen vacancies in the samples annealed in air than in those annealed in inert or reducing atmospheres. Moreover, the relative PL intensities appear affected by heat treatment duration and atmosphere. Increasing the heat treatment time, the PL relative intensity increases for the samples annealed in air while an opposite trend is seen for the samples annealed in N2 and H2/N2. The presence of graphitic carbon was suggested to enhance the photocatalytic efficiency via the conversion of long wavelengths to short ones, which significantly promoted the light utilization of the photocatalysts.66 The samples annealed in air exhibit a lower amount of graphitic carbon than the other ones. Among them, N1 and N3 exhibit a higher amount of graphitic carbon than HN1 and HN3 samples with N1 showing the highest value, while a similar content is seen for HN1 and HN3 (Table 2). The structure of the materials could cause an efficient charge separation through interfacial charge transfer between anatase nanocrystals and amorphous, defective and graphitic carbon.
In this process both the interface morphology and the compositional distribution of the C matrix play a key role because electrons can be trapped also on carbon radicals localized on the diverse C species, in agreement with EPR analysis. All these concomitant factors can affect the ability to capture the photogenerated electrons providing higher lifetimes of electron–hole pairs, and hence a higher photocatalytic activity (vide infra).
Spectroelectrochemical measurements (chronoamperometry combined with diffuse reflectance UV-vis spectroscopy) give information on the density of empty electronic states.67,68 The densities of states (DOS) calculated for the A1 and N3 samples (Fig. 5c) present similarities. One significant difference can be observed in the range from −1.0 to −1.5 V vs. Ag/Ag+, where a relatively high concentration of electronic states appears in the case of N3. A direct quantitative analysis of the concentration of electronic states for N3 and A1 materials is, however, impossible due to optical and spectral differences of the two electrodes, since the measurement is based on reflectance changes at 600 nm. Nevertheless, A1 sample seems to be richer in deep electronic states which should rather act as electron traps diminishing the photocatalytic activity of the material in reduction processes (e.g., hydrogen generation). The position of the dense electronic state with onset at about −1.5 V (corresponding to −0.7 V vs. NHE) is close to the calculated ECB edge values (vide infra, Table 3).
| Sample | Eg (eV) | EVB edge (eV) | ECB edge (eV) | EVB edge (V) | ECB edge (V) |
|---|---|---|---|---|---|
| A1 | 3.1 | −7.0 | −3.9 | 2.5 | −0.6 |
| A3 | 3.1 | −7.1 | −4.0 | 2.6 | −0.5 |
| N1 | 3.1 | −7.2 | −4.1 | 2.7 | −0.4 |
| N3 | 3.1 | −7.3 | −4.2 | 2.8 | −0.3 |
| HN1 | 3.1 | −7.3 | −4.2 | 2.8 | −0.3 |
| HN3 | 3.1 | −7.1 | −4.2 | 2.6 | −0.5 |
To highlight the effect of the annealing on the electronic structure of the studied materials, two amorphous titanium oxide samples were analyzed as reference: the TiO2-acac dried gel (HSGT) and a bare TiO2 particulate gel (SGT). The inspection of their DOS profiles (Fig. 5c) shows that SGT has a dense electronic state at ca. −1.6 V vs. Ag/Ag+, similar to A1 and N3, but it lacks the distribution of deep midgap states, which are not very evident also in HSGT, indicating that the annealing of the hybrid gel leads to the creation of numerous defects that can act as electron traps. It is interesting to note that in HSGT the presence of the Ti-acac complex strongly influences the density of states, causing a marked shift of electronic states towards higher energy with respect to the other investigated samples. Consequently, they should be attributed mainly to the presence of surface states induced by the ligand.
Photocurrent spectra allow to examine the photo-response of the materials in the UV-visible radiation spectrum. Noticeable photocurrents have been recorded in the range 400–440 nm for all samples pointing at their photoactivity upon visible light irradiation (Fig. 5d). This activity can be attributed mainly to the presence of carbon in the structure of the samples. Carbonaceous species are supposed to offer electrons of energy higher than the top of the valence band which can be excited to the conduction band with visible light.30 Significantly higher photocurrents are observed when UV light is involved, as the result of a direct VB to CB excitation.
The samples annealed in air show largely higher photocurrents induced by UV than the other samples, likely related to a larger amount of oxygen vacancies. These defects improve UV light utilization generating shallow electron states below the conducting band, then acting as trap for photogenerated electrons. Possibly the lower carbon content contributes to increase UV light absorption directly by TiO2. On the other hand, in the range of 400–440 nm A3 is almost inactive compared to A1 because of its lower amount of graphitic carbon.
Photocurrents have also been recorded upon addition of 5 wt% glycerol as a hole scavenger, since it is the sacrificial agent used in photocatalytic hydrogen generation tests (vide infra). Fig. 5e shows photocurrents recorded for the electrode covered with A1 powder measured under variable potential and wavelength of incident light. Clear anodic photocurrents (red areas) were significantly amplified (roughly by the factor of 3) when glycerol was added to the electrolyte (Fig. 5f). This confirms an efficient holes consumption by glycerol, resulting in a suppressed electron–hole recombination and improved efficiency of photocurrent generation. Moreover, the so-called photocurrent doubling effect may operate here. This phenomenon is based on the injection of additional electrons into the conduction band from photogenerated organic radicals. Photocurrent measurements confirm therefore not only the extended spectral range of photoactivity of A1 (Fig. 5e), but also its ability to efficiently oxidize organic compounds (pollutants, fuels or sacrificial reagents). Since the degradation of organics may involve either a direct attack of holes or an indirect oxidation with photogenerated reactive oxygen species, in particular hydroxyl radicals (HO˙), oxidation of terephthalic acid (TA) to hydroxyterephthalic acid (TAOH) was monitored to evaluate the efficiency of HO˙ generation. The photocatalysts were irradiated with UV-vis light (λ > 320 nm) in aqueous solutions of TA. The rate of TAOH formation (Fig. S7†) reflects the efficiency of HO˙ generation: A3 appeared the most active sample, however, A1 and HN3 materials also showed significant photoactivity. This observation can be explained by the generation of holes with relatively higher oxidative potential by these samples.
