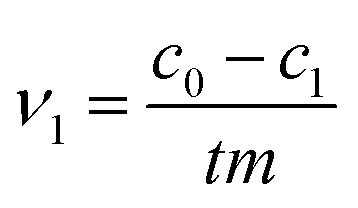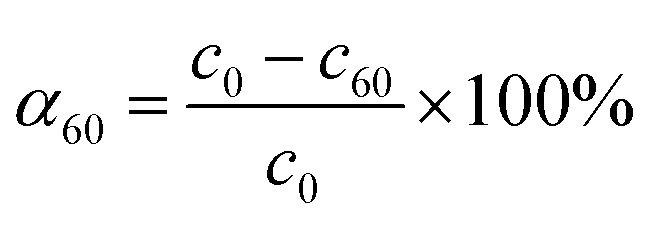DOI:
10.1039/D0RA00937G
(Paper)
RSC Adv., 2020,
10, 14441-14450
Room-temperature synthesis of nanoceria for degradation of organophosphate pesticides and its regeneration and reuse†
Received
31st January 2020
, Accepted 26th March 2020
First published on 8th April 2020
Abstract
A simple low-temperature water-based and one-pot synthesis was developed for the preparation of nanocrystalline CeO2 that was used for degradation of the toxic organophosphate pesticide parathion methyl. By changing the reaction temperature in the range from 5 °C to 95 °C, several properties (i.e., crystallinity, grain size and surface area) of nanoceria can be easily controlled. The catalytic decomposition of parathion methyl to its degradation product 4-nitrophenol was highly dependent on the CeO2 preparation temperature. It was demonstrated that at low temperature (i.e. 5 °C), CeO2 with very small crystallites (<2 nm) and high surface area can be obtained. For practical use, it was demonstrated that highly crystalline CeO2 can be prepared at room-temperature (30 °C) in at least 100 g batches. It was shown that precipitated nanoceria had high thermal stability and its post-synthesis annealing up to 400 °C did not significantly alter the material properties and hence the catalytic activity. Furthermore, as shown by the reusability tests, the sorbent can be reactivated by simply washing with water which demonstrated its durability.
Introduction
The applications of CeO2 are very diverse, though they are most often related to some type of catalysis. For example, CeO2 is used as non-active support for alumina in three-way catalysts1 due to its thermal stability which increases the lifetime of the catalyst. Nowadays, cerium oxide is also used in solid oxide fuel cells2 for the catalytic burning of hydrogen to provide electricity. CeO2 is also widely applied in scavenging of radicals3 or in drug delivery systems.4 New applications of ceria emerge in biology, biochemistry and biotechnology, thanks to the pseudoenzymatic properties5 of CeO2 such as phosphomonoesterase-like activity6 or phosphatase-like activity.7 These new directions utilize mainly nanostructured CeO2 called nanoceria. Thus, the properties and applications of nanoceria are deeply studied as documented by the abundant literature available.8–10 Its properties may be further modified either by doping11 of its structure or by depositing ceria on a conductive substrate (such as gold12). Nevertheless, only a few studies13 are devoted to the deep evaluation of the synthesis conditions such as temperature or composition of reaction atmosphere on the synthesis process.
CeO2 nanoparticles can be prepared by various procedures, such as calcination of the suitable precursor, reflux or hydrothermal synthesis in an autoclave at elevated pressure, by low-temperature synthesis at 90 °C or using photochemical synthesis. These procedures were compared for preparation of CeO2 (ref. 14 and 15) or CeO2 composites with samarium.16 Many studies are focused on the shape engineering of ceria to obtain cubic, rod-like17 and plate-like18 particles. Nowadays very popular sol–gel methods are suitable for preparation of homogeneously doped materials, for example Ce-doped TiO2 (ref. 19) or Ce-doped YAG,20 and preparation of ultrafine pure metal oxides nanoparticles of Zn, Sn, Ti, Zr, Fe, Ni, Ga, Mn, In and Ce.21 However, the calcination at high temperatures or high-pressure treatment or multi-step approach is usually necessary. Modern green chemistry favours low-temperature one-pot syntheses, especially for potential industrial production, where the energy consumption and the complexity of the synthesis and hence its price must be reduced. For example, the low-temperature synthesis was used to prepare a photocatalytic composite with titanium nanorods,22 nevertheless the effect of reaction temperature and the effect of cerium salt concentration was not discussed in detail.
Another interesting application of nanoceria is decomposition of phosphoesters,23 (e.g. organophosphate pesticides, warfare agents and others). However, its catalytic activity can be inhibited by the degradation product24 that leads to inactivation of the catalyst for further use. It is therefore highly desirable to find a way how to prevent or eliminate catalyst inactivation and find an easy process how to regenerate its function. It has been previously described25 that both CeO2 bivalent character26 (also found in Mn3O4, MnFe2O4 and other metal oxides) and surface –OH groups play an important role in its catalytic properties. It was suggested that –OH surface groups are consumed during the degradation reactions but can be easily replenished and thus the catalyst can be regenerated.
In this work, we prepared nanocrystalline cerium oxides by simple low-temperature synthesis. We demonstrated that by simple changing of the reaction temperature, the properties such as particle size, specific surface area, porosity or surface chemical composition can be modified. The catalytic activity of prepared materials was tested by decontamination of parathion methyl in non-aqueous solution. Reusing of catalyst led to the decrease of its catalytic activity although this was not observed during testing in water in our previous study.27 We combined these two procedures and demonstrated that the catalyst used in a non-aqueous solution can be reactivated by water washing for further use.
Experimental
Materials and chemicals
All chemicals were obtained from commercial sources and were used without further purification. Cerium salt, Ce(NO3)3·6H2O, was obtained from Sigma-Aldrich (Czech Rep.) with 99% purity. Organophosphate pesticide parathion methyl (PM) and its degradation product 4-nitrophenol (4-NP) were obtained from Sigma-Aldrich as chromatographic standards. Ammonium hydroxide solution (25 to 29)% was obtained from Penta (Czech Rep.) with p.a. purity. HPLC gradient grade organic solvents (acetonitrile, methanol) and deionized water were used to prepare the solutions including mobile phases for liquid chromatography.
Synthesis of CeO2
In a typical synthesis, 500 mL of 20 mM cerium(III) nitrate solution in a closed stirred beaker was purged by CO2-free air (CO2 was removed by passing through 1 M KOH solution to prevent undesired formation of carbonates) for couple of minutes and heated to the desired temperature: 5, 20, 40, 60, 80 and 95 °C. The slow addition of 10 mL ammonia solution to adjust pH above 10 led to precipitation of white cerium hydroxide. The mixture was further stirred and purged with CO2-free air until it gradually transformed to yellowish CeO2 (typically after ca. 4–5 hours). The obtained powder was washed with deionized water several times and air-dried at 60 °C. The samples were denoted as CeT_xx, where “xx” is the reaction temperature.
Sample CeS_30 was prepared analogously at room temperature (30 °C) in a larger amount. Concretely, 12 L of 60 mM cerium salt solution was used in a 15 L reactor to yield 100 g of the powder sample that was used for extensive degradation and reusability tests and for preparing samples annealed at 400, 600, 800 and 1000 °C. The samples were denoted as CeA_yyyy, where yyyy is the annealing temperature.
Methods of characterization
The diffraction patterns were collected using Bruker D2 equipped with conventional X-ray tube (Cu Kα radiation, 30 kV, 10 mA) and the LYNXEYE 1-dimensional detector. The primary divergence slit module width 0.6 mm, Soler Module 2.5, Airscatter Screen Module 2 mm, Ni-Kβ filter 0.5 mm, in the 2θ range of 5° to 90° with step 0.00808° and time per step 1.0 s were used. The crystallite size was calculated in X'Pert HighScore Plus software. XPS measurements were performed using a PHI VersaProbe II XPS system (Physical Electronics, Chanhassen, USA) with a monochromatic micro-focused Al-Kα source (15 kV, 50 W) and photon energy of 1486.7 eV. The spectra were measured in a vacuum (1.3 × 10−8 Pa) at 20 °C. The survey scan spectra were measured with a pass energy of 187.850 eV, a binding energy range of (0 to 1300) eV, and an electron-volt step of 0.8 eV, while the high-resolution spectra were measured with a pass energy of 23.500 eV and an electron-volt step of 0.2 eV. Dual-beam compensation was used for all measurements. The obtained spectra were evaluated using MultiPak software (version 9.4.0.7; Ulvac-PHI, Inc.). All the binding energy values were referenced to the carbon peak C 1s at 284.80 eV. The Raman spectra were acquired with DXR Raman microscope (Thermo Scientific) with 780 nm (10 mW) laser, under 10× objective of Olympus microscope. Setsys Evolution 1750 thermal analysis instrument from Setaram and QMG 422 quadrupole mass spectrometer from Pfeiffer. The Setaram device uses a sophisticated QMS module connection system called SuperSonic System, which accelerates releasing gas molecules and introduces them directly into MS. The crystalline structure, shape and size of nanoceria were inspected by high-resolution transmission electron microscopy (HRTEM) using a 200 kV TEM microscope FEI Talos F200X. As a specimen support for TEM investigations, a microscopic copper grid covered by a thin transparent holey carbon film was used. The sample suspensions were sonicated for several minutes, and drop casted onto copper grids. The specific surface area and porosity of the powder samples were measured using a Belsorp Max II instrument at liquid nitrogen temperature. The samples were degassed for 4 hours at 120 °C. The Brunauer–Emmett–Teller (BET) method was used for surface area calculation. The Non-Local Density Functional Theory (NLDFT) was used for pore size distribution calculation.
Parathion-methyl degradation tests
The decomposition of organophosphorus pesticide parathion-methyl (PM) in acetonitrile was studied by our well-established procedure.28 In detail, 50 mg of powder sample was weighed into a glass vial and 4 mL of 500 mg L−1 PM in acetonitrile (MeCN) solution was added. The closed vial was stirred at 1000 rpm at constant temperature (20 °C) in a thermoshaker during the degradation test. 0.1 mL of the reaction mixture was withdrawn from the vial at 0.25, 1, 2, 5, 10, 20, 30, 60 and 120 min and mixed with 0.9 mL of water. The solution was centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min and the residual concentration of PM and concentration of 4-NP formed were measured immediately by HPLC. To determine the reaction balance of the PM degradation and 4-NP formation on the sample CeS_30 after 60 min, the sample was extracted by MeOH and MeCN and measured by HPLC. The extraction was made by methanol because the reaction does not proceed in this solvent and the adsorbed PM and 4-NP are effectively released into solution. In detail, 20 mg of sorbent was weighed into a 2 mL centrifuge tube and 1.6 mL of the PM standard (500 mg L−1 in MeCN) was added. The vial was kept in thermoshaker (1000 rpm/20 °C) during the degradation test. The mixture was centrifuged and washed two times with pure MeOH and then twice more with MeCN. All centrifuged aliquots were combined into a 10 mL volumetric flask which was topped up with water and measured.
000 rpm for 2 min and the residual concentration of PM and concentration of 4-NP formed were measured immediately by HPLC. To determine the reaction balance of the PM degradation and 4-NP formation on the sample CeS_30 after 60 min, the sample was extracted by MeOH and MeCN and measured by HPLC. The extraction was made by methanol because the reaction does not proceed in this solvent and the adsorbed PM and 4-NP are effectively released into solution. In detail, 20 mg of sorbent was weighed into a 2 mL centrifuge tube and 1.6 mL of the PM standard (500 mg L−1 in MeCN) was added. The vial was kept in thermoshaker (1000 rpm/20 °C) during the degradation test. The mixture was centrifuged and washed two times with pure MeOH and then twice more with MeCN. All centrifuged aliquots were combined into a 10 mL volumetric flask which was topped up with water and measured.
To monitor parathion-methyl (PM) and its degradation product 4-nitrophenol (4-NP) high-performance liquid chromatography (HPLC) system with a diode array detector (DAD) DIONEX UltiMate 3000 (Thermo Scientific™, Palo Alto, USA) was used. Chromatographic analysis was carried out in a reverse phase system (RPLC-C18) on AccucoreTM column, 2.6 μm, PFP, 150 × 4.6 mm. Acetonitrile (MeCN), methanol (MeOH) and water (H2O) acidified with formic acid (HCOOH, 0.1%) were used as mobile phases for gradient elution. Further details are given in the electronic ESI in Table 1S.†
Results and discussion
Influence of the reaction temperature on material properties
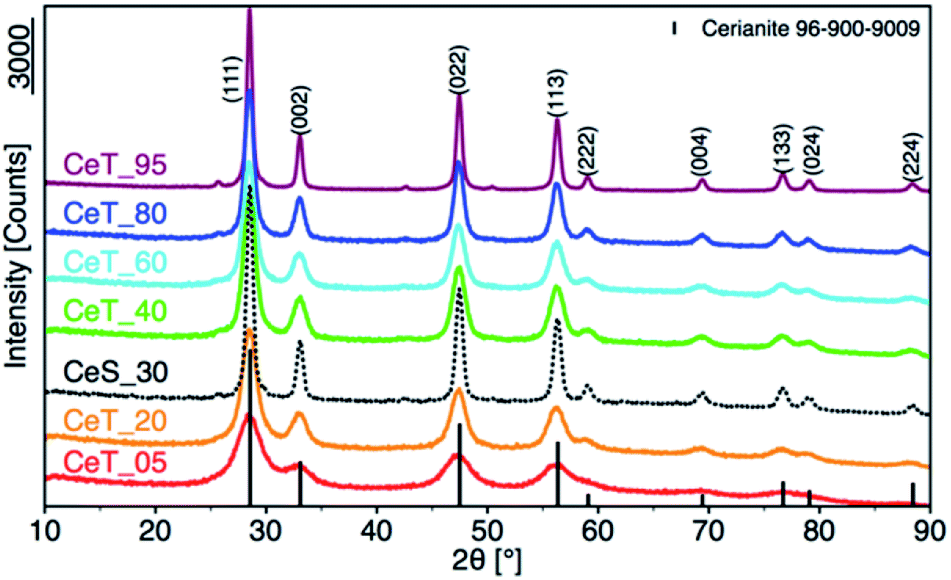
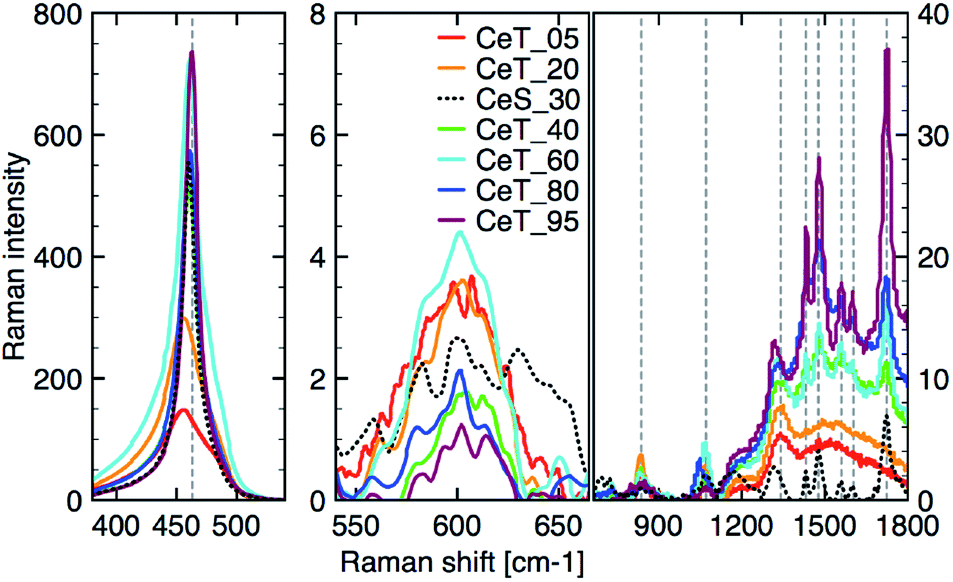
The simple precipitation of cerium nitrate water solution by ammonia under CO2-free air in the temperature range from 5 °C to 95 °C leads consequently to the formation of pure crystalline CeO2 nanostructures in all samples as show XRD data (Fig. 1). It demonstrates that the pure and well-crystallized nanoceria can be prepared even at low (5 °C) and laboratory temperature (20 to 30) °C without the need of annealing. The grain size was calculated using Williamson–Hall plot for all determined lines (Table 1). Results shows strong dependency of the grain size on the reaction temperature as also confirmed by TEM measurements (Table 1 and Fig. 2). By simple changing the reaction temperature from 5 °C to 95 °C, the nanoparticles with sizes ranging from 2 nm to 15 nm, respectively, can be easily prepared. At low temperature (5 °C), very small crystallites with indistinct grain boundaries are formed (Fig. 2a), while with increasing temperature the nanocrystals grow and form well-distinguishable loosely aggregated nanoparticles with distinct individual cubic morphology produced at a higher temperature (95 °C). Note that the nanoparticles have a crystalline structure even if they were synthesized at 5 °C though the individual grains are difficult to discern, as shown in Fig. 3a. Comparison with the sample prepared in scale-up synthesis at room temperature (CeS_30) is shown in Fig. 3S in an ESI.† The linear growth of the grain size also corresponds to the shift and increase of the intensity of the dominant F2g band related to the Ce–O bond29 in the Raman spectra of the prepared samples (Fig. 4). The band is shifted from 455 cm−1 for CeT_05 to 463 cm−1 for CeT_95.
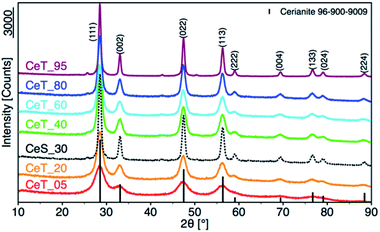 |
| | Fig. 1 X-ray powder diffraction patterns of samples prepared at different reaction temperatures. | |
Table 1 Grain size calculated from XRD and obtained by TEM, specific surface area (SSA), mean pore diameter (MPD), total pore volume (Vpore), weight loss at 1000 °C (Δm1000), content of Ce(III) and Ce(IV) by XPS, Raman intensity of oxygen vacancies band (O-vac) of the prepared samples
| Sample |
Grain size (XRD), (nm) |
Grain size (TEM), (nm) |
SSA, (m2 g−1) |
MPD, (nm) |
Vpore, (cm3 g−1) |
Δm1000, (%) |
Ce(III), (%) |
Ce(IV), (%) |
O-vac 600 cm−1, (Raman int.) |
| CeT_05 |
2 |
3 |
112 |
3.5 |
0.12 |
11.5 |
29.4 |
70.6 |
3.5 |
| CeT_20 |
3 |
5 |
128 |
4.7 |
0.17 |
8.3 |
31.1 |
68.9 |
3.5 |
| CeT_40 |
5 |
6 |
106 |
5.0 |
0.15 |
5.8 |
25.7 |
74.3 |
1.5 |
| CeT_60 |
8 |
7 |
115 |
5.6 |
0.18 |
5.0 |
19.7 |
80.3 |
4.5 |
| CeT_80 |
10 |
9 |
89 |
7.6 |
0.18 |
3.9 |
16.5 |
83.5 |
2.0 |
| CeT_95 |
14 |
13 |
83 |
14.0 |
0.18 |
2.1 |
22.2 |
77.8 |
1.0 |
| CeS_30 |
11 |
12 |
68 |
8.9 |
0.15 |
3.0 |
21.0 |
79.0 |
2.4 |
 |
| | Fig. 2 The growth of crystalline nanoparticles and the loss of the amorphous fraction is visible in a series of TEM images of samples prepared at (a) 5, (b) 20, (c) 40, (d) 60, (e) 80 and (f) 95 °C. | |
 |
| | Fig. 3 High-resolution transmission electron images of the lattice structure of oriented crystals along the axis (111) for samples prepared at (a) 5, (b) 20, (c) 40, (d) 60, (e) 80 and (f) 95 °C. | |
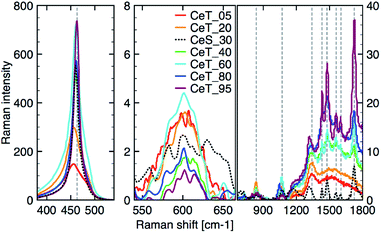 |
| | Fig. 4 Raman spectra after background correction of the three different regions from left: (350 to 550) cm−1; (550 to 700) cm−1; (700 to 1800) cm−1. | |
Furthermore, D-band centered at about 600 cm−1 suggesting a formation of the oxygen vacancies30 was found in all prepared samples, but its intensity was the highest for samples CeT_05, CeT_20 and CeT_60. The bands in the region (700 to 1800) cm−1 result from dissociative adsorption of CO2 on the CeO2 surfaces31 that is typical for samples stored at ambient air. Interestingly, the intensity of these bands increases for the samples prepared at higher temperature suggesting different reactivity and adsorption properties of the prepared samples.
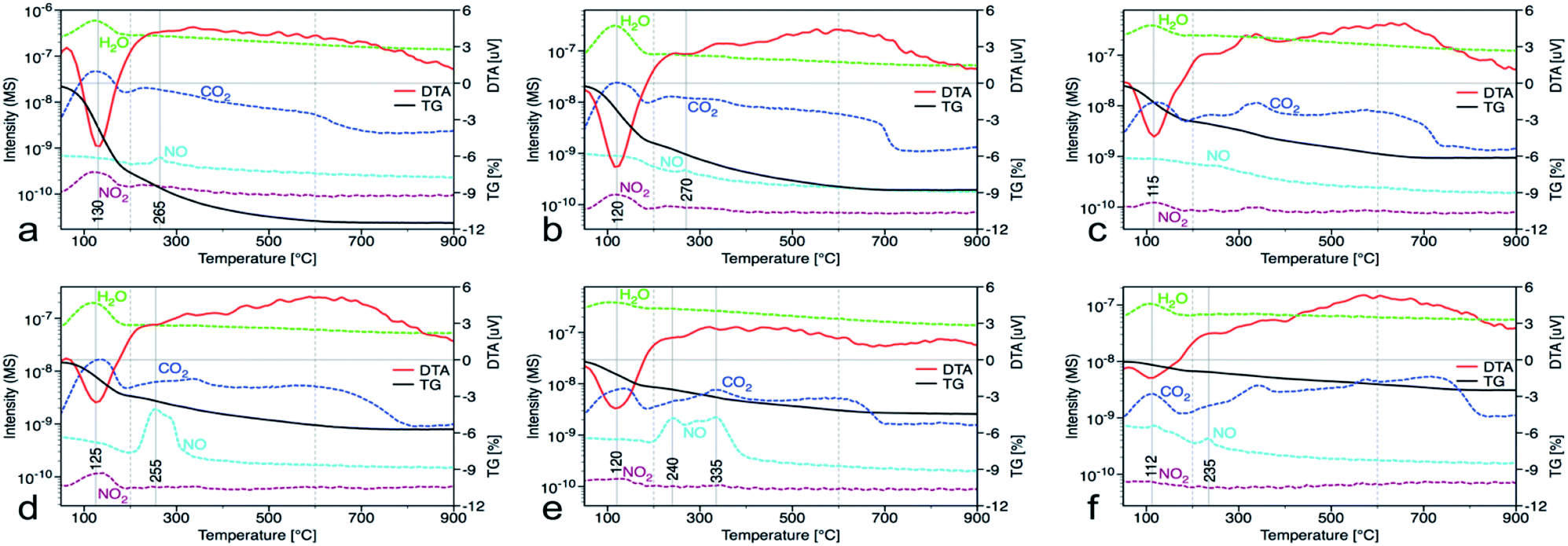
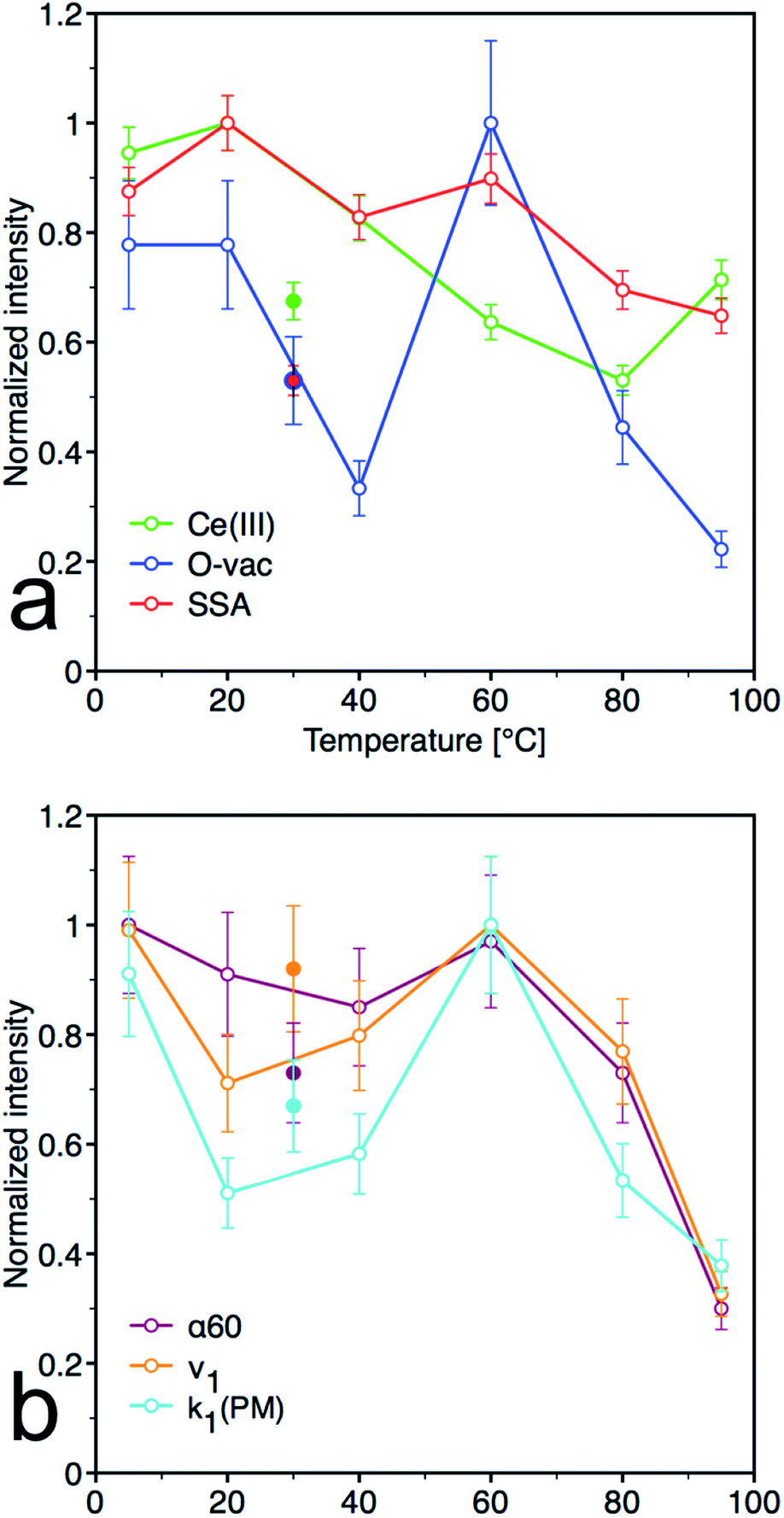
In the nanocrystalline CeO2, the surface Ce(III)/Ce(IV) ratio is one of the important parameters that can be obtained by XPS measurements. Nevertheless, it is important to mention that measurements under strong vacuum may induce the formation of Ce(III) states. The results (Table 1 and Fig. 1S†) show that higher Ce(III)/Ce(IV) ratio was in samples prepared at lower reaction temperature (up to 60 °C). The amount of Ce(III) ions is well correlated with the formation of oxygen vacancies as show Raman data. This suggests that at lower temperatures, more defectious particles are formed that can be related to the soft synthesis procedure employed. The higher amount of amorphous fraction (detected by HRTEM) was present at lower temperatures and can be due to the higher amount of precipitated Ce(OH)3 gel that has not been fully oxidized (as suggest higher amount of Ce3+ detected by XPS). Different properties among the samples were also revealed by thermal analysis with MS detection (Fig. 5) (for comparison with CeS_30 sample see Fig. 4S in an ESI†). The sample weight loss linearly decreases with increasing synthesis reaction temperature from 12% (for CeT_05) to 2% (for CeT_95). Interestingly, a significant difference was observed in the releasing of residual (NO3)− ions from samples originating probably from Ce precursor. While samples synthesized at low temperatures (5, 20 and 40 °C) release weakly bound nitrates mainly as NO2 at relatively low temperature (up to 130 °C), CeT_60 sample released both NO2 and NO but the latter at significantly higher temperature (200 to 250) °C. In the sample CeT_80, mainly NO releasing at even higher temperatures (200 to 350) °C was detected while in the sample CeT_95, only negligible amount of NO without any NO2 was registered. This suggests the different binding of (NO3)− in the crystal lattice of the samples prepared at various temperature and it also may be strongly related to the surface structure, Ce(III)/Ce(IV) ratio and oxygen vacancies in the samples. Most of the mass from samples, especially water and carbon dioxide, is released between temperatures 112 °C (for CeT_95) and 130 °C (for the CeT_05). It is also evident that CO2, resulting from surface adsorbed carboxylates and carbonates,31 is released continuously up to temperatures (650–800) °C (Table 1).
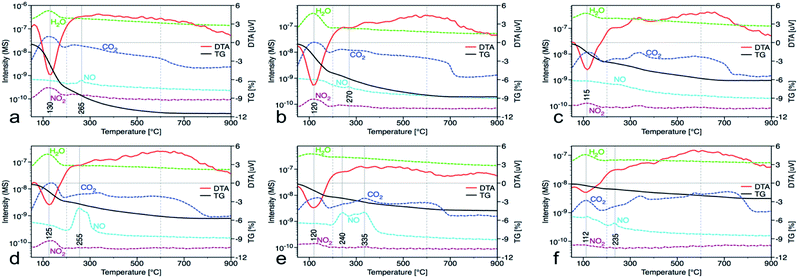 |
| | Fig. 5 Thermal gravimetric (black) and differential thermal analysis (red), along with records of released water (green), carbon dioxide (dark blue), nitrogen dioxide (dark red) and nitrous oxide (light blue) from the mass spectrometer for samples prepared at (a) 5, (b) 20, (c) 40, (d) 60, (e) 80 and (f) 95 °C. | |
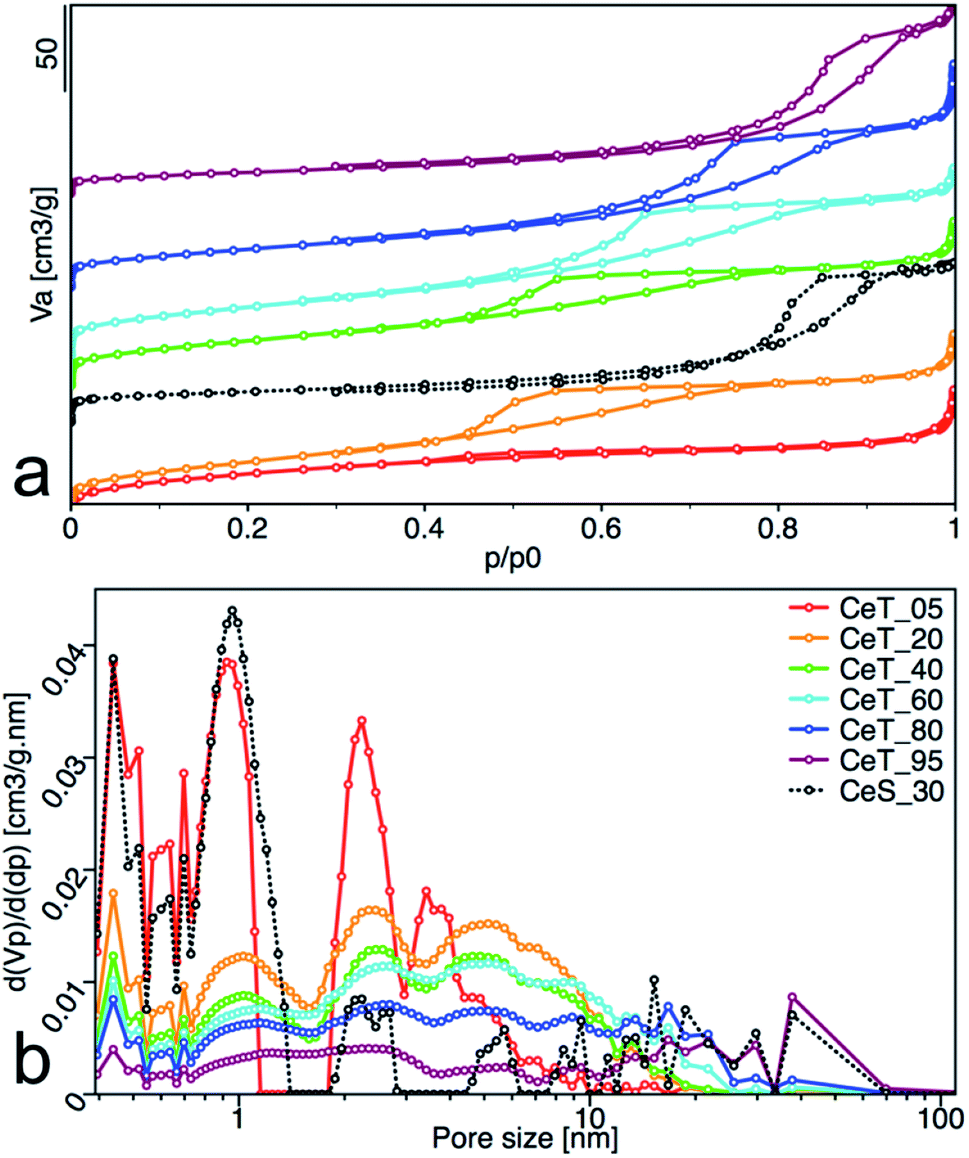
Increasing reaction temperature also leads to a decrease of the specific surface area from 112 m2 g−1 at 5 °C and 128 m2 g−1 at 20 °C, to 83 m2 g−1 at 95 °C (Table 1). All samples showed type IV isotherm with hysteresis which is characteristic for finely non-aggregated (powdered or nanoparticulate) non-porous samples with capillary condensation in the interparticle space. Interestingly, as show the changing shape of the hysteresis (Fig. 6a) and its shift to higher p/p0 values, the shape of the pores and their size distribution (Fig. 6b) is strongly dependent on the reaction temperature. While the hysteresis of the CeT_05 sample is close to the H4 type with crevice pores in the microporous region, as the temperature rises, hysteresis transforms to H2 type suggesting formation of the complex porous structure consisting mainly of interconnected porous networks with pores with different widths and shapes. NLDFT calculation (Fig. 6b) shows that all materials are mesoporous with pores between (2 to 30) nm with a significant amount of micropores that are most evident in the CeT_05 sample. Similarly, to the calculated grain size (XRD data, Table 1), the mean pore diameter of the samples linearly increases with increasing reaction temperature. This demonstrates, in accordance with XRD and TEM observations, that the particle size and porosity can be easily controlled by simple changing of the reaction temperature. The total pore volume of all samples is between (0.1 to 0.2) cm3 g−1 (Table 1).
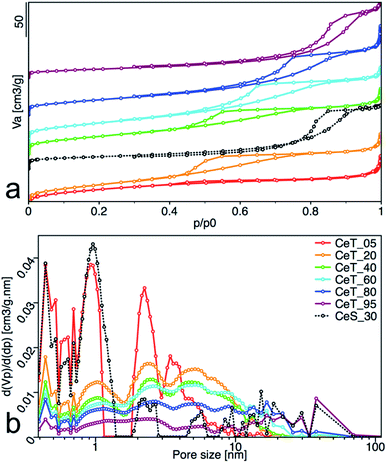 |
| | Fig. 6 (a) Nitrogen adsorption/desorption isotherms, and (b) pore-size distribution obtained from NLDFT analysis of the nanoceria samples. The adsorbed volume (Va), the partial derivative pore volume (Vp) with respect to the pressure difference (dp) and the relative pressure (p/p0) are plotted on the axes. | |
Parathion-methyl degradation
The materials properties described above determine the degradation activity of the prepared materials. To compare their activity, the catalytic decomposition of the organophosphate pesticide parathion methyl (PM) in MeCN solution used previously32 was investigated. PM degradation and formation of its product 4-nitrophenol (4-NP) can be easily monitored in a simple arrangement by HPLC-DAD. From the measured data, it was possible to determine the kinetics and rate constants for PM degradation and 4-NP formation, the initial rate of degradation and the degree of conversion for PM (Table 2).
Table 2 Kinetics rate constants k1 and k2, for PM degradation and formation of 4-NP, initial rate (ν1) and degree of conversion after 60 minutes (α60) on the prepared nanoceria samples
| Sample |
k1 (PM), (min−1) |
k2 (PM), (min−1) |
ν1, (mmol L−1 min−1 g−1) |
α60, (%) |
k1 (4-NP), (min−1) |
k2 (4-NP), (min−1) |
| CeT_05 |
0.674 |
0.038 |
10.3 |
93.7 |
0.864 |
0.050 |
| CeT_20 |
0.378 |
0.026 |
7.4 |
84.9 |
0.544 |
0.016 |
| CeT_40 |
0.431 |
0.023 |
8.3 |
80.0 |
0.476 |
0.002 |
| CeT_60 |
0.740 |
0.035 |
10.4 |
90.6 |
0.772 |
0.028 |
| CeT_80 |
0.395 |
0.009 |
8.0 |
68.0 |
0.603 |
0.009 |
| CeT_95 |
0.280 |
0.001 |
3.4 |
28.4 |
0.145 |
0.006 |
| CeS_30 |
0.496 |
0.022 |
9.6 |
68.1 |
0.378 |
0.009 |
The initial rate ν1 was calculated by formula (1). The degree of conversion α60 was calculated by formula (2):
| |
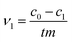 | (1) |
| |
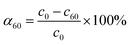 | (2) |
where
c0,
c1 and
c60 are the initial concentration and concentrations at given time
t of 1 and 60 minutes respectively, and
m is the mass of CeO
2 in the reaction.
The rate constants were calculated according to the model used in our previous work.33 Formula (3) is for PM degradation and formula (4) for 4-NP formation, respectively.
| | |
qPMt = qPM1e(−k1t) + qPM2e(−k2t) + qPM∞
| (3) |
| | |
q4NPt = 1 − (q4NP1e(−k1t) + q4NP2e(−k2t) + q4NP∞)
| (4) |
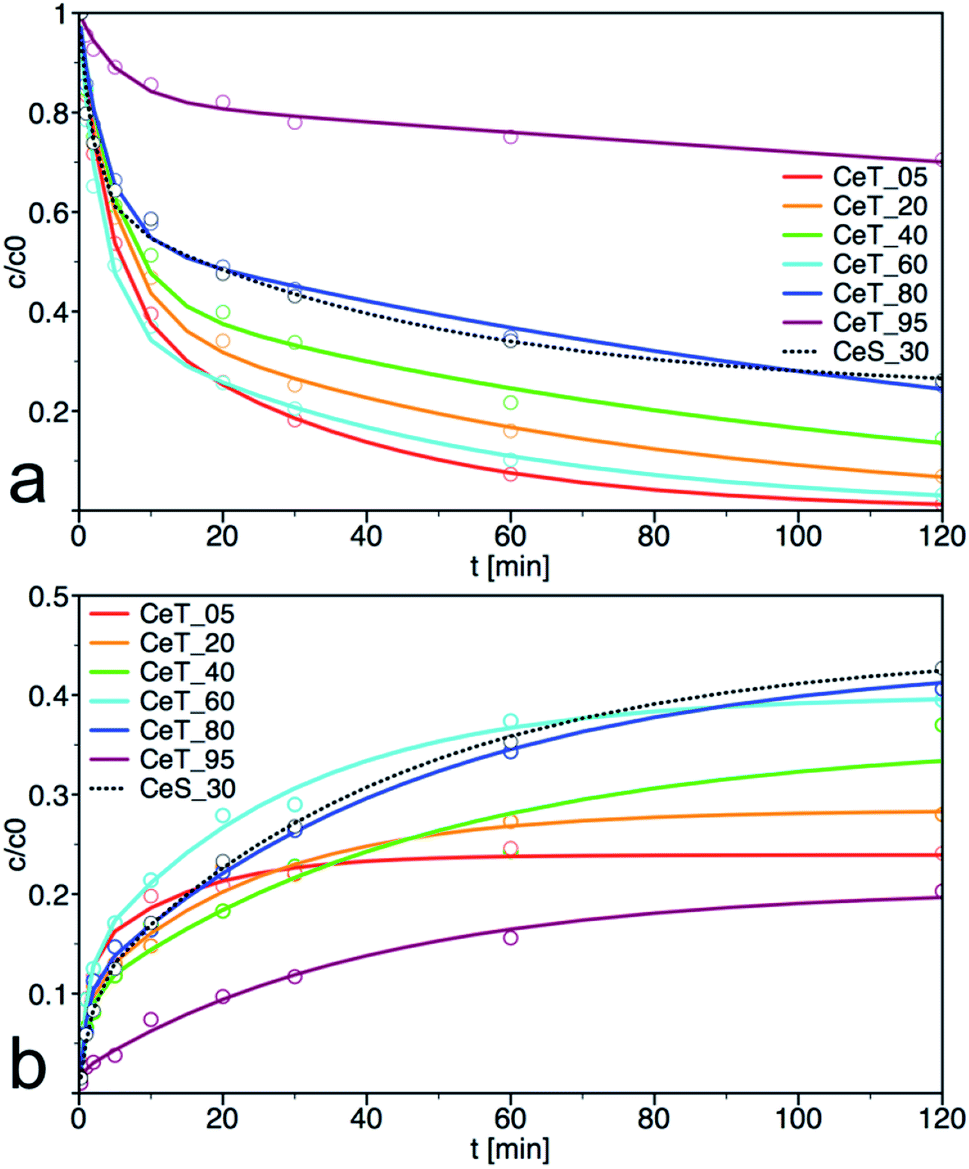
As show the kinetics curves of both PM degradation and formation of 4-NP (Fig. 7), the degradation efficiency of prepared samples differs significantly. The highest initial rate of PM decomposition was observed on the samples Ce_T05 and Ce_T60, the latter also had the highest degree of conversion at 60 min. Note that the degradation product 4-NP is after its formation partially released to the solution but also adsorbed on the surface of the catalyst and therefore, it is not possible to calculate the reaction balance in this experimental arrangement. The highest amount of the degradation product 4-NP at 120 min was in the reaction mixture catalysed by CeT_80, even though the amount of PM decomposition on this sample was not the highest. It can be argued that the amount of 4-NP released into the solution is inversely proportional to the specific surface area of the catalyst. Therefore, we investigated also the reaction balance on selected sample CeS_30 (discussed further below).
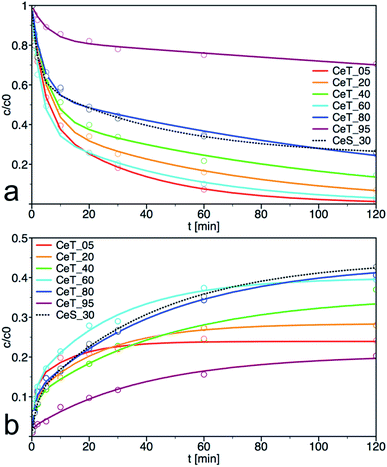 |
| | Fig. 7 Kinetic curves for (a) degradation of parathion methyl (PM) and (b) formation of 4-nitrophenol (4-NP) on prepared samples. The solid lines are least-square best fits of the data to eqn (1). | |
From the plots of the normalized materials characteristics (specific surface area, amount of oxygen vacancies and Ce(III) surface content) and normalized kinetic parameters for catalytic decomposition of PM in Fig. 8, it is clear that materials properties, mainly the amount of oxygen vacancies, significantly influence the catalytic activity. Note that the other parameters, such as content of the amorphous fraction especially in the samples prepared at low temperature, can also influence these relationships. CeT_60 sample that has the highest content of O-vac and still relatively high surface area showed the highest degree of conversion, high rate constants and the largest amount of 4NP released into solution. Therefore 60 °C seems to be the golden mean and a good compromise between the lowest and highest temperatures used in this low-temperature synthesis.
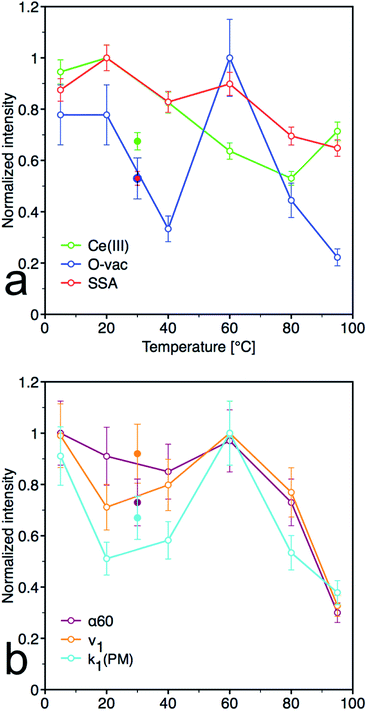 |
| | Fig. 8 Normalized pots for (a) material parameters [Ce(III) content determined by XPS, oxygen vacancies (O-vac) and specific surface area (SSA)] and (b) kinetic parameters [initial rate (ν1), kinetics rate constants k1 for decrease of PM and degree of conversion after 60 minutes (α60)]. Full dots represent sample CeS_30, empty dots represent samples CeT_xx. | |
Influence of the post-synthesis annealing temperature on material properties
To demonstrate the feasibility of the preparation procedure, we have tested scale-up of the synthesis to prepare 100 g of the sample at relatively mild laboratory temperature 30 °C (denoted as CeS_30). This sample was also used to investigate the effect of post-synthesis annealing on the structure and catalytic properties. Interestingly, the grain size (Table 3, calculated from XRD data presented in Fig. 2S†) nor BET surface area of the samples annealed at 400 °C and 600 °C do not change significantly compared to untreated sample. On the other hand, annealing at 800 °C and 1000 °C induce gradual increase of the grain size that was observed by TEM (Fig. 9) and rapid decrease of the surface area (Table 3). These results show very good thermal stability of the prepared sample and demonstrated that the grains as small as 11 nm can be maintained up to temperature 400 °C. This could be particularly beneficial for example in catalytic decomposition of gaseous pollutants of combustion products at high temperatures.
Table 3 Grain size calculated from XRD, specific surface area (SSA), mean pore diameter (MPD), total pore volume (Vpore), initial rate (ν1), degree of conversion after 60 minutes (α60), kinetics rate constants k1 and k2 for the decrease of PM for annealed samples
| Sample |
Grain size (XRD), (nm) |
SSA, (m2 g−1) |
MPD, (nm) |
Vpore, (cm3 g−1) |
ν1, (mmol L−1 min−1 g−1) |
α60, (%) |
k1 (PM), (min−1) |
k2 (PM), (min−1) |
| CeS_30 |
11 |
68 |
8.9 |
0.15 |
9.6 |
68.1 |
0.496 |
0.022 |
| CeA_400 |
11 |
67 |
9.3 |
0.16 |
7.7 |
70.7 |
0.383 |
0.023 |
| CeA_600 |
21 |
53 |
10.6 |
0.14 |
6.3 |
60.0 |
0.409 |
0.028 |
| CeA_800 |
100 |
14 |
14.1 |
0.05 |
0.8 |
14.5 |
0.090 |
0.090 |
| CeA_1000 |
186 |
<1 |
11.2 |
<0.001 |
— |
— |
— |
— |
 |
| | Fig. 9 The temperature-induced growth of nanoparticles is visible in a series of transmission electron microscope images of sample CeS_30 annealed to (a) 400, (b) 600, (c) 800 and (d) 1000 °C. | |
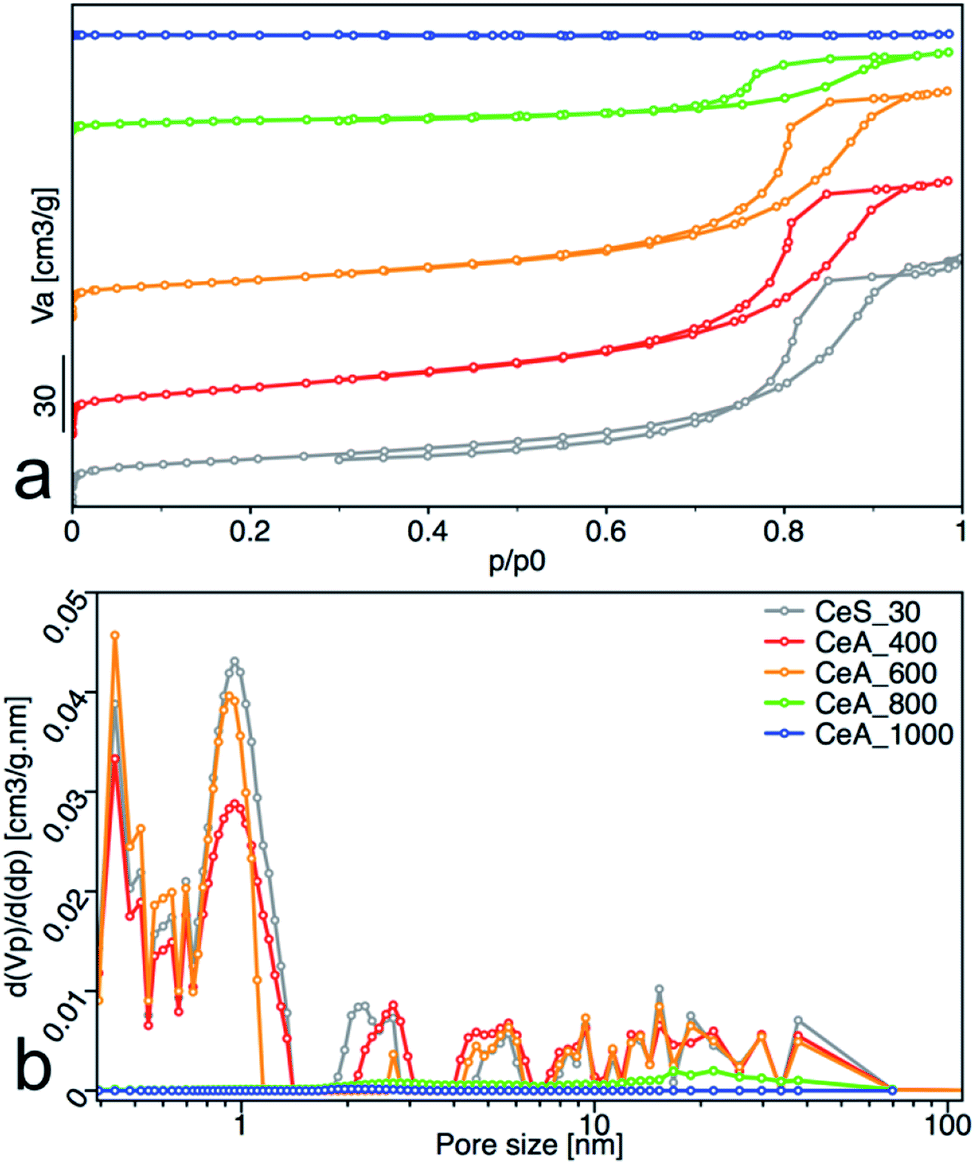
All samples showed type IV isotherms with hysteresis (Fig. 10a), except the sample annealed at 1000 °C. The samples annealed at 400 °C and 600 °C are mesoporous and have relatively high surface area (67 and 53) m2 g−1. At 800 °C, the sample surface area decreased to 15 m2 g−1 with significant loss of pore volume. Annealing to 1000 °C leads to complete loss of mesoporosity as indicates the disappearing of the hysteresis loop and negligible surface area and pore volume of the sample. Pore size distribution calculated by the NLDFT method was practically unchanged for samples annealed up to 600 °C (Fig. 10b).
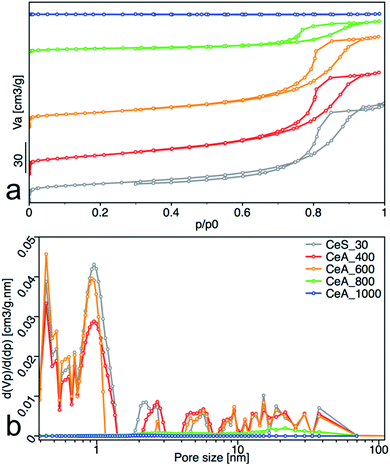 |
| | Fig. 10 (a) Nitrogen adsorption/desorption isotherms, and (b) pore-size distribution obtained from NLDFT analysis. The adsorbed volume (Va), the partial derivative pore volume (Vp) with respect to the pressure difference (dp) and the relative pressure (p/p0) are plotted on the axes. | |
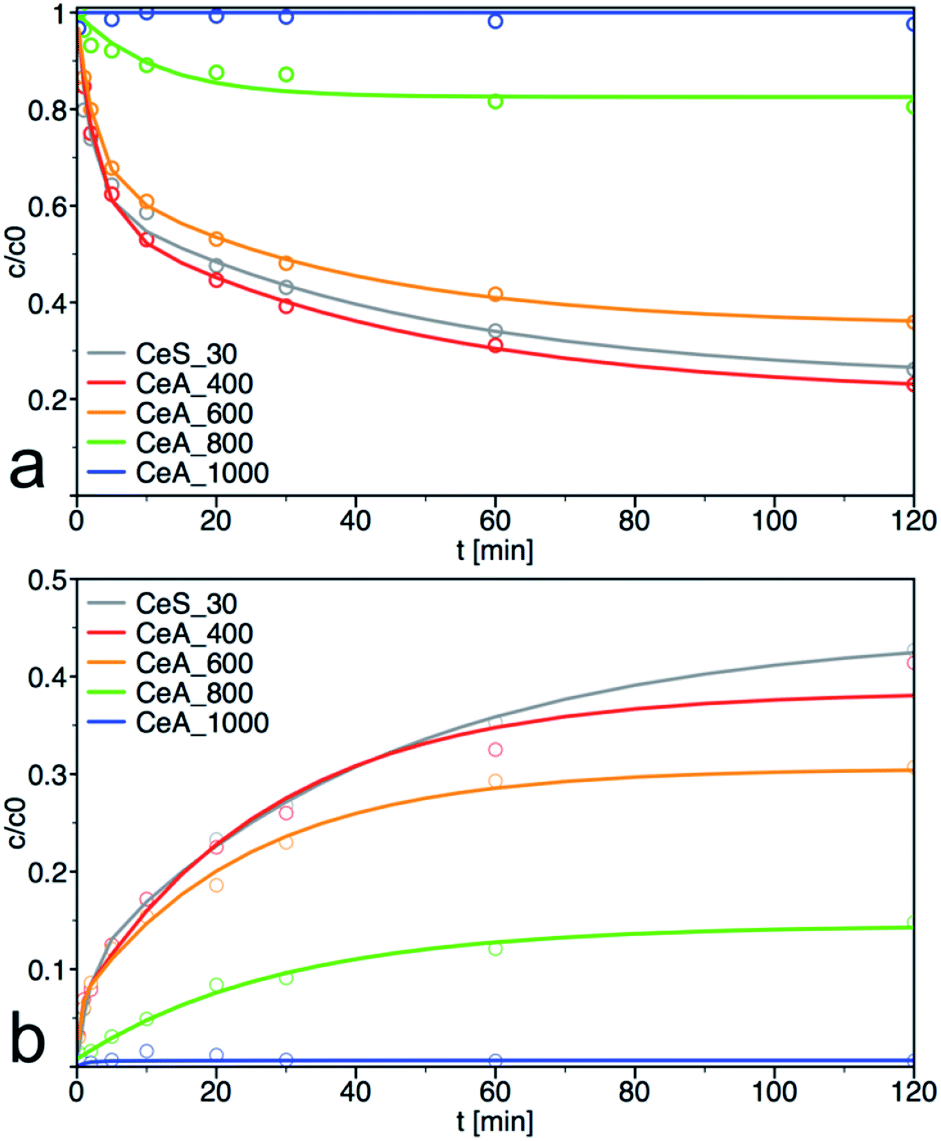
PM degradation of annealed samples
The sample CeS_30 and annealed samples were also tested in catalytic decomposition of PM. The kinetic curves of PM loss and 4-NP formation are shown in Fig. 11, the calculated initial rate of PM loss and the degree of conversion at 60 min are shown in Table 3. The initial concentration of cerium salt in synthesis of CeS_30 was three times higher than in small-scale syntheses. As a result, the grain size has increased three times and the specific surface area was two times smaller, that consequently result in lower catalytic activity. These differences can be seen in Fig. 1, 4, 6–8, S3, S4† and in Tables 1 and 2 Therefore, the synthesis parameters such as the initial salt concentration and reaction volume must be accounted in large-scale synthesis. Similar results were obtained in the synthesis with much higher initial concentration of cerium salt.13
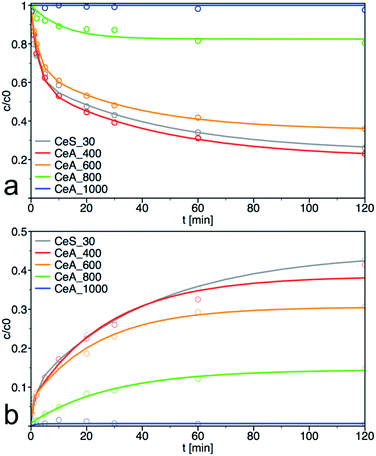 |
| | Fig. 11 Kinetic measurement of (a) degradation of parathion methyl (PM) and (b) formation of 4-nitrophenol (4-NP) on annealed samples. The solid lines are least-square best fits of the data to eqn (3) and (4). | |
As can be seen from materials characterization, the sample annealed at 400 °C retained its structural properties and thus also its catalytic activity. It also demonstrates that post-synthesis annealing of the samples to remove any residuals from the synthesis is not necessary and that the nanoceria was sufficiently crystallized during low temperature used in our synthesis. Interestingly, the samples retained very good catalytic activity even after treatment at 600 °C. At higher annealing temperatures, the specific surface area and, therefore, also the degradation activity decreased significantly.
Reaction balance, stability and regeneration of the catalyst
The CeS_30 sample was also used to verify the reaction balance and for testing of reusability of the catalyst. From the results of kinetic measurements of PM decomposition (Fig. 7), it is clear that the total balance between degraded PM and 4-NP released to the solution is not equal to one and it varies among the samples. This is because the product 4-NP is partially released to the solution but it is also adsorbed on the surface of the catalyst. Therefore, to determine total concentration of 4-NP formed and thus true degree of conversion of PM to 4-NP, the samples were extracted with MeOH and MeCN before HPLC analysis. Same procedure was also used to study the degree of conversion in reusability catalytic tests as follows:
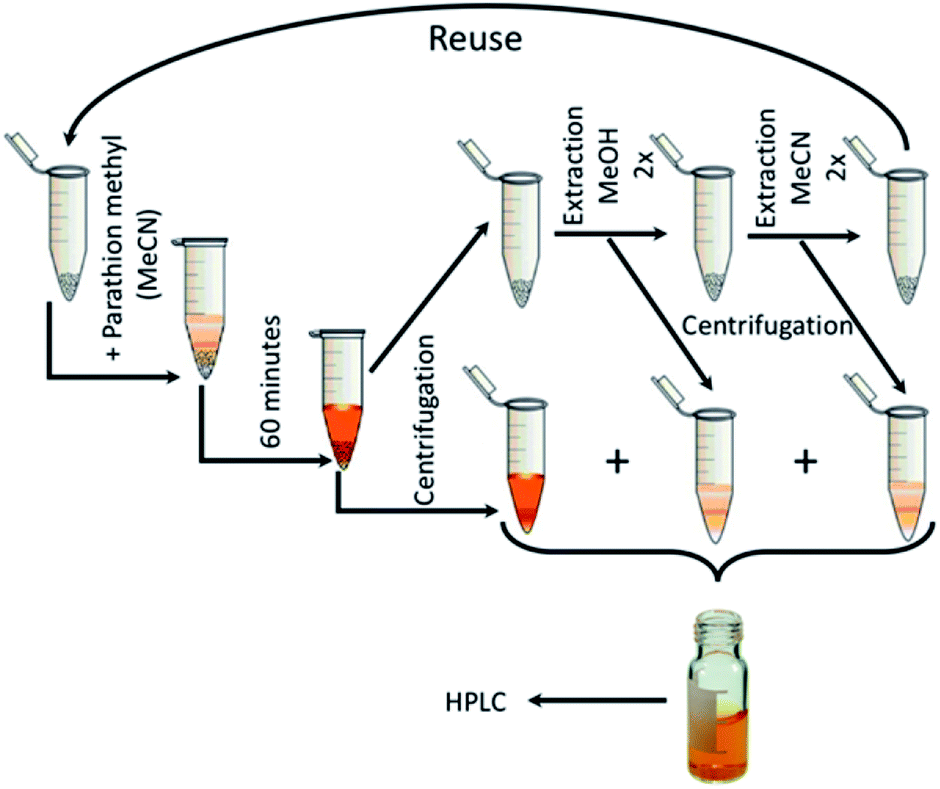
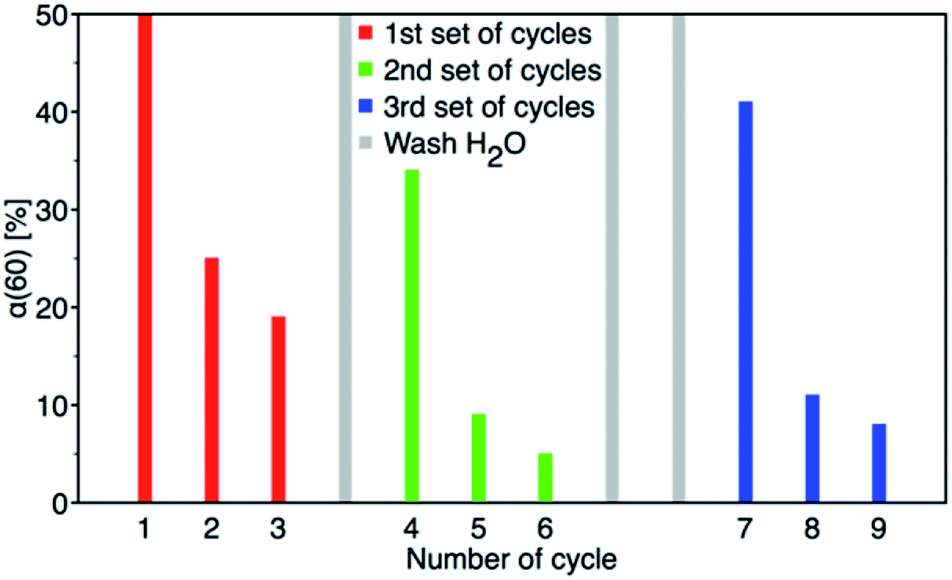
After 60 minutes of degradation in the first cycle, the sample was centrifuged and extracted twice with MeOH and then twice with MeCN; all aliquots were combined and analysed to determine the degree of conversion (α60). The catalyst was then air-dried and used in next cycle. This procedure is shown schematically in Fig. 12. The obtained c60/c0 molar ratio for PM and 4-NP are shown in Table 2S† together with degree of conversion at 60 minutes. It shows that by MeOH extraction a 1:1 molar ratio balance between PM degradation and 4-NP formation was achieved. The results of reusability experiments (9 cycles) are shown in Fig. 13 and Table 2S.† After the third cycle, the degree of conversion α60 decreased from the initial 50% to 19%. Therefore, to test a regeneration of the catalyst, the sorbent was immersed in water for 24 hours, dried and another three cycles were performed. The water regeneration of the sorbent caused improvement of the degree of conversion α60 back to 34%. After three more cycles, α60 decreased to only 9%.
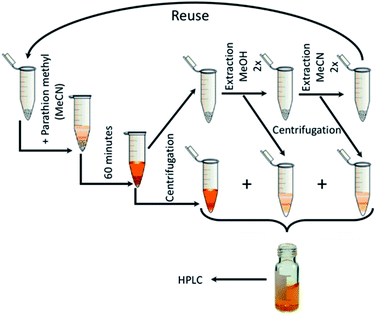 |
| | Fig. 12 Reusability measurement scheme. | |
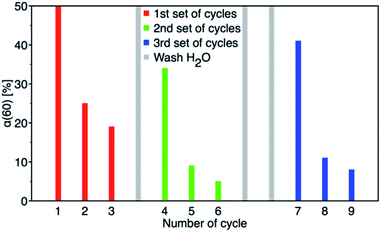 |
| | Fig. 13 Nine PM degradation cycles with two water regeneration steps (first for 24 and second for 48 hours). | |
Therefore, another catalyst regeneration using water washing was prolonged to 48 hours. Interestingly, the longer regeneration of the sorbent resulted in a substantial improvement of the degree of conversion α60 back to 41% after 6 cycles. After 9 cycles with two washing steps, the degree of conversion α60 decreased to 8%.
It is widely accepted that not only the available surface area but also its qualitative properties are important for achieving high catalytic activity in pollutant degradation. In the case of CeO2, a suitable Ce(III)/Ce(IV) ratio, as well as abundance of the surface –OH groups are the main factors. The formation of these active sites is feasible in defective nanocrystalline structures. According to our previous investigation,27 the catalytic activity of nanoceria can be preserved or recovered when the tests are performed in water, where a depletion of –OH groups can be compensated and the active sites can be regenerated. Contrary, in the non-aqueous environment, surface –OH groups that act as strong nucleophile in degradation reaction are consumed without regeneration and the catalytic activity decreases drastically after the first cycle. However, as demonstrated here, even the catalyst used in a non-aqueous solvent can be easily recovered by its simple immersion in water for a few hours. Nevertheless, we assume that this process is feasible only for nanoceria thanks to its unique properties (such as high oxygen mobility) and it cannot be simply applied to other metal oxide catalysts. Interestingly, during the degradation cycles, the colour of the catalyst gradually changes from yellow to orange and after water washing back to yellow. The orange colour was attributed to surface peroxo groups in many studies,4 but the mechanism of their formation in our catalytic test remains unclear.
Conclusions
A series of nanoceria samples was prepared at temperatures from 5 °C to 95 °C in water at ambient pressure. It was demonstrated that several CeO2 properties (i.e., crystallinity, grain size, surface area) can be easily controlled by the reaction temperature. These properties significantly affect the catalytic activity of prepared CeO2 nanoparticles in decomposition of parathion methyl to 4-nitrophenol. The lowest synthesis temperature (5 °C) can be used to prepare nanoceria with large specific surface area and very small crystallites. Conversely, high temperature can be applied to obtain loosely aggregated uniform cubic nanoparticles. For large-scale synthesis it is possible to prepare nanocrystalline CeO2 simply at room temperature. It was shown that post-synthesis annealing up to 400 °C did not significantly alter the material parameters and hence the catalytic properties. Furthermore, as shown the reusability tests, the sorbent can be reactivated by simple washing with water which demonstrates its durability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Czech Science Foundation (No. 19-07460S); Research Infrastructure NanoEnviCZ, supported by Ministry of Education, Youth and Sports of Czech Republic (No. LM2018124); and UJEP Student grant competition (UJEP-SGS-2018-44-005-3). J. Bezdičková and M. Maříková from the Institute of Inorganic Chemistry of the Czech Academy of Sciences in Řež, is thanked for measurements of the thermal gravimetric analysis and the Raman spectra of samples, respectively.
References
- A. F. Diwell, R. R. Rajaram, H. A. Shaw and T. J. Truex, Stud. Surf. Sci. Catal., 1991, 71, 139–152 CrossRef CAS.
- A. Atkinson, Solid State Ionics, 1997, 95, 249–258 CrossRef CAS.
- Y. Xue, Q. Luan, D. Yang, X. Yao and K. Zhou, J. Phys. Chem. C, 2011, 115, 4433–4438 CrossRef CAS.
- C. Li, X. Shi, Q. Shen, C. Guo, Z. Hou and J. Zhang, J. Nanomater., 2018, 4857461 Search PubMed.
- C. Xu and X. Qu, NPG Asia Mater., 2014, 6, e90 CrossRef CAS.
- M. Komiyama, Chem. Lett., 2016, 45, 1347–1355 CrossRef CAS.
- T. Yao, Z. Tian, Y. Zhang and Y. Qu, ACS Appl. Mater. Interfaces, 2019, 11, 195–201 CrossRef CAS PubMed.
- M. J. Manto, P. Xie and C. Wang, ACS Catal., 2017, 7, 1931–1938 CrossRef CAS.
- G. Wang, J. Zhang, X. He, Z. Zhang and Y. Zhao, Ceria Nanoparticles as Enzyme Mimetics, Shanghai Institute of Organic Chemistry, 2017, vol. 35 Search PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- S. Wang, M. Awano and K. Maeda, J. Ceram. Soc. Jpn., 2002, 110, 703–709 CrossRef CAS.
- D. Wang, Z. Ma, S. Dai, J. Liu, Z. Nie, M. H. Engelhard, Q. Huo, C. Wang and R. Kou, J. Phys. Chem. C, 2008, 112, 13499–13509 CrossRef CAS.
- H.-I. Chen and H.-Y. Chang, Ceram.Int., 2005, 31, 795–802 CrossRef CAS.
- I. Trenque, G. C. Magnano, J. Bárta, F. Chaput, M. A. Bolzinger, I. Pitault, S. Briançon, K. Masenelli-Varlot, M. Bugnet, C. Dujardin, V. Čuba and D. Amans, CrystEngComm, 2020, 22(10), 1725–1737 RSC.
- M. Nyoka, Y. E. Choonara, P. Kumar, P. P. D. Kondiah and V. Pillay, Nanomaterials, 2020, 10, 242 CrossRef CAS PubMed.
- N. Sutradhar, A. Sinhamahapatra, S. Pahari, M. Jayachandran, B. Subramanian, H. C. Bajaj and A. Baran Panda, J. Phys. Chem. C, 2011, 115, 7628–7637 CrossRef CAS.
- Q. Wu, F. Zhang, P. Xiao, H. Tao, X. Wang, Z. Hu and Y. Lü, J. Phys. Chem. C, 2008, 112, 17076–17080 CrossRef CAS.
- P. Janoš, J. Ederer, V. Pilařová, J. Henych, J. Tolasz, D. Milde and T. Opletal, Wear, 2016, 362–363, 114–120 CrossRef.
- Q.-Z. Yan, X.-T. Su, Z.-Y. Huang and C.-C. Ge, J. Eur. Ceram. Soc., 2006, 26, 915–921 CrossRef CAS.
- M. Veith, S. Mathur, A. Kareiva, M. Jilavi, M. Zimmer and V. Huch, J. Mater. Chem., 1999, 9, 3069–3079 RSC.
- M. Epifani, E. Pellicer, J. Arbiol and J. R. Morante, Chem. Mater., 2009, 21, 862–870 CrossRef CAS.
- Y. Ono and H. Fujii, Ceram. Int., 2015, 41, 15231–15234 CrossRef CAS.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497–501 CrossRef CAS PubMed.
- A. Tamilselvi and G. Mugesh, Chem.–Eur. J., 2010, 16, 8878–8886 CrossRef CAS PubMed.
- A. A. Vernekar, T. Das and G. Mugesh, Angew. Chem., Int. Ed., 2016, 55, 1412–1416 CrossRef CAS PubMed.
- M. M. Khan, S. A. Ansari, D. Pradhan, D. H. Han, J. Lee and M. H. Cho, Ind. Eng. Chem. Res., 2014, 53, 9754–9763 CrossRef CAS.
- P. Janoš, J. Ederer, M. Došek, J. Štojdl, J. Henych, J. Tolasz, M. Kormunda and K. Mazanec, Environ. Sci.: Nano, 2019, 6, 3684–3698 RSC.
- J. Tolasz, M. Šťastný and V. Štengl, in Materials Today: Proceedings, 2016, vol. 3 Search PubMed.
- P. Janos, P. Kuran, M. Kormunda, V. Stengl, T. M. Grygar, M. Dosek, M. Stastny, J. Ederer, V. Pilarova and L. Vrtoch, J. Rare Earths, 2014, 32, 360–370 CrossRef CAS.
- L. Li, G.-S. Hu, J.-Q. Lu and M.-F. Luo, Acta Phys.-Chim. Sin., 2012, 28, 1012–1020 CAS.
- C. Li, Y. Sakata, T. Arai, K. Domen, K. I. Maruya and T. Onishi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 929–943 RSC.
- M. Šťastný, V. Štengl, J. Henych, J. Tolasz, P. Vomáčka and J. Ederer, J. Mater. Sci., 2016, 51, 2634–2642 CrossRef.
- V. Štengl, D. Králová, F. Opluštil and T. Němec, Microporous Mesoporous Mater., 2012, 156, 224–232 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00937g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Jiří Henych
*ab,
Jiří Henych