
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrafluoroaryl azide as an N-terminal capping group for click-to-dissolve diphenylalanine hydrogels†
Sumit Dadhwal,
Jessica M. Fairhall,
Sarah Hook and
Allan B. Gamble *
*
School of Pharmacy, University of Otago, Dunedin, 9054, New Zealand. E-mail: allan.gamble@otago.ac.nz
First published on 3rd March 2020
Abstract
The synthesis of a bioorthogonal-responsive low molecular weight diphenylalanine (PhePhe)-based hydrogel that is capped with a 4-azido-2,3,5,6-tetrafluorobenzyl carbamate self-immolative linker is reported. The hydrogelator (AzF4-PhePhe) generates a stable hydrogel at 0.1 wt%, and rapidly reacts with the bioorthogonal reagent trans-cyclooctene (TCO), inducing a gel-to-solution transition. The critical gel concentration is five-fold lower than our previously synthesized non-fluorinated hydrogelator (Az-PhePhe), and the minimum concentration of TCO required for visible gel-to-solution transition in 24 hours is 1 mM. Doxorubicin can be encapsulated in the hydrogel and TCO-triggered dissolution results in 76% and 89% release after 10 and 24 hours, respectively. Compared with our non-substituted aryl azide capping group used for Az-PhePhe, the tetrafluorinated aryl azide group improves the stability of the hydrogel in unbuffered water at a lower critical gel concentration, while improving sensitivity towards the bioorthogonal reagent TCO.
1 Introduction
Short peptidic hydrogelators have been identified for their potential use in biomedical applications. In particular, the use of and scope of hydrophobic capping moieties on the N-terminus or C-terminus of the short peptides has attracted significant attention in the last few decades.1,2 These amphiphilic hydrogelators can partake in non-covalent interactions3 such as electrostatic, hydrogen bonding, π–π stacking, and hydrophobic van der Waals interactions, to form stable cross-linked fibrous gel networks.1,2,4,5 The use of halogen substituents, such as fluorine, on the aromatic rings of the hydrogelator backbone and hydrophobic N-terminal capping groups have been reported to improve the relative strengths of the hydrogel.6–11 Ryan et al.9 reported an instant gelation and the formation of a rigid hydrogel using Fmoc-protected (capped) pentafluorophenylalanine. The improved properties of the hydrogel were due to intermolecular π–π interactions between the perfluorinated phenylalanine ring and an Fmoc-group of adjacent molecules.9 In another example, Liyanage and Nilsson explored the effect of electron-donating (e.g. OH, CH3, NH2) and electron-withdrawing (e.g. NO2, CN, F) substituents on the phenylalanine group of Fmoc-phenylalanine on the gel self-assembly process.7 The authors reported rapid hydrogel formation in the presence of electron withdrawing groups, and proposed that this could be due to lower electrostatic repulsion between aromatic π-systems.7 Hsu et al.6 reported the improved mechanical properties of hydrogels via formation of thinner entangled nanofibers using the pentafluorophenyl moiety as a hydrophobic capping group on the N-terminus of the dipeptide. They also reported the gel-to-sol transition temperature of the pentafluorophenyl-capped hydrogel was higher (45 °C) than body temperature, making it a potential scaffold for tissue engineering.6 In another report from the same group,10 an N-4-fluorobenzyl-capped diphenylalanine hydrogelator exhibited a critical gel concentration at 1.5 wt%, whereas the control hydrogelator, N-benzyl-diphenylalanine formed a viscous solution at 5 wt% with a white precipitate, indicating that the presence of one fluorine atom on the benzylic capping group was sufficient to form electrostatic π–π interactions between fluorinated and non-fluorinated aromatic rings.We previously reported the synthesis and characterisation of an N-4-azidobenzylcarbamate-diphenylalanine (Az-PhePhe) 1 hydrogel that could be triggered to undergo a gel-to-solution (gel–sol) transition upon addition of a strained alkene; trans-cyclooct-4-enol/TCO (Scheme 1a).12 The critical gel concentration was found to be 0.5 wt% and hydrogel encapsulated with model drug (doxorubicin) showed controlled drug release in response to TCO addition, demonstrating that TCO-triggered hydrogels have potential for further development as stimuli-responsive drug delivery systems. However, in solution, the soluble hydrogelator 1 was found to be toxic at high μM concentrations in tumour and normal kidney cells, indicating that a lower critical gel concentration would be ideal for biological applications. The Az-PhePhe 1 hydrogel also displayed a relatively slow gel-to-sol transition in the presence of 5 mM concentrations of the trigger (TCO).
 | ||
| Scheme 1 TCO-triggered gel–sol transition of; (a) dipeptide hydrogel using hydrogelator 4-azidobenzyl carbamate-PhePhe 1 (Az-PhePhe; previous work),12 and (b) dipeptide hydrogel using hydrogelator 4-azidotetrafluorobenzyl carbamate-PhePhe 3 (AzF4-PhePhe; this work). | ||
Our lab and others, have demonstrated that the rates of bioorthogonal reactions, such as the 1,3-dipolar cycloaddition13 and Staudinger reduction,14,15 can be increased in the presence of fluorine substituents on an aryl azide group. In particular, the 1,3-dipolar cycloaddition between TCO and an azide is almost an order-of-magnitude faster when fluorine-substituents are present on the aryl azide ring.13 Therefore, we have been investigating the effect of a fluorine-substituted aryl azide N-capping group on the critical gel concentration of the dipeptide diphenylalanine (PhePhe) and sensitivity towards the bioorthogonal trigger (TCO).
Herein we report on the use of the 4-azido-2,3,5,6-tetrafluorobenzyl carbamate as a removable hydrophobic capping group for the N-terminus of PhePhe, giving hydrogelator 3 (AzF4-PhePhe; Scheme 1b). The fluorinated N-capping group improves the strength of the hydrogel and its stimuli-responsiveness towards TCO. Dissolution of the AzF4-PhePhe hydrogel is the result of a 1,3-dipolar cycloaddition, generating aniline 4; via intermediates 3a–3c and an aldehyde by-product (Fig. S1†), which can undergo 1,6-self-immolation to provide dipeptide 2 and an azaquinone methide. Evidence suggests that self-immolation of 4 to give 2, does occur during dissolution of the hydrogel (albeit slowly). However, mass spectrometry experiments show that aniline 4 is relatively stable under the acidic conditions of the hydrogel, and suggests that the strength of the hydrogel decreases (i.e. dissolution begins) upon formation of aniline 4.
2 Experimental
2.1 Materials and general experimental
Materials and general experimental techniques were purchased and conducted, respectively, as per our previous reports.12,13,16 All chemicals were purchased from commercial suppliers and used without purification. 5-Hydroxy-1-cyclooctene (cis-cyclooct-4-enol) was purchased from Carbosynth Limited, UK. Diphenylalanine and 4-amino-2,3,4,6-tetrafluoro benzoic acid were purchased from AK Scientific, USA. Doxorubicin hydrochloride (DOX) was purchased from Lancrix Chemicals, Shanghai, China. All other reagents were purchased from Sigma-Aldrich or AK Scientific. 1H and 13C NMR spectra were recorded on a 400 MHz Varian MR spectrometer. Chemical shifts are reported as δ in parts per million (ppm) and coupling constants are reported as J values in Hz. High resolution electrospray ionization mass spectra (ESI-MS) were recorded on a microTOFQ mass spectrometer. HPLC was performed using an Agilent 1200 system, equipped with a Phenomenex Synergi 4 μm Fusion-RP 80A (150 × 4.6 mm) column, and a photodiode array detector. The applied mobile phases used for kinetic studies were: A, aq. H2O + 0.1% formic acid; and B, MeCN + 0.1% formic acid. Flow speed was 1 mL min−1 and injection volumes were 50 μL. Gradient mobile phase, 80% A/20% B to 100% B in 8 minutes, 2 minutes at 100% MeCN with 0.1% formic acid and returning to starting conditions by 15 minutes.2.2 Synthesis
The synthesis of trans-cyclooct-4-enol (TCO)16,17 was conducted according to literature procedures. The synthesis of (4-azido-2,3,5,6-tetrafluorobenzyloxycarbonyl)-L-phenylalanyl-L-phenylalanine 3, and intermediates 6/7/8 from benzoic acid 5 are described in the ESI.†2.3 Hydrogel preparation
Hydrogel preparation was conducted using a similar procedure to our previous report.12 The critical gel concentration (CGC) was tested at 0.02 wt%, 0.03 wt%, 0.04 wt% 0.05 wt% and 0.1 wt%. For 0.1 wt% hydrogel, AzF4-PhePhe 3 (1 mg) was dissolved in 50 μL DMSO followed by addition of 950 μL aqueous phase. After addition of aqueous phase, the solution turned opaque, followed formation of the transparent gel. Gel formation, after addition of the aqueous phase, took approx. 1 min at 37 °C and approx. 5 min at 25 °C.2.4 Rheology
Rheology measurements were performed as per our previous report.12 Measurements were performed using 40 mm parallel plate on an Oscillatory HR-3 Rheometer (TA Instruments) at 25 °C. For 0.1 wt% hydrogels, AzF4-PhePhe 3 (1 mg) was dissolved in DMSO (50 μL) and after adding water (950 μL) or PBS (pH 7.4, 950 μL) the sample was loaded on the rheometer and left undisturbed for 10 minutes to form a fibrous gel network. The LVER measurements (Fig. S2†) were done with 0.1 to 100% oscillation strain. The storage (G′) and loss (G′′) moduli were measured using dynamic frequency sweep at a fixed strain of 1.0% in the frequency range 0.1 to 100 rad s−1. Rheology measurements were also performed at different temperatures (25 and 37 °C). For the time dependent rheology experiment, hydrogel (0.1 wt%) was prepared on the rheometer followed by addition of 0.5 mL water containing TCO (5 mM). Rheology was measured at various time points in the dark.2.5 Transmission electron microscopy (TEM)
TEM experiments were conducted using our previously reported standard conditions.12 TEM was used to investigate the morphology of 0.1 wt% gels. The sample grids were prepared by depositing 10 μL of gel onto plasma-glowed TEM grids (300 mesh, carbon-coated copper grids). Excess sample was carefully removed after one minute using blotting paper. The grids were dried completely before viewing. They were viewed on a Philips CM100 TEM (Philips Electron Optics, Eindhoven, The Netherlands) operated at an accelerating voltage of 100 keV. Images were recorded using a MegaView 3 camera (Soft Imaging System GmbH, Münster, Germany).2.6 Scanning electron microscopy (SEM)
SEM experiments were conducted using our previously reported standard conditions.12 The gel (0.1 wt%) sample was dried onto aluminum stubs overnight. After drying, the samples were sputter coated in an Emitech K575X Peltier-cooled high resolution sputter coater (EM Technologies Ltd, Kent, England). They were coated with 5 nm of gold palladium. Samples were viewed in a JEOL JSM-6700F field emission scanning electron microscope (JEOL Ltd, Tokyo, Japan) at an accelerating voltage of 5 kV.2.7 Infrared spectroscopy
Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR) was carried out using a Varian 3100 FTIR (Excalibur Series) instrument equipped with an attenuated total reflectance accessory (GladiATR, Piketech, USA). The experiments were conducted under the same conditions we previously reported.12 Following formation of the hydrogel from 3 (0.1 wt%), the sample was lyophilised and the FTIR spectrum was measured (sample prior to trigger). In parallel, TCO (5 mM) was added to a 0.1 wt% hydrogel from 3 (trigger experiment). Following an incubation period of 12 hours (at 37 °C), the sample was lyophilized and re-analysed by FTIR. For the time-dependent ATR-FTIR study, hydrogel from 3 was prepared in different vials using water and incubated at 37 °C with 0.5 mL TCO (5 mM) in water. At the stated time points, the samples were lyophilised and analysed by ATR-FTIR. For all measurements, the lyophilised gel sample was clamped directly on the ATR diamond crystal and spectra was recorded over a range of 400–4000 cm−1. The spectrum was the mean of 64 scans with a resolution of 4 cm−1. Data collected was analysed using Varian Resolution Pro, v4.1.0.101 software.2.8 HRMS analysis of TCO-triggered hydrogel
HRMS analysis was conducted using similar assay conditions to our previous report.12 Hydrogel generated from 3 (0.1 wt%) was prepared in water (5% DMSO). For the pH experiments, final pH (7.4, 6.5) was adjusted using dilute HCl and/or NaOH (measured by a pH meter S220 Seven Compact, Mettler Toledo). This was followed by addition of 0.5 mL TCO dissolved in water, and incubation at 37 °C for 12 hours (or the time indicated). The gel/solution was lyophilised and analysed by HRMS (ESI+ and/or ESI−).2.9 NMR mechanistic studies
A solution of AzF4-PhePhe 3 (4.3 mg) and internal standard (1-fluoro-2,4-dinitrobenzene; 1.4 mg) were dissolved in acetonitrile-d3 (0.75 mL; ampoule) under N2 (final concentration of 3 was 10 mM). To initiate the 1,3-dipolar cycloaddition, TCO (4.7 mg) was added to the above solution (final TCO concentration of 50 mM) and the NMR spectra (1H and 19F) were recorded at various time points. After 48 hours, 100 μL NMR sample was removed for the HRMS analysis and replaced with 100 μL D2O. After addition of D2O to the NMR tube, the sample was mixed (shaken) and spectra was recorded.2.10 HPLC kinetic study
A 6 mM stock of trans-cyclooctenol in water and a 1 mM stock of AzF4-PhePhe 3 in acetonitrile were prepared. The kinetics assay was conducted using the conditions of our previous report.12 To begin the 1,3-dipolar cycloaddition, 500 μL of trans-cyclooctenol stock was added to 500 μL of compound 3 stock. The sample was incubated at 37 °C, and at 20 min intervals, an aliquot of 50 μL was injected onto the HPLC. The absorbance for compound 3 was measured at HPLC-UV: 254 nm tR = 8.1 min. For control experiments (run in triplicate): 500 μL of AzF4-PhePhe 3 stock was added to 500 μL of water and incubated at 37 °C, an aliquot of 50 μL was injected onto the HPLC at time intervals 0 min and 24 hours. The second order rate constant, from triplicate runs, was calculated as 0.0947 ± 0.0098 M−1 s−1 (n = 3).2.11 Doxorubicin (DOX) encapsulation and release studies
Drug release studies from the hydrogel were conducted using slightly modified conditions to our previous report.12 Specifically, a 25 μL solution of AzF4-PhePhe 3 (0.5 mg) in DMSO was prepared. In parallel, a 1 mg mL−1 stock solution of DOX in water was prepared, and from this stock a 100 μL aliquot was diluted in 375 μL water. The water solution containing 100 μg mL−1 (0.17 mM) of DOX was added to the DMSO solution of 3. The gel (0.1 wt% of 3, pH 6.12) was left undisturbed for 10 min. The gel was then washed with water (3 × 0.5 mL) to remove any trace amounts of untrapped DOX. Following this, 1 mL of TCO (1 mM) in water was placed over the gel, and incubated at 37 °C. For the control experiment, water-only was added (without TCO). At the indicated time-points 0.1 mL water was removed and replaced with the same amount of water containing TCO (or water-only). Care was taken so as to not disturb the gel. The absorbance of released DOX was measured with a plate reader (PolarStar Omega, BMG Labtech) at 485 nm. The cumulative percentage of DOX released was calculated from a standard curve of DOX (Fig. S3†) in water measured at 485 nm.2.12 Cell viability study
Using the assay conditions from our previous studies,12 a stock solution of AzF4-PhePhe 3 (1 mg mL−1) was made using DMSO (2.5%) in 1 mL of cell media (Eagle's Minimum Essential Medium; EMEM), followed by sonication to dissolve 3 completely. Different concentrations of 3 were prepared using the stock solution. Madin-Darby Canine Kidney [MDCK (NBL-2)] cells [American Type Culture Collection (ATCC®), Manassas, VA, USA] were plated at a density of 7.5 × 103 cells per well. Cells were incubated with 3 at different concentrations for 24 and 48 hours, followed by addition of 10 μL of resazurin. After addition of resazurin, MDCK cells were incubated for 4 hours at 37 °C. Fluorescence was measured at 544 nm (excitation) and 590 nm (emission) in a PolarStar Omega plate reader. Cell viability was normalized against cell only (100%) and cell-free controls (0%). Data were analysed in GraphPad Prism.3 Results and discussion
3.1 Synthesis
The synthesis of AzF4-PhePhe 3 is outlined in Scheme 2. 4-Azido-2,3,5,6-tetrafluorobenzyl alcohol 7 was prepared as previously reported using the commercially available acid 5 (via 6),13 followed by conversion to the activated succinate ester 8. Activated ester 8 was reacted with diphenylalanine 2 (PhePhe) in the presence of N,N-diisopropylethylamine (DIPEA) to provide AzF4-PhePhe 3. | ||
| Scheme 2 Synthesis of hydrogelator 3 (AzF4-PhePhe). | ||
3.2 Hydrogel formulation and characterisation
Dissolving AzF4-PhePhe 3 in DMSO followed by addition of water (final DMSO conc. 5%); i.e. using the solvent switch method, resulted in hydrogel formation. After addition of water into the DMSO solution of 3 (final pH of gel = 3.7) an opaque solution was observed, that subsequently turned transparent,18–20 indicating self-assembled network formation (Fig. 1a).18 To determine the critical hydrogel concentration with 3, tube inversion at varying concentrations of 3 was conducted (0.01–0.5 wt%), and gel formation was identified at 0.05 wt%, however, a visibly superior gel at 0.1 wt% was identified (Fig. 1a). Rheology of the gel at 0.1 wt%, 0.3 wt%, and 0.5 wt% indicated that at all three concentrations, a stable hydrogel was formed (Fig. 1b–d). The linear viscoelastic region (LVER) of the hydrogel at 0.1 wt% indicated that up to 10% oscillation strain could be applied without destroying the structure of the gel (Fig. S2†). Notably, the viscoelasticity of the gel at 0.1 wt% (Fig. 1b; G′ ∼ 100 Pa and G′′ ∼ 10 Pa) was equivalent to the viscoelasticity we measured for the hydrogel formed by Az-PhePhe 1 at five-times the concentration (0.5 wt%: G′ ∼ 100 Pa and G′′ ∼ 10 Pa).12 Gel strength increased at 0.3 and 0.5 wt% (G′ ∼ 1000 Pa and G′′ ∼ 100 Pa), to ten-times the viscoelasticity reported by our original hydrogel using Az-PhePhe 1. Overall, the visual critical gel concentration and rheology studies on gel formation indicate that AzF4-PhePhe 3 can form similar strength hydrogels to the non-fluorinated Az-PhePhe 1 at five-times lower concentrations (0.1 wt% cf. 0.5 wt%).12 This supports the hypothesis that the presence of fluorine-substituents in the peptide structure improves and strengthens hydrogel formation. For stimuli-responsive studies reported herein, 0.1 wt% hydrogel was selected as this; (1) had a comparable gel viscoelasticity to Az-PhePhe 1 (providing a direct comparison to our previous studies), and (2) enabled us to work at lower concentrations of hydrogelator (preferred as we move towards biocompatible hydrogels).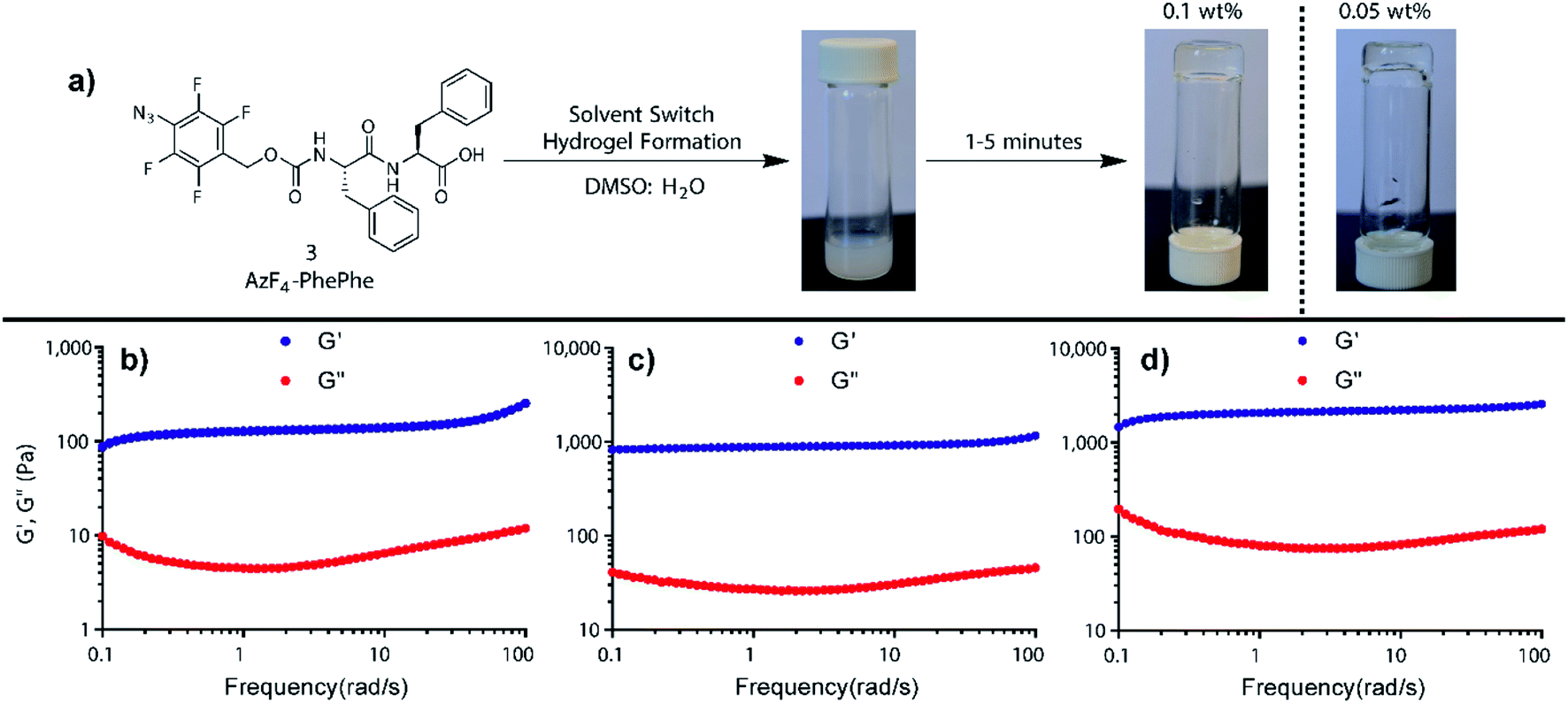 | ||
| Fig. 1 (a) Hydrogel formation using AzF4-PhePhe 3 at 0.05 and 0.1 wt%, and rheology studies conducted on the hydrogel formed at (b) 0.1 wt%, (c) 0.3 wt%, and (d) 0.5 wt%. | ||
The rate of hydrogel self-assembly from 3 was investigated at different temperatures (4, 25 and 37 °C). Hydrogels did not form at 4 °C, whereas the self-assembly process took around 5 minutes at 25 °C and 1 minute at 37 °C. Frequency sweep measurements examining the viscoelastic properties of the hydrogel from 3 at 25 °C and 37 °C indicated that stable gels form in both cases, with slightly higher viscoelasticity observed for the gel at 37 °C (Fig. S4†). Upon addition of the hydrogelator 3, the pH of the gel formed was acidic (pH 3.7), indicating the influence of the C-terminal carboxylate on the hydrogel pH. At higher temperatures, aromatic π-stacking interactions are expected to dominate self-assembly of short hydrophobic peptides,21 providing a plausible explanation for more rapid assembly of our hydrophobic hydrogelator 3 at 37 °C compared to lower temperatures. The same effect by temperature on time for gel formation was reported on a series of Fmoc-peptide-based hydrogels.22
Hydrogel formation was also attempted in PBS at 25 °C. However, there was no opaque suspension (as in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O) after addition of PBS into a DMSO solution of 3, and subsequent formation of hydrogel was not observed. Instead the addition of PBS (pH 7.4) to the DMSO solution of 3 resulted in a transparent viscous solution that contained floating fibres/precipitates (Fig. S5;† final hydrogel pH = 3.9), supporting the rheology data that showed no gel formation (Fig. S6†). Attempts to form the gel in PBS at 37 °C failed, providing the same viscous solution. Notably, the use of PBS during the solvent switch method still resulted in an acidic hydrogel (pH = 3.9), indicating that a change in pH was not responsible for the inhibition of fibril formation. The salts present in the buffer appear to be inhibiting fibril formation to some extent in the more hydrophobic fluorinated hydrogelator 3, a phenomenon not observed for the non-fluorinated hydrogelator 1.12 In support of our results, Nilsson and co-workers described the synthesis of fluorine-containing hydrogelators and noticed a transparent viscous solution and weak hydrogel with PBS.18 The authors hypothesised that solvent plays a key role in hydrogel formation, and that salts such as phosphate can shield hydrogen bonding and other non-covalent interactions between fibrils.18
:H2O) after addition of PBS into a DMSO solution of 3, and subsequent formation of hydrogel was not observed. Instead the addition of PBS (pH 7.4) to the DMSO solution of 3 resulted in a transparent viscous solution that contained floating fibres/precipitates (Fig. S5;† final hydrogel pH = 3.9), supporting the rheology data that showed no gel formation (Fig. S6†). Attempts to form the gel in PBS at 37 °C failed, providing the same viscous solution. Notably, the use of PBS during the solvent switch method still resulted in an acidic hydrogel (pH = 3.9), indicating that a change in pH was not responsible for the inhibition of fibril formation. The salts present in the buffer appear to be inhibiting fibril formation to some extent in the more hydrophobic fluorinated hydrogelator 3, a phenomenon not observed for the non-fluorinated hydrogelator 1.12 In support of our results, Nilsson and co-workers described the synthesis of fluorine-containing hydrogelators and noticed a transparent viscous solution and weak hydrogel with PBS.18 The authors hypothesised that solvent plays a key role in hydrogel formation, and that salts such as phosphate can shield hydrogen bonding and other non-covalent interactions between fibrils.18
The morphology of the hydrogel formed with 3 (0.1 wt% at pH 3.7) was investigated by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) which showed the formation of highly dense and thin fibres in the hydrogel (Fig. S7 and S8†). Morphology of AzF4-PhePhe 3 in PBS (final pH 3.9) was also investigated using TEM. The image showed the formation of non-entangled fibres which do not form a network as seen with water (Fig. S9 cf. Fig. S7†), supporting the visual observations outlined above. Incubating 3 at 37 °C in PBS for 24 hours, and subjecting the sample to electron microscopy (Fig. S9b†), revealed the same low density fibres, with no gel formation over time. Therefore, as previously reported, the data suggests that phosphate salts play some role in preventing hydrogel formation from 3.18
3.3 Triggered gel-to-solution transition
To investigate the stimuli-responsive nature of the hydrogel formed by AzF4-PhePhe 3 in water, varying concentrations of TCO (1, 3 and 5 mM) were added to the hydrogel. Visual analysis indicated that the most rapid and complete gel-to-sol transition (Fig. S10†) was observed within 4 hours at 37 °C when the AzF4-PhePhe hydrogel (0.1 wt%, pH 3.7) was incubated with 5 mM of TCO. At 3 mM and 1 mM of TCO, the gel-to-sol transition was visually evident, but the time required for complete dissolution increased from 4 hours to ∼8 and 24 hours, respectively. The gel-to-sol transition of the AzF4-PhePhe hydrogel was faster with TCO (5 mM) than our previously reported non-fluorinated azide hydrogel (Az-PhePhe 1),12 which was 10 hours for complete gel-to-sol phase transition. Overall, this result indicated that a tetrafluoro-substitution pattern on the aryl azide linker has improved the sensitivity of the hydrogel towards TCO. TEM analysis also supported the gel-to-sol transition upon addition of, and incubation with TCO (5 mM), with the entangled fibrous network being degraded (Fig. S11†). Infrared (IR) spectroscopy confirmed that a 1,3-dipolar cycloaddition had transpired via the absence of the strong asymmetric N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch of the azide group at ∼2100 cm−1 when 5 mM TCO was added (Fig. S12†).
N stretch of the azide group at ∼2100 cm−1 when 5 mM TCO was added (Fig. S12†).
To relate the visual gel–sol observations to mechanism, time-dependent rheology and IR studies on AzF4-PhePhe 3 hydrogel during the TCO-induced gel-to-sol transition were carried out (Fig. 2). The rheology data demonstrated the gradual weakening of the hydrogel after addition of TCO (5 mM) as seen by the decrease in storage (G′) and loss (G′′) moduli over time (Fig. 2a and b). Notably, after only 0.5 hours, the storage and loss moduli (and thus viscoelasticity), had decreased by ∼10-fold. Over the next 3.5 hours the G′ and G′′ continued to decrease, but only by ∼5-fold. The disappearance of the azide stretch in the time-dependent IR studies of the 1,3-dipolar cycloaddition supported the rheology data, with only a trace amount of azide remaining at 0.5 hours, and no remaining azide after 1 hour (Fig. 2c). Taken together, the results suggest that the 1,3-dipolar cycloaddition is fast, but that hydrolysis of the triazoline and imine (at least in part), is required for dissolution of the hydrogel. Based on our previous studies of the 4-azido-2,3,5,6-tertrafluorobenzyl linker,13 there is also a possibility that the self-immolative step for the AzF4-PhePhe 3 gel will be slower than the non-substituted Az-PhePhe 1 gel. However, the rheology and IR data did not provide evidence into whether or not the final 1,6-self-immolation was required for dissolution to begin in the AzF4-PhePhe hydrogel, thus further studies (NMR and mass spectrometry) were conducted (see below).
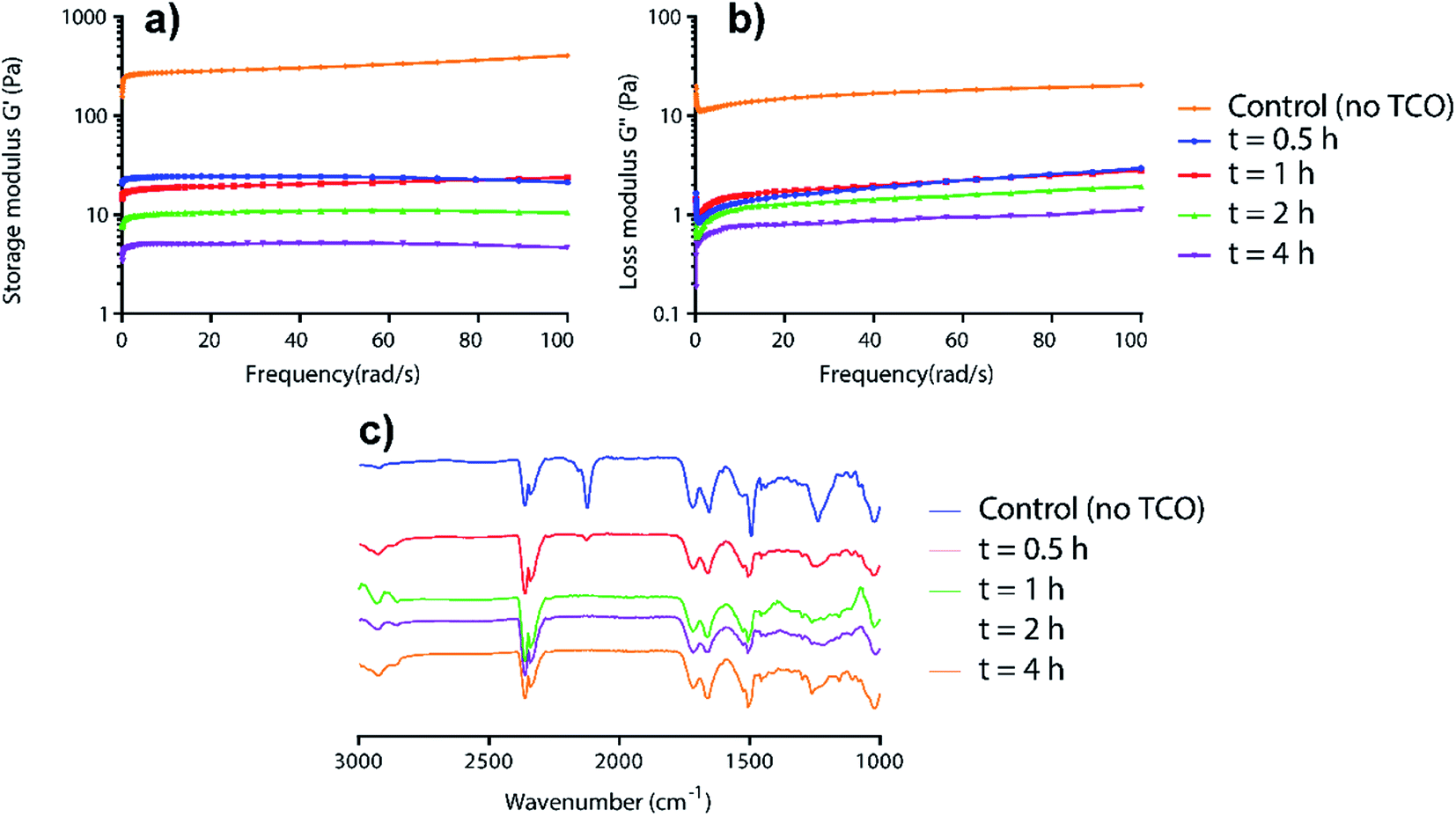 | ||
| Fig. 2 Time-dependent rheology (a and b) and IR studies (c) for the incubation of AzF4-PhePhe 3 hydrogel (0.1 wt%) and TCO (5 mM). | ||
High resolution mass spectral (HRMS) analysis of the gel-to-sol transition after 4 hours incubation at 37 °C with TCO (5 mM), followed by lyophilisation, supported the IR data above, with the absence of a molecular ion (M + H and M + Na) indicating complete consumption of the AzF4-PhePhe 3 (Fig. S13†). The major peak in the spectrum (ESI-positive) was found at m/z 556.1460 (Fig. S13†), and could be assigned to the M + Na ion of aniline, 4-amino-2,3,5,6-tetrafluorobenzylcarbamate-PhePhe dipeptide 4 (Scheme 1). Mass spectral analysis (ESI-negative) of AzF4-PhePhe hydrogel 3 incubated with TCO (5 mM) for 12 hours (lyophilised), demonstrated that self-immolation of the 4-amino-2,3,5,6-tetrafluoro linker and release of dipeptide PhePhe 2 does occur over prolonged incubation; as evidenced by the peak at m/z 311.1403 that can be assigned to PhePhe 2 [M–H], and m/z 435 that can be assigned to the imine 3e (Fig. S14†) formed by condensation of PhePhe 2 and the carbocyclic aldehyde (Fig. S1†). However, the imine (aldimine and ketamine) and/or aziridine reaction intermediates 3b–3d (Fig. S1†) were also identified by their [M–H] molecular ion at m/z 656.254 (Fig. S14;† all identical in mass). As expected, the tetrafluoro-substituent pattern led to an unstable triazoline intermediate 3a (Fig. S1†); not identified in the mass spectra (Fig. S13 and S14†). Therefore, the HRMS data for the AzF4-PhePhe 3 dipeptide hydrogel formed under relatively acidic conditions (pH 3.7) suggests that initiation of the gel–sol transition (within 4 hours when 5 mM TCO used) appears to depend on the conversion of the azide functionality to an aniline, and does not require complete 1,6-self-immolation and subsequent release of the free dipeptide.
In our previous work we demonstrated that the pKa of the imine generated after cycloaddition of TCO with 4-azido-2,3,5,6-tetrafluorobenzyl linkers was lower than the non-substituted 4-azidobenzyl linker,13 and the HRMS evidence (Fig. S14†) in this study supports the formation of a relatively stable imine intermediate, even at acidic pH (3.7). While acidic pH should promote imine hydrolysis, protonation of the subsequent product aniline 4, prior to a 1,6-self-immolation, would be expected to slow the self-immolation process. Therefore, the hydrogel of 3 was prepared at higher pH (7.4 and 6.5), to see if 1,6-self-immolation could be promoted. Once the acidity/basicity of the hydrogel was adjusted to the desired pH (using HCl and NaOH), TCO (5 mM) was added and incubated for 12 hours at 37 °C, and lyophilised. The dried sample was analysed by HRMS (ESI-negative) which showed the formation of various side products (Fig. S15 and S16†). Intermediates 3b–3d were identified, along with imine 3e, the product of condensation between PhePhe 2 and the aldehyde by-product (Fig. S1;† generated by hydrolysis of imine 3b). When compared to the HRMS experiment conducted for 12 hours at the lower pH of 3.7 (Fig. S14†), the notable difference was that the aniline 4 (Mw 533 g mol−1) was virtually absent at pH 7.4 and 6.5, suggesting that the rate of 1,6-self-immolation was faster at higher pH. There was also a peak identified at m/z 311.1424 (Fig. S15;† pH 7.4) and 311.1442 (Fig. S16;† pH 6.5), assigned as the [M–H] ion of the dipeptide (PhePhe 2). This suggests that over time, the aniline-capping group is removed from the dipeptide, with the most plausible route via a 1,6-self-immolation. We do however note that some of the self-immolation from aniline 4 may occur during the mass spectral analysis (see NMR and HRMS study below).
As final confirmation for the stability of aniline 4, and possible reduced levels of 1,6-self-immolation, a time-monitored solution phase 1H (Fig. 3) and 19F NMR (Fig. 4) study was conducted.
 | ||
| Fig. 3 1H NMR spectra of AzF4-PhePhe 3 and TCO cycloaddition in acetonitrile-d3. Int. Std. is 1-fluoro-2,4-dinitrobenzene. | ||
 | ||
| Fig. 4 19F NMR spectra of AzF4-PhePhe 3 and TCO cycloaddition in acetonitrile-d3. | ||
As AzF4-PhePhe 3 was converted to intermediates 3b–3d and aniline 4, changes around the dipeptide backbone were difficult to analyse in the 1H NMR spectrum (Fig. 3). However, the presence of an imine proton at δ 7.96 (due to aldimine 3b; structure in Fig. S1†) and the appearance of an aldehyde peak (δ 9.58) after 4 hours of reaction, were indicative of aldimine formation and subsequent hydrolysis to aniline 4. The bridging triazoline peaks (expected around δ 3.5–3.7 and 4.2–4.5)13,16 were not significant (below signal-to-noise of the spectrum), supporting the HRMS data, where the triazoline 3a was not identified. Analysis of the 19F NMR spectrum (Fig. 4) further confirmed complete reaction of AzF4-PhePhe 3 with TCO after only 1 h of incubation (loss of fluorine signals at δ −144.6, −154.0). Spectral evidence for the triazoline 3a was missing (expected around δ −144.5 and −149 ppm).13 Instead, direct evidence for formation of aldimine 3b could be observed after immediate addition of TCO (Fig. 4), lending further support to the instability of the triazoline intermediate 3a when a tetrafluoroaryl group is used as the linker. Within 1 hour, aniline 4 had started to form (δ −147.9, −163.9), along with other minor products that include the ketamine 3c, aziridine 3d, and the enamine tautomer of aldimine 3b, and by 48 hours aniline 4 was the major product. Confirmation of the major product being aniline 4, was provided by comparison of the 19F NMR at 48 hours to the 19F NMR spectra of the expected 1,6-self-immolation product 4-amino-2,3,5,6-tetrafluorobenzyl alcohol (Fig. S17†). The 19F NMR spectral comparison (Fig. S17†) clearly indicated that the immolation had not transpired under the NMR solvent conditions, with no indication of peaks belonging to the 4-amino-2,3,5,6-tetrafluorobenzyl alcohol.
HRMS analysis of the NMR reaction after 48 hours in acetonitrile-d6 (Fig. S18†) indicated a product/intermediate with a molecular weight of 657 g mol−1 (m/z 656.2418), that could be assigned to aziridine 3d. Aziridine 3d was not expected to produce aniline 4, and was identified in the 19F NMR spectrum after 48 hours incubation (δ −146.6, −156.2). Also evident was a peak at m/z 532.1520, which could be assigned to the major product of the NMR reaction, aniline 4. Although not observed in the NMR reaction mixture (Fig. 3 and 4), PhePhe 2 was present in the HRMS (m/z 311.1414), indicating that under the conditions of the mass spectrum, some of aniline 4 must be eliminating to provide the dipeptide 2.
Based on previous work in our group, the 1,6-self-immolation was faster in an aqueous environment.13,16 Therefore 100 μL of the acetonitrile-d6 solution was replaced with D2O (16% final volume). However, in this instance, no further changes to aniline 4 were observed over 24 hours (Fig. S17†). Overall, the NMR studies suggest that 1,6-self-immolation of aniline 4 does not occur under the partial aqueous conditions of the NMR experiment. While this does not directly compare to 100% aqueous conditions, the results are in contrast to the non-substituted azide/aniline linker, where the 1,6-self-immolation can be promoted by the addition of only a small amount of D2O.13,16
To summarise the experiments above, TCO addition (1–5 mM) results in relatively fast dissolution of AzF4-PhePhe derived hydrogels. The mechanistic studies suggest that self-immolation does occur in 3, but that some of the initial visual dissolution for the hydrogel of 3 could be the result of a polarity change; i.e. conversion of azide 3 to aniline 4 (Scheme 1b), especially under the acidic conditions of the hydrogel in this study. While this study provides significant mechanistic insight into the TCO-driven dissolution process, future work using the 4-azido-2,3,5,6-tetrafluorobenzyl linker and new variations of linkers, will investigate self-immolative-driven dissolution of short peptide hydrogels to polarity-driven dissolution. The use of amide-linked N-capping groups6,11 that are TCO-triggerable, but not self-immolative, will be employed.
3.4 Rate of 1,3-dipolar cycloaddition between AzF4-PhePhe 3 and TCO
The product distribution and rate of the 1,3-dipolar cycloaddition and triazoline/imine hydrolysis following reaction of TCO and 3 in an organic/aqueous solution (MeCN:H2O, 1:1) was analysed using HPLC (Fig. S19†). The rate was measured by the disappearance of AzF4-PhePhe 3 (RT = 8.1 min) over time, and was measured under pseudo first-order conditions (Fig. S20;† 6-fold excess of TCO). The second-order rate constant was calculated as 0.095 ± 0.010 M−1 s−1, a rate comparable to our previously reported bioorthogonal prodrug activation for tetrafluoro-substituted aryl azide prodrugs in MeCN:H2O,13 and approx. five-times faster than the rate of our previously reported non-substituted azido-hydrogel, Az-PhePhe 1.12
3.5 Cargo release from AzF4-PhePhe 3
Next, the AzF4-PhePhe 3 hydrogel was investigated for its drug delivery potential. We have previously reported doxorubicin (DOX) as a model drug cargo in the fibrous network of Az-PhePhe 1, whereby TCO was added at a 5 mM concentration and resulted in approximately 87% release of DOX over 10 hours.12 As discussed above, our early stimuli-responsive studies with a 0.1 wt% hydrogel of AzF4-PhePhe 3 and 5 mM TCO, resulted in a visual gel–sol transition within 4 hours, while 1 mM TCO resulted in visual gel–sol transition of ∼24 hours. As the aim of this work was; (1) to enable formation of hydrogel at a lower critical gel concentration, and (2) exhibit increased sensitivity towards the bioorthogonal trigger (TCO), a hydrogel at 0.1 wt% AzF4-PhePhe 3 encapsulating DOX and the addition of 1 mM TCO were used to study cargo encapsulation and release. DOX (100 μg) was encapsulated in 0.5 mL of hydrogel 3 and left undisturbed for 10 minutes. After addition of water containing TCO (1 mM), the hydrogel was incubated at 37 °C and at the indicated time points, 0.1 mL of sample was taken and replaced with fresh 0.1 mL water aliquot containing 1 mM of TCO (0.1 mL water without TCO for controls) and incubation continued. The cumulative release of DOX from the hydrogel was analysed by UV absorbance (Fig. 5) and quantified using a DOX-standard curve (Fig. S3†). The gel incubated with 1 mM TCO in water released 89% of DOX in 24 hours whereas control hydrogel (without TCO) released 24% of DOX in 24 hours. | ||
| Fig. 5 TCO-triggered (1 mM) drug cargo release from a 0.1 wt% hydrogel of AzF4-PhePhe 3. Absorbance of doxorubicin diffusing into the water on top of the hydrogel was measured at 485 nm. Error bars represent the SD for triplicate experiments (n = 3). | ||
When compared to the cargo release studies from the Az-PhePhe 1 hydrogel after addition of TCO (5 mM), the amount of DOX released from the AzF4-PhePhe 3 hydrogel with five-fold less TCO (1 mM) after 10 hours of incubation was 76%; comparable to the 87% DOX release for 1. This suggests that a five-fold lower critical gel concentration and five-fold lower bioorthogonal trigger can be used to release a high level of drug cargo over 10 hours, with almost complete release at 24 hours.
3.6 Cytotoxicity of AzF4-PhePhe 3
Thordarson and co-workers have previously shown that the cytotoxicity of N-Fmoc-PhePhe hydrogels is influenced by the concentration of the dissolved (soluble) peptide in the cell culture media.23 In a separate publication using a 3D cell culture they also demonstrated that the capping group on the low molecular weight hydrogel also influences cytotoxicity,24 indicating the importance of determining the cytotoxicity of the new AzF4-PhePhe 3 dipeptide and comparing it to our previously reported dipeptide Az-PhePhe 1.12 In a typical experiment, Madin-Darby Canine Kidney (MDCK) cells were exposed AzF4-PhePhe 3 for 24 or 48 hours, and cell viability determined at concentrations up to 1.7 mM (Fig. 6). The experiments were conducted under standard solution phase (see experimental section for details), therefore mimicking dipeptide that would leach from the hydrogel in a biological setting. The cell viability was compared to control cells (without 3). Substantial cytotoxicity of AzF4-PhePhe 3 (cell viability ≤ 50%) was observed at concentrations of 1.7 mM and 0.43–1.7 mM in the 24 and 48 hour incubation experiments, respectively. As a comparison, our reported hydrogel Az-PhePhe 1, showed substantial cytotoxicity to MDCK cells at a concentration of 1 mM after 24 hours of incubation.12 The data suggests that while the differences in cytotoxicity between are relatively small, the fluorinated dipeptide 3 exhibits lower cytotoxicity than the non-fluorinated dipeptide 1.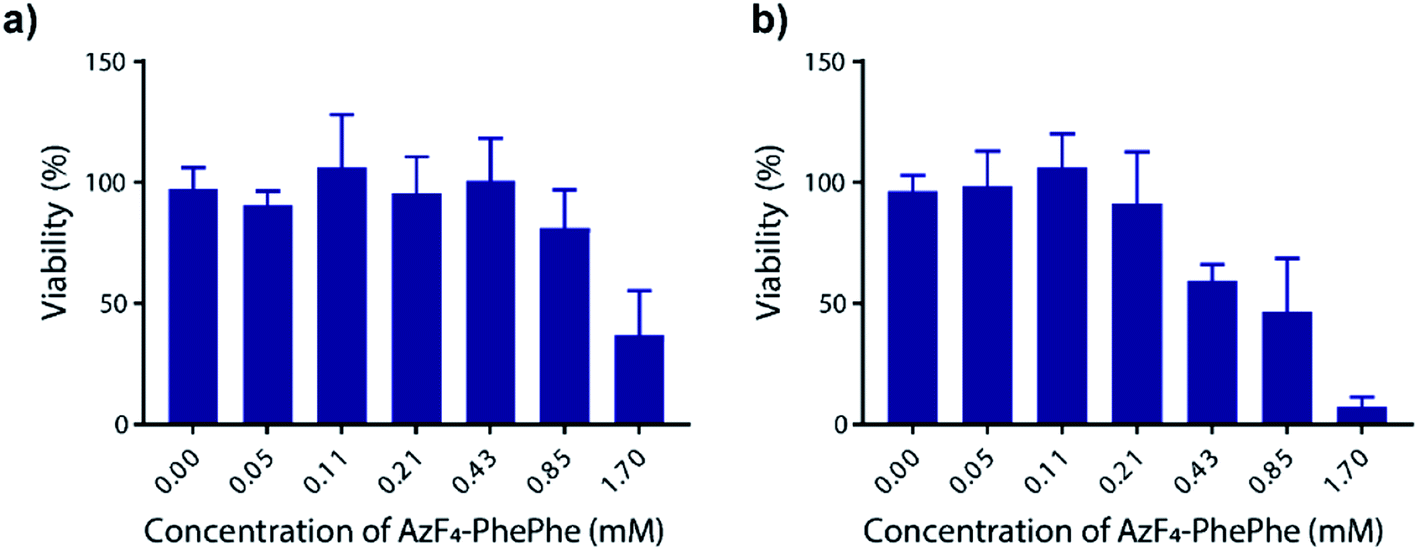 | ||
| Fig. 6 Cell cytotoxicity of AzF4-PhePhe 3 against Madin-Darby Canine Kidney (MDCK) cells. Incubation times were (a) 24 hours and (b) 48 hours, and ±SD is from triplicate runs (n = 3). | ||
4 Conclusion
In summary, the use of a self-immolative 4-azido-2,3,5,6-tetrafluorobenzyl carbamate linker to protect (cap) the N-terminus of a diphenylalanine dipeptide 2 and generate a stimuli-responsive hydrogel is reported. The capped dipeptide (AzF4-PhePhe 3) resulted in a hydrogel that formed a stable fibrous network in water, but not in phosphate buffered saline (PBS). In water (final pH of 3.7), 3 formed a weak hydrogel at 0.05 wt%, and a strong hydrogel, as evidenced by its viscoelastic properties, at 0.1 wt% of 3 (considered the critical gel concentration). The absence of a hydrogel in PBS (final pH 3.9) was likely due to disruption of H-bonding and other interactions by the phosphate salts, as reported by others.18 Reaction of the AzF4-PhePhe 3 hydrogel with TCO resulted in dissolution of the hydrogel in 4–24 hours. At the lowest concentration tested (1 mM TCO), the complete visual dissolution took 24 hours. Encapsulation of doxorubicin (DOX) as a model drug cargo was conducted at 0.1 wt% of 3, and upon addition of TCO (1 mM) dissolution resulted in 76% and 89% release of DOX after 10 and 24 hours, respectively. The rate of the 1,3-dipolar cycloaddition between an AzF4-PhePhe 3 and TCO was determined under pseudo first-order conditions in solution phase, and the second-order rate constant calculated as 0.095 ± 0.010 M−1 s−1, five-times faster than the reaction of TCO with Az-PhePhe 1.12The ability to formulate AzF4-PhePhe 3 as a stable hydrogel at 0.1 wt%, and its improved sensitivity and rate of reaction with the bioorthogonal trigger (TCO), indicates that the use of fluorine in related peptide-based hydrogels could lead to drug delivery systems with in vivo potential. Other endogenous triggers of azides, for example hydrogen sulfide, explored by others on short peptidic hydrogels,25 would also provide an alternate strategy for dissolution of hydrogels with fluorinated aryl azide-capping groups. Peptidic and polymeric hydrogels incorporating the tetrafluoroaryl azide and other azide-functionalised self-immolative linkers, and their activation mechanisms are of current interest to our research group.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported in part by a contract from the Health Research Council of New Zealand (A. B. G.). We thank the Department of Chemistry at the University of Otago for use of the NMR and HRMS instruments, and the Otago Micro and Nanoscale Imaging for use of their facilities.Notes and references
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- J. Hoque, N. Sangaj and S. Varghese, Macromol. Biosci., 2019, 19, 1800259, DOI:10.1002/mabi.201800259.
- S.-M. Hsu, Y.-C. Lin, J.-W. Chang, Y.-H. Liu and H.-C. Lin, Angew. Chem., Int. Ed., 2014, 53, 1921–1927 CrossRef CAS PubMed.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS PubMed.
- D. M. Ryan, S. B. Anderson and B. L. Nilsson, Soft Matter, 2010, 6, 3220–3231 RSC.
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475–479 RSC.
- F.-Y. Wu, S.-M. Hsu, H. Cheng, L.-H. Hsu and H.-C. Lin, New J. Chem., 2015, 39, 4240–4243 RSC.
- S.-M. Hsu, J.-W. Cheng, F.-Y. Wu, Y.-C. Lin, T.-S. Lai, H. Cheng and H.-C. Lin, RSC Adv., 2015, 5, 32431–32434 RSC.
- S. Dadhwal, J. M. Fairhall, S. K. Goswami, S. Hook and A. B. Gamble, Chem.–Asian J., 2019, 14, 1143–1150 CrossRef CAS PubMed.
- S. S. Matikonda, J. M. Fairhall, F. Fiedler, S. Sanhajariya, R. A. J. Tucker, S. Hook, A. L. Garden and A. B. Gamble, Bioconjugate Chem., 2018, 29, 324–334 CrossRef CAS PubMed.
- M. Sundhoro, S. Jeon, J. Park, O. Ramström and M. Yan, Angew. Chem., Int. Ed., 2017, 56, 12117–12121 CrossRef CAS PubMed.
- Y. Xie, L. Cheng, Y. Gao, X. Cai, X. Yang, L. Yi and Z. Xi, Chem.–Asian J., 2018, 13, 1791–1796 CrossRef CAS PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- M. Royzen, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760–3761 CrossRef CAS PubMed.
- D. M. Ryan, T. M. Doran, S. B. Anderson and B. L. Nilsson, Langmuir, 2011, 27, 4029–4039 CrossRef CAS PubMed.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Langmuir, 2011, 27, 11145–11156 CrossRef CAS PubMed.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Chem. Commun., 2011, 47, 475–477 RSC.
- S. Debnath, S. Roy, Y. M. Abul-Haija, P. W. J. M. Frederix, S. M. Ramalhete, A. R. Hirst, N. Javid, N. T. Hunt, S. M. Kelly, J. Angulo, Y. Z. Khimyak and R. V. Ulijn, Chem.–Eur. J., 2019, 25, 7881–7887 CrossRef CAS PubMed.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS PubMed.
- W. T. Truong, Y. Su, D. Gloria, F. Braet and P. Thordarson, Biomater. Sci., 2015, 3, 298–307 RSC.
- J. P. Wojciechowski, A. D. Martin, A. F. Mason, C. M. Fife, S. M. Sagnella, M. Kavallaris and P. Thordarson, ChemPlusChem, 2017, 82, 383–389 CrossRef CAS PubMed.
- R. Peltier, G. Chen, H. Lei, M. Zhang, L. Gao, S. S. Lee, Z. Wang and H. Sun, Chem. Commun., 2015, 51, 17273–17276 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01013h |
| This journal is © The Royal Society of Chemistry 2020 |