
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, crystal structures and, magnetic and photoluminescence properties of lanthanide-based metal–organic frameworks constructed with 2,5-dihydroxybenzene-1,4-dicarboxylic acid†
Sajjad Hussain *a,
Xuenian Chen*bc,
William T. A. Harrisond,
Saeed Ahmade,
Shahzad Shariff,
Jian Sug,
Shabbir Muhammadh and
Shujun Lib
*a,
Xuenian Chen*bc,
William T. A. Harrisond,
Saeed Ahmade,
Shahzad Shariff,
Jian Sug,
Shabbir Muhammadh and
Shujun Lib
aDepartment of Chemistry, Mohi-Ud-Din Islamic University, Nerian Sharif, Azad Jammu & Kashmir, Pakistan. E-mail: sajjaduet07@yahoo.com
bSchool of Chemistry and Chemical Engineering, Henan Key Laboratory of Boron Chemistry and Advanced Energy Materials, Henan Normal University, Xinxiang, 453007, China. E-mail: xnchen@htu.edu.cn
cCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, China
dDepartment of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, Scotland, UK
eDepartment of Chemistry, College of Sciences and Humanities, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia
fMaterials Chemistry Laboratory, Department of Chemistry, GC University, Lahore, 54000, Pakistan
gDepartment of Physics, Henan Normal University, Xinxiang, 453007, China
hDepartment of Physics, College of Science, King Khalid University, P. O. Box 9004, Abha 61413, Saudi Arabia
First published on 31st March 2020
Abstract
Four new lanthanide(III) coordination polymers (1–4) have been synthesized by using 2,5-dihydroxybenzene-1,4-dicarboxylate (Dhbdc, C8H6O62−) as a ligand and characterized by elemental analysis, infrared spectroscopy, TGA-DSC (Thermogravimetric Analysis-Differential Scanning Calorimetry) and single crystal X-ray diffraction studies. The isolated complexes include; [Ce(Dhbdc)(H2O)3Cl]n (1) and [Ln(Dhbdc)1.5(H2O)2]n·3n(C2H6O)·nH2O [Ln = Eu (2), Gd (3) and Tb (4)]. The structural analysis reveals that compound 1 is a two-dimensional polymer in which the cerium atoms are coordinated by six oxygen and two bridging chloride ions adopting a dodecahedral geometry. The compounds 2–4 are isomorphous and their extended structures consist of three-dimensional supramolecular frameworks encapsulating [001] channels occupied by the guest water and ethanol molecules. The metal atoms in 2–4 exhibit a square antiprism geometry. All these compounds have O–H⋯O hydrogen bonds originating from the coordinated water and solvent molecules that help to consolidate the structures. The compounds 1, 3 and 4 were investigated for magnetic properties and they exhibited weak antiferromagnetic coupling interactions between the lanthanides as per the Curie Weiss law. Anisotropic nature of 3 and 4 has been depicted as per magnetization versus field plots. Complex 4 behaves as a soft magnetic material as compared to 3. The luminescence studies indicate that compounds 2, 3 and 4 show ligand-centred fluorescence, which is significantly enhanced in the case of 4.
1. Introduction
The lanthanide-based metal–organic frameworks (MOFs) have drawn special consideration owing to their fascinating structures1–10 resulting from the flexible coordination environment of lanthanide(III) ions1,5,11 and due to their distinct magnetic and optical properties arising from the 4f electrons of lanthanides.4–6,11–16 These properties led to their prospective applications in catalysis,10,17,18 gas absorption,6,18,19 optoelectronic devices,12,13,20–25 sensors,7,26–28 contrast agents29 and molecular magnetism.5,15,16,30–32Several factors are important for the rational design of MOFs, such as the shape and binding mode of the ligands, the spacer linking the binding sites, reaction conditions and geometry of metal ions. The selection of a characteristic ligand plays a fundamental role in the formation of lanthanide-based coordination networks.2,3,10,16,33–35 Lanthanide(III) ions have high affinity towards hard bases, such as oxygen donors and in this respect multicarboxylate ligands, which possess hard base sites of high density, seem to be ideal ligands in the construction of MOFs.3–5,8,11,15,16,34–51 Among these ligands, the rigid carboxylates, benzene-1,4-dicarboxylic acid (Bdc-H2)4,34–38 and its derivatives with a centrosymmetric structure3,5,9,35,38–43 have drawn considerable attention because they possess various coordination modes such as, monodentate, bis-monodentate, bidentate, tridentate and chelating bridging; which favor the building of interesting structural patterns and packing frameworks. They can promote eminent pi stacking and help to sensitize the lanthanide centered fluorescence initiated from the ligand centered excited states.23,35
A number of lanthanide coordination compounds incorporating 2,5-dihydroxybenzene-1,4-dicarboxylate (Dhbdc) ligand have been structurally characterized and investigated for their magnetic properties.5,51–53 Their structures range from one dimensional chains to three dimensional networks. 2,5-Dihydroxybenzene-1,4-dicarboxylic acid (Dhbdc-H2) contains two sets of oxygen donors: carboxylate oxygen binds strongly to the metal center,5,51–54 whereas the hydroxyl groups may be engaged in coordination5,54 or remain uncoordinated and can form various hydrogen bonds to produce complicated supramolecular structures.5,51–54 Thus, it seems to be a suitable candidate to develop the extended lanthanide-based frameworks.
Keeping in view the continuing demand for the fabrication of novel lanthanide-based polymers to enlarge the available database and for exploration of their magnetic properties, we have used 2,5-dihydroxybenzene-1,4-dicarboxylic acid (Dhbdc-H2) as a carboxylate linker with a number of lanthanide salts to grow new functional lanthanide coordination polymers. As a result, four new mononuclear lanthanide(III) complexes (1–4) with two- or three-dimensional framework structures were prepared, which have the general formula; [Ce(Dhbdc)(H2O)3Cl]n (1) and [Ln(Dhbdc)1.5(H2O)2]n·3n(C2H6O)·nH2O [Ln = Eu (2), Gd (3) and Tb (4)]. The complexes (1–4) were characterized by thermogravimetric analysis (TGA), infrared (IR) spectroscopy, and their structures were determined by single crystal X-ray diffraction technique. The representative photoluminescent and magnetic properties were also investigated. In the previous such reports, the optical properties of the complexes were not considered.5,51–53
2. Experimental
2.1 Materials and measurements
The lanthanide salts (CeCl3·7H2O, EuCl3·6H2O, GdCl3·6H2O, TbCl3·6H2O) and ethanol were purchased from Sigma Aldrich, Johnson Matthey Company, USA. 2,5-Dihydroxybenzene-1,4-dicarboxylic acid was obtained from Alfa Aesar, China. The IR spectra were measured over the frequency range 4000–400 cm−1 using KBr pellets on PerkinElmer Spectrophotometer-100. TG-DSC analyses were performed on NETZSCH Simultaneous Thermal Analyzer 449 F5 from room temperature to 1000 °C in air with heating rate of 10 °C per minute. The percentage composition analysis for carbon and hydrogen were carried out on Vario Microcube Elementar. The photophysical properties were investigated through Fluorescence Spectrometer FLS 180 (Edinburgh Instruments). The magnetic susceptibilities were performed in the temperature range of 2–300 K by applying magnetic field of 2000 Oe (1 kOe = 7.96 × 104 A m−1) for 1, 3 and 4 as well as in the field of −90 to 90 kOe for samples 3 and 4 using a Quantum Design Dyna Cool PPMS, USA. The magnetic susceptibilities were revised for the diamagnetic corrections.55 The results are depicted as plots of χm, χm−1 and χmT versus T (χm = molar magnetic susceptibility) using Origin Pro software.56 The effective magnetic moment (μeff) was derived by using the formula μeff = 2.828 (χmT)1/2 in Bohr magneton units (μB).57,58 The Hirshfeld surfaces are generated by crystal explorer version 17.5 to analyze the interactions in the crystals structure.2.2 Crystal structures determination
Single crystal analyses of 1–4 were conducted on a Rigaku Super Nova CCD diffractometer using CuKα radiation (λ = 1.54184 Å) at 150(2) K. An empirical absorption correction was applied using SADABS.59 The crystal data were solved by direct methods with SHELXS-97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 and refined by full-matrix least-squares methods on F2 using SHELXL-2014.61 The C-bound H atoms were geometrically placed (C–H = 0.99 Å) and refined as riding atoms. The O-bound H atoms were located in difference maps and refined freely as riding atoms in their as-found relative positions or with gentle restraints. The constraint Uiso (H) = 1.2Ueq (carrier) was applied in most of the cases. The data collection and refinement details are summarized in Table S1.†
60 and refined by full-matrix least-squares methods on F2 using SHELXL-2014.61 The C-bound H atoms were geometrically placed (C–H = 0.99 Å) and refined as riding atoms. The O-bound H atoms were located in difference maps and refined freely as riding atoms in their as-found relative positions or with gentle restraints. The constraint Uiso (H) = 1.2Ueq (carrier) was applied in most of the cases. The data collection and refinement details are summarized in Table S1.†
2.3 Synthesis of complexes
Analysis for 1, C8H10O9ClCe: calculated (%): C 22.57; H 2.37. Found (%): C 22.91; H 2.51, yield ∼39%. IR peaks of 1 (cm−1, KBr): 3355 (s), 3054 (w), 1637 (vs.), 1505 (m), 1450 (m), 1363 (m), 1246 (m), 904 (w), 868 (w), 817 (m), 786 (s), 595 (w), 553 (m).