Photocatalytic hydrogen production
The hydrogen evolution rate (HER) values obtained on the annealed HSGT-v materials under UV irradiation starting from a 5 wt% glycerol solution at the spontaneous pH are shown in Fig. 6a. As expected, no H2 production was observed for a photolysis test (not reported). The highest HER values were obtained for N3 and HN3 samples, 375 and 424 μmol h−1 gcat−1, respectively, while lower H2 generation was detected for the other samples. However, all HER values, except for A3, are higher than 200 μmol h−1 gcat−1 (at 100 mW cm−2 light intensity), which is a benchmark value reported in recent research studies about the photocatalytic performances of defective TiO2 without co-catalyst (in methanol aqueous solution).69 In most cases, the H2 production from defective titania is accompanied by the use of co-catalysts based on noble metals (in particular platinum). For instance, Zhao et al. reported a HER of about 2500 μmol h−1 gcat−1 obtained from methanol aqueous solution (30 vol%) on defective TiO2 loaded with 1 wt% Pt, but reported no H2 production data without Pt.70 Other recent papers focused on the photocatalytic H2 production using defective TiO2 without co-catalyst. For example, TiO2 nanotubes were tested in the absence of noble metals, obtaining a HER of about 6 μmol h−1 cm−2,71 while for hydrogenated anatase HER values not higher than 155 μmol h−1 gcat−1 were reached under solar simulating lamp with 100 mW cm−2 light intensity.11 On the other hand, the apparent quantum efficiencies (AQE) reported in Table 2, evaluated for 4 h UV irradiation as described in the eqn (3) (Experimental), evidence values higher than 1% for N3 (1.2%) and HN3 (1.3%), which are much higher than that observed in literature for unmodified TiO2 in the photoreforming of glycerol.72 Higher AQE values were obtained only in presence of noble metal-based co-catalysts, for instance Ag2O (AQE of about 4% after 4 h of irradiation).73 Therefore, our results appear outstanding with respect to those reported in the current literature dealing with similar photocatalytic systems.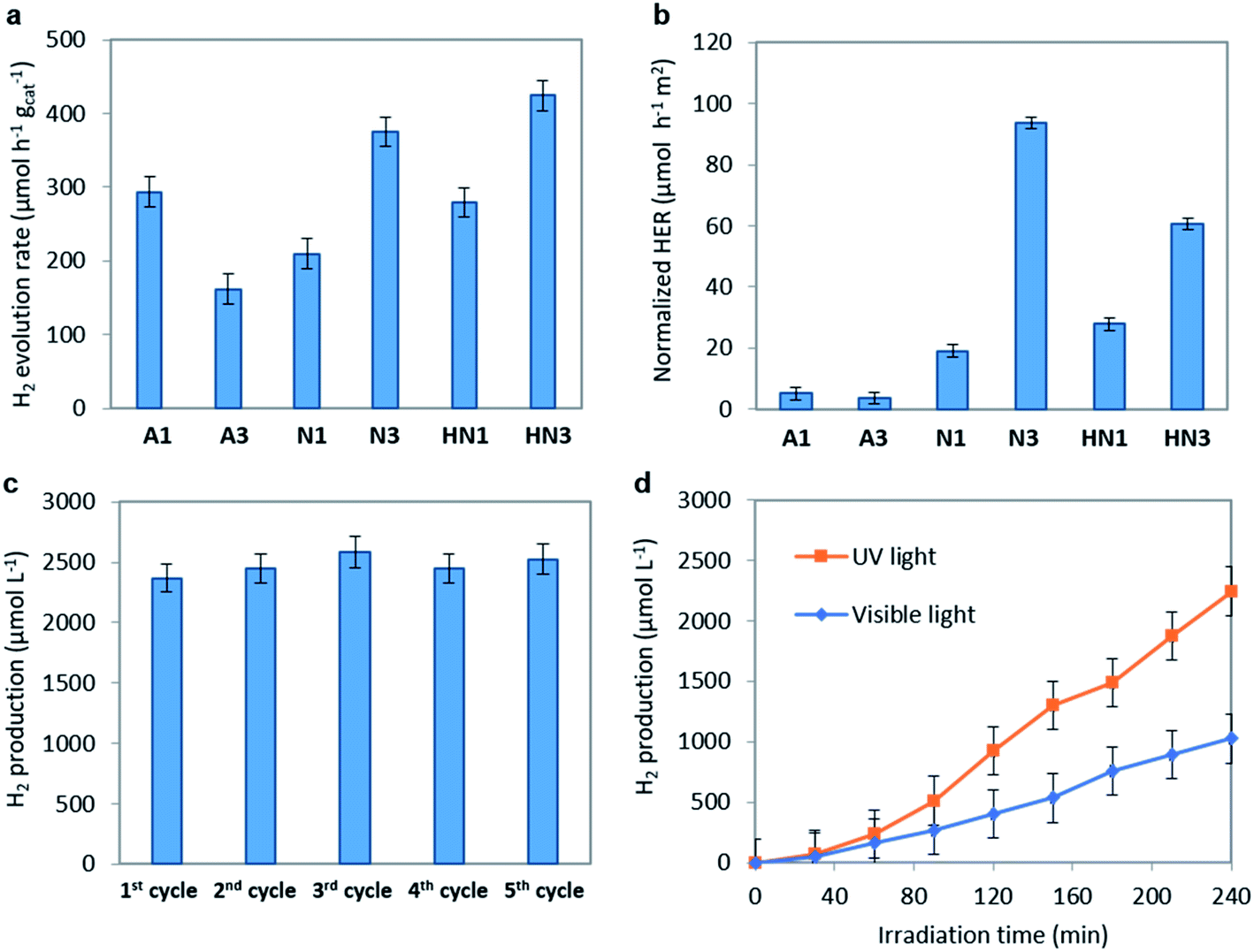 | ||
Fig. 6 (a) Photocatalytic hydrogen evolution rate (HER) and (b) normalized HER with respect to SSA of the materials derived from HSGT-v obtained under UV irradiation. Glycerol initial concentration 5 wt%; catalyst dosage: 1.5 g L−1; irradiation time: 4 h. (c) Photocatalytic hydrogen production after 4 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) hours of UV irradiation for successive reuse cycles of N3 sample; glycerol initial concentration: 5 wt%; pH = 6. (d) Photocatalytic hydrogen production during the irradiation with UV and visible light for N3 sample. Catalyst dosage: 1.5 g L−1; glycerol initial concentration: 5 wt%; pH = 6. hours of UV irradiation for successive reuse cycles of N3 sample; glycerol initial concentration: 5 wt%; pH = 6. (d) Photocatalytic hydrogen production during the irradiation with UV and visible light for N3 sample. Catalyst dosage: 1.5 g L−1; glycerol initial concentration: 5 wt%; pH = 6. | ||
The ability of a photocatalytic material to operate a reaction (e.g. water splitting) is defined by its electronic structure in terms of relative positions of valence and conduction band edges. Based on the band gap (Eg) and valence band edge (EVB) energy values it is possible to calculate the conduction band edge (ECB) energy value (vs. absolute vacuum scale, eV) according to ECB = EVB + Eg. All HSGT-v annealed samples exhibit the same Eg value (3.1 eV) as estimated by PL and UV-vis-DRS analyses. The ECB and EVB potentials (vs. NHE, V) were calculated considering the EVB values (vs. absolute vacuum scale, eV) estimated by XPS analysis and the work function of the XPS instrument (φexp = 4.0 eV vs. absolute vacuum scale), using 4.5 V as a scale factor (Table 3). It should be noted that all the EVB values result more positive than the oxidation potential O2/H2O (+1.23 V vs. NHE) and, simultaneously, all the ECB values result more negative than the reduction potential H+/H2 (0 V). Therefore, the electronic structure of all the annealed HSGT-v samples satisfies the thermodynamic requirements for the photocatalytic H2 production by water splitting reaction.74
