
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpecific ion effects of incomplete ion-exchange by electric field-induced ion polarization
Wei Du ab,
Xinmin Liub,
Rui Tianb,
Rui Lib,
Wuquan Dingc and
Hang Li*b
ab,
Xinmin Liub,
Rui Tianb,
Rui Lib,
Wuquan Dingc and
Hang Li*b
aCollege of Natural Resources and Environment, Northwest A&F University, Yangling 712100, P. R. China
bChongqing Key Laboratory of Soil Multi-Scale Interfacial Processes, College of Resources and Environment, Southwest University, Chongqing 400715, P. R. China. E-mail: lihangswu@163.com
cChongqing Key Laboratory of Environmental Materials & Remediation Technologies, Chongqing University of Arts and Science, Chongqing 402168, P. R. China
First published on 17th April 2020
Abstract
Incomplete ion-exchange results from ion interfacial reactions portray a particular scenario of interactions between ions and charged surfaces. In this study, the constant flow method was adopted to study the incomplete ion-exchange state of mono-valent cation adsorption of X+ (X+ = Cs+, Na+ and Li+) in X+/K+ exchange at the montmorillonite particle surface. The pronounced incomplete ion-exchange state and strong specific ion effects were experimentally observed. Further research found that the disparity in the activation energies for different ion exchange systems caused by electric field-induced ion polarization was responsible for the observations. Thus, a theoretical description of the incomplete ion-exchange state by taking the ion polarization into account was established and verified. Applicable new approaches to measuring the cationic diffusion coefficient in heterogeneously charged systems and the cationic actual diffuse depth in the electric double layer were also derived from the theory.
Introduction
Ion exchange adsorption is a significant physicochemical process that widely occurs at two-phase interfaces. Studies on the thermodynamics and kinetics in ion adsorption have received considerable attention in the past decades.1–3 In recent years, however, specific ion effects have opened up new fields of vision in ion-surface interactions, and the rapidly accumulating new concepts have stimulated physical chemists, biological chemists as well as colloidal chemists to recheck the availability of the well-known classical theories in describing ion-surface interactions in colloidal systems.4,5According to the law of electrical neutrality, when ion-exchange adsorption reaches an ideal equilibrium state under given conditions, the accumulated counterion amounts near the charged surface should be equal to the surface charge numbers of the adsorbent. In other words, the previously adsorbed counterions for compensating surface net charges will be entirely replaced by other exchanging ions. However, many studies have demonstrated that achieving complete ion-exchange is challenging; inversely, the pronounced incomplete ion-exchange is generally observed.6–13 Currently, the reasons for incomplete ion-exchange are as follows: the slow migration of ions arising from the immobilization of edge-interlayers or the collapse of frayed edges in clay mineral,8,9,13,14 the incompatibility of the cation size with the space available in specific sites of the materials,15 and insufficient external disturbances lead to the blocking of the diffusion of exchange ions.12,16 Although these attempts to make sense of incomplete ion exchange, systematic and theoretical explanations regarding specific ion effects are still relatively scarce.
The kinetic study is generally known as the most common method for investigating the ion-exchange adsorption process for discovering its physical mechanisms; therefore, the kinetic approach would be possibly more favorable for exploring the mechanisms and the origins of the incomplete ion-exchange state. Using kinetic and reactive-transport modeling, research from E. Tertre et al. has confirmed that incomplete Na+/Ca2+ exchange can be attributed to the kinetic origin of the too-short residence time for exchanging Ca2+ near the solid surface in comparison with the half-ion-exchange reaction time (a few seconds or minutes).16 However, the most commonly applied kinetic models for investigating the ion exchange adsorption are empirically or directly cited from simple reaction systems.17 For the exchange adsorption of cations in surface-charged systems, there is the co-existence of multi-processes, including series transport processes and exchange processes,18,19 and multi-driving forces, including electrostatic forces, induction forces as well as dispersion forces, etc.20 Therefore, obtaining insightful information from ionic adsorption data through the applications of those kinetic models is probably untenable.
By employing the miscible displacement experiment or constant flow method, some researchers have established new kinetic models based on the detailed theoretical analysis of cationic exchange adsorption in heterogeneously charged systems, for which the effects of electrostatic adsorption and/or other non-electrostatic force adsorption could be taken into account.21 According to these theories, cationic adsorption exhibits two kinetic processes and can be described by two rate equations, namely, the zero-order and first-order rate equations. Most importantly, these theories confirmed that the kinetic process should be characterized by the zero-order rate equation in the presence of strong force adsorption (either electrostatic or non-electrostatic), or by the first-order rate equation in the presence of weak force adsorption. Considering that each parameter in these two rate equations has a definite physical meaning, the application of the rate equations to deal with kinetic data would be favorable in discovering insightful information on ion adsorption at the particle surface. By extending these two rate equations to the study of cation exchange adsorptions in the presence of Hofmeister or specific ion effects in heterogeneous systems, some researchers found additional energy. This extra energy is referred to as the Hofmeister energy, apart from the classic Coulomb energy of cations at the clay mineral surface, which strongly influences the adsorption kinetics; an approach to estimating the intensity has been further established.22,23
On the other hand, in the kinetic study of cation adsorption by employing the miscible displacement experiment or the constant flow method,24 the adsorption quantity of X+ (e.g., X+ = Cs+) in X+/Y+ (e.g., Cs+/K+) exchange at equilibrium will not be limited by cation exchange equilibrium. Also, at t → ∞, all the previously adsorbed Y+ can be replaced by X+, and therefore the adsorption quantity of X+ would be equal to the surface charge numbers of the adsorbent. The reason is simple: for the constant flow method, the concentration of the X+ in bulk solution (flowing solution) is always constant but the desorbed cation Y+ is continuously taken away by the flowing solution; thus, all the previously adsorbed Y+ can theoretically be exchanged by the X+. However, if the activation energy for Y+ desorption from the negatively charged surface (or from the negative electric field of the diffuse layer) to the flowing bulk solution is tremendous, the incomplete ion-exchange state would be observed theoretically, for which the observed adsorption quantity of X+ at time t → ∞ would be lower than the surface charge numbers. Li et al.24 established an efficient approach to determining the adsorption quantity at t → ∞ based on the experimental data of cation adsorption kinetics according to the constant flow method. Therefore, by adopting the constant flow method, the kinetic study can provide a rather valid approach to discovering the incomplete ion-exchange state and its mechanisms in ion adsorption on particle surfaces.
It is well known that in aqueous solution, colloidal particles often have abundant surface charges,20,24 which, combined with the diffusively distributed counterions, could generate an electric field of up to 108 to 109 V m−1 near the particle surface.25 Undoubtedly, the adsorption process of counterions would be deeply affected by the strong electric field. Based on the Gouy–Chapman theory, some researchers have shown that the energy and state of cationic non-valence electrons could be fundamentally changed by such powerful external electric fields,5 which could result in strong ion polarization and produce non-coulombic interactions between ions and the charged surfaces of clay.26,27 Further studies have conclusively shown that the aforementioned ionic interface behavior can be enhanced with increases in ionic size.28 Thereby, the newly discovered interaction forces probably play an essential role in the incomplete ion-exchange state of cation adsorption, and dominate the possible specific ion effects. However, such important issues have scarcely been involved in the current research.
In this study, the constant flow method was employed in the mono-valent cation adsorption of X+ (X+ = Cs+, Na+ and Li+) in X+/K+ exchange at montmorillonite particle surfaces. This was done to (1) confirm the specific ion effects of incomplete ion-exchange experimentally; (2) establish a mathematical link between incomplete ion-exchange and specific ion effects with respect to ion polarization and (3) quantify and verify the validity of model predictions with experimental results.
Material and methods
Material and sample preparation
Since the surface structure of montmorillonite is relatively simple, the complex chemical bonding between ions and the surface can be ignored, except for electrostatic interactions. As such, the negatively charged montmorillonite was used as the experimental material. The sample was purchased from Wu Hua Tian Bao Mineral Resources Co. Ltd., Inner Mongolia, China, and the purity was analyzed by X-ray diffraction (XRD; Japanese Neo-Confucianism, Ultima IV) to be 98% montmorillonite and 2% quartz. The montmorillonite sample was saturated with 0.1 mol L−1 KNO3 electrolyte solution under neutral conditions to ensure that the charge-compensating ions (e.g. Na+ or Ca2+) could be readily replaced without causing obvious dissolution of clay mineral and the detachment of structural ions (e.g. Fe2+ or Mg2+).29 A more detailed preparation process of the K+-saturated sample was provided by Li et al.30 According to the combined method,31 the determined surface charge number, specific surface area, surface charge density, surface electric field strength and surface potential of the sample were 1150 mmol(−) kg−1, 725 m2 g−1, 0.1586 mmol(−) m−2, −2.164 × 107 V m−1 and −0.2855 V, respectively. The main elemental composition of the montmorillonite sample detected by X-ray fluorescence spectrometer (Axios advanced PW4400) was SiO2 (57.04%), Al2O3 (15.48%), K2O (5.01%), Fe2O3 (4.92%), MgO (4.36%), Na2O (0.57%) and CaO (0.26%) respectively.Kinetic experiments of ion adsorption
The kinetics studies on ion adsorption were carried out at 298 K. Approximately 0.5 g K+-montmorillonite was layered on the exchange column. The sample was evenly spread as thinly as possible to exclude the influence of ion diffusion caused by the longitudinal concentration gradient. The thickness of the sample was approximately 0.2–0.3 mm, and the area was 15 cm2. The CsNO3, NaNO3 and LiNO3 flowed through the sample with a constant flow rate (0.5 ml min−1). All effluents were collected by the automatic collector (DBS-100, Shang-Hai QPHX Instrument Co., LTD. China). Since the adsorption rate increases with increasing ionic concentration, the time interval was correspondingly adjusted to 20, 20, 5, 2 and 2 minutes at 0.0001, 0.001, 0.01, 0.02 and 0.03 mol L−1, respectively. Flame photometry (AP1401, Shang-Hai AP Analysis Instrument Co., LTD. China) was applied to determine the concentrations of Li+, Na+ and Cs+ in the effluents. The adsorption quantity for each cation species was calculated before and after the exchange experiments. It is worth noting that since the kinetic results of ion adsorption are continuous variables involving non-independent measurement, statistical analysis based on the average values of repetitions is unnecessary.32,33 However, the repeatability of all treatments was examined before the present research.Experimental results
Based on the constant flow experiment, the relations between cationic adsorption quantity NX(t) vs. time t for different monovalent cations of X+ (X+ = Cs+, Na+, and Li+ respectively) in X+/K+ exchanges are shown in Fig. 1. The conspicuous specificity in adsorption kinetics for Cs+, Na+ and Li+ was observed, which has been reported in our recent research.34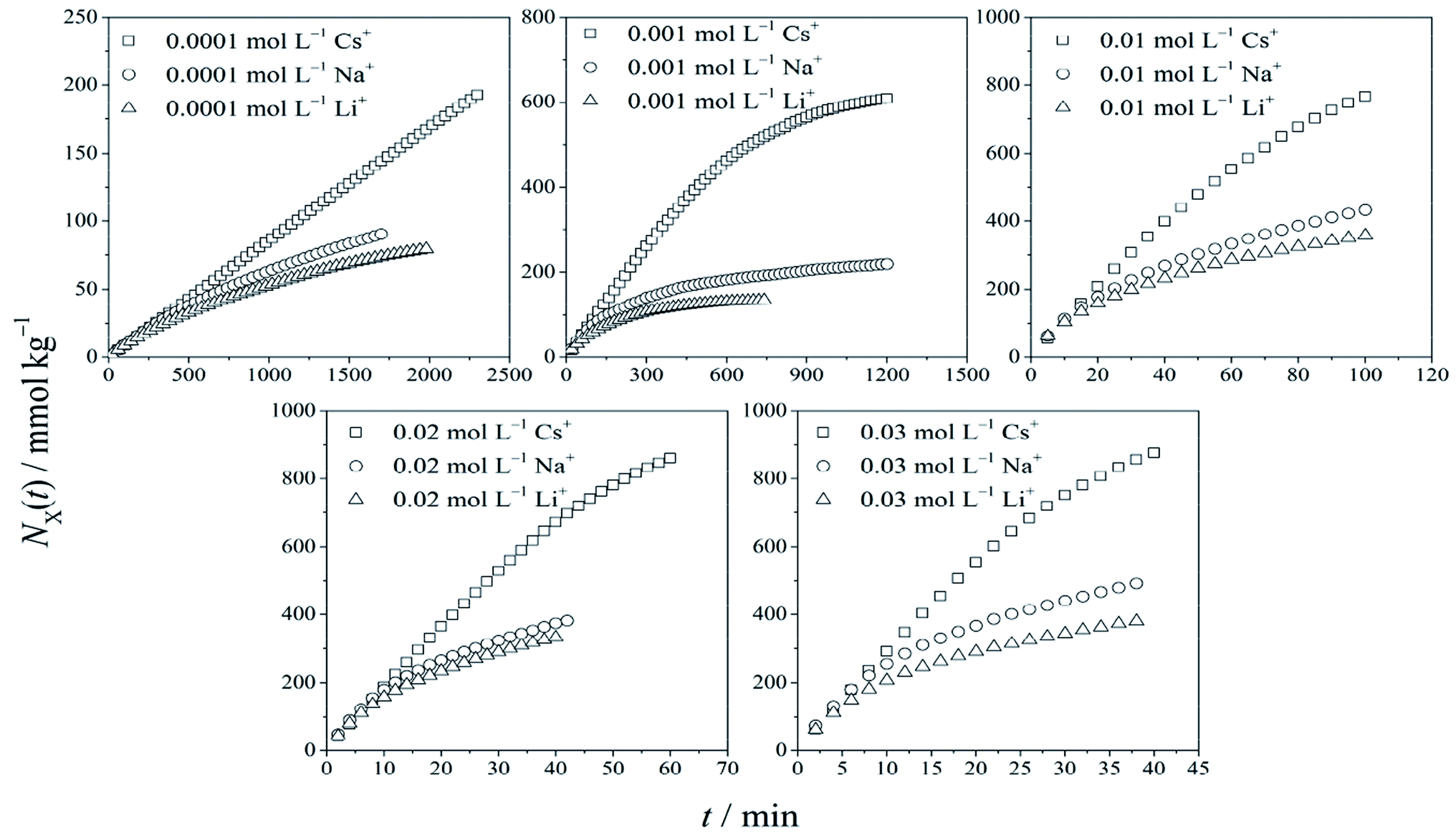 | ||
| Fig. 1 Changes in cationic adsorption quantities with time. | ||
Theoretically, cationic exchange adsorption will obey two different rate equations:
(1) The zero-order rate equation with strong force adsorption21,22
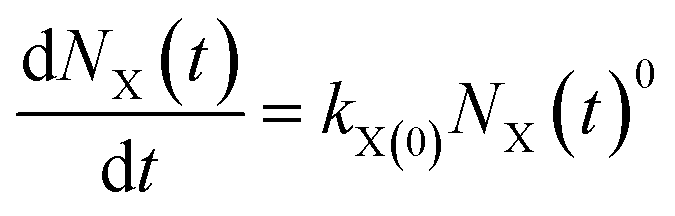 | (1) |
(2) The first-order rate equation with weak force adsorption21,22
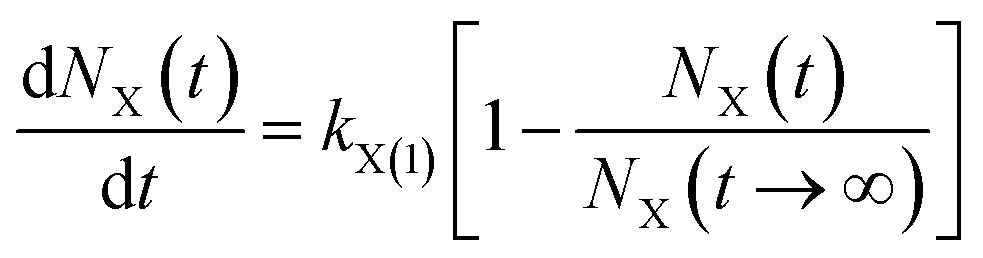 | (2) |
By employing the experimental results of NX(t) ∼ t shown in Fig. 1, the relationship of dNX(t)/dt vs. NX(t) was obtained, considering dNX(t)/dt ≈ [NX(tm+1) − NX(tm)]/(tm+1 − tm) and NX(t) ≈ NX(tm+1/2) = NX(tm) + 0.5[NX(tm+1) − NX(tm)],24 where m = 1, 2, 3,…
Fig. 2 shows that for Cs+, there were strong force adsorptions of the zero-order kinetic process in the initial stage, which changed to weak force adsorptions of the first-order kinetic process in the cationic concentration range from 0.0001 to 0.03 mol L−1. For Na+ and Li+ adsorptions, only the weak force adsorption of the first-order kinetic process appeared. According to Fig. 2, the adsorption quantities of the cations at t → ∞ under different cationic concentrations are shown in Table 1.
 | ||
| Fig. 2 The adsorption kinetics curves for Cs+, Na+ and Li+ under different concentrations. | ||
| Cation | Concentration f0(x) (mol L−1) | NX(t → ∞) (mmol kg−1) |
|---|---|---|
| Cs+ | 0.0001 | 523.0 |
| 0.001 | 647.4 | |
| 0.01 | 1098 | |
| 0.02 | 1140 | |
| 0.03 | 1152 | |
| Na+ | 0.0001 | 111.4 |
| 0.001 | 165.8 | |
| 0.01 | 380.5 | |
| 0.02 | 403.5 | |
| 0.03 | 485.8 | |
| Li+ | 0.0001 | 79.32 |
| 0.001 | 122.8 | |
| 0.01 | 348.1 | |
| 0.02 | 379.4 | |
| 0.03 | 410.8 |
The data in Table 1 show the strong specific ion effects on the adsorption kinetics for Cs+, Na+ and Li+, and under arbitrary cation concentrations, the adsorption quantities at t → ∞ were in the order of Cs+ ≫ Na+ > Li+. For example, under cationic concentrations of 0.0001 mol L−1, the adsorption quantity at t → ∞ for Cs+ was 523.0 mmol kg−1 but it decreased to 111.4 and 79.32 mmol kg−1 for Na+ and Li+, respectively. Correspondingly, under a cationic concentration of 0.01 mol L−1, the adsorption quantity at t → ∞ for Cs+ was 1098 mmol kg−1, which decreased to 380.5 mmol kg−1 for Na+ and 348.1 mmol kg−1 for Li+.
It is interesting that the adsorption quantities at t → ∞ were not constant but were dependent on cation species and their concentrations, and the adsorption quantities at t → ∞ increased with the increase in the cation concentrations. Theoretically, the adsorption quantity at t → ∞ will be the adsorption quantity at equilibrium. In this study of X+ (X+ = Cs+, Na+ and Li+) adsorption in X+/K+ exchange, the constant flow method was adopted and the adsorption quantity of X+ at equilibrium was not limited by cation exchange equilibrium. Since all the adsorbed K+ could be exchanged by Cs+, Na+ or Li+ at t → ∞, the adsorption quantity of Cs+, Na+ or Li+ would be equal to the surface charge numbers. Considering that the surface charge was 1150 mmol(−) kg−1, the adsorption quantity of Cs+, Na+ or Li+ at t → ∞ would be 1150 mmol kg−1. However, Table 1 shows that (1) for Cs+ at the concentration of 0.03 mol L−1, the adsorption quantity at t → ∞ reached the maximum value of 1150 mmol kg−1; under other concentrations for Cs+ and the other two cations, the adsorption quantities at t → ∞ were less than 1150 mmol kg−1. (2) At the given cationic concentrations, the adsorption quantities at t → ∞ were in the order of Cs+ ≫ Na+ > Li+. (3) For a given cation, with the increases in the cation concentration, the adsorption quantity at t → ∞ increased. These results imply that (1) there was an incomplete ion-exchange state for the X+ adsorption in X+/K+ exchange. (2) The incomplete ion-exchange state was determined by cationic species and their concentrations, or there were distinct specific ion effects in the incomplete ion-exchange state in the cation exchange adsorption. (3) The real equilibrium state was tough to reach even with the constant flow experiment, especially for cations with small volumes, such as Li+ and Na+.
Discussion
The experimental results show distinct specific ion effects on the incomplete ion-exchange state in cationic exchange adsorption. For example, at the cationic concentration of 0.03 mol L−1, Cs+ adsorption reached the real equilibrium but the adsorption quantities of Na+ and Li+ at t → ∞ at the same concentration merely accounted for 44.2% and 35.7% of the adsorption quantity at real equilibrium states, respectively. At the concentration of 0.0001 mol L−1, however, the adsorption quantities of Na+ and Li+ at t → ∞ were even lower and just accounted for 9.69% and 6.90% of the adsorption quantity at the real equilibrium state, respectively. Therefore, we could say that the specific ion effects strongly influenced the degree of difficulty in cation adsorption/desorption.An explanation and description of such an incomplete ion-exchange state as influenced by the specific ion effects are required. The recent investigation holds the view that the presence of the incomplete ion-exchange state is directly bound to the incompatibility between cation size and the specific sites, or the absence of sites for cation exchange.10,35 However, the observations in this study were confusing because the cation species, as well as the ion concentration profoundly influence the incomplete ion-exchange state. To answer this question, a more in-depth analysis will be required.
Theoretical analysis for the incomplete ion-exchange state in the absence of specific ion effects
For the X+ (X+ = Cs+, Na+ or Li+) adsorption in the X+/K+ exchange of the constant flow experiments, we have21,22,24
 | (3) |
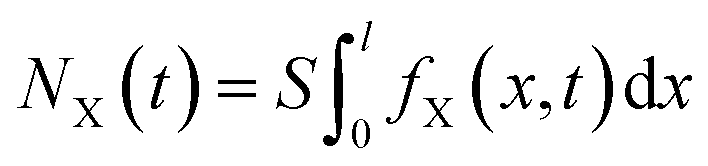 | (4) |
At equilibrium of t → ∞, the Boltzmann distribution gives
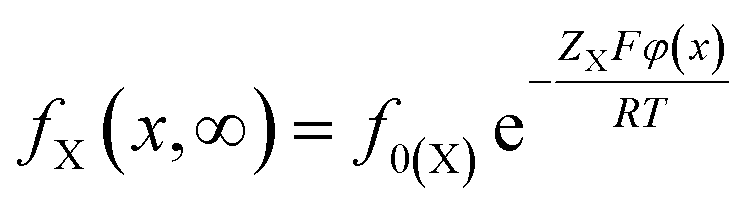 | (5) |
Thus at t → ∞, we have
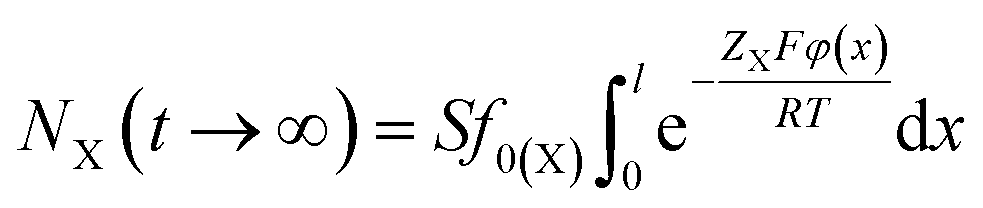 | (6) |
According to eqn (2), (3) and (6), we could have
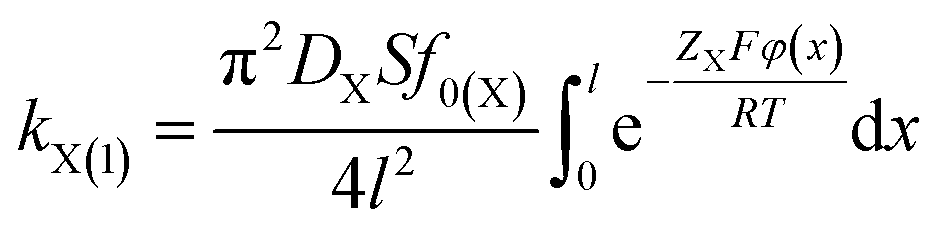 | (7) |
A comparison of eqn (6) and (7) gives
 | (8) |
Eqn (8) clearly shows that the adsorption quantity of cation X+ at t → ∞ in X+/K+ exchange will depend on the first-order rate coefficient. The higher the rate coefficient kX(1), the larger the NX(t → ∞) will be. The rate-dependent equilibrium state must be an incomplete ion-exchange state but not a real equilibrium.34 Thus, here we theoretically demonstrate that the experimentally observed equilibrium state of cationic exchange adsorption would be an incomplete ion-exchange state, other than the real exchange equilibrium, except that the adsorption rate coefficient of the first order could reach the maximum:
 | (9) |
On the other hand, according to the Arrhenius law in a chemical reaction,36 the rate coefficient of X+ adsorption in X+/K+ exchange could be expressed as
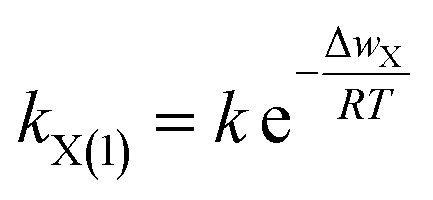 | (10) |
Obviously, when Δwactivation = 0, there will be kX(1) = max![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kX(1). Thus, from eqn (10), we have k = maxkX(1). On further considering eqn (9), we have k = [π2DX/4l2] CEC. Thus, eqn (10) changes to
kX(1). Thus, from eqn (10), we have k = maxkX(1). On further considering eqn (9), we have k = [π2DX/4l2] CEC. Thus, eqn (10) changes to
 | (11) |
Introducing eqn (11) into eqn (8), we obtain
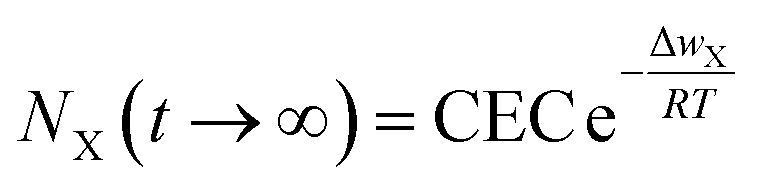 | (12) |
Eqn (12) theoretically demonstrates that as ΔwX ≠ 0, then NX(t → ∞) < CEC, indicating an incomplete ion-exchange state in the cation exchange adsorption in charged clay minerals.
In the flowing solution phase of X+ (in bulk solution), there is φ(x) → 0; thus, the potential energy for X+ in bulk solution is wX(bulk) = ZXFφ(x) → 0. On the other hand, in adsorption phase of X+, wX(adsorption) = ZXFφ(x) < 0. Therefore, the change in the potential energy for X+ in the adsorption process (from solution phase to adsorption phase), ΔwX = ZXFφ(x) < 0. Correspondingly, in the solution phase of K+ (in bulk solution), there is wK(bulk) = ZKFφ(x) → 0, and in adsorption phase, wK(adsorption) = ZKFφ(x) < 0. Therefore, the change in the potential energy for K+ in the desorption process (from the adsorption-phase to the solution-phase), would be ΔwK = ZKFφ(x) > 0. These imply that the activation energy for X+ adsorption in X+/K+ was not from the X+ adsorption process but from the K+ desorption process and thus, we have
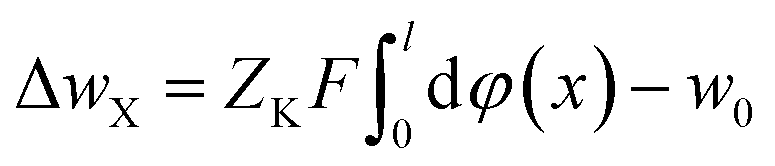 | (13) |
 | (14) |
 there is NX(t → ∞) = CEC.
there is NX(t → ∞) = CEC.
According to the classic double layer theory, under arbitrary concentrations of XNO3 (X = Cs, Na, and Li here), the φ(x) distributions in the diffuse layer in the presence of CsNO3, NaNO3 and LiNO3 will be the same; thus, according to eqn (14), under a given XNO3 concentration, the NX(t → ∞) values for X = Cs+, Na+ and Li+ will be the same. However, from the experimental data shown in Table 1, we find that they are quite different. For example, at a concentration of 0.03 mol L−1, the NX(t → ∞) values for X = Cs+, Na+ and Li+ were 1152, 485.8 and 410.8 mmol kg−1, respectively. This indicates that even though the classic theory can predict an incomplete ion-exchange state, it cannot quantitatively give a correct prediction for the incomplete ion-exchange state.
Theoretical analysis of the incomplete ion-exchange state in the presence of specific ion effects
Current studies have shown that for clay systems, the electric field-induced cation polarization could strongly influence cation exchange in both equilibrium20,37,38 and kinetics.22,23,34 Taking the electric field-induced cation polarization into account, the total potential energy of the mean force for adsorbed cations could be expressed as follows:5,22,26| w(x) ≈ γZFψ(x) | (15) |
In the absence of the electric field-induced cation polarization, eqn (15) is reduced to
| w(x) ≈ ZFφ(x) | (16) |
On the other hand, in the adsorption process of X+ in X+/K+ exchange, the potential ψ(x) in the diffuse layer might be approximately taken as solely depending on the adsorption cation of X+;39 thus, from eqn (15) and (16) we have: ψ(x) = φ(x)/γX. Therefore, in the presence of electric field-induced cation polarization, ZK and φ(x) in eqn (13) and (14) can be replaced by γKZK and φ(x)/γX, respectively. Here, γK and γX are the effective charge coefficients of K+ and X+, respectively.
Taking the electric field-induced cation polarization into account, eqn (13) and (14) are changed to
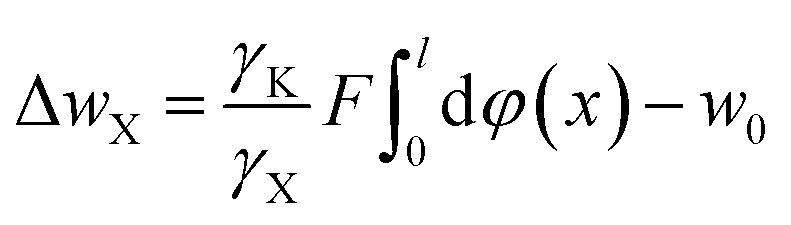 | (17) |
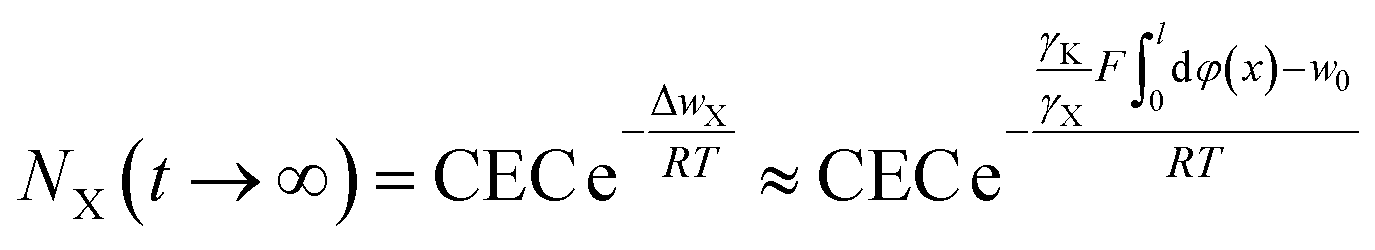 | (18) |
Here, we would like to emphasize that in eqn (16)–(18), φ(x) is the classic potential, and thus under given concentrations in bulk solution for X+ = Cs+, Na+ and Li+, the φ(x) is the same. Eqn (18) indicates that the larger the γX value for X+, the larger NX(t → ∞) will be. Considering that γX for Cs+, Na+ and Li+ were in the order of 2.404 ≫ 1.180 > 1.063,22 theoretically, the NX(t → ∞) for Cs+, Na+ and Li+ would be in the order of NCs(t → ∞) ≫ NNa(t → ∞) > NLi(t → ∞) based on eqn (18). The experimental results for NX(t → ∞) shown in Table 1 indeed exhibit the order of NCs(t → ∞) ≫ NNa(t → ∞) > NLi(t → ∞). The observed incomplete ion-exchange could be rationalized as taking the electric field-induced cation polarization into account.
According to the measured NX(t → ∞) values shown in Table 1 and eqn (18), the activation energy ΔwX could be calculated, and the results are shown in Table 2.
| Cation | Concentration f0(x) (mol L−1) | Activation energy ΔwX (RT) |
|---|---|---|
| Cs+ | 0.0001 | 0.7879 |
| 0.001 | 0.5745 | |
| 0.01 | 0.04627 | |
| 0.02 | 0.008734 | |
| 0.03 | 0 | |
| Na+ | 0.0001 | 2.334 |
| 0.001 | 1.937 | |
| 0.01 | 1.106 | |
| 0.02 | 1.047 | |
| 0.03 | 0.8617 | |
| Li+ | 0.0001 | 2.674 |
| 0.001 | 2.237 | |
| 0.01 | 1.195 | |
| 0.02 | 1.109 | |
| 0.03 | 1.029 |
For Cs+ at the concentration of 0.03 mol L−1, we have
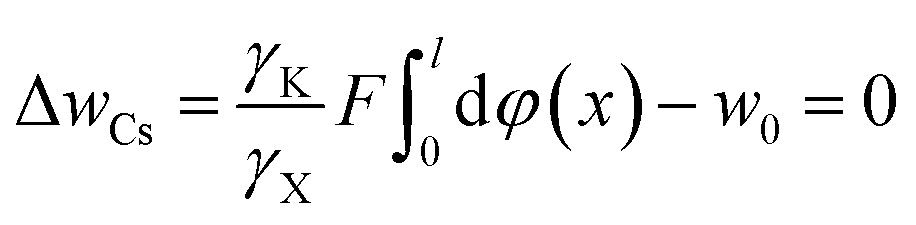 | (19) |
For Li+ under the same concentration, there is
 | (20) |
The solutions of eqn (19) and (20) give w0 = 0.8157 RT.
Here, we should keep in mind that under a given concentration of X+ (X+ = Cs+, Na+, and Li+ respectively), for different cationic species of X+ the φ(x) values are the same since φ(x) is the classic potential. Thus, under a given X+ concentration, based on eqn (17), for X+ = i and j we have
 | (21) |
Supposing w0 to be a constant of 0.8157 in advance, under other concentrations, the γCs/γNa, γNa/γLi and γCs/γLi values could be theoretically calculated from eqn (21) on employing the measured ΔwX shown in Table 2; the results are shown in Table 3.
| Cationic concentration f0(x) (mol L−1) | γCs/γNa | γNa/γLi | γCs/γLi |
|---|---|---|---|
| 0.0001 | 1.964 | 1.108 | 2.176 |
| 0.001 | 1.980 | 1.109 | 2.196 |
| 0.01 | 2.229 | 1.046 | 2.333 |
| 0.02 | 2.260 | 1.033 | 2.334 |
| 0.03 | 2.056 | 1.100 | 2.262 |
| Average | 2.098 | 1.079 | 2.260 |
On the other hand, based on the experiments of clay aggregation through dynamic light scattering and cationic exchange selectivity, the obtained γCs/γNa, γNa/γLi and γCs/γLi values were 2.037, 1.110 and 2.261, respectively.38,40 The estimated γCs/γNa, γNa/γLi, and γCs/γLi values from the measured activation energies of cationic adsorption (as shown in Table 3) meet the corresponding values from other methods very well. These results confirm that the obtained eqn (17) could be applied to describe the activation energy of cation adsorption in the presence of electric field-induced cation polarization, and the theory expressed as eqn (18) could be used to describe the incomplete ion-exchange state of cation adsorption in clay, which was produced from cation polarization in the strong electric field of clay.
Those results also confirm that w0 was indeed a constant and independent of the cationic species and their concentrations. However, currently, the physical definition of w0 is unknown.
Estimation of the cation diffusion coefficient in clay and the depth in the diffuse layer where the exchange occurs in cation exchange experiments
As discussed above, at the incomplete ion-exchange state of X+ adsorption in X+/K+ exchange, the cationic distribution in the diffuse layer could be illustrated as Fig. 3. | ||
| Fig. 3 Schematic diagram of the cationic distribution in the diffuse layer (DL) in the incomplete ion-exchange state. | ||
In the presence of electric field-induced cation polarization, the effective thickness of the diffuse layer could be expressed as follows:37
 | (22) |
Estimation of the cation diffusion coefficient in clay
For Cs+ adsorption in Cs+/K+ exchange at f0(Cs) = 0.03 mol L−1, the incomplete ion-exchange state was just the real equilibrium as discussed above; thus we have l = 1/κ = 1.12 nm (calculated from eqn (22)). At f0(Cs) = 0.03 mol L−1 the NCs(t → ∞) = 1152 mmol kg−1 and kCs(1) = 4266 mmol kg−1 min−1 (Fig. 2 and Table 1). From eqn (8), we could have DCs = 1.878 nm2 min−1. We want to emphasize that DX was not the diffusion coefficient in the bulk solution but in the porous clay system. Considering that the ion mobility for Cs+ is 1.545 or 2.000 times that of Na+ or Li+, respectively,41 and that the diffusion coefficient of an ion in solution is proportional to its mobility, we estimated the diffusion coefficients of Na+ or Li+ in the clay to be DNa = 1.216 or DLi = 0.9390 nm2 min−1 at f0(X) = 0.03 mol L−1, respectively.On the other hand, the ion mobility is the function of the ionic concentration or ionic strength in solution, and42 established an empirical equation to describe this relationship for different monovalent ions. Based on this empirical equation,
|
DX = D0Xe−0.6701I0.5035
| (23) |
According to the above obtained DX at f0(X) = 0.03 mol L−1, it could be estimated that DCs0 = 2.106 nm2 min−1, DNa0 = 1.363 nm2 min−1 and DLi0 = 1.053 nm2 min−1. Thus, DX under other cationic concentrations could be calculated from eqn (23), and the results are shown in Table 4.
| f0(x) (mol L−1) | DX (nm2 min−1) | l (nm) | 1/κ (nm) | l/(1/κ)[NX(t → ∞)/CEC] (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cs+ | Na+ | Li+ | Cs+ | Na+ | Li+ | Cs+ | Na+ | Li+ | Cs+ | Na+ | Li+ | |
| 0.0001 | 2.09 | 1.35 | 1.05 | 14.2 | 6.30 | 4.92 | 19.3 | 27.6 | 29.1 | 73.6[45.5] | 22.8[9.69] | 16.9[6.90] |
| 0.001 | 2.06 | 1.34 | 1.03 | 4.52 | 2.39 | 2.05 | 6.12 | 8.73 | 9.21 | 73.9[56.3] | 27.4[14.4] | 22.3[10.7] |
| 0.01 | 1.97 | 1.28 | 0.986 | 2.01 | 1.05 | 0.978 | 1.93 | 2.76 | 2.91 | 104 [95.4] | 38.0[33.1] | 33.6[30.3] |
| 0.02 | 1.92 | 1.24 | 0.959 | 1.36 | 0.763 | 0.706 | 1.37 | 1.95 | 2.06 | 99.3[99.1] | 39.1[35.1] | 34.3[33.0] |
| 0.03 | 1.88 | 1.22 | 0.939 | 1.12 | 0.841 | 0.619 | 1.12 | 1.59 | 1.68 | 100 [100] | 52.9[42.2] | 36.8[35.7] |
Table 4 shows that the ionic diffusion coefficient follows the order of Cs+ > Na+ > Li+ at identical ionic concentrations, which is in line with the sequence of the polarization intensities of Cs+, Na+ and Li+.22,23,43 This implies that the diffusion ability of equivalent ions is positively related to their polarization intensity. Furthermore, since the ion polarization is related to the ion size, we can, therefore, safely infer that there is also a correlation between the ion diffusion coefficient and the ion size;44 it was found that the diffusion coefficient of the cations increased with the increase in the bare diameter ratio of cation to anion. It was also demonstrated theoretically using a modified mean spherical approximation45 that the mutual diffusion coefficients of concentrated 1:1 electrolyte solutions are in the order: Cs+ > Na+ > Li+.46 The measured results of diffusion coefficients presented in Table 4 are in agreement with all the simulated and theoretically results mentioned above.
Estimation of the depth of the diffuse layer where the exchange occurs in cationic exchange adsorption experiments
On introducing the kX(1) and NX(t → ∞) shown in Table 1 and the corresponding DX estimated by eqn (23) into eqn (8), l and l/(1/κ) could be calculated, and the results are shown in Table 4.It is fascinating that the theoretically calculated l/(1/κ) data corroborated the experimentally determined NX(t → ∞)/CEC very well under relatively high cationic concentrations. This confirmed the correctness of the obtained diffusion coefficient (DX) in clay, the depth of the diffuse layer (l) where the exchange occurred, as well as the Debye–Hückel parameter (κ), taking the cation polarization into account. The data in Table 4 show that for Cs+ with concentration ≥0.01 mol L−1, the Cs+/K+ exchange occurred almost in the whole diffuse layer (l/(1/κ) → 100%) and, correspondingly, the previously adsorbed K+ was almost thoroughly exchanged by Cs+ (NX(t → ∞)/CEC → 100%). For Na+ or Li+ with concentration ≥0.01 mol L−1, the calculated l/(1/κ) also matched the determined NX(t → ∞)/CEC. The percentage of measured NX(t → ∞)/CEC was, to some degree, lower than the percentage of the calculated l/(1/κ), especially at relatively low ionic concentrations. The reason for such a difference is apparent: the cationic concentration in the inner space of x = 0 to (1/κ − l) is higher than that in the outer area of x = (1/κ − l) to l, especially at relatively low cationic concentrations. Thus, on the contrary, the percentage of the measured [CEC − -NX(t → ∞)]/CEC should be higher than the percentage of the calculated (1/κ − l)/(1/κ) especially at relatively low ionic concentrations. The data shown in Table 5 meet these predictions and under relatively high cation concentrations of ≥0.01 mol L−1, the percentage of the measured [CEC − NX(t → ∞)]/CEC still matches the percentage of the calculated (1/κ − l)/(1/κ) well.
| f0(X) (mol L−1) | (1/κ − 1)/(1/κ) {[CEC − NX(t → ∞)]/CEC} (%) | ||
|---|---|---|---|
| Cs+ | Na+ | Li+ | |
| 0.0001 | 0.264 {0.545} | 0.772 {0.903} | 0.831 {0.931} |
| 0.001 | 0.263 {0.437} | 0.726 {0.856} | 0.777 {0.893} |
| 0.01 | 0.000 {0.000} | 0.619 {0.669} | 0.664 {0.697} |
| 0.02 | 0.000 {0.000} | 0.609 {0.649} | 0.657 {0.670} |
| 0.03 | 0.000 {0.000} | 0.471 {0.577} | 0.631 {0.643} |
This study suggests possible approaches to measuring the cationic diffusion coefficient in clay and the depth of the diffuse layer where the exchange occurs in the cationic exchange adsorption experiments. Again, the comparison of the theoretically calculated l/(1/κ) and experimentally determined NX(t → ∞)/CEC values verified the correctness of the established theory for describing the incomplete ion-exchange state that was induced by the electric field-induced cation polarization.
Conclusions
In this study, the specific ion effects of incomplete ion-exchange at a montmorillonite particle surface were observed. The established theoretical framework showed that the disparity of activation energies caused by different polarized exchanging cations in the strong electric field of clay resulted in the observed specific ion effects of the incomplete ion-exchange state. Finally, a theory for describing the specific ion effects of the incomplete ion-exchange state, taking the electric field-induced cation polarization into account, was established, and the availability of the established theory has been verified independently by the effective charges of the sharply polarized cations and the depth of the diffuse layer where the exchange occurred.This study suggests applicable new approaches for measuring the cationic diffusion coefficient in clay and the actual depth in the diffuse layer where the exchange occurs in clay mineral systems in cationic exchange adsorption experiments.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 41530855 and 41371249), and the Doctoral Start-Up Research Fund of Northwest A&F University (2452018041).References
- H. Qiu, L. Lv, B.-c. Pan, Q.-j. Zhang, W.-m. Zhang and Q.-x. Zhang, J. Zhejiang Univ., Sci., A, 2009, 10, 716–724 CrossRef CAS.
- G. L. Gaines Jr and H. C. Thomas, J. Chem. Phys., 1953, 21, 714–718 CrossRef.
- R. Ezzati, Chem. Eng. J., 2019, 123705 Search PubMed.
- P. Jungwirth and P. S. Cremer, Nat. Chem., 2014, 6, 261–263 CrossRef CAS.
- C. Y. Xu, H. Li, F. N. Hu, S. Li, X. M. Liu and Y. Li, Eur. J. Soil Sci., 2015, 66, 615–623 CrossRef CAS.
- R. A. Griffin and J. J. Jurinak, Soil Sci. Soc. Am. J., 1973, 847–850 CrossRef CAS.
- W.-H. Kuan, S.-L. Lo, M. K. Wang and C.-F. Lin, Water Res., 1998, 32, 915–923 CrossRef CAS.
- R. N. J. Comans, M. Haller and P. D. Preter, Geochim. Cosmochim. Acta, 1991, 55, 433–440 CrossRef CAS.
- D. Koning and R. N. J. Comans, Geochim. Cosmochim. Acta, 2004, 68, 2815–2823 CrossRef.
- M. Jeffroy, A. Boutin and A. H. Fuchs, J. Phys. Chem. B, 2011, 115, 15059–15066 CrossRef CAS PubMed.
- S. Dultz and J. Bors, Appl. Clay Sci., 2000, 16, 15–29 CrossRef CAS.
- E. Tertre, A. Delville, D. Prêt, F. Hubert and E. Ferrage, Geochim. Cosmochim. Acta, 2015, 149, 251–267 CrossRef CAS.
- J. Sun, D. Yang, C. Sun, L. Liu, S. Yang, Y. Jia, R. Cai and X. Yao, Sci. Rep., 2014, 4, 7313 CrossRef CAS PubMed.
- W. J. Paulus, S. Komarneni and R. Roy, Nature, 1992, 357, 571–573 CrossRef CAS.
- C. Colella, Miner. Deposita, 1996, 31, 554–562 CrossRef CAS.
- E. Tertre, F. Hubert, S. Bruzac, M. Pacreau, E. Ferrage and D. Pret, Geochim. Cosmochim. Acta, 2013, 112, 1–19 CrossRef CAS.
- S. S. Gupta and K. G. Bhattacharyya, Adv. Colloid Interface Sci., 2011, 162, 39–58 CrossRef PubMed.
- L. Fischer, G. Brümmer and N. Barrow, Eur. J. Soil Sci., 2007, 58, 1304–1315 CrossRef CAS.
- S. Goldberg, L. J. Criscenti, D. R. Turner, J. A. Davis and K. J. Cantrell, Vadose Zone J., 2007, 6, 407–435 CrossRef CAS.
- X. Liu, H. Li, R. Li, D. Xie, J. Ni and L. Wu, Sci. Rep., 2014, 4 DOI:10.1038/srep05047.
- R. Li, H. Li, C. Xu, X. Liu, R. Tian, H. Zhu and L. Wu, Colloids Surf., A, 2011, 392, 55–66 CrossRef CAS.
- W. Du, R. Li, X.-M. Liu, R. Tian and H. Li, Colloids Surf., A, 2016, 509, 427–432 CrossRef CAS.
- W. Du, R. Li, X. Liu, R. Tian, W. Ding and H. Li, Appl. Clay Sci., 2017, 146, 122–130 CrossRef CAS.
- H. Li, R. Li, H. Zhu and L. Wu, Soil Sci. Soc. Am. J., 2010, 74, 1129–1138 CrossRef CAS.
- F. Hu, J. Liu, C. Xu, Z. Wang, G. Liu, H. Li and S. Zhao, Geoderma, 2018, 320, 43–51 CrossRef.
- X. R. Huang, H. Li, S. Li, H. L. Xiong and X. J. Jiang, Eur. J. Soil Sci., 2016, 67, 341–350 CrossRef CAS.
- Z. H. Yu, X. M. Liu, C. Y. Xu, H. L. Xiong and H. Li, Soil Tillage Res., 2016, 161, 63–70 CrossRef.
- Q. Li, S. Yang, Y. Tang, G. Yang and H. Li, J. Phys. Chem. C, 2019, 123, 25278–25285 CrossRef CAS.
- M. L. Rozalén, F. J. Huertas, P. V. Brady, J. Cama, S. García-Palma and J. Linares, Geochim. Cosmochim. Acta, 2008, 72, 4224–4253 CrossRef.
- S. Li, H. Li, C. Y. Xu, X. R. Huang, D. T. Xie and J. P. Ni, Soil Sci. Soc. Am. J., 2013, 77, 1563 CrossRef CAS.
- H. Li, J. Hou, X. Liu, R. Li, H. Zhu and L. Wu, Soil Sci. Soc. Am. J., 2011, 75, 2128–2135 CrossRef CAS.
- C. Hu, P. Zhu, M. Cai, H. Hu and Q. Fu, Appl. Clay Sci., 2017, 143, 320–326 CrossRef CAS.
- S. Carrick, P. Almond, G. Buchan and N. Smith, Eur. J. Soil Sci., 2010, 61, 1056–1069 CrossRef.
- W. Du, X. Liu, R. Li, R. Tian, W. Ding and H. Li, J. Soils Sediments, 2019, 19, 1839–1849 CrossRef CAS.
- Y. Khabzina, C. Laroche, C. Pagis and D. Farrusseng, Phys. Chem. Chem. Phys., 2017, 19 10.1039/C7CP02051A.
- P. Jardine and D. Sparks, Soil Sci. Soc. Am. J., 1984, 48, 39–45 CrossRef CAS.
- X. Liu, W. Ding, R. Tian, W. Du and H. Li, Soil Sci. Soc. Am. J., 2017, 81, 268–276 CrossRef CAS.
- X. Liu, H. Li, W. Du, R. Tian, R. Li and X. Jiang, J. Phys. Chem. C, 2013, 117, 6245–6251 CrossRef CAS.
- H. Li, L. Wu, H. Zhu and J. Hou, J. Phys. Chem. C, 2009, 113, 13241–13248 CrossRef CAS.
- Q. Li, Y. Tang, X. He and H. Li, AIP Adv., 2015, 5, 107218 CrossRef.
- Jilin University, Physical chemistry, People's Education Press, Beijing, 1979, vol. 2 Search PubMed.
- Y. He, X. Wu, F. Kong, C. Cao, L. Fan and H. Xiao, Chin. J. Chromatogr., 2016, 34, 625–634 CrossRef CAS.
- F. Hu, H. Li, X. Liu, S. Li, W. Ding, C. Xu, Y. Li and L. Zhu, PLoS One, 2015, 10 Search PubMed.
- H. B. Shi, G. H. Gao and Y. X. Yu, Fluid Phase Equilib., 2005, 228–229, 535–540 CrossRef CAS.
- J. F. Lu, Y. X. Yu and Y. G. Li, Fluid Phase Equilib., 1993, 85, 81–100 CrossRef CAS.
- G. H. Gao, H. B. Shia and Y. X. Yua, Fluid Phase Equilib., 2007, 256, 105–111 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |