
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured manganese oxides electrode with ultra-long lifetime for electrochemical capacitors
Madhu Gaire *a,
Kun Lianga,
Sijun Luoa,
Binod Subedia,
Shiva Adireddya,
Kurt Schroderb,
Stan Farnsworthb and
Douglas B. Chrisey*a
*a,
Kun Lianga,
Sijun Luoa,
Binod Subedia,
Shiva Adireddya,
Kurt Schroderb,
Stan Farnsworthb and
Douglas B. Chrisey*a
aDepartment of Physics and Engineering Physics, Tulane University, New Orleans, Louisiana 70118, USA
bNovaCentrix, Austin, Texas 78728, USA
First published on 29th April 2020
Abstract
We describe the instantaneous fabrication of a highly porous three-dimensional (3D) nanostructured manganese oxides-reduced graphitic oxide (MnOx-rGO) electrode by using a pulse-photonic processing technique. Such nanostructures facilitate the movement of ions/electrons and offer an extremely high surface area for the electrode/electrolyte interaction. The electrochemical performance was investigated by cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS) with 1 M KOH as the electrolyte. The as-prepared thin film electrode exhibits excellent electrochemical performance and an ultra-long lifetime by retaining 90% of the initial capacitance even after 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GCD cycles at constant areal current density of 0.4 mA cm−2. We attribute this excellent lifetime performance to the conductive reduced graphitic oxide, synergistic effects of carbon composite and the metal oxides, and the unique porous nanostructure. Such highly porous morphology also enhances the structural stability of the electrode by buffering the volume changes during the redox processes.
000 GCD cycles at constant areal current density of 0.4 mA cm−2. We attribute this excellent lifetime performance to the conductive reduced graphitic oxide, synergistic effects of carbon composite and the metal oxides, and the unique porous nanostructure. Such highly porous morphology also enhances the structural stability of the electrode by buffering the volume changes during the redox processes.
1. Introduction
Electrochemical capacitors possess a high-power density, superior cycling stability, and excellent charge/discharge characteristics, making them one of the most promising candidates for applications ranging from electronics to hybrid electric vehicles.1,2 They can be further classified into electrochemical double-layer capacitors (EDLCs), in which the energy storage is achieved through charge separation at the electrode/electrolyte interface, and pseudocapacitors, in which the energy storage is achieved through faradaic redox reactions.3 Generally, the EDLCs use carbon-based materials and the pseudocapacitors use metal oxides and conductive polymers as electrodes. For high gravimetric energy storage, metal oxides based on pseudocapacitors have attracted special attention because of their higher energy density than EDLCs due to the fast and reversible redox reactions taking place on or near the surface of the electrode material along with its fast charge/discharge processes.4,5Past work has studied several transition metal oxides, such as RuO2, Co3O4, NiO, Fe2O3, MnO2, etc.6,7 Among them, RuO2 has been the most extensively studied material due to its high theoretical capacitance, high conductivity, and long cycle life. However, its practical application has been limited by low abundance, higher cost, and toxicity.8,9 Therefore, research has been devoted to finding alternative materials that are inexpensive, abundant, and have capacitive performance comparable to RuO2.10 In this regard, manganese oxide (MnO2) has been considered as an excellent candidate because of its low cost, high theoretical capacitance and environmental compatibility.11
The most important parameters that could affect the electrochemical performance of the electrode are its nanostructure, morphology, and electrolyte. This is why the fabrication of MnO2 electrode with highly porous nanostructure is of crucial importance for achieving high capacitance12 because such structures offer a high surface area and help improve the electrochemical performance. As far as electrolytes are concerned, organic electrolytes have a wider working potential window compared to aqueous electrolytes, but their drawbacks, such as high viscosity, poor ionic transportation, lower electronic conductivity, and toxicity limit the electrode's performance.13,14 On the other hand, aqueous electrolytes are inexpensive, safe, easy to handle and have high conductivity. Therefore, in this work, we have used 1 M KOH as the electrolyte because of its high OH− ions mobility.15
In the recent past, different methods, including seeded low-temperature hydrothermal growth,16 chemical vapor deposition,17 and chemical reduction18 have been reported for the fabrication of high-performance nanostructured manganese oxide electrodes. However, the drawbacks such as poor conductivity and volume change resulting from redox reactions during the charge–discharge process limit their practical application. It is especially the volume change during redox reactions that leads to the electrode's poor structural stability, which eventually results in poor cycle life. For their application in long-term electronic devices, the electrodes' lifetime needs to be enhanced. One of the ways to improve the lifetime is to add conductive carbon materials to form a metal oxide and carbon composite electrode. In this regard, the fabrication of transition metal oxide/carbon composites electrodes for supercapacitors have been reported19–29 using various methods. However, no industrial technology for the fabrication of composites has been reported yet. Recently, our group reported a unconventional, low-cost and high-throughput pulsed photoinitiated approach to the in situ formation of three-dimensional (3D) nanostructured composited thin film composed of nanocrystalline transition metal oxide and reduced graphitic oxide through flash xenon lamp irradiation of metal–organic precursor films.30–32
Herein, we report a one-step thin-film processing method and excellent cyclic stability and ultra-long cycle life of the electrode. Firstly, we have followed a spray-coating technique, which can be accomplished in less than a minute in ambient conditions, to spray Mn-acetylacetonate precursor solution onto the Pt-coated Si substrate. To process the as-sprayed thin film, we have used a novel pulse-photonic curing technique to prepare a manganese oxide-reduced graphitic oxide (MnOx-rGO) nanocomposite thin film electrode. By following this highly scalable technique, we processed the thin film electrodes in several seconds, which is a significant reduction in processing time in comparison to the conventional techniques which would take from minutes to hours for processing. The as-prepared electrode has a highly porous 3D surface and contains dendritic structure, which increases ion/electron diffusion as well as provides a large specific surface area for the electrode/electrolyte reactions. The as-prepared MnOx-rGO composite shows pseudocapacitive behavior with the presence of redox peaks in the cyclic voltammetry (CV) curves and exhibited a specific capacitance of 11 mF cm−2 at constant areal current density of 0.4 mA cm−2. Secondly, we want to report the excellent electrochemical performance of the as-prepared electrode. Due to the combined effect of optimum processing conditions, desirable highly porous nanostructures, and the directly grown nanostructure on the substrate, which eliminates the need for a binder, as prepared electrode shows ultra-long lifetime by retaining 90% of the initial areal specific capacitance even after 100000 continuous charge–discharge cycles at a constant areal current density of 0.4 mA cm−2. Such long cyclic stability makes the as-prepared electrode a potential material for application in long term energy storage devices. These excellent results can be attributed to the increased conductivity of the binder-free electrode and highly porous morphology, which buffer the volume changes. Furthermore, such morphology helps enhance the structural stability of the electrode and allows for a large portion of the active material to take part in redox processes, resulting in high capacitance, and ultra-long cycle life.
