
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of lipid-mimetic cationic iridium complexes and their liposomal formulation for in vitro and in vivo application in luminescent bioimaging†
Julia R. Shakirova a,
Amir Sadeghib,
Alla A. Koblovaa,
Pavel S. Chelushkina,
Elisa Toropainenb,
Shirin Tavakolic,
Leena-Stiina Kontturic,
Tatu Lajunencd,
Sergey P. Tunik*a and
Arto Urtti*abc
a,
Amir Sadeghib,
Alla A. Koblovaa,
Pavel S. Chelushkina,
Elisa Toropainenb,
Shirin Tavakolic,
Leena-Stiina Kontturic,
Tatu Lajunencd,
Sergey P. Tunik*a and
Arto Urtti*abc
aSt. Petersburg State University, Institute of Chemistry, Universitetskii pr., 26, 198504 St. Petersburg, Russia. E-mail: sergey.tunik@spbu.ru
bSchool of Pharmacy, Faculty of Health Sciences, University of Eastern Finland, Yliopistonranta 1C, 70211 Kuopio, Finland. E-mail: arto.urtti@uef.fi
cDrug Research Program, Faculty of Pharmacy, University of Helsinki, Viikinkaari 5 E, 00710 Helsinki, Finland
dLaboratory of Pharmaceutical Technology, Department of Pharmaceutical Science, Tokyo University of Pharmacy & Life Sciences, 1432-1 Hachioji 192-0392, Tokyo, Japan
First published on 8th April 2020
Abstract
Two iridium [Ir(N^C)2(N^N)]+ complexes with the diimine N^N ligand containing a long polymethylene hydrophobic chain were synthesized and characterized by using NMR and ESI mass-spectrometry: N^N – 2-(1-hexadecyl-1H-imidazol-2-yl)pyridine, N^C – methyl-2-phenylquinoline-4-carboxylate (Ir1) and 2-phenylquinoline-4-carboxylic acid (Ir2). These complexes were used to prepare the luminescent PEGylated DPPC liposomes (DPPC/DSPE-PEG2000/Ir-complex = 95/4.5/1 mol%) using a thin film hydration method. The narrowly dispersed liposomes had diameters of about 110 nm. The photophysics of the complexes and labeled liposomes were carefully studied. Ir1 and Ir2 give red emission (λem = 667 and 605 nm) with a lifetime in the microsecond domain and quantum yields of 4.8% and 10.0% in degassed solution. Incorporation of the complexes into the liposome lipid bilayer results in shielding of the emitters from interaction with molecular oxygen and partial suppression of excited state nonradiative relaxation due to the effect of the relatively rigid bilayer matrix. Delivery of labeled liposomes to the cultured ARPE-19 cells demonstrated the usefulness of Ir1 and Ir2 in cellular imaging. Labeled liposomes were then injected intravitreally into rat eyes and imaged successfully with optical coherence tomography and funduscopy. In conclusion, iridium complexes enabled the successful labeling and imaging of liposomes in cells and animals.
Introduction
During the last decades liposomes have been actively studied as basic models of lipid bilayers and as carrier structures in diagnostics and drug delivery.1 Liposomes are versatile nanostructures that can be prepared with different sizes (from 40 nm to micrometer scale), lipid wall properties (rigid or fluid state) and surface functionalities (e.g. PEGylation for prolonged circulation times, antibodies for targeting).2–8 Liposomal delivery of small molecule and biomacromolecule drugs has been widely studied for many medical applications. The advantages of liposomes include their safety profile, solubility in physiological media, possibilities for controlled and induced drug release, and relatively easy surface vectorization for targeted delivery of encapsulated drug molecules.9,10Targeting of drug carrier into the target cells and tissues can be visualized non-invasively with luminescent labels and radioactive probes. In most imaging experiments, the liposomes were labelled with fluorescent dyes.11–16 The fluorescent markers display high emission intensity and they are commercially available, but these emitters show small Stokes shifts, short lifetimes and often suffer of photobleaching. These deficiencies may result in lower image resolution in luminescent microscopy and also limit the use of fluorophores in long-term experiments. On the contrary, phosphorescent emitters based on transition metal complexes display large Stokes shifts, long life-times and negligible photobleaching. Thus, phosphorescence labels are an attractive, but not widely studied, alternative to imaging of diagnostic and therapeutic nanocarriers.
Previously, phosphorescent polypyridyl ruthenium17–20 and orthometalated iridium17,21–25 complexes have been used for labeling of liposomes. Incorporation of the phosphorescent labels into liposomes can be carried out using intrinsic hydrophobicity of the ligand,17–19,21,24,25 thereby targeting the label into inner lipid layer of the liposome membrane. Alternatively, the ligand can be designed as amphiphilic moiety resembling the components of the target membrane.17,19,21,23 The first approach is based on the affinity of the labels to hydrophobic tails of the phospholipids, whereas the latter approach mimics the structural patterns of phospholipids allowing predictable localization to the liposomal membrane.
Two recent publications24,25 introduce liposome based drug delivery systems with loaded iridium complexes. These liposome formulations included iridium compounds as active components for cancer therapy and PEGylated long circulating liposomes were used for iridium complex delivery.26–29 In addition, the luminescent iridium complexes enabled visualization of the liposome localization in vitro in the cells. The results prompted us to investigate similar systems for potential applicability in ophthalmology as there is a need for long acting and targeted delivery of drugs for retinal treatment.30,31
In the present communication we describe synthesis of amphiphilic luminescent iridium complexes, which contain hydrophobic aliphatic tails and relatively hydrophilic (polar and charged) metal containing head group. These compounds were incorporated into PEGylated liposomes and the formulations were tested in in vitro and in vivo. We demonstrate the usefulness of these phosphorescent labels in imaging of liposomes in the cells and in the eye in vivo. The structure and photophysical properties of the complexes and labeled liposomes are presented and discussed.
Experimental
General comments
Solvents were used as received. Solution 1H, 1H–1H COSY, NOESY NMR spectra were recorded using a Bruker Avance 400 and AMX-400 spectrometers. Mass spectra were measured on a Bruker maXis II ESI-QTOF instrument in the ESI+ mode. Microanalyses were carried out in the analytical laboratory of the University of Eastern Finland. The bis(μ-chlorido) bridged dimeric precursor {(N^C–COOMe)2IrCl}2 (N^C–COOMe = methyl 2-phenylquinoline-4-carboxylate) were synthesized according to published procedures.32 Methyl 2-phenylquinoline-4-carboxylate, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) were purchased from Sigma-Aldrich (St. Louis, MO, USA). HEPES buffer solution contained 20 mM HEPES and 140 mM NaCl in MilliQ purified water and the pH was adjusted to 7.4 with NaOH. Calcein solution had 60 mM of calcein and 29 mM of NaCl in MilliQ purified water and the pH was set to 7.4 with NaOH.Synthesis of the diimine N^N ligand
Diimine ligand was synthesized with a slightly modified literature procedure.33 KOtBu (0.57 g, 5.12 mmol) was added to a solution of 2-(pyridin-2-yl)-1H-benzo[d]imidazole (1 g, 5.12 mmol) in 12 mL of DMF. The resulting mixture was stirred at room temperature for 30 min, and then 1-bromohexadecane (6.14 mmol) was added with constant stirring for 12 h. The reaction mixture was subsequently extracted with water and ether to remove DMF and excess KOtBu. The organic layer was isolated, dried over anhydrous Na2SO4, and filtered. The solvent was then removed under reduced pressure. The purification of the crude product was carried out by column chromatography on silica gel with hexane/DCM (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent to yield light-yellow oil that crystallized into a light-yellow solid after a week of standing into the fridge, 75%. 1H NMR (400 MHz, acetone-d6, 298 K) d 8.75 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.45 (ddd, 1H), 8.00 (dd, J = 7.8, 1.7 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.49 (ddd, J = 7.7, 4.8, 1.1 Hz, 1H), 7.37–7.31 (m, 1H), 7.30–7.25 (m, 1H), 4.98–4.85 (m, 2H), 1.49–1.22 (m, 31H). HR ESI+-MS: found 420.3376 [M + H+]; calcd. 420.3374.
:1) as the eluent to yield light-yellow oil that crystallized into a light-yellow solid after a week of standing into the fridge, 75%. 1H NMR (400 MHz, acetone-d6, 298 K) d 8.75 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.45 (ddd, 1H), 8.00 (dd, J = 7.8, 1.7 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.49 (ddd, J = 7.7, 4.8, 1.1 Hz, 1H), 7.37–7.31 (m, 1H), 7.30–7.25 (m, 1H), 4.98–4.85 (m, 2H), 1.49–1.22 (m, 31H). HR ESI+-MS: found 420.3376 [M + H+]; calcd. 420.3374.
Synthesis of iridium complexes
The synthesis of heteroleptic biscyclometallated Ir(III) complexes containing diimine ligands was slightly modified compared to the standard procedure.34:1 mixture (20 mL). The reaction mixture was refluxed for 3 h. The resulting clear red solution was cooled down to room temperature, followed by the addition of excess of KPF6 and the mixture was stirred for additional 30 minutes. The reaction mixture was evaporated to dryness, dissolved in DCM, filtered and evaporated once more. The obtained crude product was purified by flash column chromatography on silica with DCM as eluent. The pure compound was obtained as red solid (85%).1H NMR (400 MHz, acetone-d6, 298 K) d 8.91 (s, 1H), 8.60 (s, 1H), 8.55 (d, J = 8.5 Hz, 1H), 8.50 (d, J = 8.6 Hz, 1H), 8.47–8.42 (m, 2H), 8.35 (d, J = 8.0 Hz, 1H), 8.30–8.18 (m, 3H), 7.80 (d, J = 8.4 Hz, 1H), 7.78–7.71 (m, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.53–7.45 (m, 3H), 7.33–7.27 (m, 1H), 7.27–7.24 (m, 1H), 7.24–7.19 (m, 1H), 7.19–7.12 (m, 1H), 6.99–6.86 (m, 3H), 6.72 (d, J = 4.3 Hz, 1H), 6.70 (d, J = 4.4 Hz, 1H), 6.58 (d, J = 8.3 Hz, 1H), 4.88–4.75 (m, 1H), 4.75–4.60 (m, 1H), 4.15 (s, 3H), 4.06 (s, 3H), 1.49–1.36 (m, 2H), 1.31 (br. s, 29H). Anal. calculated for C62H65F6IrN5O4P: C, 58.11; H, 5.11; N, 5.47; found: C, 58.06; H, 5.26; N, 5.36 HR ESI+-MS: found 1136.4758 [M+]; calcd. 1136.4666.
1H NMR (400 MHz, DMSO-d6, 298 K) δ 8.50 (s, 1H), 8.48–8.34 (m, 3H), 8.26 (s, 1H), 8.26–8.21 (m, 2H), 8.21–8.09 (m, 2H), 7.94 (d, J = 9.0 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.77–7.69 (m, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.30 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H), 7.21–7.09 (m, 3H), 6.99 (t, J = 7.7 Hz, 1H), 6.87 (t, J = 7.7 Hz, 1H), 6.82 (t, J = 7.8 Hz, 1H), 6.72 (t, J = 7.8 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 6.48 (d, J = 7.8 Hz, 1H), 6.45 (d, J = 8.3 Hz, 1H), 4.85–4.69 (m, 1H), 4.67–4.53 (m, 1H), 1.24 (s, 31H). Anal. calculated for C60H61ClIrN5O4: C, 63.00; H, 5.38; N, 6.12; found: C, 62.89; H, 5.62; N, 6.05. HR ESI+-MS: found 1108.4347 [M+]; calcd 1108.4369.
Liposome preparation
Liposomes were prepared by thin film hydration method followed by an extrusion and purification.35 Briefly, the phospholipids in choloroform and the synthesized phosphorescent complexes (Ir1, Ir2) in MeOH were mixed together (DPPC/DSPE-PEG2000/Ir#) with molar ratios of 95/4.5/1, respectively. The organic solvent was evaporated in a rotary evaporator forming a thin film at the bottom of a glass test tube. The lipids were hydrated with 500 μL of HEPES buffer solution (+60 °C) and extruded 11 times at +60 °C through a polycarbonate membrane (100 nm pore size) with a syringe-type extrusion device (Avanti Polar Lipids). Thereafter, the liposomes were quickly cooled and stored in a refrigerator for the further use. The liposomes were purified by gel filtration through a Sephadex G-50 (Sigma-Aldrich) column with HEPES buffer elution. The final total lipid concentration of the samples was 3 μmol mL−1.Size and ζ-potential measurements
The size and ζ-potential of the liposomes were analyzed with a Zetasizer APS dynamic light scattering automated plate sampler (Malvern Instruments, Malvern, United Kingdom) and reported as hydrodynamic diameters (Dh) and polydispersity index (PDI). Dh, PDI and ζ-potentials were averaged and corresponding standard deviations (SD) were calculated based on data obtained from at least three independent liposomal formulations.Photophysical experiments
All photophysical measurements in solution were carried out in freshly distilled solvents; when appropriate the solutions were degassed by freeze–pump–thaw cycles. UV/Vis spectra were recorded using a Shimadzu UV-1800 spectrophotometer. The emission and excitation spectra in solution were measured with a Fluorolog-3 (JY Horiba Inc.) spectrofluorometer. The emission quantum yields in solution were determined by a comparative method using LED 365 nm pumping and Ru(bipy)3Cl2 in aerated water (Φr = 0.04) as the reference with the refraction coefficients of methanol and water equal to 1.331 and 1.333, respectively.36 The following equation:was used to calculate the quantum yield, where Φs is the quantum yield of the sample, Φr is the quantum yield of the reference, η is the refractive index of the solvent, As and Ar are the absorbance of the sample and the reference at the wavelength of excitation, respectively, Is and Ir are the integrated areas of emission bands. Pulse laser TECH-263 Basic (263 nm), a Tektronix (DPO2012B, band width 100 MHz) oscilloscope, Ocean Optics (Monoscan-2000, interval of wavelengths 1 nm) scanning monochromator, FASTComTec (MCS6A1T4) multiple-event time digitizer and Hamamatsu (H10682-01) Photon counting head were used for lifetime measurements.

Cell uptake studies
The cell uptake to retinal pigment epithelium cell line (ARPE-19) was imaged with Cytation 5 cell imaging multi-mode reader (BioTek Instruments, Inc., Winooski, VT, USA). The ARPE-19 cells (CRL-2302, American Type Culture Collection, ATCC, Manassas, VA, USA) were cultured in DMEM:F12 medium (Gibco 31330–038) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at +37 °C, 5% CO2. 50000 cells per well were seeded on a 24-well thin glass bottom Sensoplate (Greiner Bio-One GmbH, Kremsmünster, Austria) one day before exposing them to the liposomes. The cells were incubated for 3 h with liposomes at lipid concentrations of 1.5 μmol mL−1 (high concentration) or 0.3 μmol mL−1 (low concentration) in serum-free medium. After the incubation, the cells were washed with phosphate buffered saline (PBS) and incubated 10 min at 37 °C in 5 μg mL−1 of Hoechst 33342 label (Thermo Fisher Scientific) for cell nuclei. The cells were washed with PBS and imaged using Cytation 5 with ex/em 445 nm/685 nm for the phosphorescent liposomes and with ex/em 377 nm/447 nm for the nuclear stain.
Imaging experiments in vivo
Pigmented rats (HsdOla:LH; age 4 months; weight ∼450 g) were anesthetized with medetomidine (0.4 mg kg−1):ketamine (60 mg kg−1) mixture. The pupils were dilated with tropicamide (Oftan Tropicamid 5 mg mL−1, Santen, Finland) and the eyes were locally anaesthetized with oxybuprocaine hydrochloride (Oftan Obucain 4 mg mL−1, Santen, Finland). The intravitreal injections were performed into both eyes of the animals under direct ophthalmoscopic control through an operating microscope. The needle (34 G) was inserted, about 1 mm from the limbus, through the sclera into the vitreous, and 3 μl of the solution was injected per eye. Four eyes were used for each formulation. A topical carbomer hydrogel (Viscotears) was applied to prevent dryness of the cornea. Prior and 30 min after the intravitreal injection, imaging of each eye was performed using optical coherence tomography (OCT), and full color funduscopy (Phoenix MICRON™ MICRON IV/OCT, CA, USA) without and with the filter combination of excitation: Semrock FF01-469/35 and barrier: Semrock BLP01-488R. The transmission bands for the filters were as follows: (i) Semrock FF01-469/35: 451.5 nm to 486.5 nm; (ii) Semrock BLP01-488R: 504.7 nm to 900 nm. Due to the sensitivity limitations of the camera at near infrared region, we the maximal wavelength of detection was about 700 nm for fundus imaging.Results and discussion
Design and characterization of Ir(III) lipid–mimetic complexes
Pyridine-benzimidazole with inserted C16 aliphatic chain was chosen as diimine ligand (N^N) in the synthesis of iridium complexes to increase affinity of resulting compounds to phospholipid membrane. The N^N ligand has been synthesized according to Scheme S1 (see ESI†). The heteroleptic cationic iridium complexes of the [(N^C)2Ir(N^N)]PF6 type were synthesized according to the reaction sequence shown below in high yields (Scheme 1). The aromatic system of the N^C ligand has been modified by insertion of the ester function, which was carefully hydrolyzed at the last stage of the synthesis to increase hydrophilicity of the “head” in this amphiphilic molecule. Moreover, the presence of the electron withdrawing groups (C(O)OMe in Ir1 and C(O)OH in Ir2) gives red shift of excitation and emission bands that is useful for in vivo visualization experiments. At the last stage the ester groups of the metalating ligands in Ir1 were hydrolyzed to increase hydrophilicity of the “head” in this amphiphilic molecule.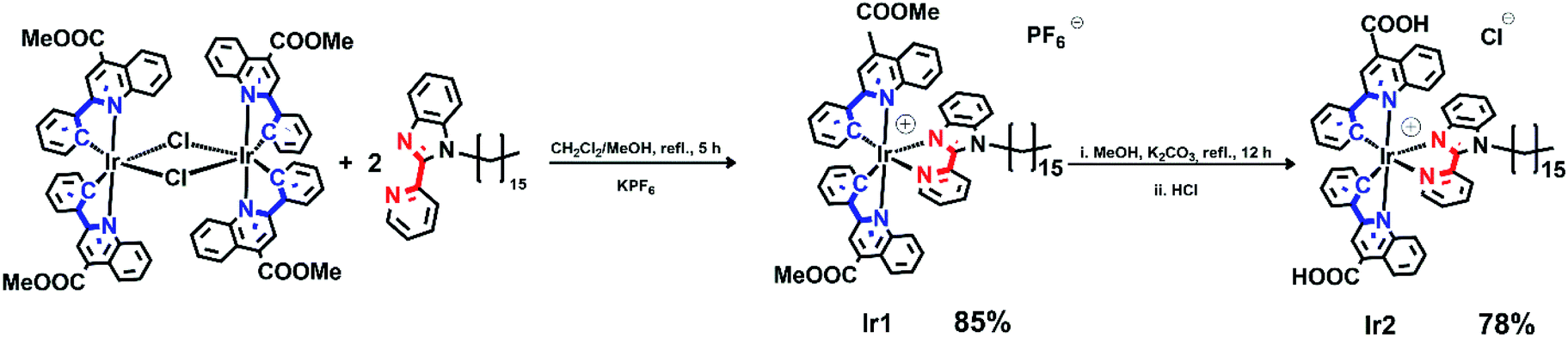 | ||
| Scheme 1 Synthesis of iridium complexes. | ||
The complexes obtained were carefully characterized using appropriate spectroscopic techniques. The HR-ESI+ mass spectra (Fig. S1 and S2†) demonstrate the dominant signals of singly charged molecular cations with m/z values equal to 1136.4758 (Ir1) and 1108.4347 (Ir2), the isotopic patterns of the corresponding signals match well the calculated ones. The 1H NMR spectra of all the complexes display three sets of signals corresponding to the N^N-ligand protons and the protons of inequivalent N^C ligands (Fig. 1 and S4†). Assignment of the signals observed in the 1H NMR spectra was done using the 1H–1H COSY, NOESY spectra (Fig. S3 and S5†). Relative intensity and multiplicity of the signals in the proton spectra fit well the suggested structures (Scheme 1).
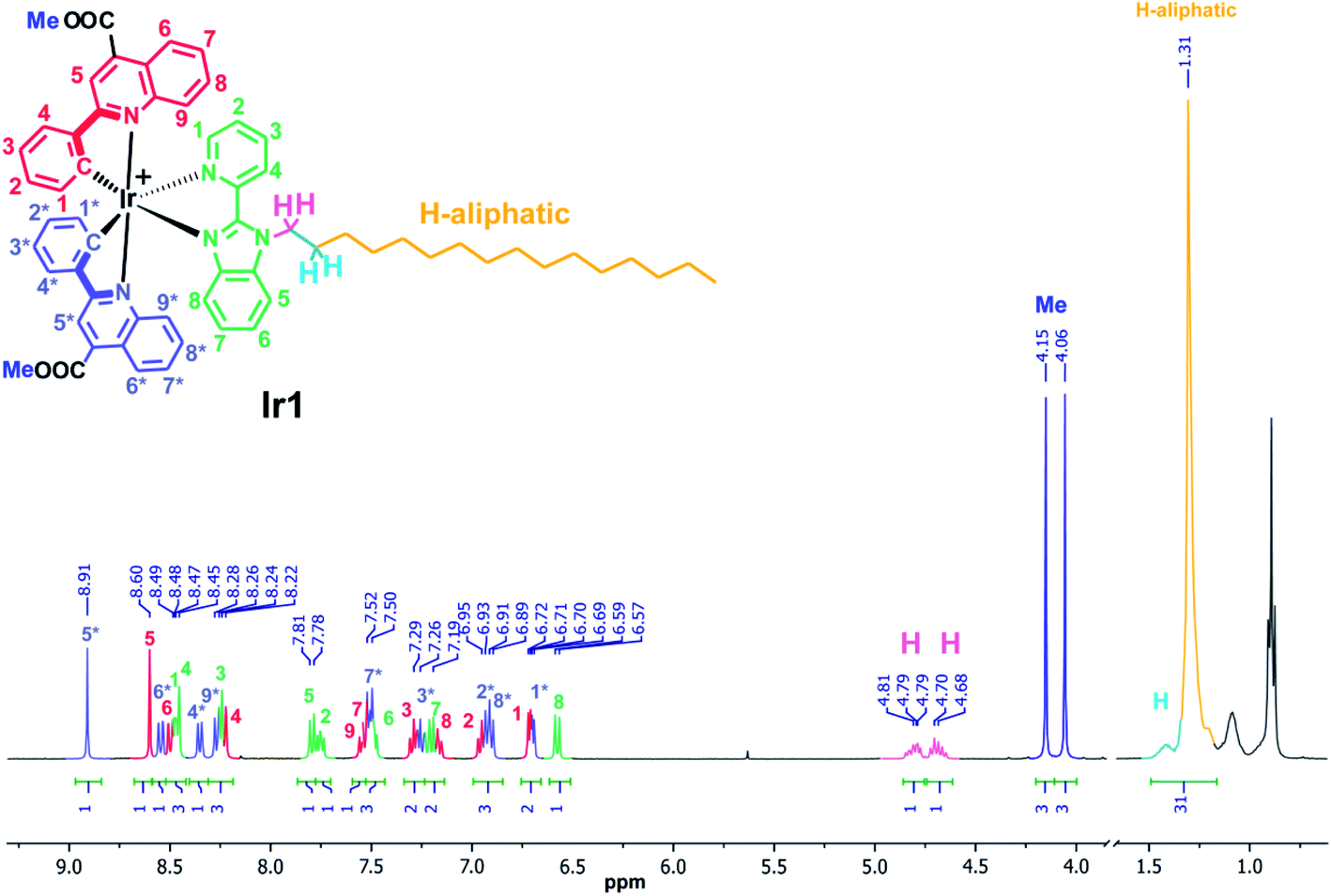 | ||
| Fig. 1 1H NMR spectrum of Ir1, acetone-d6, 298 K. | ||
Normalized electronic absorption spectra of the both complexes in methanol at 298 K are shown in Fig. S6.† Similar to the interpretation given in earlier publications32,34,37–42 for this type of iridium complexes the intense absorption bands in the 250–320 nm range could be assigned to spin allowed 1π–π* ligand centered (LC) transitions located at the cyclometalated and diimine ligands. The longer wavelength absorption bands and shoulders with lower extinction coefficients evidently originate from the mixture of metal-to-ligand (1MLCT) and ligand-to-ligand (1LLCT) charge transfer.32,42 The absorption spectra of Ir1 and Ir2 have similar features except the blue shift of the low energy absorption (1MLCT + 1LLCT transitions) in the latter complex for ca. 30 nm, which can be assigned to stronger electron withdrawing effect of carboxylic group in the metalating ligands.
Both complexes display broad and featureless emission bands (Fig. 2, Table 1), which resemble the luminescence profiles of the previously reported complexes32 with the similar cyclometalating ligand. The lifetimes in microsecond domain and a considerable quenching of emission in aerated solution clearly indicate the triplet origin of the emission observed, i.e. phosphorescence. According to the results of theoretical analysis made for the closely related heteroleptic [(N^C)2Ir(N^N)]+ complexes,32 the observed emission bands can be tentatively assigned to the mixture of 3MLCT and 3LLCT transitions with dominating contribution of the N^C ligand orbitals into the triplet excited state. The Ir2 complex containing carboxylic group in the N^C ligand displays a considerable blue shift of emission band compared to Ir1 with non-hydrolyzed ester function. This trend is essentially similar to the behavior of analogous pair of iridium complexes upon hydrolysis of ester group in the pyridyl-benzimidazole metalating ligands.18
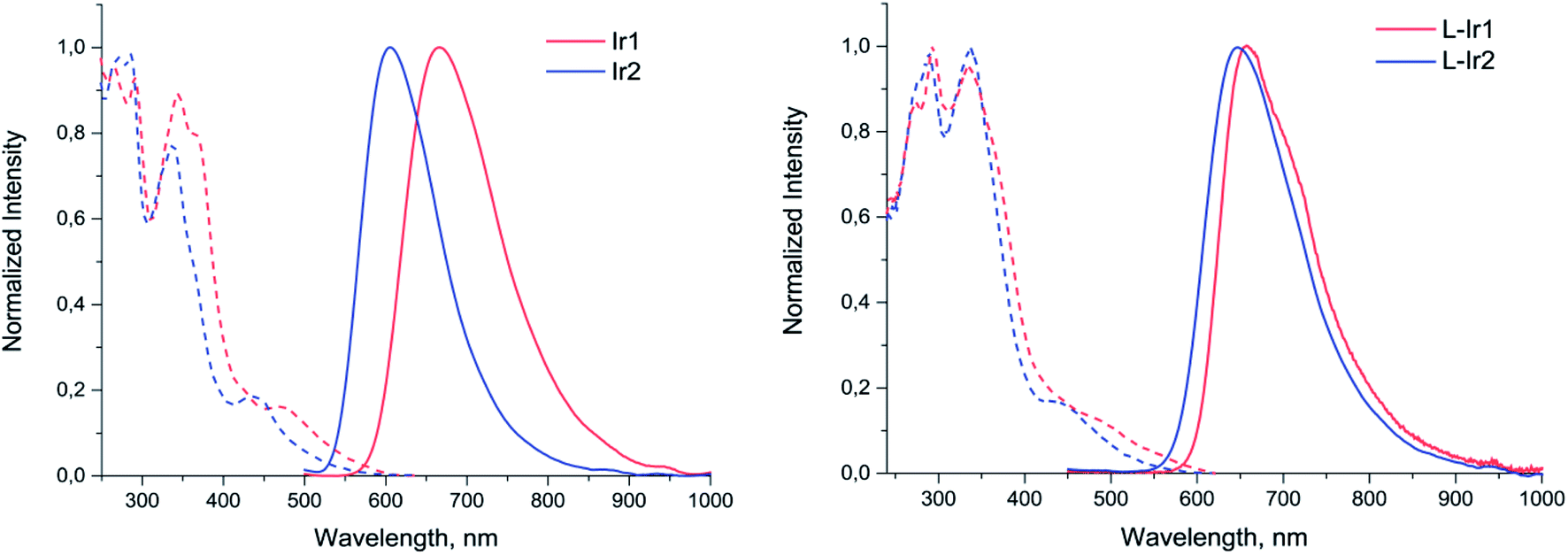 | ||
| Fig. 2 Excitation (dotted lines) and emission (solid lines) spectra of Ir1, Ir2 (methanol solution) and L-Ir1, L-Ir2 (aqueous solution). | ||
| Sample | Absorbance, λmax, nm (ε × 10−4, M−1 cm−1) | Excitation, λmax, nm | Emission, λmax, nm | τ, μs (deg/aer) | QY, % deg/aer |
|---|---|---|---|---|---|
| a These are unavailable due to strong scattering of solutions. | |||||
| Methanol solution | |||||
| Ir1 | 267(59), 293(55), 342(49), 367sh(34), 468(6) | 250–288, 344, 366, 468 | 667 | 0.19/0.12 | 4.8/2.3 |
| Ir2 | 267(57), 285(55), 335(47), 435(6) | 250–288, 338, 435 | 605 | 0.42/0.26 | 10.0/5.3 |
|
|||||
| Aqueous solution | |||||
| L-Ir1 | 332, 437 | 293, 334, 476 | 657 | 0.50/0.46 | a |
| L-Ir2 | a | 289, 337, 437 | 647 | 0.41/0.38 | a |
Preparation and photophysical characterization of liposomes labelled with Ir(III) complexes
Both Ir(III) complexes were then embedded into lipid bilayers of the PEGylated DPPC liposomes (DPPC/DSPE-PEG2000/Ir# = 95/4.5/1 mol%) by thin film hydration method. The formed liposomes were of ca. 110 nm in diameter and narrowly dispersed (Table 2). They also had slightly negative ζ-potential (Table 2) because of 4.5 mol% of anionic DSPE-PEG2000 added to sterically protect the liposomes by the PEG outer shell.| Sample | Dh ± SD, nm | PDI |
|---|---|---|
| Neat liposomes | 94 ± 25 | 0.050 |
| L-Ir1 | 118 ± 24 | 0.075 |
| L-Ir2 | 106 ± 23 | 0.153 |
Photophysical investigation of the liposomes bearing the iridium complexes (L-Ir1 and L-Ir2) revealed that they display luminescence with very similar band profiles (Fig. 2, Table 1). As compared to the starting complex L-Ir1 demonstrates slight (ca. 10 nm) hypsochromic shift, evidently due to variations in polarity of the liposomal microenvironment compared to methanol; on the contrary, L-Ir2 emission band displays pronounced (ca. 32 nm) bathochromic shift, most probably due to (at least, partial) charging of carboxyl groups of the N^C ligand at pH 7.4. The lifetime values of iridium chromophores are more characteristic with respect to microenvironment and may be considered as indirect indications of emitters' location in lipid bilayers of the liposomes. In aerated aqueous solution both L-Ir1 and L-Ir2 show longer lifetime compared to the free complexes in methanol that point to isolation of the chromophores from oxygen quenching. However, L-Ir1 displays much stronger increase in lifetime, which may be considered as deeper immersion of the complex into the liposome bilayer compared to the L-Ir2 counterpart. However, substantial increase in lifetime (L-Ir1 cf. Ir1) in degassed solution also indicates considerable effect of the matrix rigidity (suppression of nonradiative vibrational relaxation), which also plays important role in the observed lifetime variations. Nearly equal lifetime values for L-Ir2 and Ir2 in degassed solutions point to the absence of “rigidity effect”, whereas relatively small lifetime growth (L-Ir2 vs. Ir2) in aerated solutions indicate ineffective shielding of the chromophore from interaction with molecular oxygen. The both observations are very probably indicative of the iridium emitter location in the “near-surface” area of the liposome that is a natural consequence of hydrophilic character of the carboxylic groups at the N^C ligands.
Both types of phosphorescent liposomes were then evaluated for their application potential in both in vitro and in vivo imaging.
Evaluation of phosphorescent liposomes in bioimaging
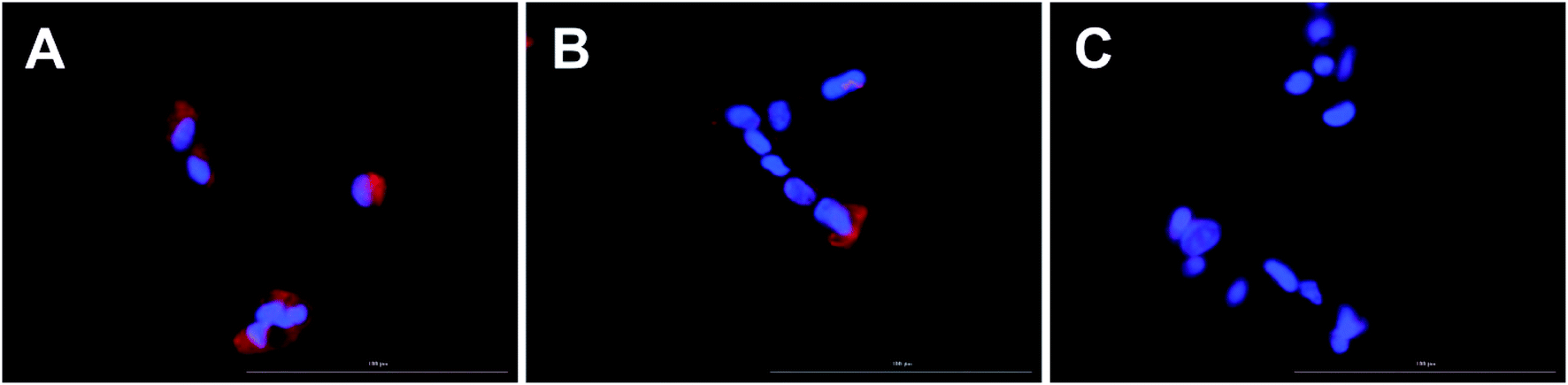 | ||
| Fig. 3 Uptake of liposomes labeled with Ir1 (panel A) and Ir2 (panel B), and unlabeled control liposomes (panel C) into ARPE-19 cells. The Ir# labeled liposomes are shown in red and cell nucleic labeled with Hoechst in blue. Images were acquired using the Cytation 5 fluorescence microscope. Lipid concentration: 0.3 μmol mL−1. Magnification: 40×. Scale bar: 100 μm. | ||
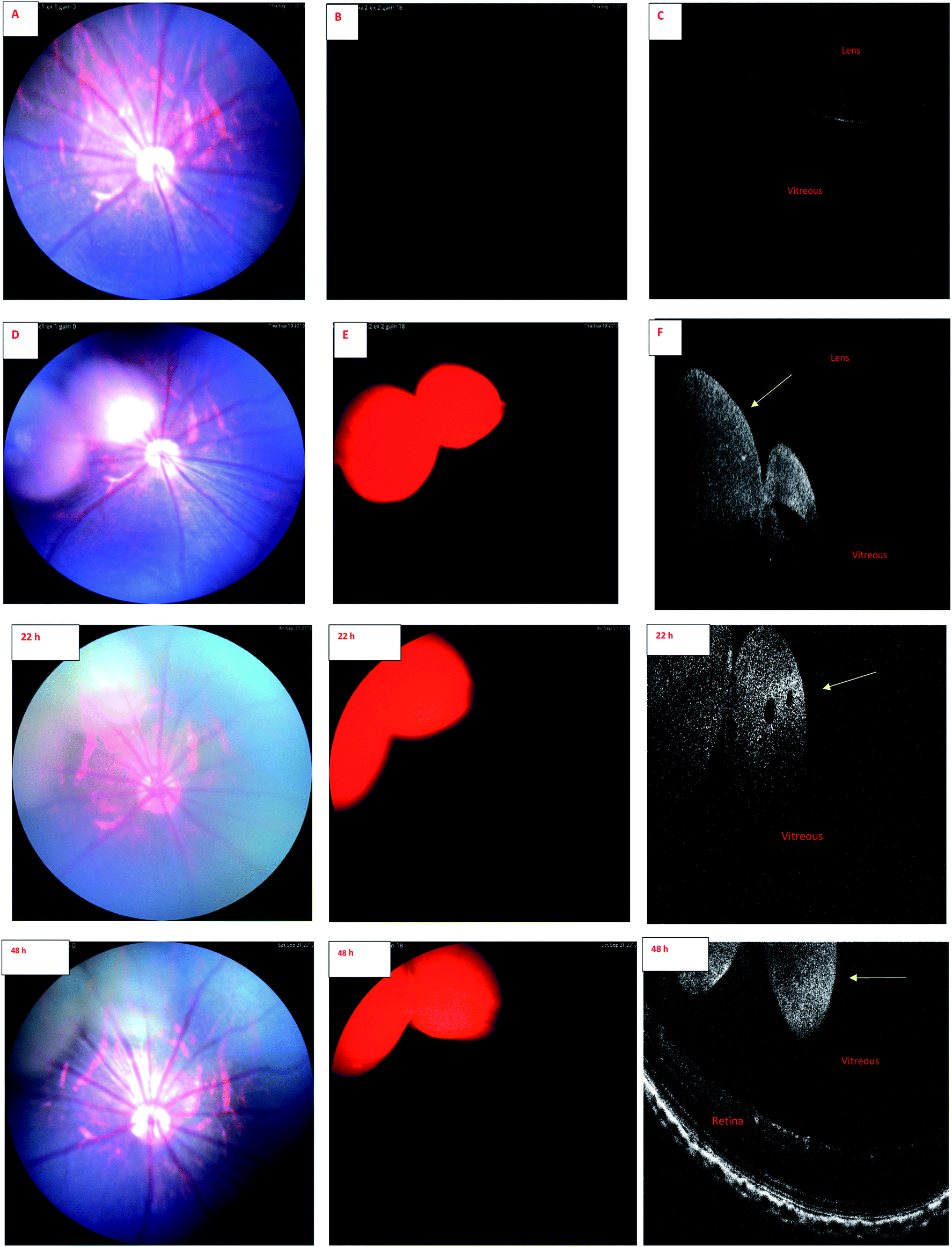 | ||
| Fig. 4 IR-2 Fundus and OCT images of rat eye. Upper panel. (A) Color fundus image before injection of liposomes containing IR-2 compound. (B) Fundus image with the filter combination of excitation: Semrock FF01-469/35 and barrier: Semrock BLP01-488R, before injection. (C) OCT image before injection. (D) Color fundus image 30 min after injection of liposomes containing IR-2 compound. (E) Fundus image 30 min after injection with the filter combination of excitation: Semrock FF01-469/35 and barrier: Semrock BLP01-488R. (F) OCT image 30 min after injection. The liposomes are visible in the vitreous after the injection (yellow arrow). Lower panel. Following images are from same eye and same set of filters 22 and 48 h after intravitreal injection. | ||
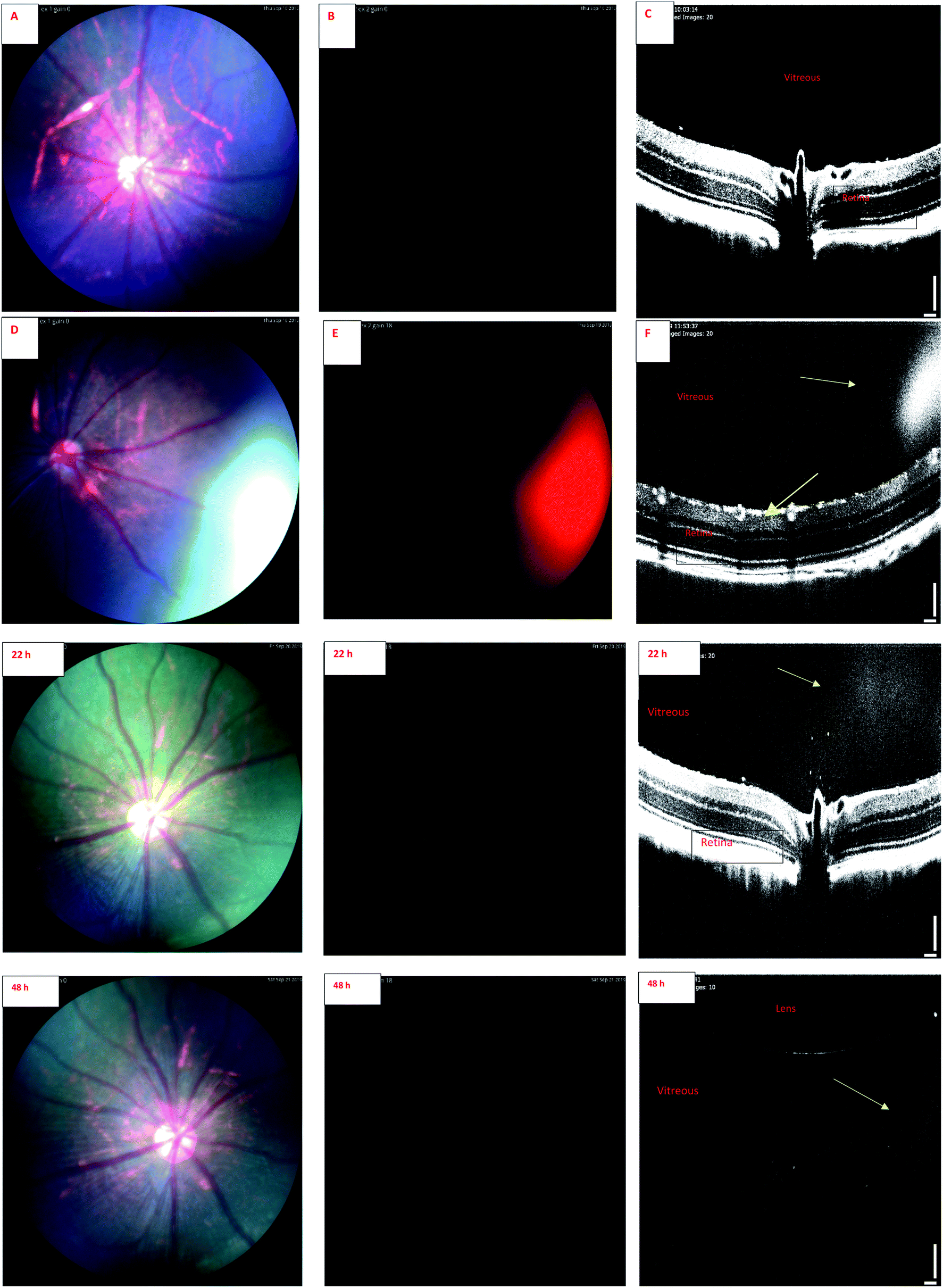 | ||
| Fig. 5 IR-1 Fundus and OCT images of rat eye. Upper panel. (A) Color fundus image before injection of liposomes containing IR-1 compound. (B) Fundus image with the filter combination of excitation: Semrock FF01-469/35 and barrier: Semrock BLP01-488R, before injection. (C) OCT image before injection. (D) Color fundus image 30 min after injection of liposomes containing IR-1 compound. (E) Fundus image with the filter combination: excitation Semrock FF01-469/35 and barrier Semrock BLP01-488R, 30 min after injection. (F) OCT image 30 min after injection. The liposomes are visible in the vitreous after the injection (yellow arrow). Lower panel. Following images are from same eye and same set of filters 22 and 48 h after injection. | ||
Wavelength of the light in ocular OCT imaging is in the near infra-red region. Therefore, the molecules that can absorb or emit light could cause contrast in the OCT images. Previous study has demonstrated OCT-based detection of microspheres in vivo in liver tissue.44 The contrast mechanisms of OCT are based on the absorption or back scattering of light. Endogenous melanin44 and exogenous indocyanine green45 are examples of contrast agents in OCT. Since the wavelength of light in OCT is in the near infrared region, and IR-1 and IR-2 do not have strong emission in this range, it is expected that the OCT signals of the labeled liposomes stem from physical light scattering. Interestingly, the signals of fluorescence fundus images and OCT overlap. This shows that the liposomes are detectable by both OCT and fluorescence imaging. Colocalization of the signals also indicates that the labels are stably embedded in the liposomes after intravitreal injections.
Combination of OCT and fluorescence imaging simultaneously may provide means to monitor simultaneously tissue microstructures (OCT) and molecular processes (fluorescence signals).46 Such approach may be interesting in the experiments of ocular drug safety and pharmacodynamics in vivo. Fluorescence signals may demonstrate the distribution of compound or drug delivery system, while OCT could reveal the distribution of materials and ocular microstructures, such as retinal layers.47
Fig. 6 shows the fundus images of the eyes of a rat at the age of more than one year. The images were captured by BLP01-488R and FF01-469/35 set of filters. Green autofluorescence, mostly originating from anterior ocular tissues (e.g. lens), is seen in this old rat. It is known that ocular green autofluorescence increases with aging.48 The rats in our liposome studies were 4 months old and they showed low natural autofluorescence in the eyes. With IR-1 and IR-2 dyes green autofluorescence background of the eyes can be eliminated, because IR-1 and IR-2 show red signals with this set of filters.
 | ||
| Fig. 6 Fundus image of both eyes of an one-year old rat (excitation: Semrock FF01-469/35 and barrier: Semrock BLP01-488R). Green natural autofluorescence is seen. | ||
Endogenous autofluorescence of posterior ocular tissues, like retina, is also an important factor that may affect the quality of fluorescence imaging. Marmorstein et al.49 elucidated the spectral profile of natural autofluorescence of human retina. They studied retinas of healthy and diseased (age-related macular degeneration) human subjects using different excitation and emission wavelengths. When excitation was performed at 488 nm, the emitted signal at 655 nm wavelength was minimal. It is noteworthy that we used excitation wavelength of 488 nm and monitored the IR-1 and IR-2 dyes at 657 nm and 647 nm, respectively. Therefore, the background noise from the retinal autofluorescence should be minimal for these dyes. This may open potential applications of these dyes in retinal research.
Another potential application of these dyes involves their simultaneous detection with green dyes, like fluorescein, in a single image. This is feasible, because BLP01-488R and FF01-469/35 filter sets are ideal for fluorescein angiography of the retina. For example, if we inject intravitreally compounds or formulations labeled with IR-1 or IR-2, and perform retinal angiography by systemic administration of fluorescein, we could still detect the intravitreally injected materials with the same set of filters in a single image. Retinal angiography is a valuable tool in the evaluation of retinal vessels, an important part in preclinical safety and drug efficacy studies. These new labels may broaden the information gain in such investigations.
Our results suggest that phosphorescence labeling of drug delivery systems can be used as a tool to follow intracellular and intraocular particle distribution. To the best of our knowledge, this is the first report of the applicability of a phosphorescent sensor for simultaneous use in in vivo fundoscopy and OCT. Compared to fluorescent labels, the phosphorescent labels are expected to show improved signal-to-noise ratio and negligible quenching in long-term experiments with prolonged action ocular drug delivery systems. Despite the preliminary character of these in vivo results this research allows to conclude that the developed iridium complexes have high potential for in vitro and in vivo bioimaging applications. Wider in vivo use, particularly in clinical settings, would require more thorough exploration of the safety aspects of these materials.
Conflicts of interest
There are no conflicts of interest to declare.Ethical statement
The animal experiments were approved by the national Animal Experiment Board (ELLA, Regional State Administrative Agency for Southern Finland). The experiments were performed under project license (ESAVI/8621/04.10.07/2017) and in compliance with 3Rs principle (replacement, reduction, refinement). The animal-welfare body of University of Eastern Finland Lab Animal Center (UEF LAC) monitored the use of the animals and followed the development and outcome of the project.Acknowledgements
Grant support from Russian Government Mega-Grant 14.W03.031.0025 “Biohybrid technologies for modern biomedicine” is acknowledged. The NMR, photophysical and analytical measurements were performed using the following core facilities at St. Petersburg State University Research Park: Centre for Magnetic Resonance and Centre for Chemical Analysis and Materials Research. We would like to thank the Bioactivity Screening core facility of HI-LIFE, University of Helsinki (Dr Polina Ilina) for enabling the use of Cytation 5 instrument.References
- H. Daraee, A. Etemadi, M. Kouhi, S. Alimirzalu and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 381–391 CrossRef CAS PubMed.
- Z. Amoozgar and Y. Yeo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 219–233 CAS.
- T. Lajunen, K. Hisazumi, T. Kanazawa, H. Okada, Y. Seta, M. Yliperttula, A. Urtti and Y. Takashima, Eur. J. Pharm. Sci., 2014, 62, 23–32 CrossRef CAS PubMed.
- H. Talsma, A. Y. Özer, L. van Bloois and D. J. A. Crommelin, Drug Dev. Ind. Pharm., 1989, 15, 197–207 CrossRef CAS.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS PubMed.
- O. Meyer, D. Kirpotin, K. Hong, B. Sternberg, J. W. Park, M. C. Woodle and D. Papahadjopoulos, J. Biol. Chem., 1998, 273, 15621–15627 CrossRef CAS PubMed.
- K. Maruyama, T. Takizawa, N. Takahashi, T. Tagawa, K. Nagaike and M. Iwatsuru, Adv. Drug Delivery Rev., 1997, 24, 235–242 CrossRef CAS.
- F. Szoka and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef CAS PubMed.
- A. Gomez-Hens and J. Fernandez-Romero, TrAC, Trends Anal. Chem., 2006, 25, 167–178 CrossRef CAS.
- T. Lajunen, R. Nurmi, L. Kontturi, L. Viitala, M. Yliperttula, L. Murtomäki and A. Urtti, J. Controlled Release, 2016, 244, 157–166 CrossRef CAS PubMed.
- P. S. Uster and R. E. Pagano, in Methods in Enzymology, 1989, pp. 850–857 Search PubMed.
- V. Torchilin and T. Levchenko, Curr. Protein Pept. Sci., 2003, 4, 133–140 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2005, 57, 95–109 CrossRef CAS PubMed.
- G. J. Strijkers, E. Kluza, G. A. F. Van Tilborg, D. W. J. van der Schaft, A. W. Griffioen, W. J. M. Mulder and K. Nicolay, Angiogenesis, 2010, 13, 161–173 CrossRef CAS PubMed.
- B. C. Roy, R. Peterson, S. Mallik and A. D. Campiglia, J. Org. Chem., 2000, 65, 3644–3651 CrossRef CAS PubMed.
- X. Yue and Z. Dai, Curr. Med. Chem., 2018, 25, 1397–1408 CrossRef CAS PubMed.
- A. Mechler, B. D. Stringer, M. S. H. Mubin, E. H. Doeven, N. W. Phillips, J. Rudd-Schmidt and C. F. Hogan, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2939–2946 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- F. R. Svensson, M. Li, B. Nordén and P. Lincoln, J. Phys. Chem. B, 2008, 112, 10969–10975 CrossRef CAS PubMed.
- S. Bonnet, B. Limburg, J. D. Meeldijk, R. J. M. Klein Gebbink and J. A. Killian, J. Am. Chem. Soc., 2011, 133, 252–261 CrossRef CAS PubMed.
- Y. Hisamatsu, A. Shibuya, N. Suzuki, T. Suzuki, R. Abe and S. Aoki, Bioconjugate Chem., 2015, 26, 857–879 CrossRef CAS PubMed.
- W. Jiang, Y. Gao, Y. Sun, F. Ding, Y. Xu, Z. Bian, F. Li, J. Bian and C. Huang, Inorg. Chem., 2010, 49, 3252–3260 CrossRef CAS PubMed.
- K. K.-W. Lo, P.-K. Lee and J. S.-Y. Lau, Organometallics, 2008, 27, 2998–3006 CrossRef CAS.
- C. Liao, D. Xu, X. Liu, Y. Fang, J. Yi, X. Li and B. Guo, Int. J. Nanomed., 2018, 13, 4417–4431 CrossRef CAS PubMed.
- W.-Y. Zhang, F. Du, M. He, L. Bai, Y.-Y. Gu, L.-L. Yang and Y.-J. Liu, Eur. J. Med. Chem., 2019, 178, 390–400 CrossRef CAS PubMed.
- T. M. Allen, Adv. Drug Delivery Rev., 1994, 13, 285–309 CrossRef CAS.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- A. G. Kohli, P. H. Kierstead, V. J. Venditto, C. L. Walsh and F. C. Szoka, J. Controlled Release, 2014, 190, 274–287 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- A. Urtti, Adv. Drug Delivery Rev., 2006, 58, 1131–1135 CrossRef CAS PubMed.
- E. M. del Amo, A. K. Rimpelä, E. Heikkinen, O. K. Kari, E. Ramsay, T. Lajunen, M. Schmitt, L. Pelkonen, M. Bhattacharya, D. Richardson, A. Subrizi, T. Turunen, M. Reinisalo, J. Itkonen, E. Toropainen, M. Casteleijn, H. Kidron, M. Antopolsky, K. S. Vellonen, M. Ruponen and A. Urtti, Prog. Retinal Eye Res., 2017, 57, 134–185 CrossRef CAS PubMed.
- R. A. Smith, E. C. Stokes, E. E. Langdon-Jones, J. A. Platts, B. M. Kariuki, A. J. Hallett and S. J. A. Pope, Dalton Trans., 2013, 42, 10347–10357 RSC.
- C. D. Sunesh, G. Mathai and Y. Choe, ACS Appl. Mater. Interfaces, 2014, 6, 17416–17425 CrossRef CAS PubMed.
- K. K. W. Lo, C. K. Chung, T. K. M. Lee, L. H. Lui, K. H. K. Tsang and N. Zhu, Inorg. Chem., 2003, 42, 6886–6897 CrossRef CAS PubMed.
- T. Lajunen, L. S. Kontturi, L. Viitala, M. Manna, O. Cramariuc, T. Róg, A. Bunker, T. Laaksonen, T. Viitala, L. Murtomäki and A. Urtti, Mol. Pharm., 2016, 13, 2095–2107 CrossRef CAS PubMed.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- K. J. Lee, W.-K. Oh, J. Song, S. Kim, J. Lee and J. Jang, Chem. Commun., 2010, 46, 5229–5231 RSC.
- K. Hasan, L. Donato, Y. Shen, J. D. Slinker and E. Zysman-Colman, Dalton Trans., 2014, 43, 13672–13682 RSC.
- B. Tong, P. Ma, Q. Mei and Z. Hua, Inorg. Chim. Acta., 2014, 421, 405–409 CrossRef CAS.
- B. Tong, J. Qiang, Q. Mei, H. Wang, Q. Zhang and Z. Han, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 213–218 CAS.
- Q. Zhao, M. Yu, L. Shi, S. Liu, C. Li, M. Shi, Z. Zhou, C. Huang and F. Li, Organometallics, 2010, 29, 1085–1091 CrossRef CAS.
- J. R. Shakirova, O. A. Tomashenko, E. E. Galenko, A. F. Khlebnikov, P. Hirva, G. L. Starova, S.-H. Su, P.-T. Chou and S. P. Tunik, Inorg. Chem., 2018, 57, 6853–6864 CrossRef CAS PubMed.
- K. C. Dunn, A. E. Aotaki-Keen, F. R. Putkey and L. M. Hjelmeland, Exp. Eye Res., 1996, 62, 155–170 CrossRef CAS PubMed.
- T. M. Lee, A. L. Oldenburg, S. Sitafalwalla, D. L. Marks, W. Luo, F. J.-J. Toublan, K. S. Suslick and S. A. Boppart, Opt. Lett., 2003, 28, 1546–1548 CrossRef CAS PubMed.
- J. P. Ehlers, S. McNutt, S. Dar, Y. K. Tao and S. K. Srivastava, Br. J. Ophthalmol., 2014, 98, 1588–1591 CrossRef PubMed.
- S. Yuan, C. A. Roney, J. Wierwille, C.-W. Chen, B. Xu, G. Griffiths, J. Jiang, H. Ma, A. Cable and R. M. Summers, Phys. Med. Biol., 2009, 55, 191 CrossRef PubMed.
- C. Yang, Photochem. Photobiol., 2005, 81, 215–237 CrossRef CAS.
- F. Holz, A. C. Bird, S. Schmitz-Valckenberg and R. Spaide, Atlas of Fundus Autofluorscence Imaging, Springer, 2007, vol. 1 Search PubMed.
- A. D. Marmorstein, L. Y. Marmorstein, H. Sakaguchi and J. G. Hollyfield, Invest. Ophthalmol. Visual Sci., 2002, 43, 2435–2441 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01114b |
| This journal is © The Royal Society of Chemistry 2020 |