
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sea-shell-like B31+ and B32: two new axially chiral members of the borospherene family†
Ling Peiab,
Miao Yana,
Xiao-Yun Zhaoa,
Yue-Wen Mu *a,
Hai-Gang Lu*a,
Yan-Bo Wua and
Si-Dian Li*a
*a,
Hai-Gang Lu*a,
Yan-Bo Wua and
Si-Dian Li*a
aInstitute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: ywmu@sxu.edu.cn; luhg@sxu.edu.cn; lisidian@sxu.edu.cn
bDepartment of Chemical Engineering and Safety, Binzhou University, Binzhou 256603, China
First published on 10th March 2020
Abstract
Since the discovery of the cage-like borospherenes D2d B40−/0 and the first axially chiral borospherenes C3/C2 B39−, a series of fullerene-like boron clusters in different charge states have been reported in theory. Based on extensive global minimum searches and first-principles theory calculations, we present herein two new axially chiral members C2 B31+ (I) and C2 B32 (VI) to the borospherene family. B31+ (I) features two equivalent heptagons on the top and one octagon at the bottom on the cage surface, while B32 (VI) possesses two equivalent heptagons on top and two equivalent heptagons at the bottom. Detailed bonding analyses show that both sea-shell-like B31+ (I) and B32 (VI) follow the universal σ + π double delocalization bonding pattern of the borospherene family, with ten delocalized π bonds over a σ skeleton, rendering spherical aromaticity to the systems. Extensive molecular dynamics simulations show that these novel borospherenes are kinetically stable below 1000 K. The IR, Raman, and UV-vis spectra of B31+ (I) and B32 (VI) are computationally simulated to facilitate their future experimental characterizations.
1. Introduction
As the lighter neighbour of carbon in the periodic table, boron is a typical electron-deficient element which shares with carbon the rare ability to form stable covalently bonded molecular frameworks with multicentre–two-electron bonds (mc–2e bonds) in both polyhedral molecules and bulk allotropes.1,2 Persistent joint photoelectron spectroscopy (PES) experimental and first-principles theory investigations by Lai-Sheng Wang and co-workers in the past two decades on size-selected negatively-charged boron clusters Bn− (n = 3–42) have revealed a rich landscape for boron nanoclusters from planar or quasi-planar (2D) structures (n = 3–38, 41, and 42) to cage-like borospherenes (n = 39, 40).3–8 The first all-boron fullerenes D2d B40−/0, dubbed borospherenes, were discovered in 2014, marking the onset of borospherene chemistry.5 The spherically aromatic borospherene D2d B40 is found to be composed of twelve interwoven boron double chains with two hexagons at the top and bottom and four heptagons on the waist. It features a unique bonding pattern of σ + π double delocalization, with twelve delocalized π bonds spherically distributed over a σ skeleton. The axially chiral B39− appears to be the only boron cluster monoanion observed in experiments to date which has a cage-like global minimum (GM).4 The spherically aromatic borospherene family has been expanded by our group at first-principles theory level to the cage-like Bnq series (n = 36–42, q = n − 40) in different charge states which are all composed of twelve interwoven double chains with a σ + π double delocalization bonding pattern.4,5,9–12 Two lowest-lying cage-like Cs B39+ isomers in the same bonding pattern were also predicted in theory.13 Sea-shell-like C2 B28−/0 and Cs B29− with nine delocalized π bonds over a σ-skeleton were later observed as minor isomers in PES experiments.14,15 Following the same structural motif, our group predicted the possibility of sea-shell-like Cs B29+, B34, and B35+ at first-principles theory levels16,17 which also appear to follow the σ + π double delocalization bonding pattern of the borospherene family. Ion mobility measurements in combination with density-functional theory (DFT) calculations, on the other hand, indicate that Bn+ monocations possess double-ring tubular structures in the size-range between n = 16–25, showing another important structural domain for boron.18 However, the geometrical and electronic structures of Bn+ monocations in the size range between n = 30–38 has remained unknown to date, except the sea-shell-like B35+ previously predicted by our group.17In this work, we perform a theoretical investigation on the structures and bonding patterns of B31+ and B32 via extensive global minimum searches and first-principles theory calculations. Sea-shell-like C2 B31+ (I) and C2 B32 (VI) are found to be the well-defined GMs of B31+ and B32, respectively, presenting two new axially chiral members to the borospherene family. Both B31+ (I) and C2 B32 (VI) appear to follow the universal σ + π double delocalization bonding pattern of the borospherene family, with ten delocalized π bonds over a σ skeleton, rendering spherical aromaticity to these novel borospherenes.