To analyze the kinetic aspects, the HER values were normalized with respect to the specific surface area (SSA) of each sample (n-HER, Fig. 6b). The samples annealed in N2 or H2/N2 exhibit much higher n-HER values than those annealed in air, with N3 showing the best performance. This result can be explained considering the concurrent effects related to the defective structure and the morphology of these materials formed by TiO2 anatase nanocrystals incorporated in an amorphous and graphitic carbon matrix (Fig. 1). In agreement with TG/DTA data the annealing in reducing atmospheres results in the formation of a larger (about three times) amount of carbon than the annealing in air. This causes a dramatic reduction of the SSA values reaching 4 m2 g−1 in N3 (see Table 2). Therefore, to analyze the trend of n-HER values we separately compare the samples annealed in air and the ones annealed in N2 or H2/N2. A3 shows a n-HER value lower than that of A1. A3 contains more total carbon, but insignificant graphitic carbon on the surface (see Raman spectra, Fig. 2b), indicating the relevance of the graphite phase and a likely contribution of oxygen vacancies. Considering the trend of n-HER of the samples annealed in reducing atmospheres, besides the effects of the graphitic phase and of the oxygen vacancies, another contribution should be considered. Under UV irradiation electrons localized on carbon radicals, whose density appear enhanced in the EPR spectra of these samples, can be directly injected into CB. N3 exhibits the highest n-HER and the larger carbon content and radical density, while HN3 shows a higher n-HER than HN1 despite their similar carbon content, suggesting that in H2/N2 annealed samples the oxygen vacancies have a greater influence on the photocatalytic activity. It should be noted that the trend of n-HER values well agrees with the PL emission intensities under the same annealing atmosphere, since a lower PL intensity is usually related to the lower recombination rate of the e−/h+ pairs (see Fig. 5b).
Based on the above observations N3 was chosen to investigate the influence of operating conditions on the photocatalytic H2 generation.
The optimal photocatalyst dosage was evaluated exploring the 0.75–2.0 g L−1 concentration range under UV light irradiation for 4 hours (Fig. S8†). It was found that the photocatalytic efficiency increases with the catalyst dosage up to 1.5 g L−1 (2540 μmol L−1) and then does not change increasing the dosage up to 2.0 g L−1, keeping about the same value of H2 production. This shows that the whole amount of catalyst used in the test is well exposed to light radiation at 1.5 g L−1 dosage. At higher concentration the lack of improvement can be explained assuming that the excess of photocatalyst does not participate in the reaction probably due to both an increased turbidity of the suspension and to the scattering effects of catalyst particles.75
The recyclability is one of the most important aspects to be considered in the formulation of a new photocatalytic material.76 To check the recyclability of N3 sample, the H2 production tests were repeated up to 5 cycles (Fig. 6c). At the end of each test, the catalyst was recovered by centrifugation and dried at room temperature for 48 hours. No regeneration step was carried out on the recovered catalyst. It is worthwhile to note that almost the same amount of H2 was obtained in all the reuse cycles, being in the range 2300–2500 μmol L−1. These results are consistent with the photocatalytic data reported in literature for black TiO2 with and without the use of a co-catalyst16 and emphasize the stability of the N3 photocatalyst in the H2 production from glycerol aqueous solution.
The H2 production activity of the N3 photocatalyst under visible light irradiation was also evaluated, reaching a value of 1204 μmol L−1 after 4 hours of irradiation (corresponding to 200 μmol h−1 g−1) (Fig. 6d). This result is very interesting since it confirms the ability of the optimized N3 photocatalyst to produce H2 in the absence of a co-catalyst also under visible light. In literature few works report the photocatalytic activity of black TiO2 and hydrogenated TiO2 under visible light irradiation without noble metals, and in most cases the photocatalytic activity is reported in the presence of simulated solar light, which includes a certain fraction of UV radiation.11,16 In a recent paper, the H2 production from only water splitting reaction, using black TiO2 without co-catalyst and under visible light, has been reported as 0.045 μmol H2 g−1 after 18 hours.77 The visible light activity of the N3 photocatalyst may be explained considering the above discussion and recent literature findings which underlined that the presence of paramagnetic Ti3+ states could act as recombination centers for charge carriers, suppressing the visible light photoactivity.11 The XPS analysis excludes the presence of Ti3+ on the studied samples and the EPR and PL spectra display signals that can be related to trapped electrons localized either on oxygen vacancies or on carbon radicals. Moreover, the strong absorption in the whole visible light region observed despite the absence of band gap narrowing appears to benefit the photocatalytic performances under visible light irradiation.78 Therefore, the visible light driven H2 evolution is related to the presence of specific defect centers acting as catalytic reactive sites: the carbon species and the oxygen vacancies. These defects allow to obtain enhanced photoactivity also in the absence of Ti3+ ions by two synergistic effects: reducing the electrons–holes recombination rate and increasing the visible light absorption.
Conclusions
Band gap narrowing and Ti3+-self doping are considered two essential electronic features for TiO2-based materials, which allow visible light harvesting and hydrogen production by a photocatalytic route, respectively. In this work, we have obtained TiO2/C bulk heterostructures which exhibit remarkable visible light photoresponsivity and impressive H2 evolution rates under both UV and visible light irradiation. They are formed by defective anatase TiO2 nanocrystals, characterized by the same band gap of pristine TiO2 (3.1 eV) and whose structure contains oxygen vacancies without Ti3+, embedded in a carbonaceous matrix. These materials were synthesized under mild conditions, by heating a hybrid TiO2-acetylacetonate gel at 400 °C for 1 or 3 h under different atmospheres (air, N2, H2/N2). Regardless of atmosphere the pyrolysis of the organic phase belonging to the dried gel results in the formation of a carbonaceous matrix, formed by amorphous and crystalline sp2 carbon atoms, in which the anatase TiO2 nanocrystals are incorporated. The graphitic phase, the carbon radicals and the oxygen vacancies with a trapped electron (F+-centers) play a key and synergistic role in determining the ability to give outstanding and stable H2 production under UV and visible-light irradiation without metallic co-catalysts. These performances were obtained using glycerol as electron donor and represent a promising step in the development of the exploitation of this biomass-derived compound. Furthermore, photocurrent and HO˙ photogeneration tests evidenced also an interesting oxidative ability of some of the materials, particularly those calcined in air, suggesting their potential for other photocatalytic processes.Experimental
Synthesis of the materials
The TiO2 materials were synthesized by a hydrolytic sol–gel route at room temperature. Titanium(IV) n-butoxide (97+%), acetylacetone (Hacac) (99+%), 1-propanol (99.80+%) and hydrochloric acid (37 wt%) were provided by Sigma-Aldrich and used as received. From a solution with molar ratios Ti:Hacac:propanol:water 1:0.4:5:10 a homogeneous chemical gel was obtained, as described in a previous work.23 The gel dried at 30 °C in an air-ventilated oven for a few days was named HSGT (hybrid sol–gel titania), while a wet gel dried in a vacuum oven was named HSGT-v. The xerogels were ground and finally annealed at 400 °C in a tubular furnace in different conditions, as reported in Table 1. It is relevant to note that the SA1 and SA3 samples were obtained by heating the HSGT dried gel at 400 °C for 1 or 3 hours, respectively, in a furnace with the ends of the quartz tube open to air (static air conditions), while the A1 and A3 samples were obtained by heating the HSGT-v dried gel at 400 °C for 1 or 3 hours in laminar air flow, by analogy with the treatments carried out in N2 (N1 and N3 samples) and in 3% H2/N2 mixture (HN1 and HN3 samples).
Characterization
X-ray diffraction (XRD) measurements were performed with a Philips X'PERT-PRO diffractometer by using monochromatized CuKα radiation (40 mA, 40 kV) with a step width of 0.013° 2θ. The average size of TiO2 anatase nanocrystals was evaluated by Scherrer's equation using the FWHM of the (101) peak, centred at 25.3° 2θ.Field emission scanning electron microscopy images were obtained with a FEI Nova NanoSEM 450 emission SEM at an accelerating voltage of 5 kV (range of acceleration voltage: 50 V to 30 kV) equipped with an Everhart Thornley detector (ETD) and a Through-Lens-Detector (TLD). The samples were first sputter coated with a thin layer of Au–Pd alloy in order to prevent charging of the surface.
TEM micrographs were collected using a FEI TECNAI G2 S-twin apparatus operating at 200 kV (LaB6 source). The powder samples were transferred on carbon-coated copper grids by dispersing them in ethanol and then adding one drop on the copper grid and evaporating the solvent.
Raman spectra were acquired with a Dispersive MicroRaman spectrometer (Invia, Renishaw) with a 514 nm diode-laser. The laser power was set at 25 mW and each sample was exposed to the laser beam with accumulation time of about 1 min. Curve fitting analysis of the experimental D and G Raman lineshapes to a set of Gaussian functions was performed using Origin 8.5 software with non-linear least squares fitting based on the Levenberg–Marquardt method.
Thermogravimetric and differential thermal analysis (TG/DTA) were performed by a TA Instrument simultaneous thermoanalyser (SDT Q600), heating in air at 10 °C min−1 rate.
X-ray photoelectron spectroscopy (XPS) experiments were carried out using a PHI QuanteraSXM spectrometer (ULVAC PHI, Chanhassen, MN, USA) equipped with a monochromatic Al Kα X-ray source. Photoelectron spectra were recorded in fixed analyser transmission mode; the pass energy value was set at 69 eV and 280 eV for the narrow scan and the survey spectra, respectively. The binding energies were referred to C 1s peak at 285.0 eV. More details on the instrument setup, calibration and data processing are provided elsewhere.23
Electron paramagnetic resonance (EPR) spectra were recorded on the solid samples using an X-band (9 GHz) Bruker Elexys E−500 spectrometer (Bruker, Rheinstetten, Germany). The capillary containing the sample was placed in a standard 4 mm quartz sample tube. Measurements were performed at 25 °C. The instrumental settings were as follows: sweep width, 140 G; resolution, 1024 points; modulation frequency, 100 kHz; modulation amplitude, 1.0 G; time constant, 20.5 ms, attenuation, 10 dB. The g-factor value and the spin density of the samples were evaluated by means of an internal standard, Mn2+-doped MgO and calibrated with reference to a diphenylpicrylhydrazyl (DPPH) standard solution. For power saturation experiments, microwave power was gradually incremented from 0.001 to about 100 mW.26 Elaboration and curve fitting of the spectra were performed using Xepr software (Bruker).
Terahertz Time Domain Spectroscopy (THz-TDS) transmission measurements have been carried out by means of a femtosecond laser driven all fiber coupled THz spectrometer (Menlo Systems). Photoconductive antennas are used for both THz generation and detection, and TPX (Polymethylpentene) lenses to collimate and focus the broad THz frequency range onto the samples. The time domain signal passing through the sample at normal incidence and the free space (reference) signal are acquired separately. Once the time domain signals are transferred in the frequency domain by applying a Fast Fourier Transform (FFT), the material transfer complex function T(ω) can be extracted from the ratio of the electric field transmitted through the sample, Es(ω), and the reference signal, Er(ω).
T(ω) correlates with the amplitude and phase changes in the transmitted signal due to refraction (n) and absorption (k) inside each sample. Once the complex index of refraction ñ(ω) = n(ω) + ik(ω) is extracted from the transfer function, the complex dielectric function ![[small epsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0de.gif) = ε′ + ε′′, where ε′ = n2 − k2 and ε′′ = 2nk, can be evaluated. All samples were investigated in pressed pellets having 1 cm in diameter. Pellets of HSGT-derived samples (SA1 and SA3) are prepared from pure powders, whereas HSGT-v-derived samples, due to their highly absorptive nature, are first mixed with pure KBr with a weight ratio 1:5 and then pressed into disk form, following the procedure successfully used in a previous work.55 Considering the possible dishomogeneities due to the KBr mixing, the measurements on HSGT-v-derived pellets are carried out in 5 different spots for each sample. Multi-spot measurements have shown a 10% standard deviation in the transfer function, confirming a high degree of homogeneity.
= ε′ + ε′′, where ε′ = n2 − k2 and ε′′ = 2nk, can be evaluated. All samples were investigated in pressed pellets having 1 cm in diameter. Pellets of HSGT-derived samples (SA1 and SA3) are prepared from pure powders, whereas HSGT-v-derived samples, due to their highly absorptive nature, are first mixed with pure KBr with a weight ratio 1:5 and then pressed into disk form, following the procedure successfully used in a previous work.55 Considering the possible dishomogeneities due to the KBr mixing, the measurements on HSGT-v-derived pellets are carried out in 5 different spots for each sample. Multi-spot measurements have shown a 10% standard deviation in the transfer function, confirming a high degree of homogeneity.
The dielectric response of the mixture is retrieved from the frequency domain analysis by using a commercial software (TeraLyzer) which utilizes an algorithm based on the iterative minimization of the discrete peaks originated from the multiple reflections inside the target due to periodic Fabry–Perot oscillations.79 With the same algorithm, one can determine the thickness for each pellet with high precision. Then, the intrinsic dielectric properties of the mixed pellets are extracted by using the Landau–Lifshitz–Looyenga equation model under the mean field theory assumption.80
The dielectric behaviour of the samples can then be described invoking the standard Drude–Smith (DS) theory, commonly used to model carriers undergoing restricted motion.59 Assuming a reasonably strong charge carrier localization, the formula for (ω) in the DS model under single-scattering approximation is given by the eqn (1):
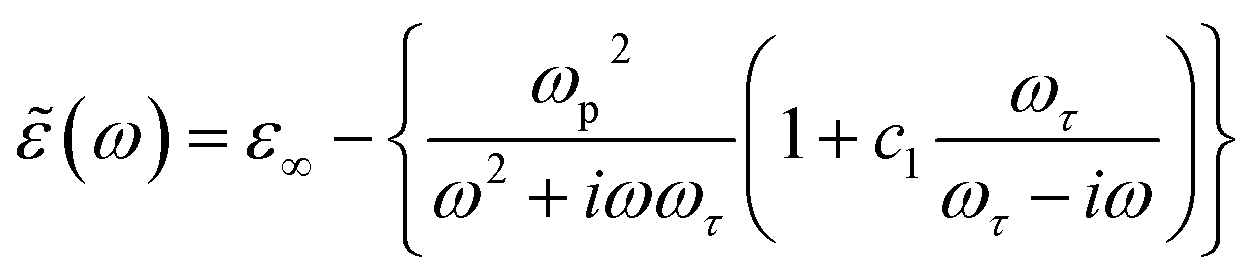 | (1) |
The corresponding complex conductivity can be obtained using the expression: (ω) = −i[(ω)−ε∞]ε0ω, where ε0 is the free space permittivity.
The surface impedance (ZS) for an ideal system was determined by the eqn (2):84
 | (2) |
Ultraviolet-visible light diffuse reflectance (UV-vis DRS) spectra were recorded in the 200–800 nm range using a double beam Jasco spectrophotometer. Samples were mixed with BaSO4, used as standard. The measured intensity was expressed as the value of the Kubelka–Munk function F(R).
Steady-state photoluminescence (PL) spectra were recorded exciting the sample by a continuous light emission He–Cd laser at 325 nm (KIMMON Laser System). PL was collected at normal incidence respect to the surface of samples through an optical fiber, dispersed in a spectrometer (Princeton Instruments, SpectraPro 300i), and detected using a Peltier cooled charge coupled device (CCD) camera (PIXIS 100F). A long pass filter with a nominal cut-on wavelength of 350 nm was used to remove the laser line at monochromator inlet.
The specific surface area (SSA) determination was performed using BET method by N2 adsorption at −196 °C with a Costech Sorptometer 1042 after a pretreatment of the sample at 150 °C for 4 hours in He flow (99.9990%).
Redox properties and density of electronic states (DOS) of the materials were determined using a combination of electrochemical measurements and UV-vis diffuse reflectance spectroscopy. The spectroelectrochemical measurements were carried out in a three-electrode system using Pt as counter and Ag/Ag+ (10 mmol L−1 AgNO3 in 0.1 mol L−1 Bu4NClO4 in acetonitrile) as reference electrodes, respectively. As a working electrode platinum foil (ca. 3 cm2) covered by the studied materials was used. The electrodes were placed in a cuvette with a quartz window filled with 0.1 mol L−1 LiClO4 acetonitrile solution as the electrolyte. The cuvette was then placed in front of the integrating sphere, with working electrode oriented toward the light beam. Prior to (15 min) and during the experiment argon was purged in order to remove dissolved oxygen. The electrochemical part of measurements was performed with a Bio-Logic, SP-150 potentiostat. The applied potential was lowered every 10 min by 25 mV. The relative reflectance was collected by PerkinElmer UV-Vis Lambda 12 spectrophotometer equipped with a 5 cm diameter integrating sphere. The relative reflectance was converted to the Kubelka–Munk function changes (ΔKM). The density of states was calculated as a difference in the Kubelka–Munk function between each applied potential at 600 nm.
Photocurrent measurements were performed with a photoelectric spectrometer (Instytut Fotonowy) and a three electrodes configuration with Ag/AgCl as the reference electrode and platinum wire as the counter electrode. The working electrode – a thin layer of the material – was deposited at the surface of ITO-coated transparent foil (60 Ω sq−1 resistance, Sigma-Aldrich). The electrolyte (0.1 mol L−1 KNO3, pH = 6.1) was purged with argon for 15 min prior to and during the measurement. The working electrodes were irradiated by xenon lamp in the range of 330–500 nm with 10 nm step. The irradiation was carried out from the backside through the ITO layer. The incident photon-to-current conversion efficiency (IPCE) was calculated based on photocurrents and incident light power. In order to determine the effect of glycerol additive on the value of photocurrents, the measurement was carried out twice with the same working electrode, adding 5 wt% glycerol to the electrolyte before the second measurement.
Photocatalytic experiments
The photocatalytic experiments for hydrogen production were carried out in a pyrex cylindrical reactor (ID = 2.6 cm) equipped with an N2 distributor device (Q = 0.075 N L min−1) to assure the absence of O2 during the tests. The photoreactor was irradiated by four UV lamps (Philips, nominal power: 8 W each) with main emission peak at 365 nm or by four visible lamps (Philips, nominal power: 8 W each) with spectral emission in the range 400–650 nm. The lamps surrounded the photoreactor external surface and were positioned at an equal distance from it (about 30 mm) in order to uniformly irradiate the suspension. The photon flux at reactor external surface was about 25 and 26 mW cm−2 for UV and visible lamps, respectively. The value of photon flux was obtained through actinometrical technique by means of a spectro-radiometer (StellarNet Inc).85 To ensure the complete mixing of the suspension in the reactor, a peristaltic pump (Watson Marlow) was used. In a typical photocatalytic test, a glycerol aqueous solution with initial concentration equal to 5 wt% was used. The treated solution volume was 80 mL. The photocatalytic hydrogen evolution rate was evaluated at the spontaneous pH of the solution (pH = 6) and at different photocatalyst dosages (in the range 0.75–2 g L−1). Before the irradiation, the suspension was stirred in the dark for 2 hours, in order assure the adsorption–desorption equilibrium of glycerol on the photocatalyst surface, and then the reaction was started under UV or visible light up to 4 hours. The analysis of the gaseous phase coming from the photoreactor was performed by using a continuous analyzer (ABB Advance Optima AO2020 equipped with a thermal conductivity detector, TCD) to measure the H2 concentration during the irradiation time. The effect of catalyst dosage, reusability of the optimized photocatalyst as well as the influence of the light source (UV or visible light) was evaluated. The apparent quantum efficiency (AQE) was calculated according to the eqn (3):86
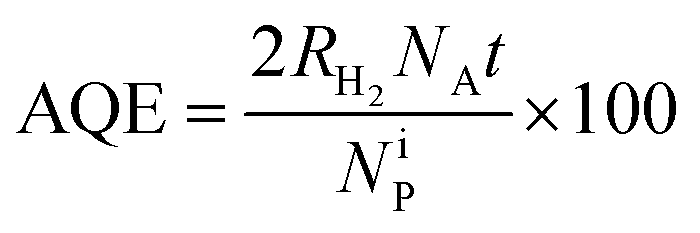 | (3) |
Authors contribution
The project was conceived by A. Ar. and C. I., they performed materials synthesis, XRD, TG/DTA and UV-vis DRS measurements; A. R. and M. F. conducted, processed, and interpreted XPS experiments; G. D. E. and C. I. carried out the EPR measurements; A. An. and C. K. designed and carried out THz spectroscopy measurements and data analysis; L. D. S. and I. R. performed PL experiments; R. D. G. performed SEM and TEM measurements; W. M., M. K. and C. I. conducted the photocurrent, spectroelectrochemical and hydroxyl radical generation analyses and elaborated the data; V. V. and G. I. designed, performed and interpreted the photocatalytic hydrogen production tests and carried out Raman and BET analyses. All the authors discussed the results, contributed to the writing of the manuscript and approved its final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to express their deep gratitude to Prof. N. D. Spencer (Laboratory for Surface Science and Technology. ETH Zürich, Switzerland) for access to the XPS facilities. Giovanni Cossu is acknowledged for technical assistance. This work was undertaken with financial support from the University of Cagliari and the Italian Ministry of Education, University and Research (MIUR). A. R. thanks Fondazione Banco di Sardegna and Regione Autonoma della Sardegna Progetti Biennali di Ateneo Annualità 2016, Fondazione Sardegna CUP F72F16003070002 for the financial support. W. M. and M. K. acknowledge the support by the Foundation for Polish Science (FNP) within the TEAM POIR.04.04.00-00-3D74/16 project.Notes and references
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- L. Li, J. Yan, T. Wang, Z. J. Zhao, J. Zhang, J. Gong and N. Guan, Nat. Commun., 2015, 6, 5881 CrossRef PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- X. Liu, G. Zhu, X. Wang, X. Yuan, T. Lin and F. Huang, Adv. Energy Mater., 2016, 6, 1–29 CAS.
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601 RSC.
- K. Zhang and J. H. Park, J. Phys. Chem. Lett., 2017, 8, 199–207 CrossRef CAS PubMed.
- S. Selcuk, X. Zhao and A. Selloni, Nat. Mater., 2018, 17, 923–928 CrossRef CAS PubMed.
- S. H. Li, S. Liu, J. C. Colmenares and Y. J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- G. Wang, L. Zhang, B. Yan, J. Luo, X. Su and F. Yue, ChemCatChem, 2019, 11, 1057–1063 CAS.
- S. Bai, N. Zhang, C. Gao and Y. Xiong, Nano Energy, 2018, 53, 296–336 CrossRef CAS.
- N. Liu, X. Zhou, N. Nguyen, K. Peters, F. Zoller, I. Hwang, C. Schneider, M. E. Miehlich, D. Freitag, K. Meyer, D. Fattakhova-Rohlfing and P. Schmuki, ChemSusChem, 2017, 10, 62–67 CrossRef CAS PubMed.
- B. Liu, K. Cheng, S. Nie, X. Zhao, H. Yu, J. Yu, A. Fujishima and K. Nakata, J. Phys. Chem. C, 2017, 121, 19836–19848 CrossRef CAS.
- S. K. Cushing, F. Meng, J. Zhang, B. Ding, C. K. Chen, C. J. Chen, R. S. Liu, A. D. Bristow, J. Bright, P. Zheng and N. Wu, ACS Catal., 2017, 7, 1742–1748 CrossRef CAS.
- W. Fang, M. Xing and J. Zhang, J. Photochem. Photobiol., C, 2017, 32, 21–39 CrossRef CAS.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2017, 9, 1523–1544 CrossRef CAS.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, S. Kment, R. Zboǐil and P. Schmuki, ACS Catal., 2019, 9, 345–364 CrossRef CAS PubMed.
- J. Wang, Z. Xu, C. Zhuang, H. Wang, X. Xu, T. Li and T. Peng, Dalton Trans., 2018, 47, 14556–14565 RSC.
- J. Liu, Q. Zhang, J. Yang, H. Ma, M. O. Tade, S. Wang and J. Liu, Chem. Commun., 2014, 50, 13971–13974 RSC.
- L. W. Zhang, H. B. Fu and Y. F. Zhu, Adv. Funct. Mater., 2008, 18, 2180–2189 CrossRef CAS.
- S. Lettieri, V. Gargiulo, D. K. Pallotti, G. Vitiello, P. Maddalena, M. Alfè and R. Marotta, Catal. Today, 2018, 315, 19–30 CrossRef CAS.
- C. Chen, M. Long, H. Zeng, W. Cai, B. Zhou, J. Zhang, Y. Wu, D. Ding and D. Wu, J. Mol. Catal. A: Chem., 2009, 314, 35–41 CrossRef CAS.
- K. Zhang, L. Wang, J. K. Kim, M. Ma, G. Veerappan, C. L. Lee, K. J. Kong, H. Lee and J. H. Park, Energy Environ. Sci., 2016, 9, 499–503 RSC.
- A. Aronne, M. Fantauzzi, C. Imparato, D. Atzei, L. De Stefano, G. D'Errico, F. Sannino, I. Rea, D. Pirozzi, B. Elsener, P. Pernice and A. Rossi, RSC Adv., 2017, 7, 2373–2381 RSC.
- Q. Wu, F. Huang, M. Zhao, J. Xu, J. Zhou and Y. Wang, Nano Energy, 2016, 24, 63–71 CrossRef CAS.
- J. Seo, H. Lee, H. J. Lee, M. S. Kim, S. W. Hong, J. Lee, K. Cho, W. Choi and C. Lee, Appl. Catal., B, 2018, 225, 487–495 CrossRef CAS.
- F. Sannino, P. Pernice, C. Imparato, A. Aronne, G. D'Errico, L. Minieri, M. Perfetti and D. Pirozzi, RSC Adv., 2015, 5, 93831–93839 RSC.
- J. Lee, Z. Li, L. Zhu, S. Xie and X. Cui, Appl. Catal., B, 2018, 224, 715–724 CrossRef CAS.
- V. G. Kessler, G. I. Spijksma, G. A. Seisenbaeva, S. Håkansson, D. H. A. Blank and H. J. M. Bouwmeester, J. Sol-Gel Sci. Technol., 2006, 40, 163–179 CrossRef CAS.
- D. Pirozzi, C. Imparato, G. D'Errico, G. Vitiello, A. Aronne and F. Sannino, J. Hazard. Mater., 2020, 387, 121716 CrossRef PubMed.
- C. Lettmann, K. Hildenbrand, H. Kisch, W. Macyk and W. F. Maier, Appl. Catal., B, 2001, 32, 215–227 CrossRef CAS.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- S. Claramunt, A. Varea, D. López-Díaz, M. M. Velázquez, A. Cornet and A. Cirera, J. Phys. Chem. C, 2015, 119, 10123–10129 CrossRef CAS.
- T. Lee, H. T. Bui, J. Yoo, M. Ra, S. H. Han, W. Kim and W. Kwon, ACS Appl. Mater. Interfaces, 2019, 11, 41196–41203 CrossRef CAS PubMed.
- W. Kiciński, M. Norek and M. Bystrzejewski, J. Phys. Chem. Solids, 2013, 74, 101–109 CrossRef.
- K. K. W. Jiang, G. Nadeau and K. Zaghib, Thermochim. Acta, 2000, 351, 85–93 CrossRef.
- M. Berrettoni, M. Ciabocco, M. Fantauzzi, M. Giorgetti, A. Rossi and E. Caponetti, RSC Adv., 2015, 5, 35435–35447 RSC.
- M. Carrus, M. Fantauzzi, F. Riboni, M. Makosch, A. Rossi, E. Selli and J. van Bokhoven, Appl. Catal., A, 2016, 519, 130–138 CrossRef CAS.
- G. Ou, Y. Xu, B. Wen, R. Lin, B. Ge, Y. Tang, Y. Liang, C. Yang, K. Huang, D. Zu, R. Yu, W. Chen, J. Li, H. Wu, L. M. Liu and Y. Li, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- X. Zou, J. Liu, J. Su, F. Zuo, J. Chen and P. Feng, Chem.–Eur. J., 2013, 19, 2866–2873 CrossRef CAS PubMed.
- C. Imparato, M. Fantauzzi, C. Passiu, I. Rea, C. Ricca, U. Aschauer, F. Sannino, G. D'Errico, L. De Stefano, A. Rossi and A. Aronne, J. Phys. Chem. C, 2019, 123, 11581–11590 CrossRef CAS.
- D. Atzei, M. Fantauzzi, A. Rossi, P. Fermo, A. Piazzalunga, G. Valli and R. Vecchi, Appl. Surf. Sci., 2014, 307, 120–128 CrossRef CAS.
- R. Liu, H. Li, L. Duan, H. Shen, Y. Zhang and X. Zhao, Ceram. Int., 2017, 43, 8648–8654 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara, S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem., 2000, 161, 205–212 CrossRef CAS.
- J. M. Cho, J. M. Seo, J. K. Lee, H. Zhang and R. Lamb, Phys. B, 2009, 404, 127–130 CrossRef CAS.
- N. Guskos, A. Guskos, G. Zolnierkiewicz, J. Typek, P. Berczynski, D. Dolat, B. Grzmil, B. Ohtani and A. W. Morawski, Mater. Chem. Phys., 2012, 136, 889–896 CrossRef CAS.
- H. Zhang, J. Cai, Y. Wang, M. Wu, M. Meng, Y. Tian, X. Li, J. Zhang, L. Zheng, Z. Jiang and J. Gong, Appl. Catal., B, 2018, 220, 126–136 CrossRef CAS.
- B. K. Vijayan, N. M. Dimitrijevic, D. Finkelstein-Shapiro, J. Wu and K. A. Gray, ACS Catal., 2012, 2, 223–229 CrossRef CAS.
- M. Alfè, D. Spasiano, V. Gargiulo, G. Vitiello, R. Di Capua and R. Marotta, Appl. Catal., A, 2014, 487, 91–99 CrossRef.
- D. Dvoranová, M. Mazúr, I. Papailias, T. Giannakopoulou, C. Trapalis and V. Brezová, Catalysts, 2018, 8, 47 CrossRef.
- E. Konstantinova, A. Minnekhanov, A. Beltiukov, V. Ivanov, A. J. Sutherland and O. Boytsova, New J. Chem., 2018, 42, 15184–15189 RSC.
- S. W. Sweeney, G. Roseman, C. P. Deming, N. Wang, T. A. Nguyen, G. L. Millhauser and S. Chen, Int. J. Hydrogen Energy, 2016, 41, 18005–18014 CrossRef CAS.
- S. O. Baumann, M. J. Elser, M. Auer, J. Bernardi, N. Husing and O. Diwald, Langmuir, 2011, 27, 1946–1953 CrossRef CAS PubMed.
- G. P. Papari, B. Silvestri, G. Vitiello, L. De Stefano, I. Rea, G. Luciani, A. Aronne and A. Andreone, J. Phys. Chem. C, 2017, 121, 16012–16020 CrossRef CAS.
- E. Hendry, M. Koeberg, B. O'Regan and M. Bonn, Nano Lett., 2006, 6, 755–759 CrossRef CAS PubMed.
- H. Němec, Z. Mics, M. Kempa, P. Kužel, O. Hayden, Y. Liu, T. Bein and D. Fattakhova-Rohlfing, J. Phys. Chem. C, 2011, 115, 6968–6974 CrossRef.
- M. Janus, M. Inagaki, B. Tryba, M. Toyoda and A. W. Morawski, Appl. Catal., B, 2006, 63, 272–276 CrossRef CAS.
- N. V. Smith, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 155106 CrossRef.
- P. Tiwana, P. Parkinson, M. B. Johnston, H. J. Snaith and L. M. Herz, J. Phys. Chem. C, 2010, 114, 1365–1371 CrossRef CAS.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS.
- B. Liu, L. M. Liu, X. F. Lang, H. Y. Wang, X. W. Lou and E. S. Aydil, Energy Environ. Sci., 2014, 7, 2592–2597 RSC.
- J. Tian, H. Gao, H. Deng, L. Sun, H. Kong, P. Yang and J. Chu, J. Alloys Compd., 2013, 581, 318–323 CrossRef CAS.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- D. K. Pallotti, L. Passoni, P. Maddalena, F. Di Fonzo and S. Lettieri, J. Phys. Chem. C, 2017, 121, 9011–9021 CrossRef CAS.
- S. Feizpoor, A. Habibi-Yangjeh, I. Ahadzadeh and K. Yubuta, Adv. Powder Technol., 2019, 30, 1183–1196 CrossRef CAS.
- M. Kobielusz, K. Pilarczyk, E. Świętek, K. Szaciłowski and W. Macyk, Catal. Today, 2018, 309, 35–42 CrossRef CAS.
- E. Świętek, K. Pilarczyk, J. Derdzińska, K. Szaciłowski and W. Macyk, Phys. Chem. Chem. Phys., 2013, 15, 14256 RSC.
- N. Liu, C. Schneider, D. Freitag, U. Venkatesan, V. R. R. Marthala, M. Hartmann, B. Winter, E. Spiecker, A. Osvet, E. M. Zolnhofer, K. Meyer, T. Nakajima, X. Zhou and P. Schmuki, Angew. Chem., Int. Ed., 2014, 53, 14201–14205 CrossRef CAS PubMed.
- Z. Zhao, X. Zhang, G. Zhang, Z. Liu, D. Qu, X. Miao, P. Feng and Z. Sun, Nano Res., 2015, 8, 4061–4071 CrossRef CAS.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Müller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309–3313 CrossRef CAS PubMed.
- C. Wang, X. Cai, Y. Chen, Z. Cheng, X. Luo, S. Mo, L. Jia, P. Lin and Z. Yang, Chem. Eng. J., 2017, 317, 522–532 CrossRef CAS.
- C. Wang, X. Cai, Y. Chen, Z. Cheng, X. Luo, S. Mo, L. Jia, R. Shu, P. Lin, Z. Yang, S. Sun, E. Pu and Y. Shen, Int. J. Hydrogen Energy, 2017, 42, 17063–17074 CrossRef CAS.
- R. M. Navarro Yerga, M. C. Álvarez Galván, F. del Valle, J. A. de la Mano and J. L. G. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef CAS PubMed.
- V. Vaiano, M. A. Lara, G. Iervolino, M. Matarangolo, J. A. Navio and M. C. Hidalgo, J. Photochem. Photobiol., A, 2018, 365, 52–59 CrossRef CAS.
- G. Iervolino, V. Vaiano, D. Sannino, L. Rizzo, A. Galluzzi, M. Polichetti, G. Pepe and P. Campiglia, Int. J. Hydrogen Energy, 2018, 43, 2184–2196 CrossRef CAS.
- L. Sun, J. Xie, Q. Li, F. Wang, X. Xi, L. Li, J. Wu, R. Shao and Z. Chen, J. Solid State Electrochem., 2019, 23, 803–810 CrossRef CAS.
- S. Chen, Y. Wang, J. Li, Z. Hu, H. Zhao, W. Xie and Z. Wei, Mater. Res. Bull., 2018, 98, 280–287 CrossRef CAS.
- M. Scheller, C. Jansen and M. Koch, Opt. Commun., 2009, 282, 1304–1306 CrossRef CAS.
- H. Looyenga, Mol. Phys., 1965, 9, 501–511 CrossRef CAS.
- J. Lloyd-Hughes and T. I. Jeon, J. Infrared, Millimeter, Terahertz Waves, 2012, 33, 871–925 CrossRef CAS.
- G. P. Papari, V. Gargiulo, M. Alfè, R. Di Capua, A. Pezzella and A. Andreone, J. Appl. Phys., 2017, 121, 145107 CrossRef.
- G. Papari, C. Koral, T. Hallam, G. S. Duesberg and A. Andreone, Materials, 2018, 11, 1613, DOI:10.3390/ma11091613.
- S. Lucyszyn, PIERS Online, 2007, 3, 554–559 CrossRef.
- V. Vaiano, M. Matarangolo, O. Sacco and D. Sannino, Appl. Catal., B, 2017, 209, 621–630 CrossRef CAS.
- J. Yu, Y. Hai and B. Cheng, J. Phys. Chem. C, 2011, 115, 4953–4958 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM images, XRD patterns, Raman spectra fitting data, additional XPS spectra and data, EPR data and power saturation curves, impedance plot from THz spectroscopy, UV-vis-DRS Tauc plot, photoactivity results. See DOI: 10.1039/d0ra01322f |
| This journal is © The Royal Society of Chemistry 2020 |