Analysis for 2, C18H30O15Eu: calculated (%): C 33.86; H 4.74. Found (%): C 33.91; H 4.82, yield ∼40%. IR data of 2 (cm−1, KBr): 3418 (s), 1568 (w), 1503 (m), 1452 (s), 1357 (m), 1237 (vs.), 1117 (w), 1040 (w), 902 (w), 872 (m), 817 (m), 812 (s), 787 (s) 786 (s), 605 (w), 549 (m).
Analysis for 3, C18H30O15Gd: calculated (%): C 33.58; H 4.70. Found (%): C 33.81; H 4.75, yield ∼45%. IR data of 3 (cm−1, KBr): 3393 (m), 1573 (m), 1503 (s), 1448 (vs.), 1361 (s), 1239 (vs.), 1116 (w), 1043 (w), 870 (m), 812 (w), 787 (m), 605 (w), 546 (m).
Analysis for 4, C18H30O15Tb: calculated (%): C 33.50; H 4.68. Found (%): C 33.65; H 4.77, yield ∼43%. IR data of 4 (cm−1, KBr): 3402 (m), 1570 (m), 1503 (s), 1448 (vs.), 1357 (s), 1239 (vs.), 1116 (w), 1041 (w), 871 (m), 812 (w), 787 (m), 605 (w), 548 (m).
3. Results and discussion
3.1 Synthesis
The compounds 1–4 were obtained by treating the respective metal chlorides with 2,5-dihydroxybenzene-1,4-dicarboxylic acid (Dhbdc-H2) in 1:1 or 1:2 molar ratio in water–ethanol mixture maintained at a pH of 4–5 by using 0.1 M NaOH solution. In all cases, the carboxylate groups were fully deprotonated, while the hydroxyl groups remained protonated. The presence of water and ethanol molecules within the structures demonstrate that the mixture solvent was crucial for the formation of complexes. Except for complex 1, the chloride ion did not participate in coordination to metal ions. The chloride binding was also not detected in the previously reported structures of lanthanide–Dhbdc complexes prepared using chloride salts.52 All four prepared complexes exist as mononuclear polymers. The X-ray analyses revealed that these compounds could be divided into two different families. Complex 1, belongs to family 1 and features a two-dimensional layer structure. Complexes 2–4 belong to family 2 and possess a three-dimensional framework. It is reported that mononuclear, dinuclear or non-cluster polynuclear complexes are usually formed at pH range 3–5,2,62 while the polynuclear lanthanide–hydroxido complexes are obtained under physiological pH conditions.63
3.2 IR analysis
The IR spectrum of complex 1 is displayed in Fig. 1, while those of complexes 2–4 are shown in Fig. S1–S3† respectively. The spectral pattern of all complexes is nearly similar because of having the same ligands, particularly for 2–4, which are isostructural. The IR spectra of the complexes show the characteristic absorptions for the asymmetric and symmetric stretches of carboxylate functions in the regions of 1600 and 1500, and near 1350 cm−1 respectively,5,53 which are not found in the spectrum of Dhbdc-H2. In the IR spectrum of Dhbdc-H2, these stretching frequencies are observed around 1700 and 1400 cm−1 respectively. Coordination involving the carboxylate groups for all the complexes is supported by the shifts of their bands towards lower wavenumbers as compared to free ligand. The absence of peaks at about 1700 cm−1 indicates that the carboxylic groups in the complexes are deprotonated. The broad signals due to O–H stretching vibrations of water and hydroxyl group appear at about 3420–3300 cm−1 in all these compounds. The broadening represents the presence of hydrogen bonding in the complexes, particularly in 2–4. The rocking vibrations of coordinated H2O (ρ(H2O)) were found near 800 cm−1. The medium intensity bands in the vicinity of 1240 cm−1 can be assigned to the carboxylate (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretches. The carboxylate bending vibrations, δ(O–C–O) are observed around 900 cm−1.49,50 The spectra also give characteristic ν(CC) modes of benzene ring near 1450 cm−1. The weak bands appearing around 3000 cm−1 are assigned to the C–H stretching vibration of the ligand. The C–H stretch of ethanol solvate in 2 was detected at 2873 cm−1.
O) stretches. The carboxylate bending vibrations, δ(O–C–O) are observed around 900 cm−1.49,50 The spectra also give characteristic ν(CC) modes of benzene ring near 1450 cm−1. The weak bands appearing around 3000 cm−1 are assigned to the C–H stretching vibration of the ligand. The C–H stretch of ethanol solvate in 2 was detected at 2873 cm−1.
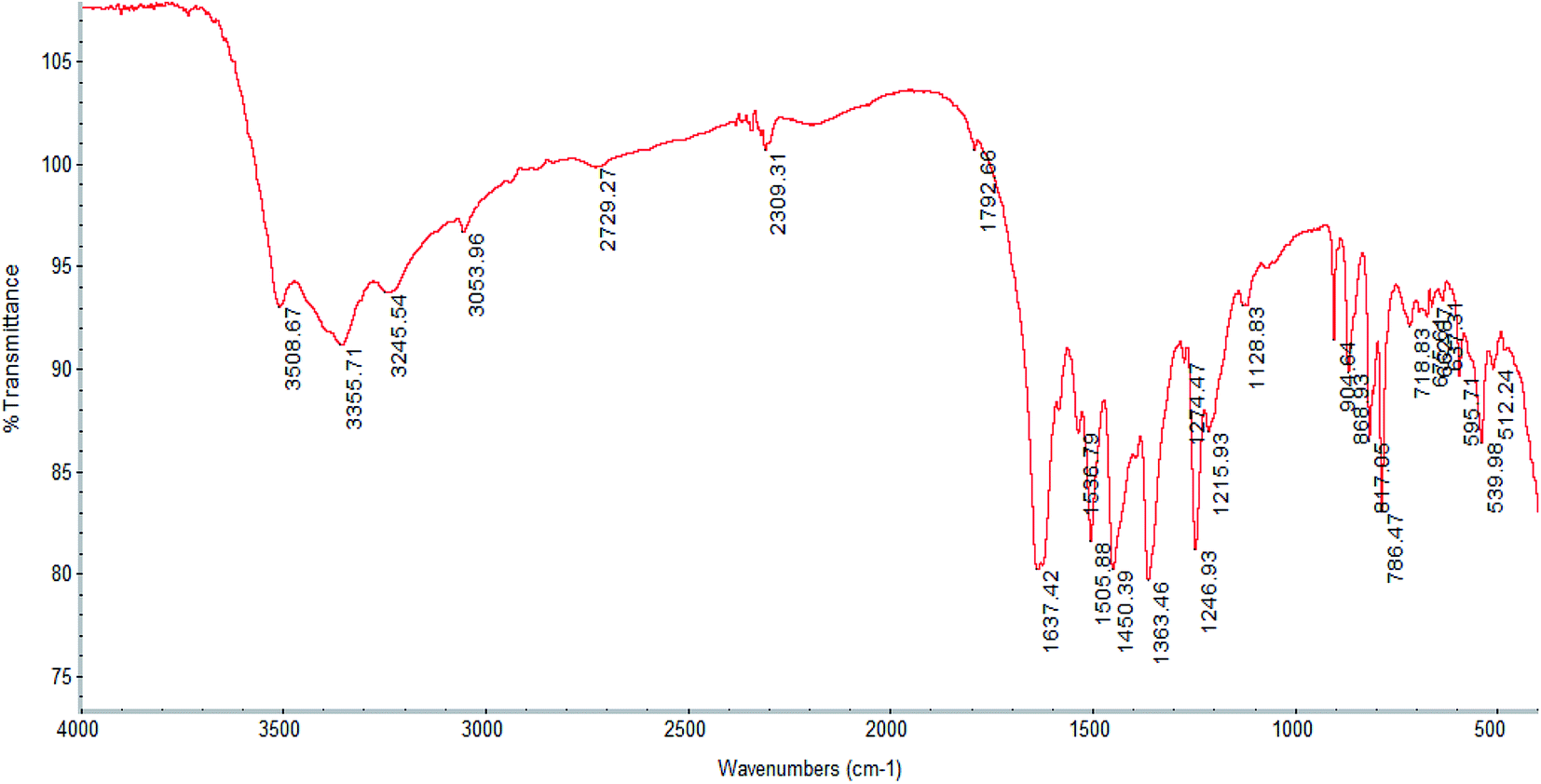 | ||
| Fig. 1 FTIR spectrum of [Ce(C8H4O6)Cl(H2O)3]n, 1. | ||
3.3 Thermal analysis
The TG-DSC plots of complex 1 is depicted in Fig. S4.† The first weight loss of 11.5% (calculated = 12.7%) that occurs at 100–160 °C, attributes to the removal of three coordinated aqua ligand. The loss of water is accompanied by an endothermic transition at 110 °C in the DSC curve. The dehydrated complex remains stable up to 225 °C. Above 225 °C, a continuous decomposition occurs until 825 °C in two overlapping steps. In the first stage two molecules of CO2 along with ½O2 and in the later, chloride ions are lost with a combined weight loss of 32.5% (theoretical 24.4 + 8.3 = 32.7%). The DSC plot shows an endothermic dip at 260 °C corresponds to the release of CO2. The total weight loss is 44% (calculated = 45.4%). The final residue probably corresponds to the presence of cerium–phenolate species with a percentage 56% (calculated = 55%).The compound 2 is selected as a representative from the isostructural series of 2, 3 and 4. The TG-DSC curves of 2 are depicted in Fig. S5.† The first weight loss of 24% occurs between 60 °C and 125 °C that corresponds to the non-coordinated water and ethanol molecules (calculated = 24.6%). The second weight loss of 5.8% (calculated = 5.4%) observed in the temperature range 130–190 °C is attributed to the removal of two aqua ligand. The next decomposition step falling in the region 190–925 °C indicates the removal of benzene–dicarboxylate ligands (with minus 1.5 oxygen atoms) with the weight loss of 43.2% (calculated = 42.3%) leaving behind ½Eu2O3 as a residue (experimental = 27%, calculated = 27.4%). The DSC curve depicts three endotherms at 100 °C, 175 °C and 320 °C corresponding to the above three weigh losses respectively.
3.4 Description of crystal structures
Complex 1, [Ce(Dhbdc)Cl(H2O)3]n shows a 2D layered framework, in which the asymmetric unit consists of one Ce(III) cation, a doubly-deprotonated Dhbdc2− (C8H4O62−) anion, a chloride ion and three coordinated water molecules as exhibited in Fig. 2. The important bond parameters are presented in Table 1. The Ce atom in 1 is coordinated by three oxygen atoms from three different Dhbdc ligands, two chloride ions and three water oxygen atoms. The resulting CeO6Cl2 coordination polyhedron approximates quite well to a dodecahedron as shown in Fig. 3. The Dhbdc2− anion adopts a monodentate (η1) coordination mode towards the Ce(III) ion from the C7/O1/O2 carboxylate group and bridging monodentate (μ2-η1,η1) to two symmetry related Ce(III) ions from the C8/O3/O4 group. The dihedral angles between C7/O1/O2 and C8/O3/O4 groups and the central C1–C4 benzene ring are 11.01 (9) and 13.30 (17)°, respectively. The OH groups at the 2- and 5-positions of the benzene ring form intramolecular O–H⋯O hydrogen bonds to adjacent carboxylate O atoms to generate S(6) rings. The Ce–O bond lengths vary from 2.402(3) to 2.573(3) Å (mean Ce–O = 2.478) and fall in the range of already reported Ce(III) carboxylate complexes.3,46,64–66 The Ce–O bond lengths for carboxyl groups are shorter than those of water molecules indicating their stronger binding. The mean Ce–Cl bond distance (2.878 Å) is considerably longer. The chloride ions are also connected with a neighboring metallic center, forming the four-membered rings as depicted in Fig. 2. Crystal symmetry results in a centrosymmetric Ce2Cl2 diamond with a Ce⋯Cei (i = −x, 1 − y, 2 − z) = 4.6668 (4) Å and Ce–Cl–Cei = 108.32 (4)°.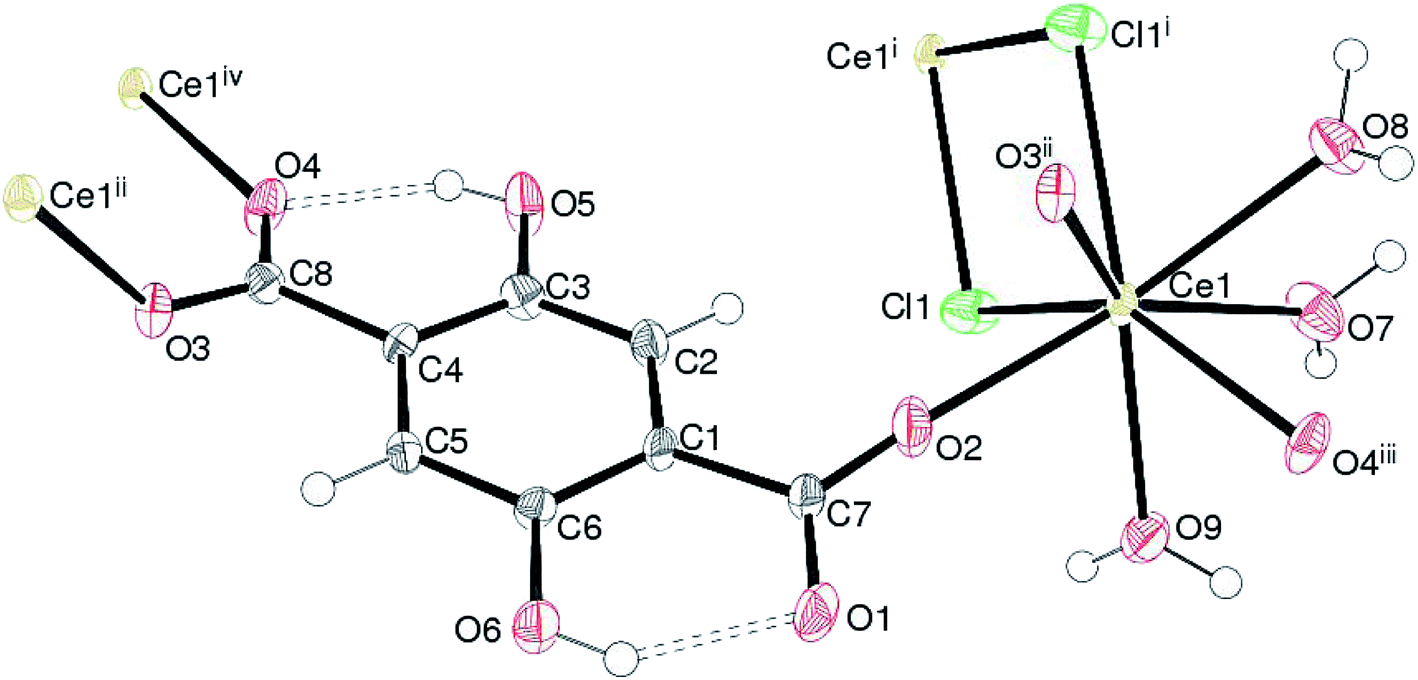 | ||
| Fig. 2 The asymmetric unit of 1 showing 50% displacement ellipsoids expanded to show adjacent Ce ions. The H-bonds are shown by dashed lines. Symmetry codes: (i) −x, 1 − y, 2 − z; (ii) −x, 1 − y, 1 − z; (iii) x, y − 1, z + 1; (iv) x, y + 1, z − 1. | ||
| 1 | |||
| Ce1–O2 | 2.402 (3) | Ce1–O3i | 2.451 (3) |
| Ce1–O4ii | 2.457 (3) | Ce1–O7 | 2.490 (3) |
| Ce1–O9 | 2.493 (3) | Ce1–O8 | 2.573 (3) |
| Ce1–Cl1iii | 2.8734 (11) | Ce1–Cl1 | 2.8832 (11) |
|
|||
| 2 | |||
| Eu1–O1 | 2.343 (2) | Eu1–O4i | 2.367 (3) |
| Eu1–O10 | 2.402 (3) | Eu1–O3ii | 2.407 (2) |
| Eu1–O11 | 2.421 (3) | Eu1–O2iii | 2.429 (3) |
| Eu1–O7 | 2.435 (3) | Eu1–O8 | 2.540 (3) |
|
|||
| 3 | |||
| Gd1–O1 | 2.3310 (19) | Gd1–O4i | 2.3607 (19) |
| Gd1–O3ii | 2.3936 (19) | Gd1–O10 | 2.395 (2) |
| Gd1–O11 | 2.416 (2) | Gd1–O2iii | 2.418 (2) |
| Gd1–O7 | 2.426 (2) | Gd1–O8 | 2.526 (2) |
|
|||
| 4 | |||
| Tb1–O1 | 2.310 (2) | Tb1–O4i | 2.341 (2) |
| Tb1–O3ii | 2.372 (2) | Tb1–O10 | 2.374 (2) |
| Tb1–O11 | 2.398 (2) | Tb1–O2iii | 2.402 (2) |
| Tb1–O7 | 2.408 (2) | Tb1–O8 | 2.517 (2) |
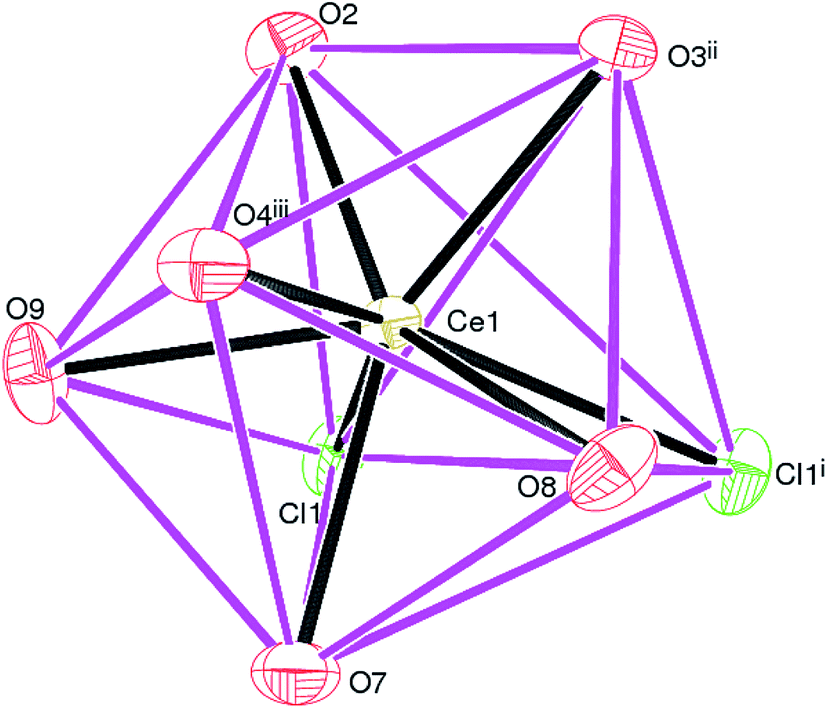 | ||
| Fig. 3 The CeO6Cl2 dodecahedron in 1; symmetry codes are same as given in Fig. 2. | ||
In the extended structure of 1, the combination of the edge-sharing (via the chloride-ion bridges) between the CeO6Cl2 polyhedra and the bridging Dhbdc ligands lead to (011) polymeric layers without any pores or free space. Within the crystal structure, the intra- and inter-layer O–H⋯O hydrogen bonding interactions connect the coordinated water molecules (O7W, O8W and O9W) of one polymer to the carboxylate oxygen atoms of an adjacent chain or layer. These H-bonds help to consolidate the 2D layers to a 3D network. The details of H-bonds are given in Table S2.†
The structural pattern of complex 1 is not similar to any of the previously reported structures of the lanthanide(III) complexes. However, its composition matches with two related hydrated compounds, {[Ln(malonate)(H2O)3Cl]·0.5H2O}n (Ln = Ce67 and Pr68). The metal ions in both complexes are 10-coordinated to three H2O molecules, six oxygen from carboxylates and a chloride ion. The carboxylate groups behave as symmetric chelating and monoatomic bridging ligands. In 1, the cerium atom exhibits a coordination number of eight and the carboxylate ligand adopts a monodentate (η1) and bridging monodentate (μ2-η1,η1) coordination mode towards Ce(III) ions. The chloride ions act as bridging ligands between the two metal atoms, while in the above two complexes they are bound to one metal ion only. The chloride ions in 1 along with the bridging carboxylate ligands crosslink the Ce(III) chains into a two-dimensional double layer structure. The structure of another cerium(III) complex of Dhbdc ligand, [Ce2(Dhbdc)3(dmf)4)]·dmf is also known.52 In each dinuclear unit, two nine-coordinated metal ions are bridged by four carboxylate groups of the four Dhbdc ligands. The binuclear complex exists as a 3D MOF.
Compounds 2, 3 and 4 crystallize in the monoclinic space group P21/n and are isostructural. The structure of 2 (containing europium) is discussed in details as a representative of the series with highlighting significant differences for 3 (gadolinium complex) and 4 (terbium complex), wherever appropriate. Selected bond distances are listed in Table 1. As illustrated in Fig. 4, the asymmetric unit of 2 consists of a Eu(III) ion, one and a half Dhbdc2− anion and two coordinated water molecules (O10 and O11). The structure is completed by one water molecule of crystallization (O12) and three uncoordinated ethanol solvent molecules: the overall formula being [[Eu(Dhbdc)1.5(H2O)2]·3(C2H6O)·H2O]n. The Eu(III) ion attains a coordination number of eight by binding with six carboxylate oxygen atoms from three Dhbdc ligands and two water molecules, adopting a square antiprism coordination environment (Fig. 5). The square faces of an EuO8 polyhedron are defined by O1/O2i/O3ii/O4iii ((i) = 1 − x, 1 − y, 1 − z; (ii) = ½ + x, ½ − y, ½ + z; (iii) = ½ − x, ½ + y, ½ − z) and O7/O8/O10/O11 sets of oxygen atoms. The dihedral angle between these is 3.89 (14)° and the metal ion is displaced from them by −1.0574 (13) and 1.5255 (15) Å, respectively. The Dhbdc ligands display two kinds of coordination modes; the C7/O1/O2 and C8/O3/O4 carboxylate groups are bis(monodentate) bridging (μ2-η1,η1), whereas C12/O7/O8 is η2 (O,O′) chelating. The dihedral angles for C7/O1/O2 and C8/O3/O4 with their attached aromatic ring are 6.6(6)° and 4.9(6)° respectively; while the equivalent value for C12/O7/O8 with respect to its ring is 2.3(6)°. In each case, the hydroxyl groups of the Dhbdc2− anions form intramolecular O–H⋯O hydrogen bonds to an adjacent carboxylate oxygen atom to generate an S(6) ring.
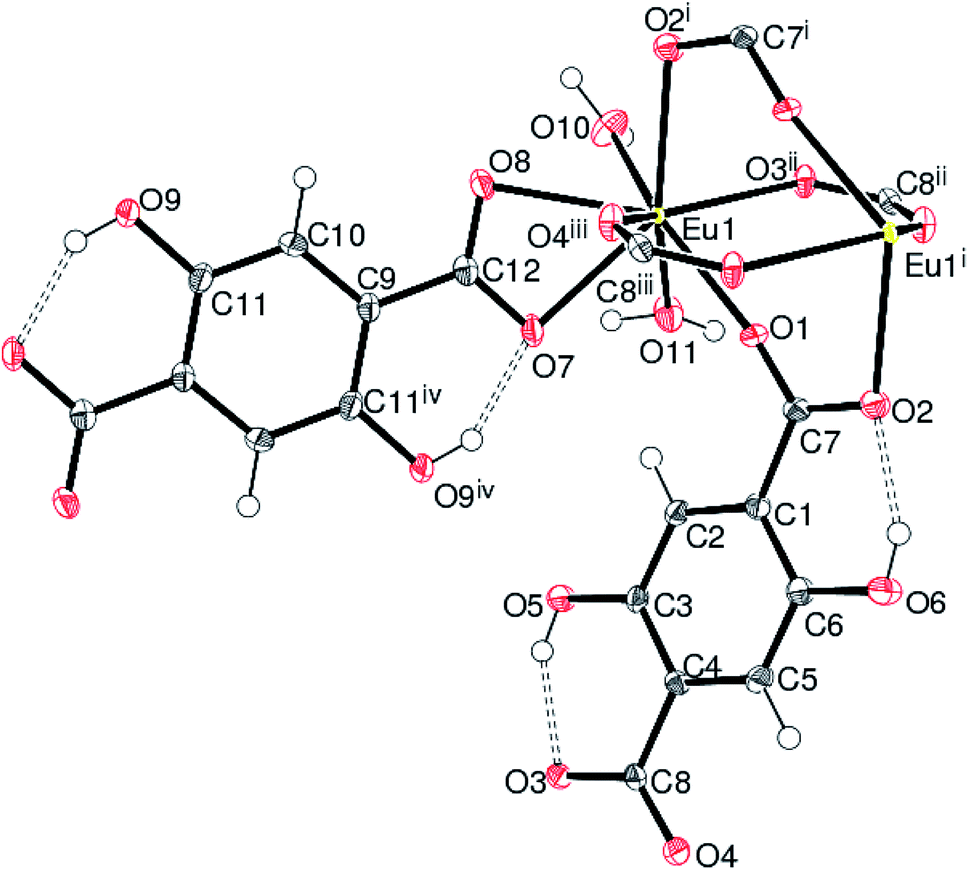 | ||
| Fig. 4 The asymmetric unit of 2 expanded to show the complete C9 ligand and adjacent Eu ion with extra-framework species omitted for clarity. Symmetry codes: (i) 1 − x, 1 − y, 1 − z; (ii) x + ½, ½ − y, z + ½; (iii) ½ − x, ½ + y, ½ − z; (iv) −x, 1 − y, 1 − z. | ||
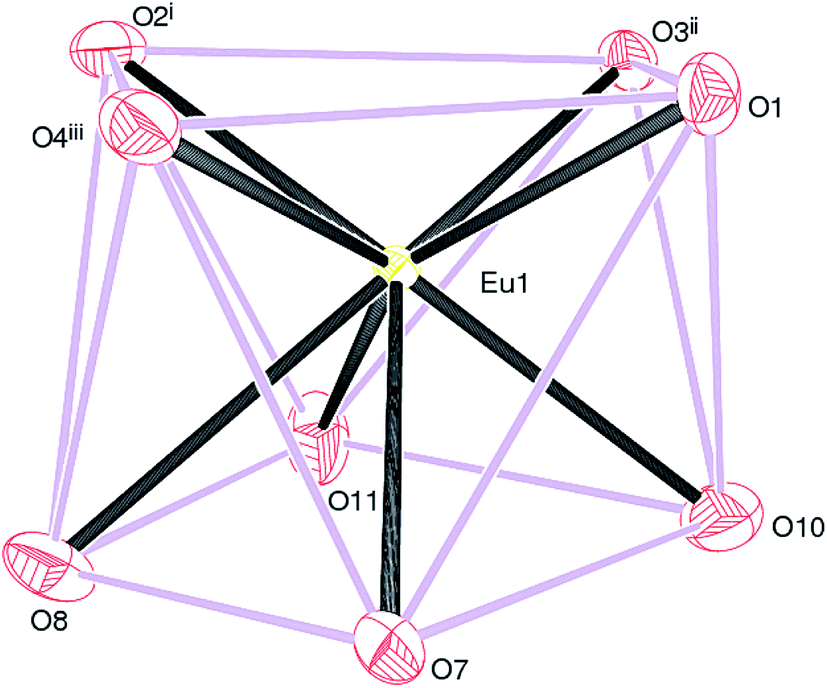 | ||
| Fig. 5 The EuO8 square antiprism in 2: symmetry codes as in Fig. 4. | ||
The mean Ln–O bond distances of 2.417, 2.408 and 2.389 Å in 2, 3 and 4, respectively, are close to those reported in other Ln(III) complexes with carboxylic acids.3–5,35,49,50,69–71 This bond length order is consistent with the expected trend based on the lanthanide contraction effect, which describes that the ionic radii decrease as the atom number increases, that lead average Ln–O bond lengths decrease accordingly.3,4,72 It may be noted that in each case, the Ln–O bonds for the chelating C12/O7/O8 carboxylate group are relatively longer due to formation of strained four-membered rings.
The (μ2-η1,η1) bridging carboxylate groups are bound to the adjacent metal atoms to form two 8-membered chelate rings. The pairs of metal atoms are separated with Eu⋯Eui distance (i = 1 − x, 1 − y, 1 − z) of 4.1317 (3) Å. The equivalent metal–metal separations for 3 and 4 are 4.1317 (4) and 4.1329 (4) Å, respectively. The dimers thus formed are extending through monodentate bridging carboxylate ligands into 2D sheets. Then the sheets are bridged by chelating dicarboxylate ligand on either side into a three-dimensional framework. The extended three-dimensional metal–organic framework encapsulates the [001] channels occupied by the guest water and ethanol molecules (Fig. 6). In each case, a network of O–H⋯O hydrogen bonds involving both the coordinated water molecules and ethanol molecules helps to stabilize the structure. The geometrical parameters of the hydrogen bonds involved in the structure are explained in Tables S3–S5.†
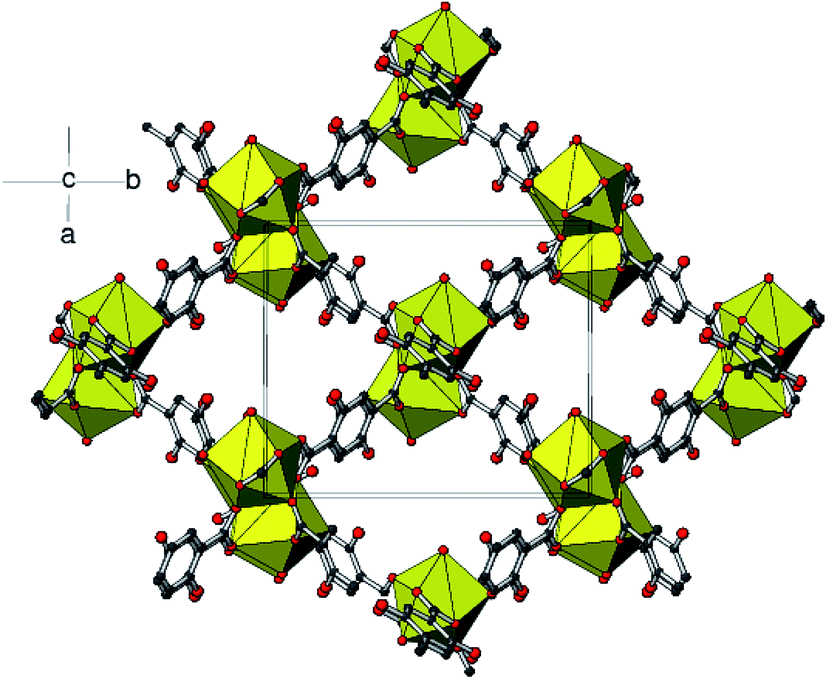 | ||
| Fig. 6 Polyhedral view of the metal–organic framework in 2 showing the formation of [001] channels. | ||
Crystal structures of two lanthanide complexes with the similar composition, [Ln(Dhbdc)1.5(H2O)2]n (Ln = La and Pr)5 have been reported but they are not isomorphous to the present series (complexes 2–4). In those two complexes metal ions are nine-coordinated and Dhbdc2− ion acts as a tridentate ligand coordinating to three Ln(III) ions with a monodentate and a bridging bis(monodentate) carboxylate groups, and one monodentate hydroxyl group.5 In the related dinuclear complexes, [Ln2(Dhbdc)3(dmf)4]·2dmf (Ln = La, Nd, Sm, Gd, and Er), the coordination modes are same as in 2.51 Later, another similar type of complexes with one dmf solvent of crystallization, [Ln2(Dhbdc)3(dmf)4)]·dmf (Ln = La, Ce, Pr, Nd and Gd), was reported, in which three different coordination modes are realized by the carboxylate groups of the Dhbdc linkers, described as: μ2-η1,η2, μ2-η1,η1, and μ2-η2.52 All these complexes yield the 3D MOFs. The crystal structures of analogous lanthanide-terephthalate (benzene-1,4-dicarboxylate, Bdc) compounds consist of 3D molecular frameworks and contain no crystallization water molecules; [Ln2(Bdc)3(H2O)4]n with Ln = La, Ce, Er, Tb.4,73 The Ln(III) ions are eight coordinated; bound with six O atoms from carboxylate and two O atoms from water molecules. The Bdc ligand exhibits only the μ2-η1,η1 bonding mode.
3.5 Magnetic properties
The magnetic behaviors of 1, 3 and 4 are represented in the forms of χm, χm−1 and χmT vs. T (χm molar magnetic susceptibility) are shown in Fig. S6, S7† and 7 respectively. The best fit for 1 gives the Curie constant C = 45.796 ± 4.404 cm3 K mol−1 and the Weiss constant θ = −36.325 K. At a temperature of 300 K, the product of χmT is 0.86596 cm3 K mol−1 with an effective magnetic moment μeff of 2.631 μB, which is a little higher than the calculated value of 2.543 μB for one independent Ce(III) ion (gJ = 0.85) in 2F5/2 ground state. At the lower end of temperature (2 K) the product of χmT decreases as the temperature is dropped to reach a final value of 0.34347 cm3 K mol−1 with an effective magnetic moment of 1.657 μB (Fig. S6†). For compound 3, the Curie constant and Weiss constant values are calculated as: C = 0.203 ± 0.022 cm3 K mol−1 and θ = −1.55 K respectively. At 300 K, χmT = 7.61 cm3 K mol−1 with an effective magnetic moment μeff of 7.77 μB, which to some extent is smaller than the calculated value of 7.94 μB per formula for one independent Gd(III) ion (gJ = 2.0) in 8S7/2 ground state. The χmT decreases to 7.41 cm3 K mol−1 at 2 K with an effective magnetic moment of 7.69 μB (Fig. S7†). For 4, Curie constant C = 0.992 ± 0.042 cm3 K mol−1 and the Weiss constant θ = −11.54 K were calculated. At 300 K, χmT = 11.24 cm3 K mol−1 gives an effective magnetic moment μeff of 9.50 μB, which is a little less than the calculated value of 9.72 μB per formula for one independent Tb(III) ion (gJ = 1.5) in 7F6 ground state. The product χmT decreases as the temperature is lowered to reach a final value of 5.38 cm3 K mol−1at 2 K with an effective magnetic moment of 6.56 μB (Fig. 7). Such behaviors and shapes of the plots of 1, 3, and 4 indicate weak antiferromagnetic exchange coupling interactions between the Ln(III) ions within complexes. The small difference between the calculated and experimental χmT value is due to the reduction in orbital angular momentum by the presence of ligands with respect to the free metal ion and the depopulation of excited MJ states of the Ln(III) ions.74,75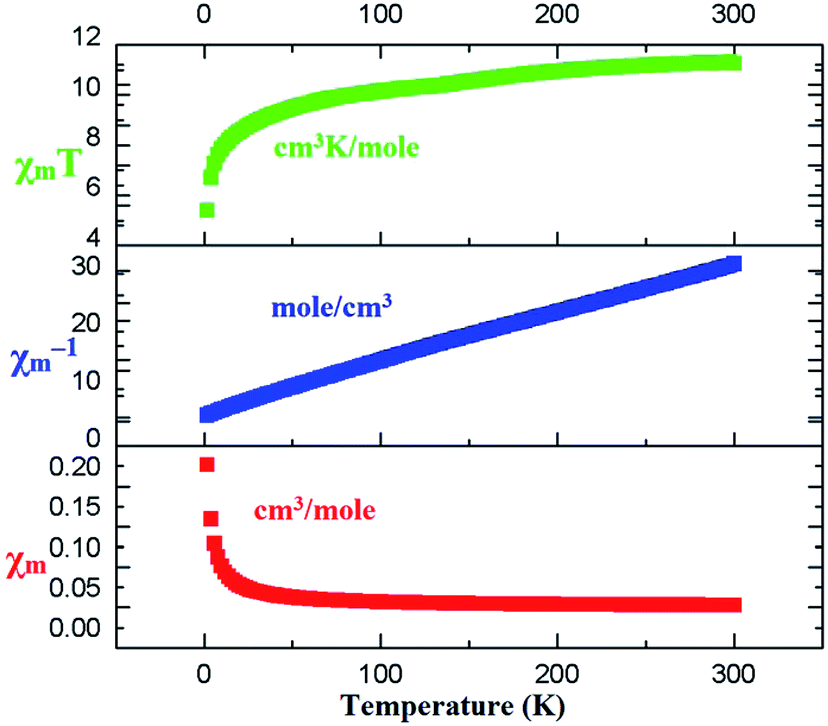 | ||
| Fig. 7 Temperature dependence of χm, χm−1 and χmT for 4 at an applied field of 2 KOe. | ||
Magnetization (M) data were measured against applied field (H) in the range of 0–90 kOe at 2–10 K and 6–50 K for complexes 3 and 4 as shown in Fig. 8 and S8† respectively. The curves show steady increase with increasing field (H). The plots of M versus H for complexes show nonsuperposition of curves, indicating magnetic anisotropic nature of 3 and 4.76–78 Magnetization versus field (−90 to 90 kOe) plots of 3 and 4 (Fig. 9) at 4 K is showing relatively more hysteresis loop for 3 and hence presence of more coercive forces. As the quantum regime is relatively fast for 4 as shown in Fig. 9, compound 4 does not exhibit hysteresis effect above 4 K and can be categorized as soft magnetic materials as compare with 3.79
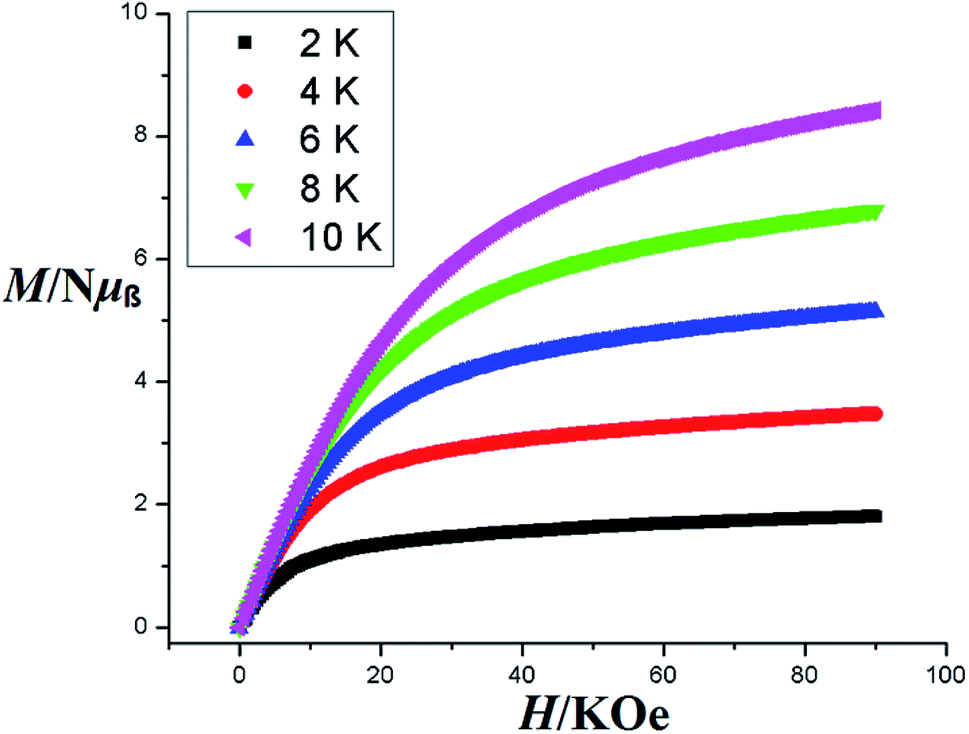 | ||
| Fig. 8 Magnetization M versus field H of 3 in the range 0–90 kOe. | ||
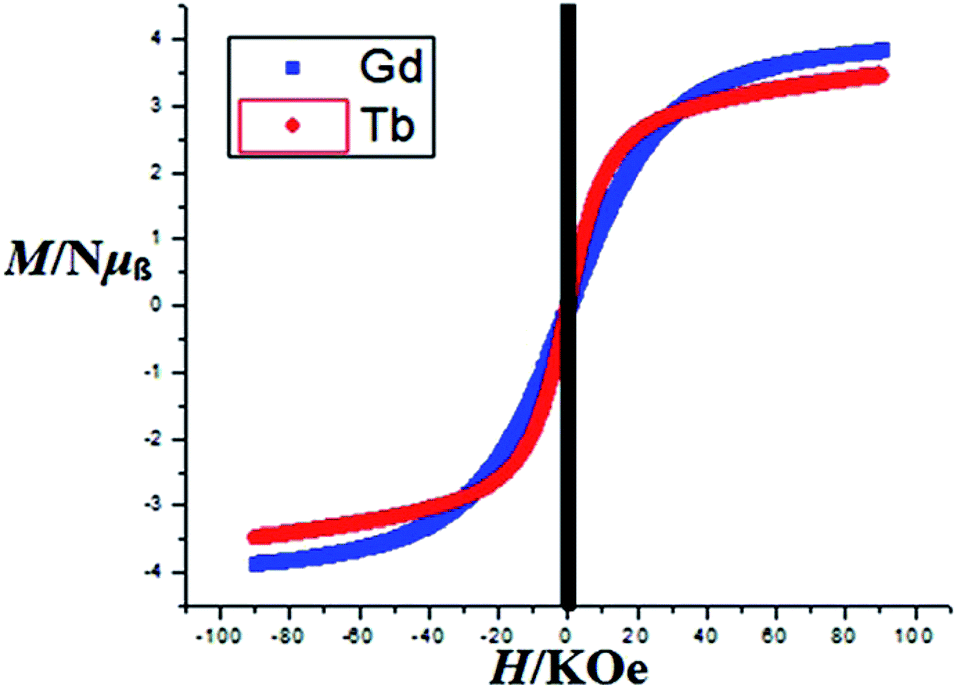 | ||
| Fig. 9 Magnetization M vs. H of 3 and 4 at 4 K. | ||
3.6 Photophysical properties
The conjugated ligands having aromatic ring are considered as sensitive reagents to modify the lanthanide fluorescence. The fluorescence emission of lanthanide complexes is due to adequate energy transfer from the lowest triplet state of ligand to the lanthanide ions' resonance energy level. The solid-state photoluminescent properties of lanthanide complexes of 2,5-dihydroxybenzene-1,4-dicarboxylate are not explored so far. The solid-state photoluminescence spectra of compounds 2, 3 and 4 were recorded, which exhibited ligand centered emission as shown in Fig. 10. Upon excitation at 340 nm the ligand gives an emission peak at ∼480 nm. The emission bands in all these complexes are almost similar to that in the free ligand, which demonstrate that they have acquired the emission wavelength of the ligand. The significant luminescence enhancement might be due to the reason that lanthanide ions are coordinated by the ligand that increase the rigidity and reduce the loss of energy by radiationless decay.80 The metal to ligand charge transfer might be another possible reason for the luminescence enhancement. The characteristics emission peaks of lanthanides are not observed in the spectra of complexes, which might be due to energy gap between the lowest triplet state of ligand to the resonance energy level of lanthanide ions.81 This energy barrier is significant that may not support the energy transfer process from the ligand triplet state to lanthanide ions. The luminescence quenching of coordinated water molecules is another possible reason to deprive these complexes from characteristics emission of lanthanides. The luminescence emission intensity of 4 (Tb) is more as compared to that of 2 (Eu) and 3 (Gd), which might be due to overlapping of 5D4 → 7F6 transitions originating from terbium. Thus, complex 4 efficiently enhances the luminescence emission intensity of the ligand and can be used as promising photoluminescent materials.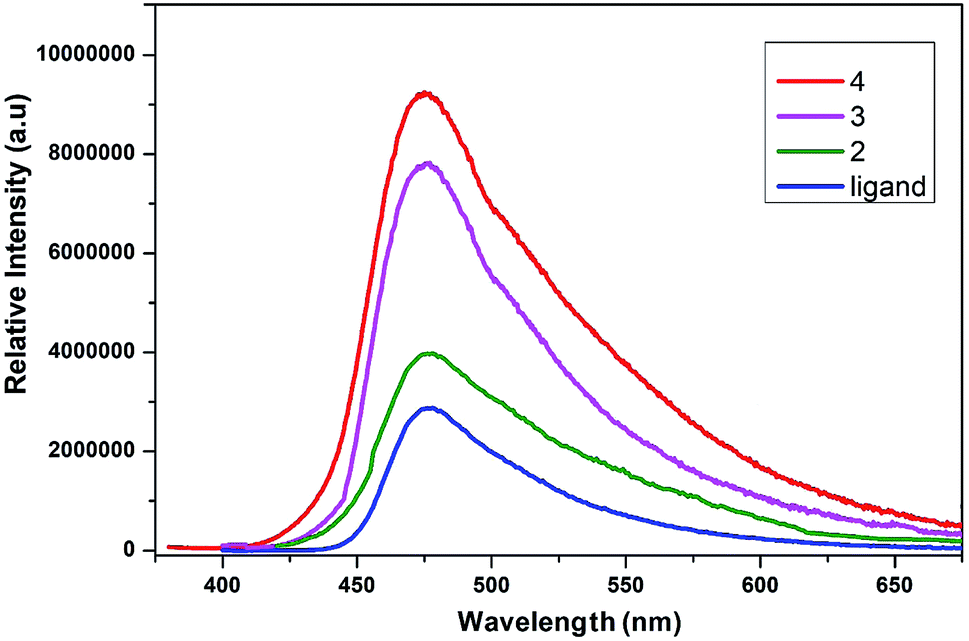 | ||
| Fig. 10 Emission spectra of the ligand and its complexes excited at 340 nm. | ||
3.7 Hirshfeld surface analysis
Over the past few decades, the study of crystal structures based on Hirshfeld surfaces have received significant importance. The Hirshfeld surface defines the space of a molecule inside the crystal for describing the crystal electron density into the molecular fragments. Usually, the other molecular surfaces are defined only by the molecule itself, whereas the Hirshfeld surface is well-defined by the molecule and the proximity of its nearest neighbors, and hence encodes the information about intermolecular interactions. In present investigation, we have chosen two representative compounds, 1 and 3 for the purpose of studying their Hirshfeld surfaces analysis. Based on Hirshfeld techniques, we have represented the dnorm, di and de maps as shown in the Fig. 11. Among the dnorm surfaces, the sharp red regions appear on the surface are due to the short interatomic contacts. The adjoining red blue shade signifies the weak C–H⋯O interactions over the surface. The di and de surfaces indicates that inner intermolecular contacts are more towards the groups bearing O atoms. The stronger intermolecular interactions are visible with red spots on Hirshfeld surfaces of compounds 1 and 3, which mostly indicate H⋯O, O⋯O and O⋯metal atom interactions. For example, in case of compound 1, two sharp peaks are visible in 2D figure print region for O atom interactions with all other atoms outside (Fig. S9 in ESI†). These sharp symmetric spikes constitute the prominent contributions of 29.5% for inside O atom and all outer atoms, while 20% for O atom outside all inside atoms at de + di ≈ 2.4 Å. The 2D figure print regions of compound 1 for all atomic interactions (100%) and specifically partial contributions for O atoms are shown in Fig. S9.†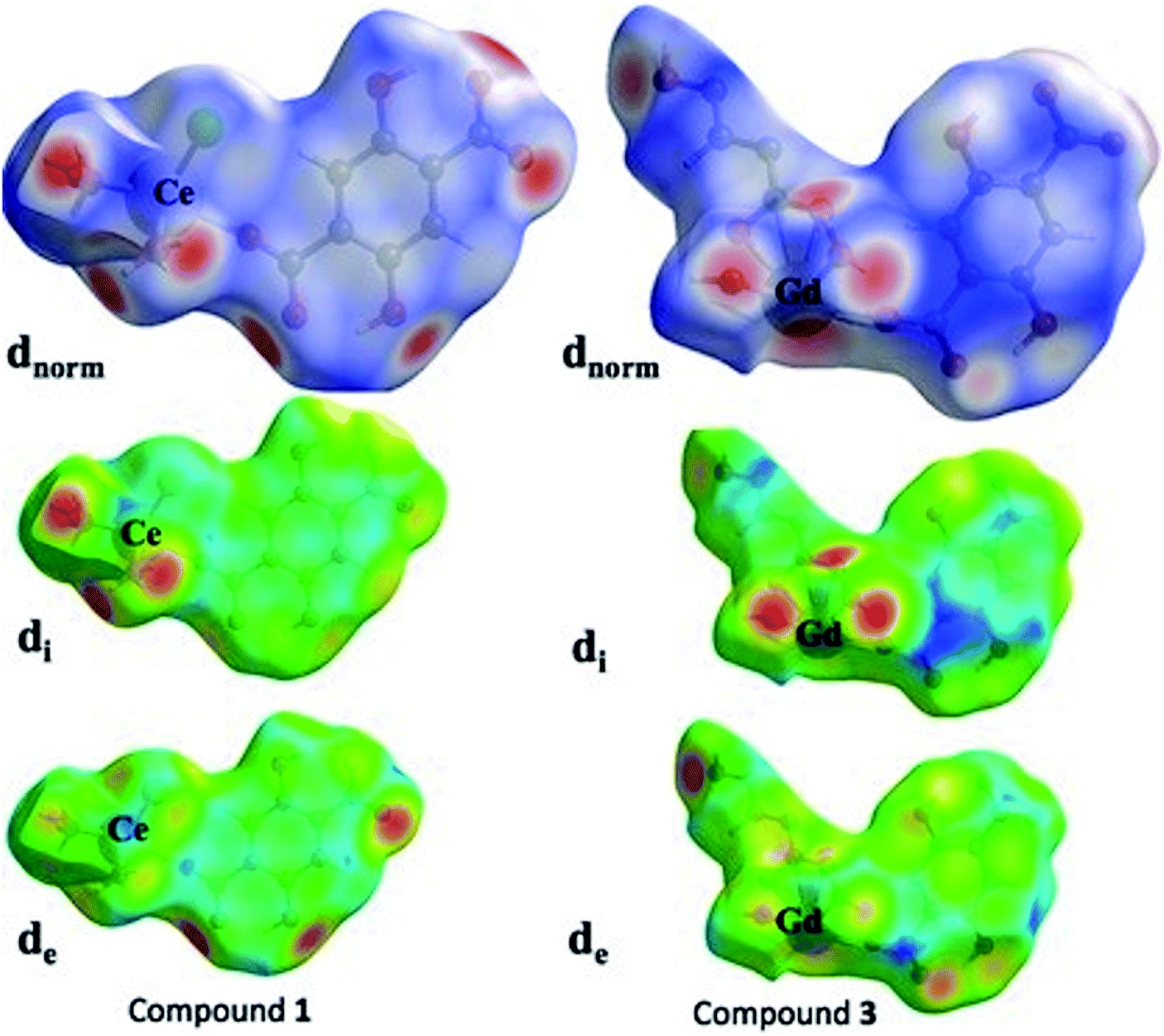 | ||
| Fig. 11 The Hirshfeld surfaces of 1 and 3 with their dnorm, di and de maps. | ||
4. Conclusions
In the present study, four new lanthanide coordination polymers based on 2,5-dihydroxybenzene-1,4-dicarboxylate ligand have been synthesized and structurally characterized by X-ray crystallography. The results indicate that 2,5-dihydroxybenzene-1,4-dicarboxylate ligand is a suitable candidate to generate a variety of coordination polymers through two carboxylic groups. The two hydroxyl groups on the benzene ring can also modify the final structure through hydrogen bonding. The Ce(III) complex (compound 1) has two-dimensional layered structure without any pores or free space, while those of Eu(III) (2), Gd(III) (3) and Tb(III) (4) have three dimensional frameworks with one dimensional channels propagating along [001] plane occupied by guest water and solvent molecules. The Curie Weiss law and Curie constant are satisfying for the weak antiferromagnetic interactions between lanthanide centers for complexes 1, 3 and 4. Separation of M versus H plots is an indication of magnetic anisotropic nature of 3 and 4. Complex 4 can be regarded as soft magnetic materials as compared to 3 due to less hysteresis effect at 4 K. The fluorescence studies show a significant enhancement of complexes' fluorescence when compared with the ligand's fluorescence. The Hirshfeld surface analysis for selected compounds (1 and 3) showed the maps of dnorm, di and de with stronger intermolecular interactions through the O atoms dominantly as visible on Hirshfeld surfaces. We believe that our study will be interesting for the scientific community engaged in designing lanthanide-based materials for the potential application in magnetic and photoluminescent devices.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Prof X. Chen acknowledges National Natural Science Foundation of China (Grants No. 21771057 and U1804253) for the financial support. S. Hussain is thankful to the Henan Normal University for postdoctoral support. The author from King Khalid University also acknowledge the financial and technical support of Deanship of Scientific Research (DSR) at King Khalid University of Saudi Arabi.References
- J.-C. G. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 CrossRef PubMed.
- I. M. Rosa, M. C. Costa, B. S. Vitto, L. Amorim, C. C. Correa, C. B. Pinheiro and A. C. Doriguetto, Cryst. Growth Des., 2016, 16, 1606–1616 CrossRef CAS.
- C.-K. Xia, W. Sun, Y.-Y. Min, K. Yang and Y.-L. Wu, Polyhedron, 2018, 141, 377–384 CrossRef CAS.
- C. Daiguebonne, N. Kerbellec, O. Guillou, J.-C. Bünzli, F. Gumy, L. Catala, T. Mallah, N. Audebrand, Y. Gérault and K. Bernot, Inorg. Chem., 2008, 47, 3700–3708 CrossRef CAS PubMed.
- Y.-L. Wang, Y.-L. Jiang, Z.-J. Xiahou, J.-H. Fu and Q.-Y. Liu, Dalton Trans., 2012, 41, 11428–11437 RSC.
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273, 139–164 CrossRef.
- C. M. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512–2527 CrossRef CAS.
- Z. Min, M. A. Singh-Wilmot, C. L. Cahill, M. Andrews and R. Taylor, Eur. J. Inorg. Chem., 2012, 2012, 4419–4426 CrossRef CAS.
- Z. Wang, Y.-H. Xing, C.-G. Wang, L.-X. Sun, J. Zhang, M.-F. Ge and S.-Y. Niu, CrystEngComm, 2010, 12, 762–773 RSC.
- C. Pagis, M. Ferbinteanu, G. Rothenberg and S. Tanase, ACS Catal., 2016, 6, 6063–6072 CrossRef CAS.
- A.-H. Yang, H.-L. Gao, J.-Z. Cui and B. Zhao, CrystEngComm, 2011, 13, 1870–1876 RSC.
- M. H. Werts, Sci. Prog., 2005, 88, 101–131 CrossRef CAS PubMed.
- J. Heine and K. Müller-Buschbaum, Chem. Soc. Rev., 2013, 42, 9232–9242 RSC.
- R.-C. Gao, F.-S. Guo, N.-N. Bai, Y.-L. Wu, F. Yang, J.-Y. Liang, Z.-J. Li and Y.-Y. Wang, Inorg. Chem., 2016, 55, 11323–11330 CrossRef CAS PubMed.
- Y.-C. Ou, X. Gao, Y. Zhou, Y.-C. Chen, L.-F. Wang, J.-Z. Wu and M.-L. Tong, Cryst. Growth Des., 2016, 16, 946–952 CrossRef CAS.
- Z.-H. Zhang, T.-A. Okamura, Y. Hasegawa, H. Kawaguchi, L.-Y. Kong, W.-Y. Sun and N. Ueyama, Inorg. Chem., 2005, 44, 6219–6227 CrossRef CAS PubMed.
- M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2210 CrossRef CAS PubMed.
- H. He, H. Ma, D. Sun, L. Zhang, R. Wang and D. Sun, Cryst. Growth Des., 2013, 13, 3154–3161 CrossRef CAS.
- H. He, D. Yuan, H. Ma, D. Sun, G. Zhang and H.-C. Zhou, Inorg. Chem., 2010, 49, 7605–7607 CrossRef CAS PubMed.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- S. Xiang, D.-X. Bao, J. Wang, Y.-C. Li and X.-Q. Zhao, J. Lumin., 2017, 186, 273–282 CrossRef CAS.
- J.-C. G. Buenzli, Coord. Chem. Rev., 2015, 293, 19–47 CrossRef.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- H. A. Azab, A. Duerkop, Z. Anwar, B. H. Hussein, M. A. Rizk and T. Amin, Anal. Chim. Acta, 2013, 759, 81–91 CrossRef CAS PubMed.
- R. F. Bogale, Y. Chen, J. Ye, S. Zhang, Y. Li, X. Liu, T. Zheng, A. Rauf and G. Ning, New J. Chem., 2017, 41, 12713–12720 RSC.
- R. F. Bogale, J. Ye, Y. Sun, T. Sun, S. Zhang, A. Rauf, C. Hang, P. Tian and G. Ning, Dalton Trans., 2016, 45, 11137–11144 RSC.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369–2388 CrossRef CAS PubMed.
- W.-H. Zhu, S. Li, C. Gao, X. Xiong, Y. Zhang, L. Liu, A. K. Powell and S. Gao, Dalton Trans., 2016, 45, 4614–4621 RSC.
- J. Tian, B. Li, X. Zhang, X. Li, X. Li and J. Zhang, Dalton Trans., 2013, 42, 8504–8511 RSC.
- C.-G. Wang, Y.-H. Xing, Z.-P. Li, J. Li, X.-Q. Zeng, M.-F. Ge and S.-Y. Niu, J. Mol. Struct., 2009, 921, 126–131 CrossRef CAS.
- J.-L. Wang, K.-L. Hou, F.-Y. Bai, Y.-H. Xing and Z. Shi, Struct. Chem., 2012, 23, 275–285 CrossRef CAS.
- C. Daiguebonne, N. Kerbellec, K. Bernot, Y. Gérault, A. Deluzet and O. Guillou, Inorg. Chem., 2006, 45, 5399–5406 CrossRef CAS PubMed.
- F. Le Natur, G. Calvez, S. Freslon, C. Daiguebonne, K. Bernot and O. Guillou, J. Mol. Struct., 2015, 1086, 34–42 CrossRef CAS.
- A. Thirumurugan and S. Natarajan, Eur. J. Inorg. Chem., 2004, 2004, 762–770 CrossRef.
- L. Canadillas-Delgado, J. Pasán, O. Fabelo, M. Hernández-Molina, F. Lloret, M. Julve and C. Ruiz-Pérez, Inorg. Chem., 2006, 45, 10585–10594 CrossRef CAS.
- W.-M. Zhang, T.-F. Dong, Z.-M. Lu and Y. Ding, Z. Kristallogr. - New Cryst. Struct., 2013, 228, 171–172 CrossRef CAS.
- B. Yan, Y. Bai and Z. Chen, J. Mol. Struct., 2005, 741, 141–147 CrossRef CAS.
- Y. Luo, K. Bernot, G. Calvez, S. Freslon, C. Daiguebonne, O. Guillou, N. Kerbellec and T. Roisnel, CrystEngComm, 2013, 15, 1882–1896 RSC.
- L. Cañadillas-Delgado, J. Pasán, O. Fabelo, M. Julve, F. Lloret and C. Ruiz-Pérez, Polyhedron, 2013, 52, 321–332 CrossRef.
- L. Cañadillas-Delgado, J. Pasán, O. Fabelo, M. Hernández-Molina, F. Lloret, M. Julve and C. Ruiz-Pérez, Inorg. Chem., 2006, 45, 10585–10594 CrossRef PubMed.
- T. K. Maji, G. Mostafa, H.-C. Chang and S. Kitagawa, Chem. Commun., 2005, 2436–2438 RSC.
- J.-J. Yang, X.-Y. Yu, Y.-H. Luo, H. Zhang and W.-P. Gao, Inorg. Chem. Commun., 2015, 61, 16–20 CrossRef CAS.
- S. C. Manna, E. Zangrando, A. Bencini, C. Benelli and N. R. Chaudhuri, Inorg. Chem., 2006, 45, 9114–9122 CrossRef CAS PubMed.
- F. S. Delgado, P. Lorenzo-Luís, J. Pasán, L. Cañadillas-Delgado, O. Fabelo, M. Hernández-Molina, A. D. Lozano-Gorrín, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2016, 18, 7831–7842 RSC.
- S. Hussain, X. Chen, W. T. Harrison, M. R. J. Elsegood, S. Ahmad, S. Li and D. Awoyelu, Front. Chem., 2019, 7, 728 CrossRef PubMed.
- S. Hussain, X. Chen, W. T. Harrison, S. Ahmad, M. Elsegood and S. Muhammad, Front. Chem., 2019, 7, 260 CrossRef CAS PubMed.
- Y.-L. Wang, Y.-L. Jiang, Q.-Y. Liu, Y.-X. Tan, J.-J. Wei and J. Zhang, CrystEngComm, 2011, 13, 4981–4987 RSC.
- S. Nayak, H. P. Nayek, C. Pietzonka, G. Novitchi and S. Dehnen, J. Mol. Struct., 2011, 1004, 82–87 CrossRef CAS.
- K. Gurunatha, S. Mohapatra, P. Suchetan and T. K. Maji, Cryst. Growth Des., 2009, 9, 3844–3847 CrossRef CAS.
- Q. Gao, F.-L. Jiang, M.-Y. Wu, Y.-G. Huang, W. Wei, Q.-F. Zhang and M.-C. Hong, Aust. J. Chem., 2010, 63, 286–292 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- OriginPro 8 SRO Software (version 8.0724 (B724)), Northampton, MA (USA), 2007, available online via www.originLab.com Search PubMed.
- S. Blundell, Condensed Matter Physics, Oxford Series Publications, 2001 Search PubMed.
- B. Want, F. Ahmad and P. Kotru, J. Alloys Compd., 2008, 448, L5–L6 CrossRef CAS.
- G. Sheldrick, SADABS v. 2014/5, Bruker AXC Inc., Madison, WI, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- D. Weng, X. Zheng, L. Li, W. Yang and L. Jin, Dalton Trans., 2007, 4822–4828 RSC.
- Y. Li, J.-W. Yu, Z.-Y. Liu, E.-C. Yang and X.-J. Zhao, Inorg. Chem., 2015, 54, 153–160 CrossRef CAS PubMed.
- S. Hussain, I. U. Khan, M. R. Elsegood, N. Jabeen, M. N. Tahir, S. Ahmad and S. Mutahir, Polyhedron, 2018, 151, 452–457 CrossRef CAS.
- S. Hussain, I. U. Khan, W. T. Harrison, M. N. Tahir and S. Ahmad, J. Struct. Chem., 2015, 56, 126–133 CrossRef CAS.
- N. Rahahlia, B. Benmerad, A. Guehria-Laïdoudi, S. Dahaoui and C. Lecomte, J. Mol. Struct., 2007, 833, 42–48 CrossRef CAS.
- P. Silva, J. A. Fernandes and F. A. Almeida Paz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1514–m1515 CrossRef CAS PubMed.
- K. E. Chrysomallidou, S. P. Perlepes, A. Terzis and C. P. Raptopoulou, Polyhedron, 2010, 29, 3118–3124 CrossRef CAS.
- G.-H. Cui, J.-R. Li, R.-H. Zhang and X.-H. Bu, J. Mol. Struct., 2005, 740, 187–191 CrossRef CAS.
- M. G. Lahoud, R. C. Frem, L. F. Marques, G. Arroyos, P. Brandão, R. A. Ferreira and L. D. Carlos, J. Solid State Chem., 2017, 253, 176–183 CrossRef CAS.
- T. Zhu, K. Ikarashi, T. Ishigaki, K. Uematsu, K. Toda, H. Okawa and M. Sato, Inorg. Chim. Acta., 2009, 362, 3407–3414 CrossRef CAS.
- J.-C. G. Bünzli, J. Coord. Chem., 2014, 67, 3706–3733 CrossRef.
- T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651–1657 CrossRef CAS.
- J. E. Huheey, E. A. Keiter, R. L. Keiter and O. K. Medhi, Inorg. Chem.: Principles of Structure and Reactivity, Pearson Education India, 2006 Search PubMed.
- B. Hussain, D. Savard, T. J. Burchell, W. Wernsdorfer and M. Murugesu, Chem. Commun., 2009, 1100–1102 RSC.
- L. Zhao, S. Xue and J. Tang, Inorg. Chem., 2012, 51, 5994–5996 CrossRef CAS PubMed.
- I. A. Gass, B. Moubaraki, S. K. Langley, S. R. Batten and K. S. Murray, Chem. Commun., 2012, 48, 2089–2091 RSC.
- L. Zhang, S. Lu, C. Zhang, C. Du and H. Hou, CrystEngComm, 2015, 17, 846–855 RSC.
- Y.-N. Guo, G.-F. Xu, P. Gamez, L. Zhao, S.-Y. Lin, R. Deng, J. Tang and H.-J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538–8539 CrossRef CAS PubMed.
- Y.-X. Chi, Y.-J. Liu, Y. Li, R. Wang, J. Jin, G.-N. Zhang and S.-Y. Niu, J. Mol. Struct., 2012, 1018, 122–130 CrossRef CAS.
- X. Guo, G. Zhu, Q. Fang, M. Xue, G. Tian, J. Sun, X. Li and S. Qiu, Inorg. Chem., 2005, 44, 3850–3855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The IR spectra, TG-DSC plot, crystal data tables, H-bond parameters, and computational details. CCDC 1937367–1937370 contain the crystallographic data for 1–4 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01294g |
| This journal is © The Royal Society of Chemistry 2020 |