2. Theoretical procedure
Extensive GM searches were performed on B31+ and B32 using the TGmin program,19–21 in conjunction with manual structural constructions based on the previously reported low-lying isomers of B31−/0 and B32−/0.22 About 5500 and 4500 trial points were generated on the potential energy surface for B31+ and B32 at the PBE/TZP level of theory. Frozen core approximation was used for the inner shells of [1s2] for B. The low-lying isomers for B31+ and B32 were then fully optimized at PBE0 and TPSSh levels23,24 with the 6-311+G(d) basis set,25 with vibrational frequencies checked to make sure all the low-lying isomers obtained were true minima. All these calculations were implemented using the Gaussian 16 program.26 To obtain more accurate relative energies, the top five lowest-lying isomers of B31+ and B32 were further refined at the single-point CCSD(T)/6-311G(d) level27,28 at their PBE0/6-311+G(d) geometries with the zero-point energy (ZPE) corrections included at PBE0. The obtained GMs C2 B31+ (I) and C2 B32 (VI) and their degenerated enantiomers C2 B31+ (I′) and C2 B32 (VI′) are shown in Fig. 1 and more low-lying isomers are listed in Fig. S1 (ESI).† Chemical bonding analyses on B31+ (I) and B32 (VI) (Fig. 1) were conducted using the Adaptive Natural Density Partitioning (AdNDP) method29,30 at the PBE0/6-31G level. Nucleus-independent chemical shifts (NICS)31,32 were calculated at the cage centres to assess the spherical aromaticity of B31+ (I) and B32 (VI). The IR and Raman spectra of B31+ (I) and B32 (VI) were simulated at PBE0/6-311+G(d) level and UV-vis absorption spectra calculated using the time-dependent DFT approach (TD-PBE0)33,34 implemented in Gaussian 16. Extensive Born–Oppenheimer molecular dynamics (BOMD) simulations were performed for B31+ (I) and B32 (VI) at 500 K, 700 K, and 1000 K for 30 ps (Fig. S2, ESI†) using the CP2K software,35 with the GTH-PBE pseudopotential and the DZVP-MOLOPT-SR-GTH basis set adopted.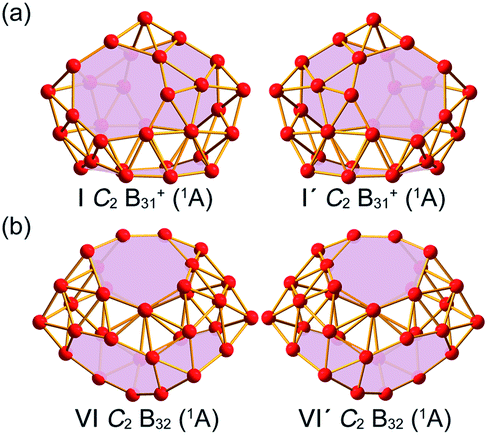 | ||
| Fig. 1 Optimized global minimum structures of the axially chiral borospherenes (a) C2 B31+ (I) and (b) C2 B32 (VI) and their degenerate enantiomers C2 B31+ (I′) and C2 B32 (VI′), with the B7 heptagons and B8 octagons on the cage surfaces highlighted in pink. | ||
3. Results and discussion
3.1. Structures and stabilities
As shown in the configurational energy spectrum of B31+ at the CCSD(T)/6-311G(d) level in Fig. 2a, the axially chiral sea-shell-like C2 B31+ (I) is the well-defined GM of B31+ with the lowest vibrational frequency of 66.2 cm−1. It consists of twenty-six triangles and eight quadrilaterals on the cage surface, two equivalent B7 heptagons on the waist, and one B8 octagon at the bottom (shaded in pink in Fig. 1a), following the Euler's rule in this case which reads: E (66 edges) = F (26 triangular + 8 quadrilaterals + 2 heptagonal + 1 octagonal faces) + V (31 vertices) − 2. The second, third, fourth, and fifth lowest-lying cage-like C1 B31+ (II), C1 B31+ (III), Cs B31+ (IV), and Cs B31+(V) lie 0.22, 0.24, 0.34, and 0.36 eV higher in energy than C2 GM at CCSD(T) level, respectively (Fig. 2a), though B31+ (III) lying 0.04 eV lower than the C2 GM at the less accurate PBE0 level (Fig. S1a, ESI†). The sea-shell-like low-symmetry C1 B31+ (II) possesses three heptagons on the surface, while C1 B31+ (III) contains two hexagons and one heptagon. We notice that most of the low-lying B31+ isomers are cage-like, whereas the first close-packed quasi-planar isomer C1 B31+ lies much higher (by 0.38 eV) than the GM at PBE0 (Fig. S1a, ESI†). The quasi-planar C1 B31+ with a hexagonal hole at the centre which corresponds to the GM of quasi-planar B31− appears to be 0.63 eV higher than C2 GM at PBE0, well illustrating the charge-induced structural transition from planar B31− reported in ref. 22 to cage-like B31+ obtained in this work due to two valence electrons' difference (Fig. S1a, ESI†).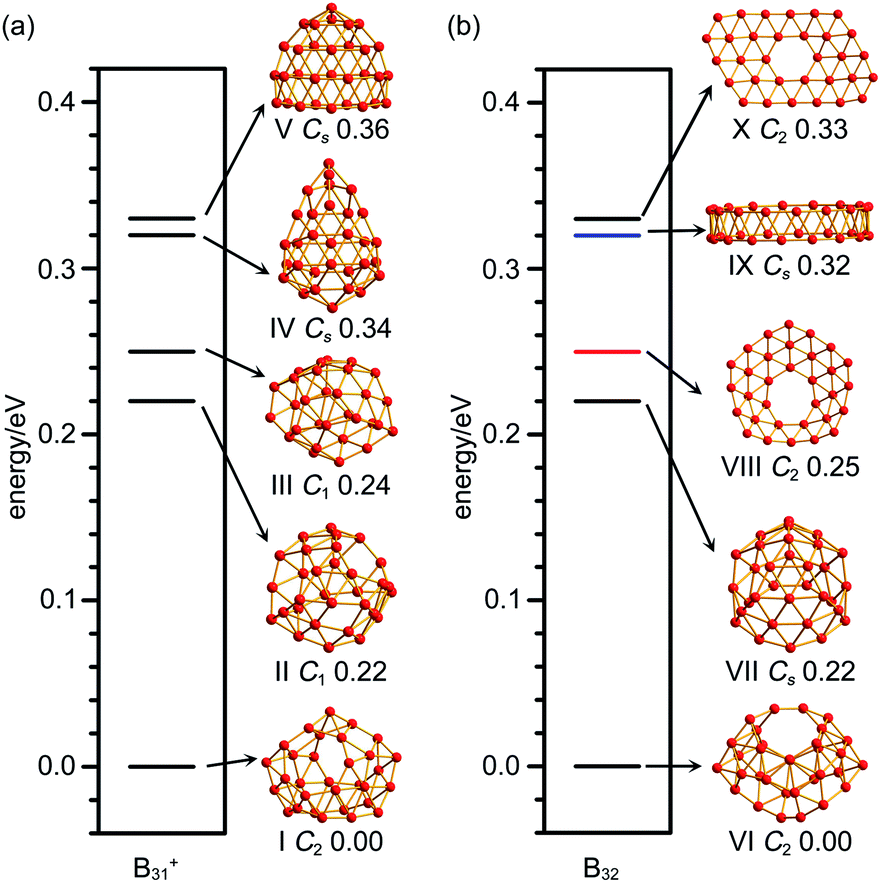 | ||
| Fig. 2 Configurational energy spectra of (a) B31+ and (b) B32 at CCSD(T)/6-311G(d)//PBE0/6-311+G(d) level, with the relative energies indicated in eV. Black, red and blue horizontal lines represent cage-like, quasi-planar and tubular structures, respectively. | ||
C2 B32 (VI), the well-defined GM of neutral B32 at CCSD(T) level, also possesses an axially chiral sea-shell-like structure with the lowest vibrational frequency of 141 cm−1. It contains two equivalent heptagons on the top and two equivalent heptagons at the bottom on the cage surface (shaded in pink in Fig. 1b) and follows the Euler's rule which in this case reads: E (72 edges) = F (36 triangular + 2 quadrilaterals + 4 heptagonal faces) +V (32 vertices) − 2. The second lowest-lying Cs B32 (VII) possesses two hexagons on the waist and one octagon at the bottom (Fig. S1b, ESI†). It is worth noticing that the quasi-planar Cs B32 (VIII) predicted by Nguyen et al.36 and the double-ring tubular D16d B32 (IX) proposed by Zhao's group37 in Fig. 2b turned out to be the third and fourth lowest-lying isomers of neutral B32 lying 0.25 eV and 0.32 eV higher in energy than our C2 GM (VI) at CCSD(T), respectively (Fig. 2b). B32 (VI) thus has the lowest energy in all the structures obtained to date for neutral B32. The much-concerned quasi-planar C2 B32 (X) with a B6 hexagon at the center which corresponds to the experimentally observed third isomer of C2 B32− (ref. 22) appears to be 0.33 eV less stable than B32 (VI) at CCSD(T) (Fig. 2b).
Extensive molecular dynamics (MD) simulations were performed on B31+ (I) and B32 (VI) to check the dynamical stabilities of these axially chiral borospherenes. As shown in Fig. S2 (ESI),† both B31+ (I) and B32 (VI) appear to be dynamically stable between 500–1000 K. The calculated average root-mean-square-deviations (RMSD) and maximum bond length deviations (MAXD) of B31+ (I) are RMSD = 0.07, 0.11, and 0.13 Å and MAXD = 0.28, 0.46 and 0.58 Å at 500 K, 700 K, and 1000 K, respectively. The corresponding values of B32 (VI) turn out to be RMSD = 0.07, 0.08 and 0.10 Å and MAXD = 0.21, 0.27 and 0.36 Å at 500 K, 700 K, and 1000 K, respectively. No high energy isomers are observed in these MD simulation processes.
3.2. Bonding pattern analyses
The high thermodynamic and dynamic stabilities of these axially chiral borospherenes originate from their unique electronic structures and bonding patterns. We choose to use the widely used AdNDP approach developed by Boldyrev and co-workers to analyse both the localized and delocalized bonding interactions in these novel species.29,30 Detailed AdNDP analyses indicate that B31+ (I) possesses 8×2c–2e σ bonds, 24×3c–2e σ bonds and 4×4c–2e σ bonds on the cage surface with the occupation numbers of ON = 1.80–1.94 |e|, 1.72–1.96 |e|, and 1.79–1.81 |e|, respectively. The remaining 20 valence electrons form 10 delocalized π bonds spherically distributed over the σ-skeleton, including 6×4c–2e π bonds and 4×5c–2e π bonds with ON = 1.68–1.83 |e|, in an overall bonding symmetry of C2 (Fig. 3a). B32 (VI) possesses a similar bonding pattern with B31+ (I) (Fig. 3b). It contains 2×2c–2e σ bonds, 32×3c–2e σ bonds, and 4×4c–2e σ bonds on the cage surface. There exist 10 delocalized π bonds spherically distributed over the σ-skeleton, including 6×4c–2e π bonds and 4×5c–2e π bonds with ON = 1.78–1.90 |e|, in an overall symmetry of C2. Both B31+ (I) and B32 (VI) thus possess 10 delocalized π bonds over a σ-skeleton and follow the universal σ + π double delocalization bonding pattern of the borospherene family.4,5,9–16 Detailed bonding analyses further indicate that Cs B32 (VII), the second lowest-lying isomer of B32, also matches the σ + π double delocalization bonding pattern, with 10 delocalized π bonds over a σ skeleton (Fig. S3, ESI†).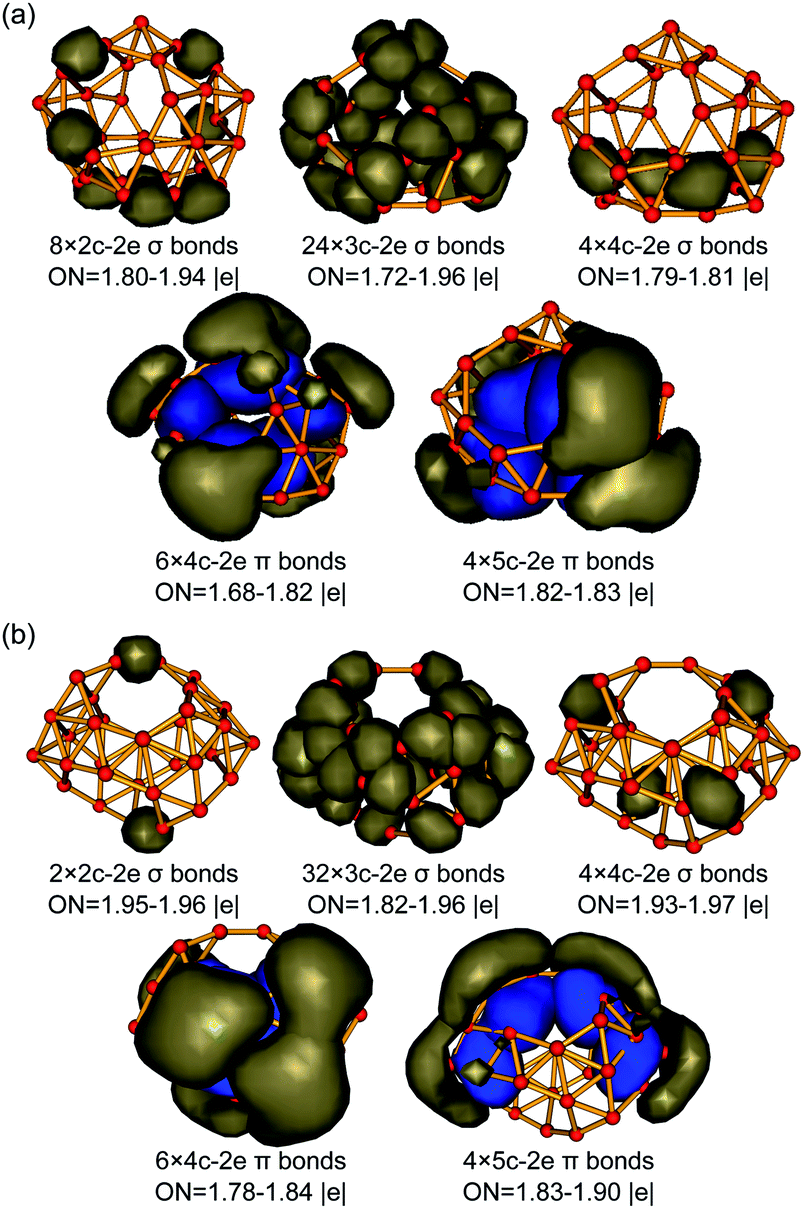 | ||
| Fig. 3 σ and π AdNDP bonding patterns of (a) C2 B31+ (I) and (b) C2 B32 (VI), with the occupation numbers (ONs) indicated. | ||
The 10 delocalized π bonds on the cage surfaces renders spherical aromaticity to sea-shell-like B31+ (I) and B32 (VI), as evidenced by their calculated negative NICS values of NICS = −29 ppm and −24 ppm at the cage centers. We tabulate the numbers of σ bonds, π bonds, and calculated NICS values of the borospherene family reported so far in Table 1. It can be seen that the numbers of σ bonds increase monotonously with the number of valence electrons of the systems, while the numbers of π bonds increase in a stepwise pattern, with C2 B28, Cs B29+, and Cs B29− possessing 9 delocalized π bonds, C2 B31+ and C2 B32 having 10 delocalized π bonds, C2 B34 and C2 B35+ containing 11 delocalized π bonds, and Th B364−, Cs B373−, Cs B382−, Cs B39+, C3 B39−, C2 B39−, D2d B40, C1 B41+, and C2 B422+ in different charge states possessing 12 delocalized π bonds, respectively. These borospherenes all appear to be spherically aromatic with the negative calculated NICS values of NICS = −21 to −43 ppm. It is these delocalized π bonds that help to maintain the cage-like structures of the borospherene family and render spherical aromaticity to the systems.
| nσ | nπ | NICS/ppm | |
|---|---|---|---|
| C2 B28 (ref. 14) | 33 | 9 | −40 |
| Cs B29+ (ref. 16) | 34 | 9 | −34 |
| Cs B29− (ref. 15) | 35 | 9 | −21 |
| C2 B31+ | 36 | 10 | −29 |
| C2 B32 | 38 | 10 | −24 |
| C2 B34 (ref. 17) | 40 | 11 | −40 |
| C2 B35+ (ref. 17) | 41 | 11 | −38 |
| Th B364− (ref. 12) | 44 | 12 | −36 |
| Cs B373− (ref. 10) | 45 | 12 | −33 |
| Cs B382− (ref. 11) | 46 | 12 | −37 |
| Cs B39+ (ref. 13) | 46 | 12 | −40 |
| C3 B39− (ref. 4) | 47 | 12 | −38 |
| C2 B39− (ref. 4) | 47 | 12 | −39 |
| D2d B40 (ref. 5) | 48 | 12 | −43 |
| C1 B41+ (ref. 9) | 49 | 12 | −41 |
| C2 B422+ (ref. 9) | 50 | 12 | −40 |
3.3. Spectral simulations
Infrared photodissociation (IR-PD) spectra in combination with first-principles theory calculations have proven to be an effective approach in characterizing novel clusters.38 B31+ (I) and B32 (VI) possess 87(43a + 44b) and 90(46a + 44b) vibrational modes, respectively. The simulated IR, Raman and UV-vis spectra of B31+ (I) and B32 (VI) are shown in Fig. 4. The major IR peaks of the two borospherenes appear to lie between 1100 and 1400 cm−1, with two major IR active peaks at 1254 cm−1 (b) and 1298 cm−1 (b) in B31+ (I) and two major peaks at 1277 cm−1 (b) and 1315 cm−1 (b) in B32 (VI). All the other IR vibrational modes appear to have much lower intensities. The major Raman active peaks occur at 374 cm−1 (a), 669 cm−1 (a) and 1394 cm−1 (a) in B31+ (I) and 491 cm−1 (a), 1181 cm−1 (a) and 1308 cm−1 (a) in B32 (VI), with main contributions originating from the symmetric vibrational modes. The Raman vibrational modes at 374 cm−1 (a) in B31+ (I) and 491 cm−1 (a) in B32 (VI) correspond to the typical “radial breathing modes” (RBMs) of the two borospherenes which can be used to characterize the hollow structures of single-walled boron nanoclusters in experiments.39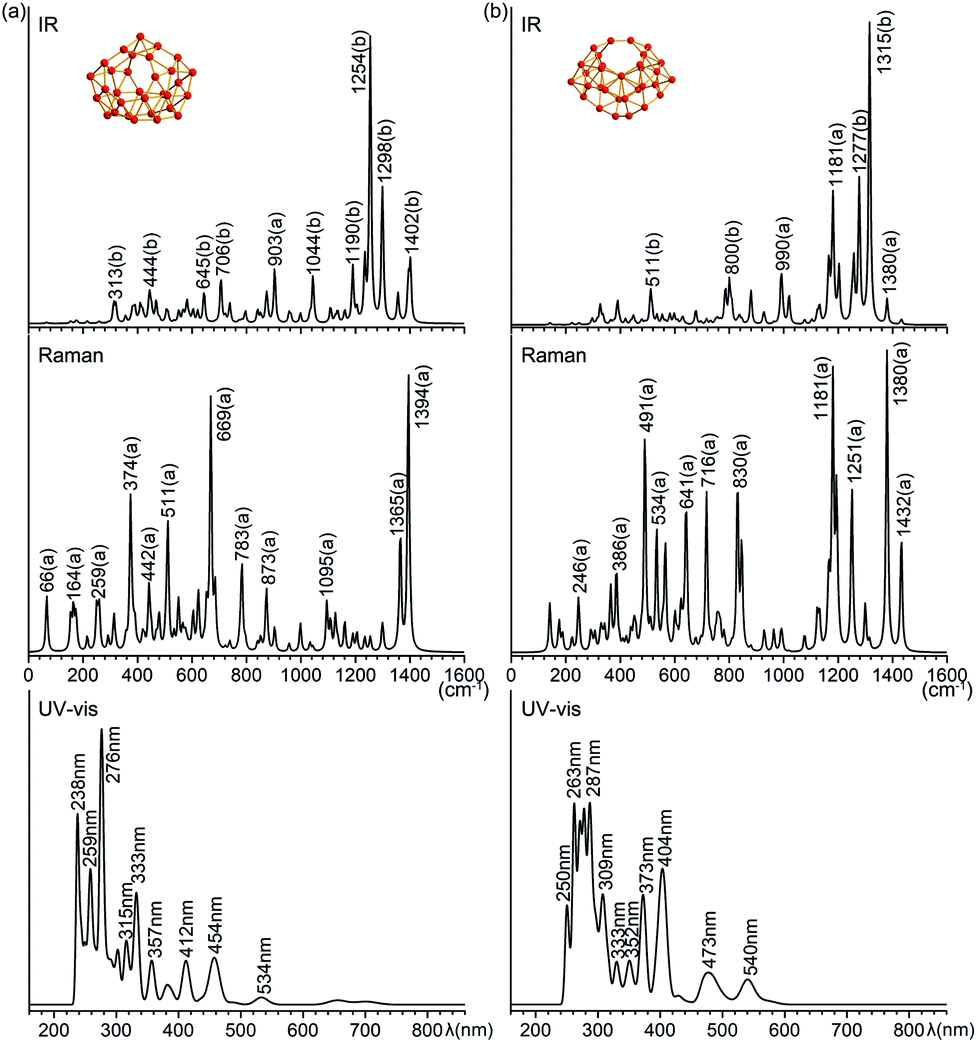 | ||
| Fig. 4 Simulated IR, Raman, and UV-vis absorption spectra of (a) C2 B31+ (I) and (b) C2 B32 (VI) at PBE0/6-311+G(d) level. | ||
The simulated UV-vis spectra of B31+(I) and B32 (VI) lie between 200–550 nm, with the main absorption peaks lying at 238 nm, 276 nm, 333 nm, 412 nm, 454 nm, and 534 nm in B31+(I) and at 263 nm, 287 nm, 373 nm, 404 nm, 473 nm, and 540 nm in B32 (VI), respectively (Fig. 4). The strong UV-vis peaks originate from electronic excitations from the deep inner shells to the high-lying unoccupied molecular orbitals of the systems, while the weak absorption bands above 500 nm are attributed to electronic excitations from the occupied frontier orbitals (HOMO and HOMO−1) to the unoccupied frontier orbitals (LUMO, LUMO+1, and LUMO+2).
4. Summary
We have performed in this work an extensive first-principles theory investigation on sea-shell-like C2 B31+ (I) and C2 B32 (VI), presenting two new axially chiral members to the borospherene family. These novel borospherenes follow the universal σ + π double delocalization bonding pattern of the borospherene family, with 10 delocalized π bonds over an σ skeleton on the cage surface, rendering spherical aromaticity to these borospherene species. B31+ (I) may be characterized in gas-phases IR-PD spectral measurements, while B32 (VI) may be detected in matrix isolation infrared spectroscopy.40 More investigations on cage-like Bn+/0 clusters are currently in progresses to further expand the borospherene family and enrich borospherene chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21720102006 to S.-D. Li).Notes and references
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley & Sons, Inc., 1999, p. 131 Search PubMed
.
- W. N. Lipscomb, Science, 1977, 196, 1047 CrossRef CAS PubMed
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811 CrossRef CAS
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754 CrossRef CAS PubMed
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727 CrossRef CAS PubMed
- H. Bai, T. T. Chen, Q. Chen, X. Y. Zhao, Y. Y. Zhang, W. J. Chen, W. L. Li, L. F. Cheung, B. Bai, J. Cavanagh, W. Huang, S. D. Li, J. Li and L. S. Wang, Nanoscale, 2019, 11, 23286 RSC
- L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69 Search PubMed
- T. Jian, X. Chen, S. D. Li, A. I. Boldyrev, J. Li and L. S. Wang, Chem. Soc. Rev., 2019, 48, 3550 RSC
- Q. Chen, S. Y. Zhang, H. Bai, W. J. Tian, T. Gao, H. R. Li, C. Q. Miao, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2015, 54, 8160 CrossRef CAS PubMed
- Q. Chen, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 14186 RSC
- Q. Chen, H. R. Li, C. Q. Miao, Y. J. Wang, H. G. Lu, Y. W. Mu, G. M. Ren, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 11610 RSC
- W. J. Tian, Q. Chen, H. R. Li, M. Yan, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 9922 RSC
- X. Y. Zhao, Q. Chen, H. R. Li, Y. W. Mu, H. G. Lu and S. D. Li, Phys. Chem. Chem. Phys., 2017, 19, 10998 RSC
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed
- H. R. Li, T. Jian, W. L. Li, C. Q. Miao, Y. J. Wang, Q. Chen, X. M. Luo, K. Wang, H. J. Zhai, S. D. Li and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 29147 RSC
- L. Pei, H. R. Li, M. Yan, Q. Chen, Y. W. Mu, H. G. Lu, Y. B. Wu and S. D. Li, Phys. Chem. Chem. Phys., 2018, 20, 15330 RSC
- H. Liu, Q. Chen, H. R. Li, X. Y. Zhao, X. X. Tian, Y. W. Mu, H. G. Lu and S. D. Li, Phys. Chem. Chem. Phys., 2018, 20, 15344 RSC
- E. Oger, N. R. Crawford, R. Kelting, P. Weis, M. M. Kappes and R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503 CrossRef CAS PubMed
- X. Chen, Y. F. Zhao, L. S. Wang and J. Li, Comput. Theor. Chem., 2017, 1107, 57 CrossRef CAS
- Y. Zhao, X. Chen and J. Li, Nano Res., 2017, 3407 CrossRef CAS
- X. Chen, Y. F. Zhao, Y. Y. Zhang and J. Li, J. Comput. Chem., 2019, 40, 1105 CAS
- Q. Chen, T. T. Chen, H. R. Li, X. Y. Zhao, W. J. Chen, H. J. Zhai, S. D. Li and L. S. Wang, Nanoscale, 2019, 11, 9698 RSC
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS
- M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed
- K. Raghavachari and G. W. Trucks, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC
- D. Y. Zubarev and A. I. Boldyrev, J. Org. Chem., 2008, 73, 9251 CrossRef CAS PubMed
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed
- Z. F. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS
- T. B. Tai and M. T. Nguyen, Chem. Commun., 2015, 51, 7677 RSC
- X. W. Yang, X. Wu, L. W. Sai, M. D. Chen and J. J. Zhao, The Second International Conference on Boron Chemistry, ICBC-II, Taiyuan, 2019, p. 139 Search PubMed
- G. Wang, M. Zhou, J. T. Goettel, G. J. Schrobilgen, J. Su, J. Li, T. Schloder and S. Riedel, Nature, 2014, 514, 475 CrossRef CAS PubMed
- D. Ciuparu, R. F. Klie, Y. Zhu and A. L. Pfefferle, J. Phys. Chem. B, 2004, 108, 3967 CrossRef CAS
- X. Wu, L. Zhao, J. Jin, S. Pan, W. Li, X. Jin, G. Wang, M. Zhou and G. Frenking, Science, 2018, 361, 912 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01087a |
| This journal is © The Royal Society of Chemistry 2020 |