2. Experimental section
2.1. Thin film deposition
Organometallic precursor Mn(III)-acetylacetonate powder (Sigma Aldrich) was dissolved in acetone (Sigma Aldrich) to prepare a saturated solution. The substrate was cleaned before spray-coating by ultrasonic treatment with acetone and deionized water. The precursor solution was then spray coated in ambient condition on Pt-coated silicon wafer (University Wafer, USA) with the help of an air spray gun (Paasche Airbrush, Chicago). After the solvent (acetone) evaporated, the precursor solution remained as a uniform precursor film on the substrate.2.2. Synthesis of highly porous 3D nanostructured manganese oxide
The as spray-coated precursor film was processed by using a pulsed-photonic curing system (PulseForge 1300, NovaCentrix Corp., Austin, Texas) with an output spectrum between 220–1000 nm and peaked at 450 nm. The pulsed light irradiation parameters (e.g., number of pulses, applied voltage, pulse length, etc.) were controlled with the help of a computer, and the energy density of a pulse sequence onto the samples was measured by an integrated bolometer before starting the irradiation. The photonic curing parameters used were: 600 V, envelope width: 1950 μs, μpulses: 5, fire rate: 1.2 Hz, pulse fluence: 5.6 J cm−2, number of pulses: 2. The pulsed-photonic curing system uses the flashlight from a xenon lamp to process the thin films in ambient conditions. It is well-known that transition metal acetylacetonate (acac) complexes such as Co-acac, Ti-acac, Fe-acac, and Mn-acac absorb the visible light. Our previous work found that the pulsed light irradiation results in the photoinitiated chemical reaction of the metal-acac molecules for the in situ formation of metal oxide-reduced graphitic oxide nanocomposite thin films.30–32 In this work, it was also found that the Mn-acac precursor absorbs the visible light during the pulsed light irradiation, resulting in the formation of a nanostructured composite thin film electrode. The first pulse irradiation produced a sharp and loud sound, possibly because all the Mn-acac molecules transformed into MnOx-reduced graphitic oxide (rGO) after the absorption of enough light energy. The second pulse irradiation worked similarly as the traditional post-photothermal treatment of MnOx-rGO. After the pulsed-photonic processing, the spray-coated Mn-acac precursor film was transformed into a solid nanostructured conductive thin film.2.3. Photonic processing, nucleation, and growth
Nucleation and growth of the oxide material on the substrate determines nanostructure and as such is crucial to the electrochemical performance of the electrode. By controlling the film growth, which is a highly non-equilibrium process, we can improve the mobility of the charge, that in turn determines the performance of the electrode. The pulsed photonic processing rapidly heats the film and in a concomitant manner, it rapidly cools after the pulse. The rapid cooling will trap metastable phases and nanostructures. Initially, the film cools via conduction on the substrate and subsequently through the porous MnOx-rGO nanostructure. The film growth and nanostructure formation occur during the irradiation by the 2 pulses as previously described.The film growth is preceded by the photonic pyrolysis of the precursor. During the photothermal treatment of the as-sprayed films, as we overcome the surface-volume energy barrier, nucleation, and growth of oxide material starts. As opposed to the conventional laser-based chemical vapor deposition (LCVD) which has a small exposure area with short pulse length and a fixed wavelength, photonic processing with the PulseForge can be done in a quick and roll-to-roll manner (in case of flexible substrates) because of its extremely short processing time, large exposure area, and broadband spectrum. Furthermore, the pulsed light in photonic curing has higher fluence, which means that higher processing temperature can be attained without damaging the substrate. At the initial stage of the nucleation, the thermal energy from the photonic processing causes atoms or molecules to condense and, undergo surface diffusion and migration to form a continuous film. Thin-film growth by conventional vapor deposition methods typically occurs via one of three different mechanisms: island growth, layer-by-layer growth and mixed growth.33 For high conductivity films, it is desired to have layer-by-layer growth, which is beneficial to the performance of the electrodes, as opposed to 3D-like island growth that gives rise to voids in the film. The two most important parameters for nucleation and growth are the energy barrier to surface diffusion (Ediff) and the energy barrier to desorption (Edes).34 The nucleation density of the films depends directly on these energy barriers. Most importantly, a smaller diffusion barrier is advantageous for the nucleation and growth as this results in higher surface mobility, which results in more chances for molecules to interact with one another and form a stable cluster. The smoothness of both the substrate and the as-sprayed metallic film is of crucial importance as the rough surface limits the electrical conductivity due to the electron scattering at grain boundaries.35
During the photonic curing process, we propose that the heterogeneous nucleation takes place (i) at the interface between the substrate and the precursor film, (ii) throughout the precursor film and (iii) between nanoparticles at the top of the film. The irradiation of the precursor film with the first pulse results in the formation of the bottom dense layer whereas the subsequent pulses result in the nucleation of nanoparticles and eventually the necking of the nanoparticles starts, as shown in nucleation and growth schematics Fig. 1. Further pulsed irradiation removes organic matter that hinders the sintering. Additionally, with the increasing number of pulses the temperature gets higher, which results in activation of the nanoparticles, the rapid growth of nanoparticle necking and the densification of the sintering neck. Finally, we get a conductive sintered thin film with a dendritic top layer and a dense bottom layer. Along with being nearly instantaneous, the other advantage of photonic synthesis is that as-cured metal oxide films show a higher surface roughness than thermally annealed ones due to the aforementioned rapid introduction of thermal energy that minimizes re-crystallization by not allowing self-diffusion of the metal oxide.36 This provides more active sites for electrode–electrolyte interaction which enhances the electrochemical performance of the electrode.
 | ||
| Fig. 1 Schematic illustration of nucleation and growth of the nanoparticles during photonic processing (“R” refers to the organic component in the film). | ||
2.4. Characterization methods
The surface and cross-sectional morphologies of the as-prepared MnOx-rGO electrode were studied by using a field-emission scanning electron microscopy (SEM) (Hitachi S-4800, Hitachi Corp., Tokyo, Japan). X-ray diffraction (XRD) patterns were collected using a powder diffractometer (Rigaku Smart Lab, USA) configured with a Cu Kα radiation at 40 kV voltage and 40 mA current. Raman spectrum measurement was carried out using a DXR Raman Microscope (Thermo Scientific, Massachusetts, USA) with a 532 nm laser as the excitation source.The electrochemical properties were studied by cyclic voltammetry (CV), galvanostatic charge–discharge cycling (GCD) and electrochemical impedance spectroscopy (EIS) in a three-electrode experimental setup using potentiostat/galvanostat (Model M204, Autolab, Metroohm, The Netherlands) with a Hg–HgO (CH Instruments, cat. no.152), platinum wire and MnOx-rGO being reference, counter and working electrodes, respectively. The CV and GCD tests were done in the potential range of 0–0.6 V vs. Hg–HgO and EIS measurements were carried out with a sinusoidal signal of 10 mV over the frequency range from 1 MHz to 10 mHz. All the measurements were carried out in 1 M KOH aqueous electrolyte solution at room temperature (23 °C).
3. Results and discussion
3.1. Nanostructure characterization
The surface morphology of the as-prepared MnOx-rGO was observed by SEM, which is shown in Fig. 2. In supercapacitors, the surface morphology of the electrode plays a significant role in its performance as the electrochemical reactions take place on (or near) the active material surface.4,6 From surface SEM images, we can see that the as-prepared MnOx-rGO electrode has a highly porous 3D structure, which is desirable for a supercapacitor electrode as capillary force mechanism in such morphology helps enhance the electrochemical performance due to the increased mass transport and diffusion of the electrolyte into the electrode.37 The porous morphology contributes to improving charge storage capacity because it provides a large specific surface area for the active material and electrolyte interaction. Therefore, most of the material is accessible to the electrolyte that in turn results in lower series resistance due to open transport paths. As such, it enhances ion diffusion in the charging and discharging processes that eventually contributes to the better electrochemical performance of the electrode. Furthermore, such porous morphology is advantageous to enhance the exploitation of the active site of the electrode material through intercalation and extraction of electrolyte ions, resulting in better electrochemical behavior.6 From the cross-sectional SEM image, it can be easily seen that the electrode has a dense bottom layer and a porous and dendritic top layer, with each dendrite of size around 50 nm. Such dendritic structures increase ion/electron diffusion, resulting in better performance. | ||
| Fig. 2 (a–c) Low to high magnification SEM surface images showing porous morphology and (d) cross-sectional image showing dendritic top layer of MnOx-rGO electrode. | ||
3.2. XRD analysis
The phase and composition of the as-prepared electrode was analyzed by X-ray diffraction (XRD), which is shown in Fig. 3. It is noted that three strong peaks can be found in Fig. 3, which can be associated with Pt from the substrate. Two characteristic peaks located at 35.2° and 43.8° are well indexed with randomly oriented grains of Ramsdellite, which is an orthorhombic MnO2.38 Additionally, two more peaks at 59.1° and 66.1° are also observed due to randomly oriented grains of Mn3O4.39 The above analysis demonstrates that the as-prepared electrode exists as a composite of MnO2 and Mn3O4, which can be directly used as an electrode without any organic binders in supercapacitor. | ||
| Fig. 3 XRD pattern of MnOx-rGO electrode. | ||
3.3. Raman spectroscopy analysis
Raman spectroscopy is an effective and nondestructive technique to investigate the structural characteristics and the chemical composition of carbon-based materials. Fig. 4 shows the Raman spectrum of the as-prepared nanostructured MnOx-rGO electrode. From the figure, Raman bands from 200 to 1000 cm−1 are present due to the Mn–O vibration of manganese oxides. Three peaks centered at 311, 364 and 653 cm−1 are in good agreement with the literature.40,41 The peaks at 311 cm−1 and 364 cm−1 are attributed to the out of plane bending modes of Mn–O and the asymmetric modes of bridge oxygen species (Mn–O–Mn), while a sharp peak 653 cm−1 corresponds to A1g mode due to the Mn–O breathing vibration of divalent Mn ions in tetrahedral coordination.41 The presence of D (1350 cm−1) peak, which is associated with the structural defects of carbon, and G (1580 cm−1) peak, which is associated with in-plane vibration of sp2 hybridized carbon atoms, further confirm that the carbon material present in the electrode is graphitic oxide in nature.32 The well-defined Raman spectrum signifies the good crystallinity of the as-prepared MnOx-rGO electrode. | ||
| Fig. 4 Raman spectrum of MnOx-rGO electrode. | ||
3.4. Electrochemical properties
To investigate the electrochemical performance of the as-prepared MnOx-rGO electrode, electrochemical measurements, such as CV, GCD and EIS, were carried out in 1 M KOH. CV measurements were carried out in the potential range between 0.0 to 0.6 V at various scan rates ranging from 10 to 100 mV s−1. Fig. 5 (a) compares the CV curves at 50 mV s−1 scan rate for the Pt–Si substrate and the as-synthesized MnOx-rGO electrode. It can be clearly seen from the figure that the substrate has no contribution to the electrochemical performance of the electrode. Fig. 5(b) shows the CV curves for MnOx-rGO electrode at various scan rates.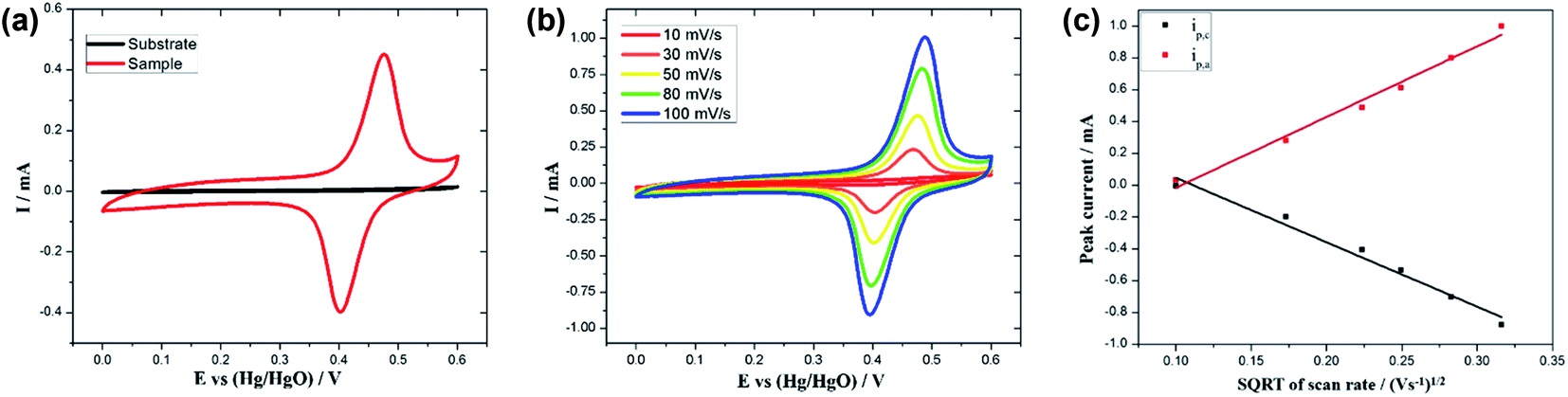 | ||
| Fig. 5 (a) CV curves at 50 mV s−1 scan rate for the Pt–Si substrate and the MnOx-rGO electrode, (b) CV curves at various scan rates for MnOx-rGO electrode, and (c) Randles–Sevcik plot for the MnOx-rGO electrode, where ip,c and ip,a refer to cathodic and anodic peak currents, respectively. | ||
The presence of the well-defined redox peaks in the CV curves at all scan rates indicates that the electrochemical capacitance of the electrode results from the pseudocapacitive charge storage mechanism. As obtained CV curves are similar to those previously reported42,43 with a slight difference in the redox peak potentials, which could be attributed to the unique morphology and nanostructure of the electrode. The increase in the area of the CV curves with increasing scan rates suggests the higher charge storage capability and good reversibility of the electrode along with fast electrons and ion diffusion rate in the electrode. Anodic peak around 0.45 V and cathodic peak around 0.40 V mainly come from the reversible faradaic redox reactions of active material and the electrolyte. With the increase in scan rate, anodic peak current has increased, and cathodic peak current has decreased, suggesting relatively low resistance of the electrode and fast redox reaction at the interface of the electrode and the electrolyte.44 Additionally, the anodic and cathodic peaks show slight positive and negative shifts, respectively, with increasing scan rates, due to the internal resistance of the electrode.
The capacitance of the electrode mainly depends on the two possible charge storage mechanisms in the electrode. The first one is the intercalation of alkali metal cations (C+), such as K+, during reduction and de-intercalation during the oxidation in the electrode;45
| MnO2 + C+ + e− ↔ MnOOC+ | (1) |
The second one is that the cations in the electrolyte are adsorbed at the manganese oxide electrode:
| (MnO2)surface + C+ + e− ↔ (MnO2−C+) surface | (2) |
Also, possible pseudo-capacitive mechanism for Mn3O4 electrode in KOH electrolyte can be given by following relations;46
| 3Mn(OH)2 + 2OH− ↔ Mn3O4·2H2O + 2e− + 2H2O | (3) |
| Mn3O4·2H2O + OH− ↔ 2MnOOH + Mn(OH)3 + e− | (4) |
Rapid transfer of K+ cation and electrons help accelerate the above charge storage reactions. Because of the high porosity, a greater surface area of the electrode is accessible for the electrolyte. Moreover, the porous structure shortens the ion diffusion length that helps facilitate the transfer of electron and ions involved in the redox reactions.47,48 Also, the minimal change in the shape of the CV curves, which can be attributed to the porous nanostructure that allows for greater interaction between the active material and the electrolyte, at higher scan rates suggests a small equivalent series resistance (ESR),49 excellent rate performance and electrochemical reversibility of the electrode.
Fig. 5(c) shows a plot of anodic and cathodic peak currents vs. square root of scan rates, known as the Randles–Sevcik plot, for the as-prepared MnOx-rGO electrode. For both the anodic and cathodic peak current densities, straight lines can be seen in the figure, which suggests the diffusion-controlled rate kinetics is the dominating mechanism of the redox processes.13,50
In order to further evaluate the electrochemical performance of the electrode, GCD measurements were performed in the potential range from 0.0 to 0.6 V (vs. Hg/HgO). Fig. 6(a) shows the schematic of a typical GCD curve for pseudocapacitors, with a characteristic internal resistance (IR) drop at the start of the discharge process. We performed charge–discharge measurements at various current densities for the MnOx-rGO electrode, as shown in Fig. 6(b). It can be seen that all the curves are nonlinear, which further indicates the pseudocapacitive behavior of the electrode, and contain a very small IR drop, indicating the low ESR of the electrode.
 | ||
| Fig. 6 (a) Schematic of a GCD curve for a pseudocapacitor and (b) GCD curves for MnOx-rGO electrode at various areal current densities. | ||
It is well known that calculating capacitance from a discharge curve is the most commonly used method. Therefore, the areal specific capacitance at various current densities of the electrode was calculated from the GCD curves by using the following equation;51
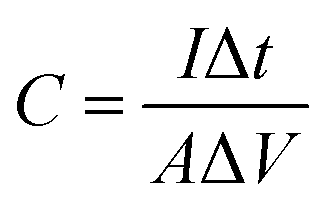 | (5) |
Initially, the specific capacitance is calculated to be 11 mF cm−2 and as the number of cycles increased, a slight decrease in the capacitance is recorded.
Cycling stability is another important requirement for supercapacitor electrodes. To test the cycling stability of the MnOx-rGO electrode, the GCD measurements were carried out for 100,000 continuous cycles at constant areal current density of 0.4 mA cm−2. Fig. 7(a) shows the trend of areal specific capacitance versus the number of GCD cycles at constant areal current density of 0.4 mA cm−2. Finally, even after 100000 GCD cycles, the capacitance is calculated to be 10 mF cm−2. This shows the excellent life cycle and the electrochemical stability of the MnOx-rGO electrode. The percentage of specific capacitance retention is calculated after a various number of cycles and are plotted in Fig. 7(b). It can be seen that the MnOx-rGO electrode has excellent capacitance retention as it retains 90% of initial capacitance even after 100000 GCD cycles, exhibiting excellent electrochemical and cycling stability as well as ultra-long cycle life. Such excellent electrochemical performance of the electrode can be attributed to the pseudocapacitive behavior of the electrode as well as the highly porous 3D morphology that allows for easy intercalation and de-intercalation of high-mobility K+ ions. Additionally, such porous morphology contributes to the electrochemical performance of the electrode by providing a buffer during volume changes. The number of GCD cycles we performed is significantly greater than what is reported in the literature for most of the manganese-oxide based supercapacitor electrodes.10,43,45,50,52–54 The excellent performance of the as-prepared electrode in terms of long-term cycling stability is superior to the metal-oxide based supercapacitor electrodes previously reported. The slight decrease in capacitance after 100000 GCD cycles can be attributed to irreversible faradaic redox reactions or the physical change in nanostructures in the process of OH− insertion/extraction during oxidation/reduction. These excellent electrochemical performances demonstrate the MnOx-rGO electrode's potential for application in long-term electronics.
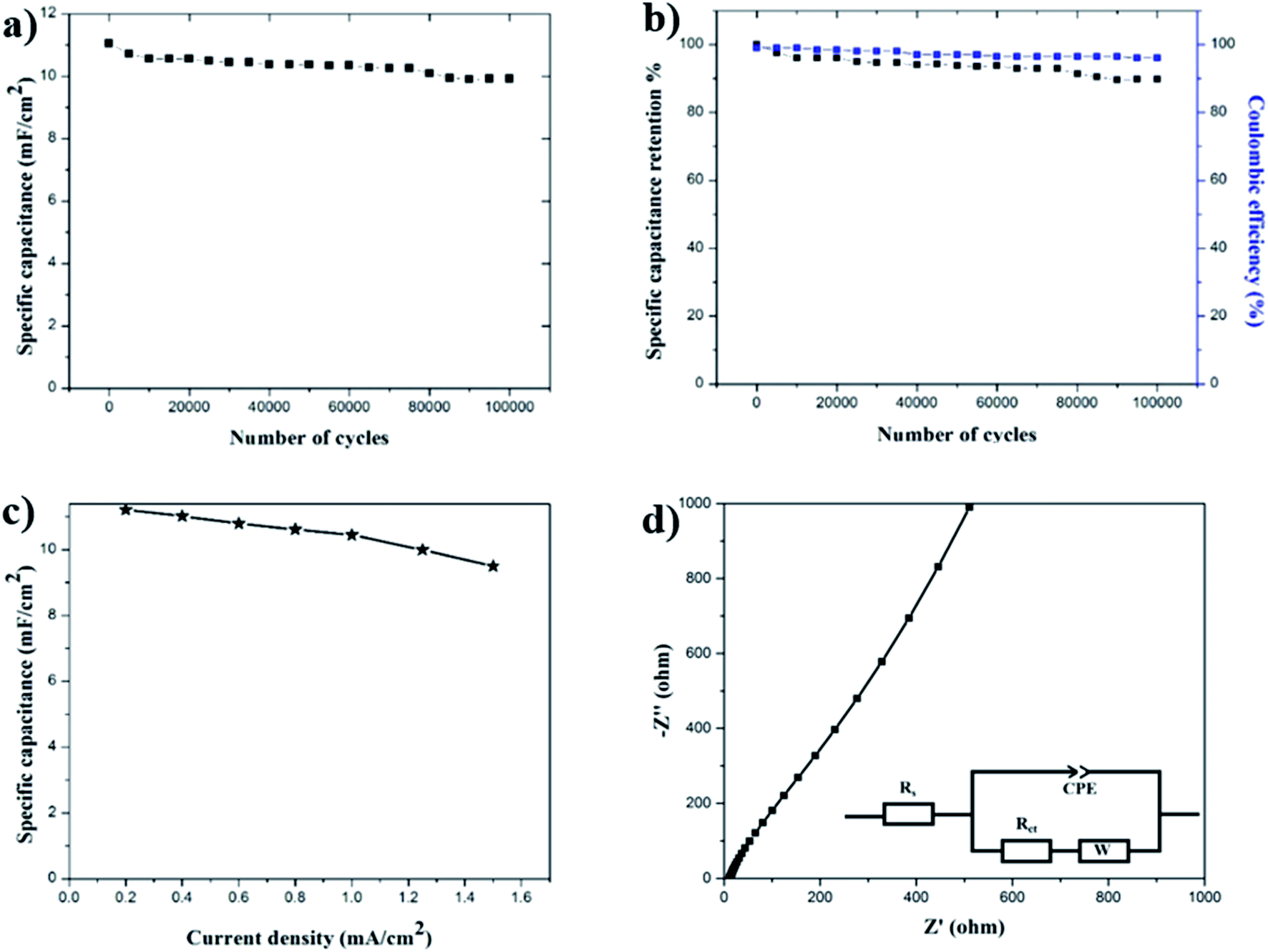 | ||
| Fig. 7 (a) Specific capacitance at 0.4 mA cm−2 constant areal current density, (b) capacitance retention and coulombic efficiency, (c) specific capacitance at various current densities, and (d) Nyquist plot for EIS measurements with equivalent circuit for MnOx-rGO electrode. | ||
Another important parameter of the electrochemical measurement is the rate capability measurement, which is related to the rate of ion diffusion in the electrode.33 To test the rate capability of the electrode, GCD measurements were performed at various current densities and specific capacitance at various current densities was calculated, which is shown in Fig. 7(c). We can see from the figure that even at 1.5 mA cm−2 current density, the electrode maintains a specific capacitance of 8.5 mF cm−2, exhibiting a good rate capability. At lower current densities, electrolyte ions get enough time to fully access the bulk of active material and an increased rate of intercalation/deintercalation takes place, which results in higher capacitance. On the other hand, at high current densities, the ions suffer from a low diffusion and cannot penetrate deep into the active material and only the outer surface of the electrode could take part in the redox reactions, thereby decreasing the effective interaction between the active material and the electrolyte. This is why the electrode shows low capacitance at higher current densities.
The coulombic efficiency of the electrode was also evaluated from the GCD curves by using the following equation;55
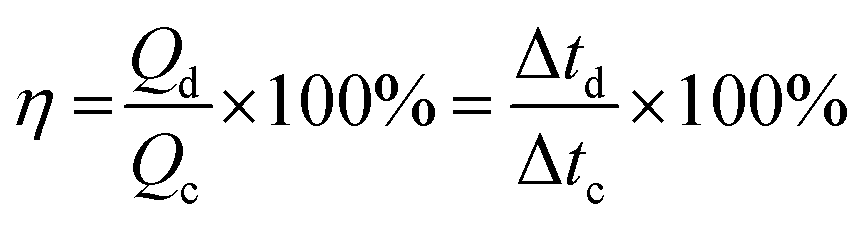 | (6) |
000 GCD cycles, the electrode maintains a coulombic efficiency of 95%, indicating the excellent performance of the electrode.
To further understand the electrochemical behavior of the MnOx-rGO electrode, EIS was performed. This is an effective method to study the fundamental behavior of electrode materials and the relationship between capacitive properties and electronic as well as ionic resistance of the electrode.56 An EIS plot is presented in the form of a Nyquist plot, where imaginary and real impedance are plotted against each other over a wide frequency range. A typical Nyquist plot contains a semicircle at higher frequencies that corresponds to the limited charge-transfer process, and a linear region at lower frequencies due to Warburg resistance (W), which is a result of the frequency dependence of ionic diffusion/transport in the electrolyte and to the surface of the electrode.57–59 For ideal capacitive behavior, the plot in low frequency region is supposed to be parallel to the imaginary Z-axis. Moreover, the intercept in the x-axis of a Nyquist plot gives the total internal resistance (Rs) that is the combination of the ionic resistance of the electrolyte, intrinsic resistance of the substrate and contact resistance between the active material and the current collector.60 The diameter of the semicircle at the high frequency region gives the charge transfer resistance (Rct), resulting from the faradaic reactions and the double-layer capacitance (Cdl) on the electrode surface.53 For the as-prepared MnOx-rGO electrode, we performed EIS measurements at room temperature over a frequency range of 1 MHz to 10 mHz, as shown in Fig. 7(d). From this measurement, the series resistance of the MnOx-rGO electrode is calculated to be 12.85 Ω. This could be because of the relatively poor conductivity of the electrode. Additionally, as can be seen from the figure, the EIS plot does not contain a semicircle at high frequency region, which is indicative of a very low charge transfer resistance of the electrode.61 This could also be another factor contributing to excellent electrochemical performance of the electrode.
4. Conclusions
In summary, we presented an extremely convenient and cost-effective one-step method to synthesize highly porous 3D nanostructured manganese oxide-reduced graphitic oxide (MnOx-rGO) thin film electrode. By following the photonic curing technique, we have significantly reduced the typically hours long processing time (in case of conventional techniques) to several seconds. To our knowledge, such a quick processing technique has not been reported for the fabrication of metal oxide supercapacitor electrodes. The specific capacitance of the as-prepared MnOx-rGO oxide electrode was 11 mF cm−2 at a constant current density of 0.4 mA cm−2. Even after 100000 GCD cycles, the electrode retained 90% of its initial capacitance, exhibiting excellent electrochemical stability and ultra-long lifetime. These excellent results can be attributed to the increased conductivity resulting from the conductive rGO component, the synergistic effect of carbon composite and the metal oxide, optimum processing conditions and a highly porous nanostructure, which offers enhanced ions/electron movement and high electrolyte-accessible surface area. The capacitance calculated at various current densities shows that the electrode exhibits high rate capability. In consideration of the excellent electrochemical performance, simple and quick preparation method, and very short processing time (several seconds), as prepared manganese oxide-reduced graphitic oxide (MnOx-rGO) electrode shows promising potential for application as supercapacitor electrodes. More importantly, the ultra-long lifetime of the electrode signifies its potential in long-term electronics applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Louisiana Board of Regents, Industrial Ties to Research Subprogram, Grant Number LEQSF (2017-20)-RD-B-04.References
- Z. Gao, W. Yang, J. Wang, N. Song and X. Li, Nano Energy, 2015, 13, 306–317 CrossRef CAS
.
- K. Liang, T. Gu, Z. Cao, X. Tang, W. Hu and B. Wei, Nano Energy, 2014, 9, 245–251 CrossRef CAS
- Y. Wu, S. Liu, H. Wang, X. Wang, X. Zhang and G. Jin, Electrochim. Acta, 2013, 90, 210–218 CrossRef CAS
- C. H. Ng, H. N. Lim, S. Hayase, Z. Zainal, S. Shafie and N. M. Huang, Ind. Eng. Chem. Res., 2018, 57, 2146–2154 CrossRef CAS
- K. Liang, X. Tang, B. Wei and W. Hu, Mater. Res. Bull., 2013, 48, 3829–3833 CrossRef CAS
- E. Mitchell, R. K. Gupta, K. Mensah-Darkwa, D. Kumar, K. Ramasamy, B. K. Gupta and P. Kahol, New J. Chem., 2014, 38, 4344–4350 RSC
- K. Liang, K. Marcus, Z. Yang, L. Zhou, H. Pan, Y. Bai, Y. Du, M. H. Engelhard and Y. Yang, Small, 2018, 14, 1702295 CrossRef PubMed
- B. Pandit, D. P. Dubal and B. R. Sankapal, Electrochim. Acta, 2017, 242, 382–389 CrossRef CAS
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS
- M. S. Wu and H. H. Hsieh, Electrochim. Acta, 2008, 53, 3427–3435 CrossRef CAS
- Z. S. Wu, W. Ren, D. W. Wang, F. Li, B. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed
- Y. T. Wang, A. H. Lu, H. L. Zhang and W. C. Li, J. Phys. Chem. C, 2011, 115, 5413–5421 CrossRef CAS
- R. Manikandan, C. J. Raj, M. Rajesh, B. C. Kim, J. Y. Sim and K. H. Yu, ChemElectroChem, 2018, 101 CrossRef CAS
- X. Lin, H. Lou, W. Lu, F. Xu, R. Fu and D. Wu, Chin. Chem. Lett., 2018, 29, 633–636 CrossRef CAS
- H. Wu, X. Wang, L. Jiang, C. Wu, Q. Zhao, X. Liu, B. Hu and L. Yi, J. Power Sources, 2013, 226, 202–209 CrossRef CAS
- C. J. Raj, M. Rajesh, R. Manikandan, J. Y. Sim, K. H. Yu, S. Y. Park, J. H. Song and B. C. Kim, Electrochim. Acta, 2017, 247, 949–957 CrossRef CAS
- S. Ortaboy, J. P. Alper, F. Rossi, G. Bertoni, G. Salviati, C. Carraro and R. Maboudian, Energy Environ. Sci., 2017, 10, 1505–1516 RSC
- M. Sawangphruk, P. Srimuk, P. Chiochan, A. Krittayavathananon, S. Luanwuthi and J. Limtrakul, Carbon, 2013, 60, 109–116 CrossRef CAS
- B. Wang, J. Park, C. Wang, H. Ahn and G. Wang, Electrochim. Acta, 2010, 55, 6812–6817 CrossRef CAS
- R. Amade, E. Jover, B. Caglar, T. Mutlu and E. Bertran, J. Power Sources, 2011, 196, 5779–5783 CrossRef CAS
- H. Jiang, J. Ma and C. Li, Adv. Mater., 2012, 24, 4197–4202 CrossRef CAS PubMed
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, J. Mater. Chem., 2011, 21, 16197–16204 RSC
- Y. Hou, Y. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS PubMed
- J. M. Ko and K. M. Kim, Mater. Chem. Phys., 2009, 114, 837–841 CrossRef CAS
- R. K. Sharma, H. S. Oh, Y. G. Shul and H. Kim, J. Power Sources, 2007, 173, 1024–1028 CrossRef CAS
- W. Meng, W. Chen, L. Zhao, Y. Huang, M. Zhu, Y. Huang, Y. Fu, F. Geng, J. Yu, X. Chen and C. Zhi, Nano Energy, 2014, 8, 133–140 CrossRef CAS
- G. Lota, K. Fic and E. Frackowiak, Energy Environ. Sci., 2011, 4, 1592–1605 RSC
- Z. Li, Y. Mi, X. Liu, S. Liu, S. Yang and J. Wang, J. Mater. Chem., 2011, 21, 14706–14711 RSC
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 767–784 RSC
- S. Luo, S. Zhang, B. B. Bourgeois, B. C. Riggs, K. A. Schroder, Y. Zhang, J. He, S. Adireddy, K. Sun, J. T. Shipman, M. M. Oguntoye, V. Puli, W. Liu, R. Tu, L. Zhang, S. Farnsworth and D. B. Chrisey, J. Mater. Res., 2017, 32, 1701–1709 CrossRef CAS
- B. Bourgeois, S. Luo, B. Riggs, Y. Ji, S. Adireddy, K. Schroder, S. Farnsworth, D. Chrisey and M. Escarra, Nanotechnology, 2018, 29, 315401 CrossRef PubMed
- B. B. Bourgeois, S. Luo, B. C. Riggs, S. Adireddy and D. B. Chrisey, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 588 CrossRef
- Y. Wang, W. Chen, B. Wang and Y. Zheng, Materials, 2014, 7, 6377–6485 CrossRef PubMed
- A. A. Virkar, S. Mannsfeld, Z. Bao and N. Stingelin, Adv. Mater., 2010, 22, 3857–3875 CrossRef CAS PubMed
- X. Yang, P. Gao, Z. Yang, J. Zhu, F. Huang and J. Ye, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed
- K. Tetzner, Y. H. Lin, A. Regoutz, A. Seitkhan, D. J. Payne and T. D. Anthopoulos, J. Mater. Chem. C, 2017, 5, 11724–11732 RSC
- P. Iamprasertkun, A. Krittayavathananon, A. Seubsai, N. Chanlek, P. Kidkhunthod, W. Sangthong, S. Maensiri, R. Yimnirun, S. Nilmoung, P. Pannopard, S. Ittisanronnachai, K. Kongpatpanich, J. Limtrakul and M. Sawangphruk, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed
- I. Stoševski, A. Bonakdarpour, F. Cuadra and D. P. Wilkinson, Chem. Commun., 2019, 55, 2082–2085 RSC
- S. Hirai, A. M. Dos Santos, M. C. Shapiro, J. J. Molaison, N. Pradhan, M. Guthrie, C. A. Tulk, I. R. Fisher and W. L. Mao, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 014417 CrossRef
- F. Xiao and Y. Xu, Int. J. Electrochem. Sci., 2012, 7, 7440–7450 CAS
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS
- H. U. Shah, F. Wang, A. M. Toufiq, A. M. Khattak, A. Iqbal, Z. A. Ghazi, S. Ali, X. Li and Z. Wang, Int. J. Electrochem. Sci., 2016, 11, 8155–8162 CrossRef CAS
- J. Shin, D. Shin, H. Hwang, T. Yeo, S. Park and W. Choi, J. Mater. Chem. A, 2017, 5, 13488–13498 RSC
- W. Li, B. Zhang, R. Lin, S. M. Ho-Kimura, G. He, X. Zhou, J. Hu and I. P. Parkin, Adv. Funct. Mater., 2018, 28, 1705937 CrossRef
- M. Feng, Q. Du, L. Su, G. Zhang, G. Wang, Z. Ma, W. Gao, X. Qin and G. Shao, Sci. Rep., 2017, 7, 1–8 CrossRef CAS PubMed
- L. He, G. Zhang, Y. Dong, Z. Zhang, S. Xue and X. Jiang, Nano-Micro Lett., 2014, 6, 38–45 CrossRef
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC
- R. Wang and X. Yan, Sci. Rep., 2014, 4, 3712 CrossRef PubMed
- T. Zhai, F. Wang, M. Yu, S. Xie, C. Liang, C. Li, F. Xiao, R. Tang, Q. Wu, X. Lu and Y. Tong, Nanoscale, 2013, 5, 6790–6796 RSC
- A. N. Naveen and S. Selladurai, RSC Adv., 2015, 5, 65139–65152 RSC
- H. An, Y. Wang, X. Wang, N. Li and L. Zheng, J. Solid State Electrochem., 2010, 14, 651–657 CrossRef CAS
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915–2921 RSC
- W. Yang, Z. Gao, J. Wang, B. Wang, Q. Liu, Z. Li, T. Mann, P. Yang, M. Zhang and L. Liu, Electrochim. Acta, 2012, 69, 112–119 CrossRef CAS
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L. C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS
- A. Mirmohseni, M. S. S. Dorraji and M. G. Hosseini, Electrochim. Acta, 2012, 70, 182–192 CrossRef CAS
- S. Afriyanti, F. C. Yao, W. Xu and L. P. See, Adv. Mater., 2013, 25, 2809–2815 CrossRef PubMed
- E. Lohrasbi and M. Asgari, Adv. Anal. Chem., 2015, 5, 9–18 CAS
- S. Dhibar, P. Bhattacharya, G. Hatui and C. K. Das, J. Alloys Compd., 2015, 625, 64–75 CrossRef CAS
- J. Yan, J. Liu, Z. Fan, T. Wei and L. Zhang, Carbon, 2012, 50, 2179–2188 CrossRef CAS
- L. Wang, H. Ji, S. Wang, L. Kong, X. Jiang and G. Yang, Nanoscale, 2013, 5, 3793–3799 RSC
- N. Bundaleska, J. Henriques, M. Abrashev, A. M. Botelho do Rego, A. M. Ferraria, A. Almeida, F. M. Dias, E. Valcheva, B. Arnaudov, K. K. Upadhyay, M. F. Montemor and E. Tatarova, Sci. Rep., 2018, 8, 12595 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |