
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel thermoplastic shape memory polymer with solid-state plasticity derived from exchangeable hydrogen bonds†
Xu Zhang ab,
Guangping Sun*a and
Xuequan Zhang*b
ab,
Guangping Sun*a and
Xuequan Zhang*b
aCollege of Materials Science and Engineering, Jilin University, Changchun 130022, People's Republic of China. E-mail: sungp@jlu.edu.cn
bCAS Key Laboratory of High-Performance Synthetic Rubber and Its Composite Materials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, People's Republic of China. E-mail: xqzhang@ciac.ac.cn
First published on 4th March 2020
Abstract
A novel thermoplastic shape memory polymer (SMP) was synthesized by partly modifying the double bonds of trans-1,4-polybutadiene (TPB) and subsequently introducing mono-isocyanate as pendant groups. The comb-like chains consisted of abundant hydrogen bonds on their side groups, which could readily assemble into dynamic non-covalent polymer networks. The physical crosslink net-points formed by the hydrogen bonds were utilized to anchor the permanent shape of PBTP, while the soft trans-1,4-polybutadiene segments served as a switching phase to afford the temporary shape, and the glass transition temperature (Tg) was taken as the switching temperature. The optimal result was obtained with the sample with 30% modification, which showed an excellent shape fixity ratio and inferior shape recovery ratio of 100% and 89.57%, respectively. According to the results of the stress relaxation test, the relaxation time for this sample is 27 s when heated at 70 °C, implying that the dynamic network inside the sample can undergo topological rearrangement rapidly while maintaining the network integrity during the process. The high density of exchangeable hydrogen bonds constitutes a highly crosslinked non-covalent network, the dynamic nature of which is responsible for this outstanding network topological structure rearrangement behaviour, and endows this SMP with solid-state plasticity to reconfigure its permanent shape in the absence of catalysts or moulds. The highly complex permanent shape reconfiguration at a moderate temperature within a rational period will greatly promote the practical use of SMPs, and enable a wide variety of future engineering applications.
Introduction
Shape memory polymers (SMPs) are a class of intelligent materials, which possess the ability to fix a temporary shape and recover to its memorized original shape under specific stimuli.1,2 SMPs have been known for over half a century and have applications in many fields including bio-medical devices and aerospace applications.3,4 In the last decade, breakthrough technologies such as triple-shape, multiple-shape and reversible-shape memory behaviours, which are superior to the classical one-way dual-shape mode, have greatly enriched the performances of SMPs.5–7 Besides the progress that mainly focused on temporary shape memory behaviours, some attention has also been paid to permanent shape reconfiguration.8,9T. Xie and coworkers firstly reported a new class of SMPs called “thermadapt shape memory polymers” (TASMPs), which can change their permanent shapes in the solid state by utilizing the exchangeable nature of the dynamic covalent bond.10 TASMPs show adaptability by rearranging the topological structure of their network to adopt a neo-thermodynamically stable state, which is determined by environmental conditions or external stimuli (e.g. light, temperature and stretching), without loss of their integrity. The underlying mechanism of this particular adaptability is known as the “covalent adaptable network” (CAN). A CAN commonly contains a sufficient number of dynamic covalent bonds, which make up the network joints and can reshuffle with each other to adapt to environmental changes, while keeping its initial crosslinking density unchanged, and the network maintains a three-dimensional (3D) architecture throughout the rearrangement.11 CAN endows TASMPs with the ability to achieve permanent-shape 3D structure reconfiguration in the solid state in the absence of moulds, thus making them conspicuously superior to the traditional thermoplastic and thermoset SMPs. To date, a series of TASMPs have been reported such as thermoset shape memory polyurethane (SMPU) networks, crosslinked poly(caprolactone) networks, polysulfide networks, and epoxy resin networks.12–23 These TASMPs share a common root in that they all originate from chemical cross-linked networks based on dynamic covalent bonds.
However, despite the diversity of these dynamic covalent bonds, more attention should be paid to the dynamic exchange behaviour of bonds, which endow materials with unique performances or functions. Dynamic bonds, both covalent and non-covalent, can undergo bond breaking and reforming in response to environmental changes. Thus, it can be predicted that as the inherent counterpart of thermosets SMPs composed by dynamic covalent bonds, thermoplastic SMPs with physical crosslinked networks composed by non-covalent bonds (e.g. hydrogen bonds), will share the capability of topological reconfiguration in the solid state equally with thermoset SMPs. However, to date, no thermoplastic SMPs with “non-covalent adaptable networks” have been reported or generalized in TASMPs.
Covalent bonds lead to thermoset SMPs with outstanding shape memory abilities, whereas limit material reshaping and reprocessing simultaneously. However, the development of TASMPs has greatly alleviated this dilemma by endowing polymers with solid-state plasticity, but because of their inherent high binding energy, dynamic covalent bonds require intense stimuli to activate bond exchange, e.g. high temperature is generally needed, and in some circumstances, to accomplish the equilibration in a reasonable timescale, catalysts are usually of great significance.13 However, sluggish bond exchange is unsatisfactory for practical purposes and high temperature may result in catalyst instability or thermal degradation of the matrix. In addition, the plasticity originating from transcarbamoylation and transesterification is cumulative, and new permanent shapes are based on the prior permanent shape, thus the reprocessability of thermoset SMPs is still inferior to that of thermoplastic SMPs.8,12
Equipped with intrinsic low bonding energies, non-covalent bonds can be readily dissociated when subjected to external stimuli, and thus thermoplastic SMPs composed of non-covalent networks may achieve thermodynamic equilibrium much faster than thermoset SMPs. However, the weak interaction between polymer chains leads to thermoplastic SMPs having compromised shape fixity and low recovery stress, even for systems equipped with quadruple hydrogen bonds such as ureidopyrimidine (UPy).24,25 All of these drawbacks severely reduce the reliability of thermoplastic SMPs, and limit their application in real-world devices. In addition, despite their intrinsic reprocessability, thermoplastic SMPs can only be processed in the high viscosity state, and thus these materials have to be heated to flow or casted from their solution afresh. In this process, the material loses its integrity, and the new shape has to be redefined through moulds; thus, extremely sophisticated permanent shapes cannot be facilely acquired.
To solve these imperfections of thermoplastic SMPs and expand their potential range, a facile and valid approach is presented in the present study to engineer an SMP equipped with an ideal combination of reprocessability and solid-state plasticity, while maintaining it satisfactory shape memory performance. A novel thermoplastic SMP, which is denoted as PBTP-X (X representing the modification degree), was synthesized from trans-1,4-polybutadiene by partly modifying its double bonds into pendent carbamate groups through a three-step reaction. In addition to the widely known reprocessing techniques, such as remolding and solution casting, we innovatively introduced solid-state plasticity into thermoplastic SMPs. The abundant hydrogen bond donors and acceptors on the pendent carbamate groups can readily pair with each other to form hydrogen bonds, which are typical dynamic non-covalent bonds. The high density of hydrogen bonds link the linear polymer chains into dynamic non-covalent networks, where the netpoints composed of hydrogen bonds anchor the permanent shape, while the soft trans-1,4-polybutadiene segments serve as a switching phase to afford the temporary shape, and Tg is taken as the switching temperature to realize shape memory behaviour. By adjusting the modification degree of the double bonds, the Tg of PBTP could be altered in a wide range, and with its optimized network structure adjustment, satisfactory shape memory parameters of shape fixity (100%) and shape recovery ratio (89.57%) were obtained in PBTP-30 (Tg is 33.81 °C according to the DMA). The 1H NMR and variable-temperature FTIR results reveal that there are three types of hydrogen bonds constructing the polymer networks, and the hydrogen bonds are formed in a novel single donator-multi acceptor mode, which greatly elevates the pair bonding rate. The high density and low bonding energies of the hydrogen bonds in PBTP-30 allow rapid bond exchange throughout the entire network, and thus topological network rearrangement could be realized under a moderate temperature and in a reasonable timescale. To the best of our knowledge, introducing hydrogen bonds to form a thermoplastic SMP composed of highly crosslinked dynamic non-covalent networks and then realizing permanent shape reconfiguration through topologically rearrangement in the solid state have not been attempted before.
Experimental
Materials
trans-1,4-Polybutadienes (TPBs, Mn = 32.4 kg mol−1, Mw/Mn = 1.56, trans-1,4 content ≥ 95%, laboratory made), m-chloro-peroxybenzoic acid (m-CPBA, 85%), m-(trifluoromethyl)phenyl isocyanate (TFPI, 96%), m-tolyl isocyanate (TI, 99%), tin(II) 2-ethylhexanoate (97%), chloroform-d (99.8 atom%), 1,2-dichlorobenzene-d4 (99.5 atom%), super dry methyl alcohol (99.9%) and HCl (36.7%) were purchased from Aladdin. All the above mentioned chemicals were used as received without further purification. Toluene, dichloromethane (DCM) and tetrahydrofuran (THF) were purified by standard methods and stored under a nitrogen atmosphere. Pendant hydroxyl group-functionalized TPB (TPB-OH) was synthesized according to the literature procedure.26Synthesis of PBTP and PBTI
Taking PBTP-30 as an example, in a 200 mL one-neck round-bottom flask equipped with a magnetic stirrer, 0.697 g of 1 × 10−2 mol TPB-OH (with 30 mol% hydroxyl groups) was dissolved in 80 mL CH2Cl2, and then 0.5145 mL TFPI (3.6 × 10−3 mol) and 0.36 mL tin(II) 2-ethylhexanoate (1 × 10−3 mol) were added to the solution in sequence, and the system was stirred at 38 °C for 30 min under a nitrogen atmosphere. After the reaction, the mixture was poured into large volume of cooled methanol to precipitate the final product, and the solid was collected by filtration and subsequently dried at room temperature (25 °C) for 24 h to allow the methanol to evaporate. The production yield was about 96%, and the precise modification degree was calculated using the 1H NMR results, as described in the literature.27 TPBs in different TFPI or TI modification degrees were also prepared according to the above process. The TPB modified by TI was denoted as PBTI-X (X denoting the modification degree). The testing sample was prepared as follows, PBTP was first dissolved in CH2Cl2 and then cast onto a polytetrafluoroethylene mould, and after the solvent evaporated, a plane translucence rectangle sample was obtained, which was used directly.Characterization
FTIR was performed on a Bruker Vertex 70 FTIR spectrometer equipped with a heating cell. The samples for FTIR analysis were first dissolved in CH2Cl2 to form a homogeneous concentrated solution and then cast onto a KBr window, and after solvent evaporation, a transparent film was obtained and used in situ, and the result was recorded at different temperatures.
Differential scanning calorimetry (DSC) analysis was performed on a Q20 DSC system (TA Instruments) under a nitrogen atmosphere in the temperature range of −30 °C to 80 °C at a heating rate of 5 °C min−1. The samples were heated twice to eliminated thermal history, and the transition temperature was taken from the second heating curve.
Dynamic mechanical analysis (DMA) of the specimen was performed on a TA Instrument DMA Q800 system in a film tension clamp using “multi-frequency strain mode” with a frequency of 1 Hz. The temperature was increased from −30 °C to 75 °C at a heating rate of 5 °C min−1. Rectangular specimens (7.2 × 7 × 0.2 mm3) were prepared for analyses.
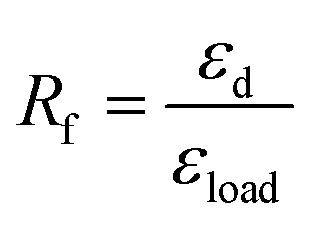 | (1) |
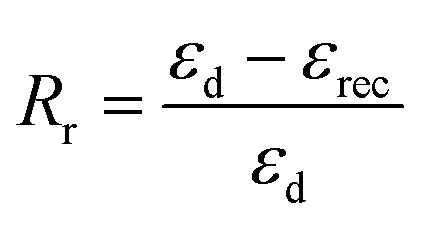 | (2) |
Results and discussion
Modification of TPBs
Modification of the TPBs was carried out via a three-step reaction, according to Scheme 1. The proton signals on the oxirane (2.74 ppm) ring and that adjacent to the hydroxyl group (3.73 ppm) were monitored by 1H NMR to verify the extent of the reaction (Fig. 1). Taking PBTP-10 as an example, the epoxidation of TPBs by m-CPBA was highly efficient, and the modification degree of the final product was quantitatively controlled by this step. The ring-opening reaction of TPB was triggered by adding hydrochloric acid, and the proton signal on the oxirane ring (2.74 ppm) transformed from a singlet to a doublet (3.96 ppm, 3.73 ppm), indicating that the oxirane ring was successfully cleaved. After TFPI was added to the system, the peak at 3.73 ppm, which was assigned to the proton approaching the carbon of the C–O groups, shifted to 4.96 ppm and overlapped with the proton signal of the vinyl on 1,2-polybutadiene. Four new signals appeared at 7.18, 7.72, 7.52 and 7.38 ppm, which indicate the presence of a benzene ring. An active hydrogen resonance appeared at 6.81 ppm, which is ascribed to the proton on the N–H groups, and this resonance signal serves as indirect evidence to prove the existence of hydrogen bonding in PBTPs.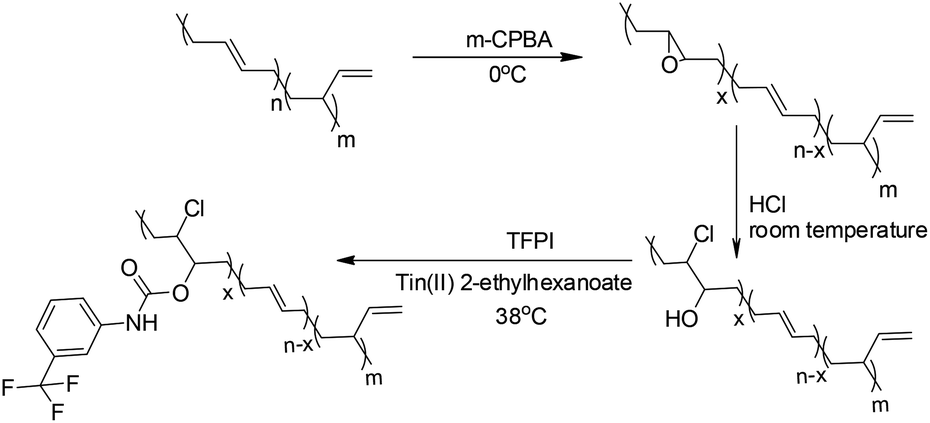 | ||
| Scheme 1 Route for the modification of TPBs. | ||
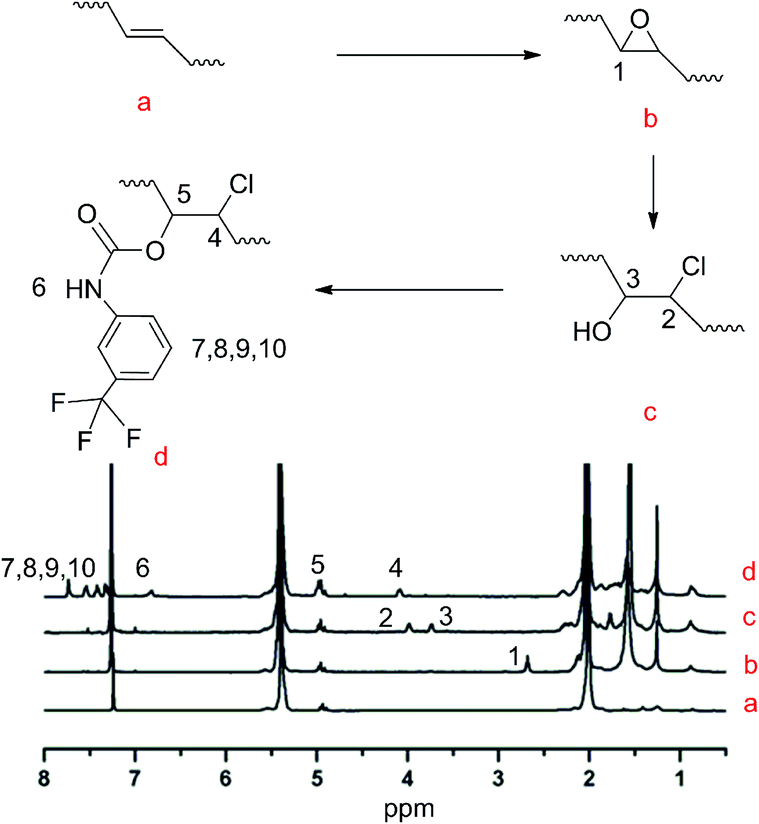 | ||
| Fig. 1 1H NMR spectra of PBTP-5 in CDCl3. | ||
Characterization of the hydrogen bonds
The intermolecular hydrogen bonding strength varies inversely with temperature. Thus, an increase in temperature will certainly break the equilibrium of hydrogen bonding association and drive it to the direction of dissociation. The proton on the N–H groups will be released from the hydrogen bonding state and recover its extra-nuclear electron cloud density, which will resume its shielding effect, thus leading to a decrease in chemical shift. By utilizing this sensitivity of hydrogen bonds to temperature, the existence of hydrogen bonds was verified by the variable-temperature 1H-NMR spectra of PBTPs. Fig. 2 shows that the signal of the proton on the N–H groups exhibited great temperature dependency. With an increase in temperature, this signal shifted to a high magnetic field, while the other signals remained at the initial value, which is solid proof for the existence of intermolecular hydrogen bonds.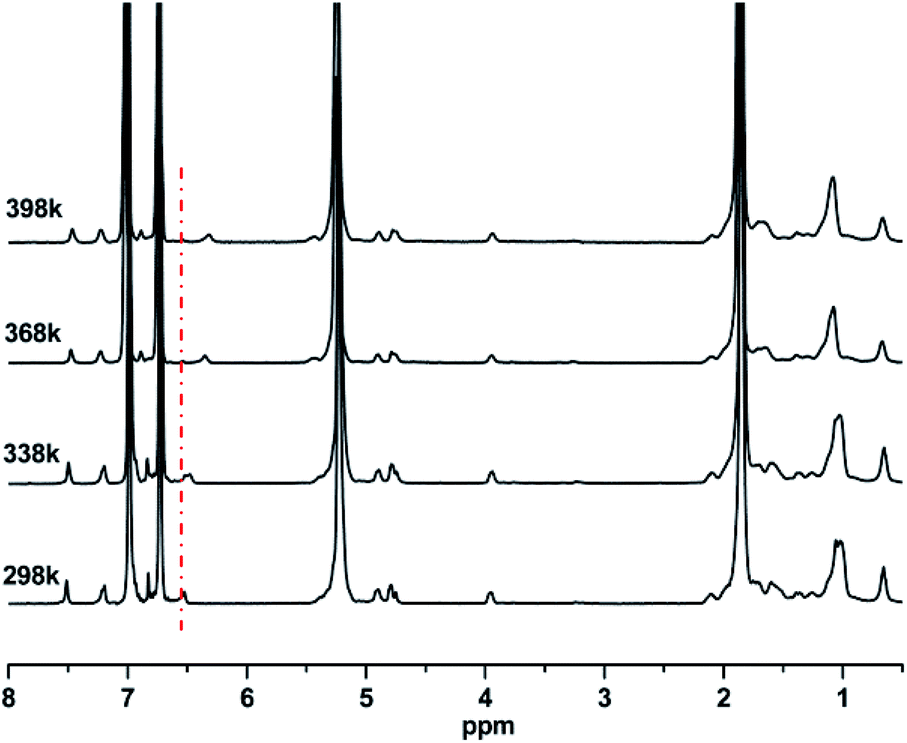 | ||
| Fig. 2 Variable temperature 1H NMR spectra of PBTP-10 in 1,2-dichlorobenzene-d4. | ||
However, although the variable-temperature 1H NMR spectra prove the existence of intermolecular hydrogen bonds, questions still remain, such as which atom forms the hydrogen bonds with the proton since fluorine atom in TPFI can also function as a hydrogen bond acceptor. Considering this, further investigation was performed to identify the hydrogen bond species. We choose TI as the contrast structure, which substitutes the trifluoromethyl group of TPFI to a methyl group while keeping its other portion unchanged, and their comparison is shown in Fig. 3. The proton signal of the secondary amine in PBTP-10 was located at 6.80 ppm, whereas in the case of PBTI-10, the peak with the same profile was observed at 6.62 ppm. This difference indicates that the proton of the N–H groups in the two samples were in different chemical environments. Since the proton signal of N–H in PBTP-10 was located at a higher magnetic field, it can be inferred that, compared with PBTI, PBTP possess stronger hydrogen bonding association, which means that fluorine acts as a hydrogen bond acceptor in the formation of hydrogen bonds. In addition, trifluoromethyl is the most lipophilic substituent group, and thus its presence can improve the solubility of PBTP in CH2Cl2. However, without the trifluoromethyl groups, the modified PBTI with a higher modification degree (>20%) is insoluble in CH2Cl2, which is due to the abundant m-methylphenyl carbamate side groups. This insolubility made PBTI a poor choice as a thermoplastic shape memory polymer, and thus, the PBTP series was mainly investigated in the subsequent experiments.
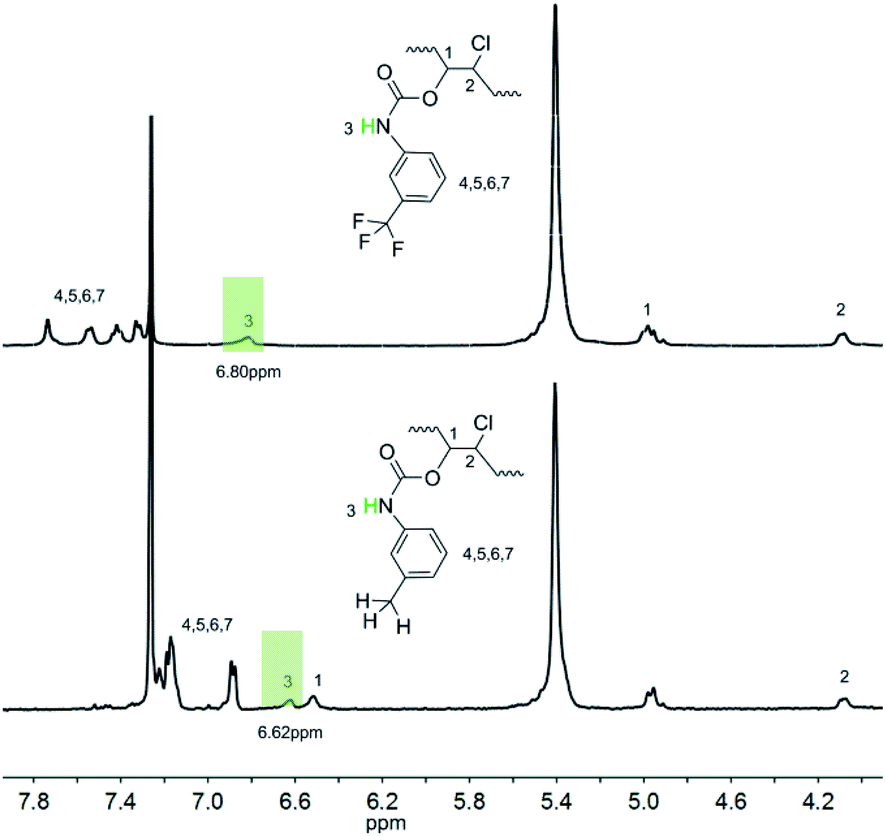 | ||
| Fig. 3 1H NMR spectra of PBTP-10 and PBTI-10 in CDCl3. | ||
To further clarify the hydrogen bond species, a variable temperature FTIR experiment was carried out, and the spectra of the region that drastically varied with temperature are shown in Fig. 4 (full range spectra can be found in the ESI, Fig. S1†).
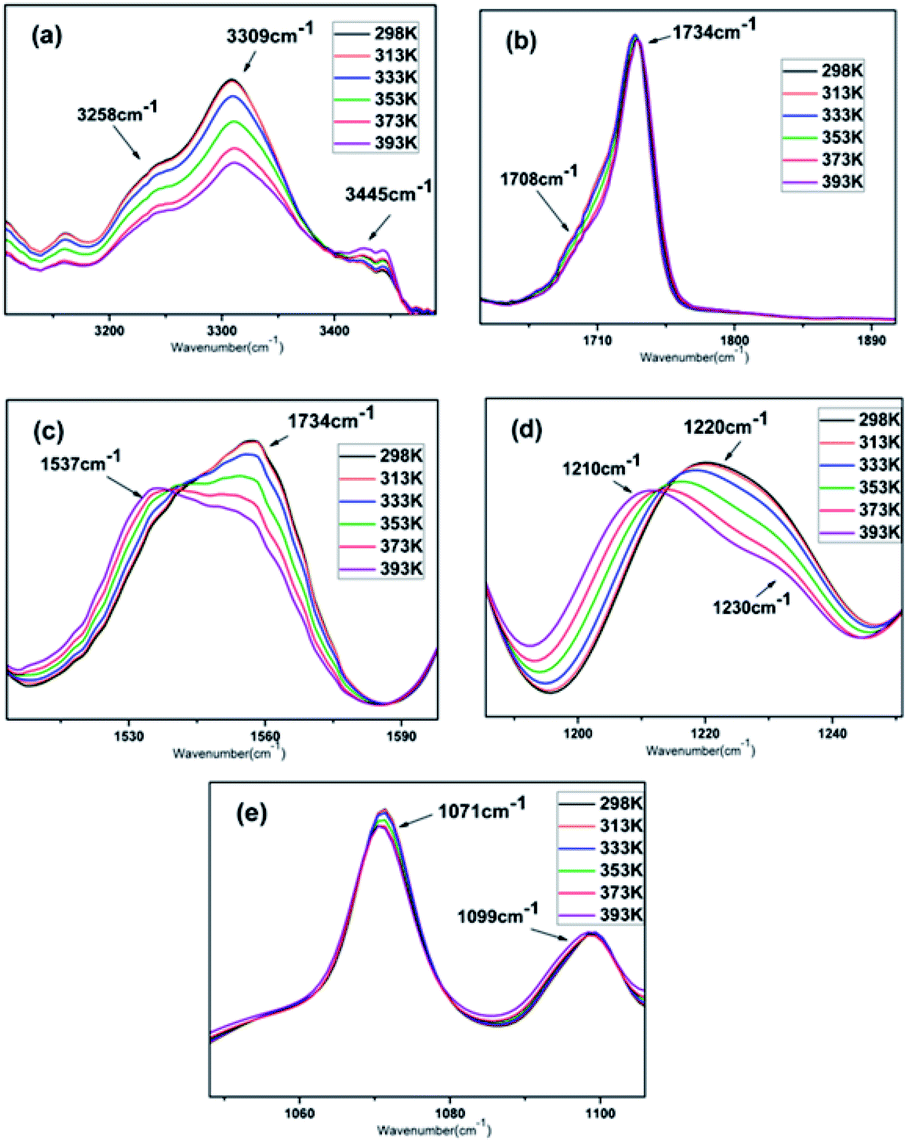 | ||
Fig. 4 Characteristic absorption peaks of (a) N–H stretching vibration, (b) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, (c) N–H bending vibration, (d) C–F stretching vibration and (e) C–O stretching vibration in the variable temperature FTIR spectra of PBTP-30. O stretching vibration, (c) N–H bending vibration, (d) C–F stretching vibration and (e) C–O stretching vibration in the variable temperature FTIR spectra of PBTP-30. | ||
Various FTIR studies on the influence of temperature on hydrogen bonding indicate that the absorption coefficient of a particular band is a strong function of frequency, and an increase in temperature will lead to a decrease in the average strength of the hydrogen bonds, and thus a reduction in absorbance intensity.28–32 Once involved in hydrogen bonding, the stretching vibrations of the proton donating and accepting groups should show shifts in their absorption frequencies towards lower wavenumbers, meanwhile the bending vibrations exhibit the opposite trend. The magnitude of the shift depends on the type and strength of the hydrogen bonds.
Assignments for the polyurethane absorption bands were reported in several publications.33–35 The FTIR spectra of the N–H, CO, C–F and C–O–C stretching regions in the temperature range of 25–120 °C are shown in Fig. 4, which are in good agreement with the literature. The broad band observed in the N–H stretching region (3200–3350 cm−1) is mainly a reflection of the distribution of hydrogen-bonded groups at different distances and geometries. The band at 3347 cm−1 is attributed to the hydrogen-bonded N–H stretching vibration, where the N–H groups are hydrogen bonded with CO (Fig. 4a). Another obvious shoulder appeared at 3258 cm−1, which is assigned to the hydrogen-bonded NH stretching vibration, where the N–H groups are hydrogen-bonded with the oxygen in the C–O–C groups, the “free” NH stretching vibration appears as a weak shoulder at about 3445 cm−1. The area of the hydrogen-bonded N–H band decreased with an increase in temperature, with a corresponding increase in the area of the “free” N–H band. The imbalance in the decrease and increase in the integrated absorbance of the N–H stretching vibration is understandable since the extinction coefficient of the hydrogen-bonded N–H is three to four times that of the “free” N–H.36 The carbonyl stretching region is dominated by a relatively sharp contribution located in the region of 1640 cm−1 to 1740 cm−1. The single peak with a peak maximum at 1734 cm−1 is assigned to the stretching of the “free” carbonyl groups (Fig. 4b), whose band absorbance is independent of temperature over the investigated temperature range. A second component appeared at about 1708 cm−1, which can be attributed to the hydrogen-bonded carbonyl. Its intensity slightly decreased as the temperature increased, implying the dissociation of the hydrogen bonds. The absorbance located in 1557 cm−1 is mainly attributed to the hydrogen-bonded N–H bending vibration (Fig. 4c), where, as the temperature increased, the absorbance gradually vanished as a weak shoulder. In contrast, the absorbance peak at 1537 cm−1, which was weak before, became much more visible, which is attributed to the free N–H bending vibration.35 The stretching vibration absorbance of C–F appeared at 1220 cm−1, where its maximum shifted to 1210 cm−1 and it exhibited a significant reduction in intensity as the temperature increased from 298 K to 393 K. Furthermore, another notable shoulder appeared at 1230 cm−1, which is assigned to stretching of the “free” C–F, and the absorbance profile changed from unimodal to a doublet (Fig. 4d).37 The C–O band assignments are 1099 cm−1 and 1071 cm−1 for the isolated and hydrogen-bonded C–O groups, respectively, which were barely affected by the temperature (Fig. 4e). It is worth noting that the curves at 298 K and 313 K were almost the same in all the regions, and obvious changes were only observed at or above 353 K, but even when the temperature was further increased to 393 K, hydrogen bonds still survived according to the curves.
Based on the FTIR analysis and the 1H NMR results, we propose that three different types of hydrogen bonds may exist in the PBTP matrix, considering N–H as the donor, and CO, C–F and C–O–C as acceptors, as shown in Scheme 2. One hydrogen bonding donor can randomly associate with one of the three acceptors, this single-donator multi-acceptor model offers the donors abundant accessible acceptors, which can certainly increase the pairing rate of hydrogen bonds, thus resulting in an increase in the cross-linking density. Since the network consisted of multi-hydrogen bonds, and considering the high flexibility of the polybutadiene chains, it is a truly complex amorphous matrix, and the bond energy of a single hydrogen bond was not only controlled by the intrinsic hydrogen bond type, but also affected by the distance between the donor and acceptor, which is quite random. Thus, it is difficult to determine the breaking sequence of the hydrogen bonds by heating or stretch; however, hydrogen bonds in PBTP-30 can persist up to 393 K. Thus, we maintain that the hydrogen bonds in the PBTP-30 sample are under rapid dynamic forming and breaking equilibrium, which is driven by thermal motion. The hydrogen bonds exchange donors and acceptors with each other through this equilibrium, and the equilibrium gradually moves to the breaking direction as the temperature increases. Accordingly, the bonds possessing a lower bond energy are sacrifices first, leaving a reduction in the number of hydrogen bonds, which is, nevertheless, still sufficient to ensure the integrity of the material. After cooling, the equilibrium moves back and the hydrogen bond reforms, and the whole network regains its crosslinking density with no loss in the number of hydrogen bonds.
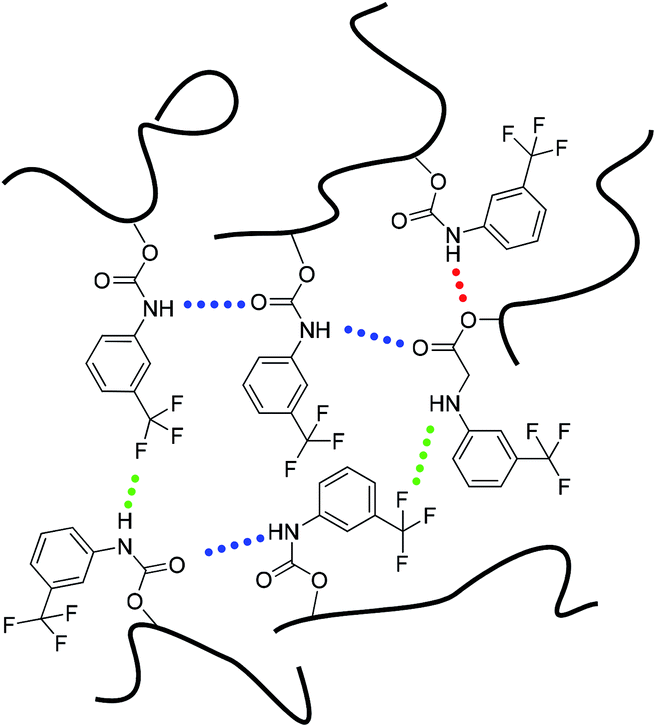 | ||
| Scheme 2 Representation of three possible intermolecular hydrogen-bonding complexes in the PBTP matrix. | ||
It should be noted that the hydrogen bonding density we discussed herein can be readily interpreted as the molar ratio of N–H groups to butadiene monomer units, unlike the expression of carbamate contents that is generally employed in polyurethane, which is the molar ratio of the carbamate groups to the soft segment chains. That means that the crosslink density in PBTPs is much higher than that in the conventional polyurethane, which was confirmed by the results from the FTIR calculations (ESI, Fig. S1†).
Thermodynamic properties of PBTP
The polybutadiene synthesized in this study is basically made up of trans-1,4-units, which is a white crystalline powder with a crystalline melting temperature (Tm) at 48.8 °C. For PBTP-10, its transition temperature (Tg) appears at −32.7 °C and Tm changes to 31.3 °C, which remarkable decreased when compared with that of the raw material. The crystallization was totally suppressed upon 20 mol% modification (Fig. 5). A possible explanation for this is that the introduction of a quantity of side chains disturbed the well-ordered trans-1,4 crystalline phases, and thus the imperfect crystals melted at a lower temperature. Upon further modification, the abundance of hydrogen bonds restricted the chain mobility of the soft segments, and to activate this mobility, a higher temperature was required, hence Tg shifted. It is important to note that Tg increased from −32.7 to 42.9 °C upon 40 mol% modification, turning the crystalline powder into a transparent elastomer and then rigid plastomer. Since the Tg of the soft segments was designed as the shape memory switching temperature, now it can be flexible manipulate by tuning the modification degree of the double bonds.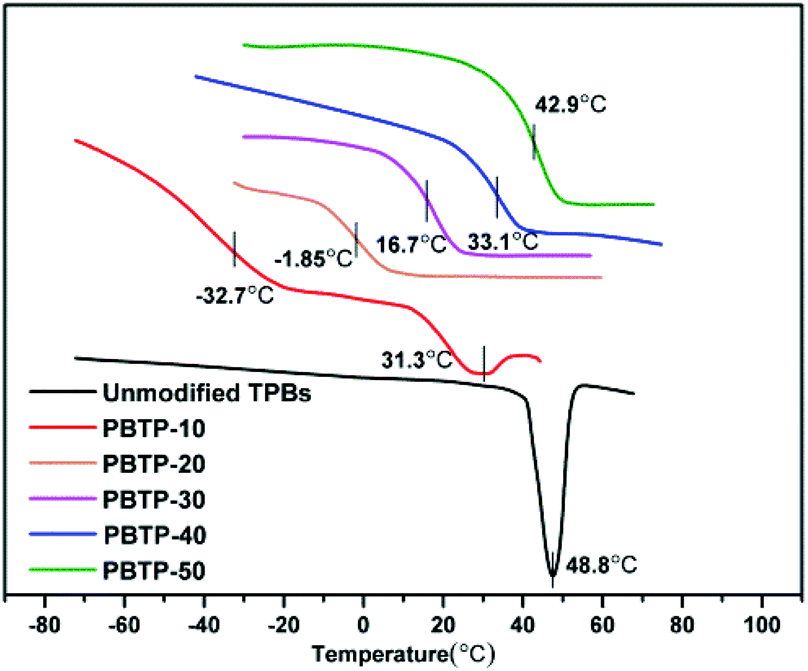 | ||
| Fig. 5 DSC curves of PBTP with different modification degrees. | ||
DMA was used as an independent method to probe the thermodynamic behaviour of PBTPs. Among the samples with different modification degrees, the Tg of PBTP-20 was slightly low for an SMP, while high modification (greater or equal to 40%) caused the material to become. Thus, hereafter we focused on PBTP-30 due to its balanced properties to explore the elasticity and plasticity caused by chain conformation changes and network topological rearrangement, respectively (Fig. 6). The temperature corresponding to the peak of the loss tangent was employed as Tg (33.81 °C). PBTP-30 had a rubbery platform above Tg, and the flow did not start until 72 °C. The value of the loss tangent for PBTP-30 was 1.68, and this significant difference between E′ below and above the Tg of a material is a critical factor in attaining the shape memory effect.35
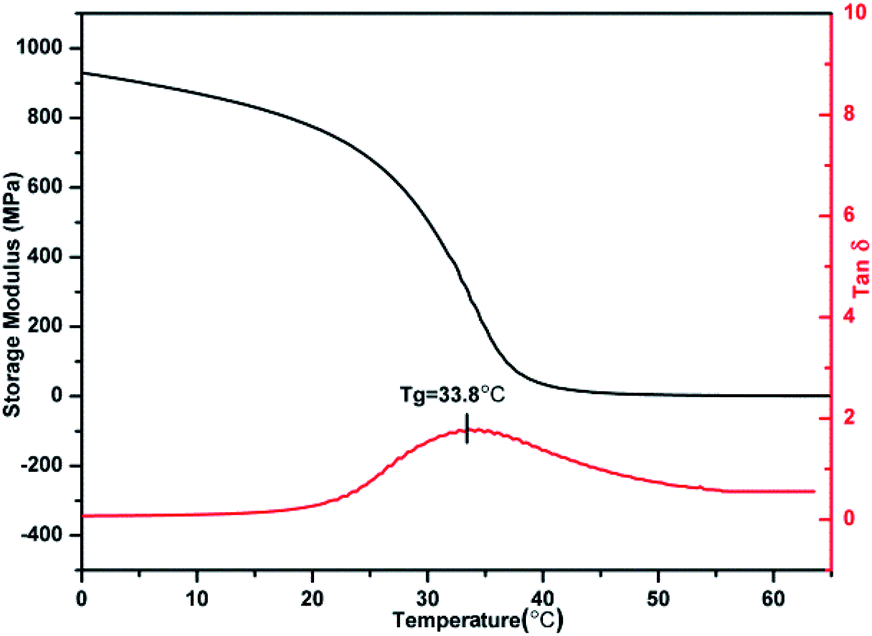 | ||
| Fig. 6 DMA curve of PBTP-30. | ||
The thermally-induced solid-state plasticity of the dynamic non-covalent network was investigated by stress relaxation analysis in a DMA strain rate module. With the strain maintained at 50%, the stress relaxation extent (σ/σ0, where σ and σ0 represent the instantaneous stress and the initial stress, respectively) was monitored. The stress relaxation curves for PBTP-30 at various temperatures between 20 °C and 60 °C are shown in Fig. 7. The relaxation time (τ*, defined as the time required for 63% stress relaxation) as a function of temperature follows the Arrhenius law (ESI, Fig. S2†), and the activation energy (Ea) of 77.59 kJ mol−1 was calculated for the PBTP-30 networks using eqn (3), as follows:
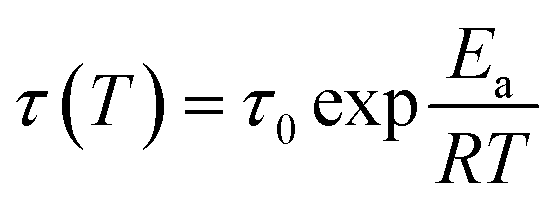 | (3) |
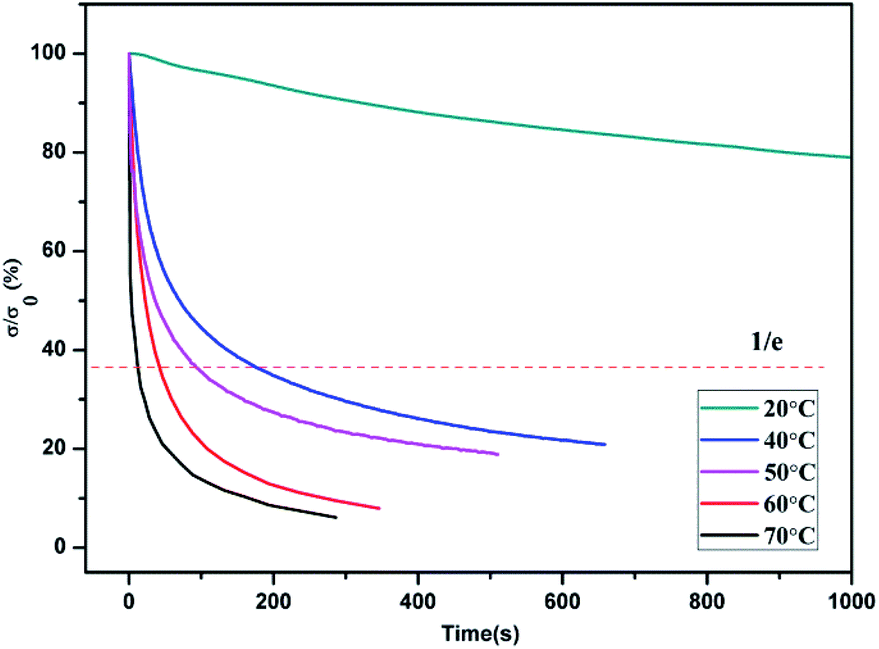 | ||
| Fig. 7 Stress relaxation curves for PBTP-30 at various temperatures. | ||
According to the stress relaxation test results, τ* was 26.52 s at 70 °C, which gradually increased as the temperature decreased, when the temperature decreased to 40 °C, τ* was 89.33 s, and once the temperature decreased to 20 °C, which is below the transition temperature range, the relaxation time bar seemed endless. The rapid stress relaxation at 60 °C indicates that permanent shape plastic deformation can be realized under a moderate temperature with a rational timescale. Thus, combined with the results from the variable temperature FTIR study, it can be inferred that the dynamic association and dissociation equilibrium of the hydrogen bonds shift toward the dissociated state with an increase in temperature. With a reduction in the number of hydrogen bonds, the polymer becomes soft and elastic, displaying a much lower mechanical strength. When subjected to an external stress, the hydrogen bonds could rapidly exchange with each other, by which their molecular chains alter their relative positions, and thus the polymer network accomplished topological rearrangement. The hydrogen bonds re-associated after cooling and restored a neo-topology structure, thereby the PBTP-30 accomplished shape reconfiguration in its solid state without melting. The topological rearrangement was in the absence of entropy changes, without an entropic driving force, and this deformation cannot be recovered, which means it is permanent. Although hydrogen bonding is a weak interaction, the fast stress relaxation at a temperature as low as 40 °C is quite surprising since this temperature is commonly known as the “stabilize region” of hydrogen bonds. A possible explanation for this is that the dynamic equilibrium allowed both the association and dissociation of the hydrogen bonds within the network despite the fact that the equilibrium strongly prefers the association of the hydrogen bonds.
Shape memory test
The shape memory behaviours of PBTP-30 were tested using a DMA Q800 (Fig. 8). PBTP-30 exhibited a high Rf of nearly 100%, where the molecular segments of trans-1,4-polybutadiene act as the switching phase, the segment motion of the polymer chains was totally suppressed at 20 °C according to the DMA curve, and the polymer network is in the frozen state, endowing high shape fixing of the matrix. Compared with the high Rf, Rr was slightly lower at 89.57%. This imperfect Rr is owing to the stress relaxation, which occurred during the cooling process from 45 °C to 20 °C in the DMA machine. Even using the fastest cooling rate in the DMA machine (20 °C min−1), it still took at least 2 min or more to stay above the 20 °C, meanwhile the loading was maintained, and the stress relaxation occurred simultaneously with the shape fixation, which induced significant unintended plasticity.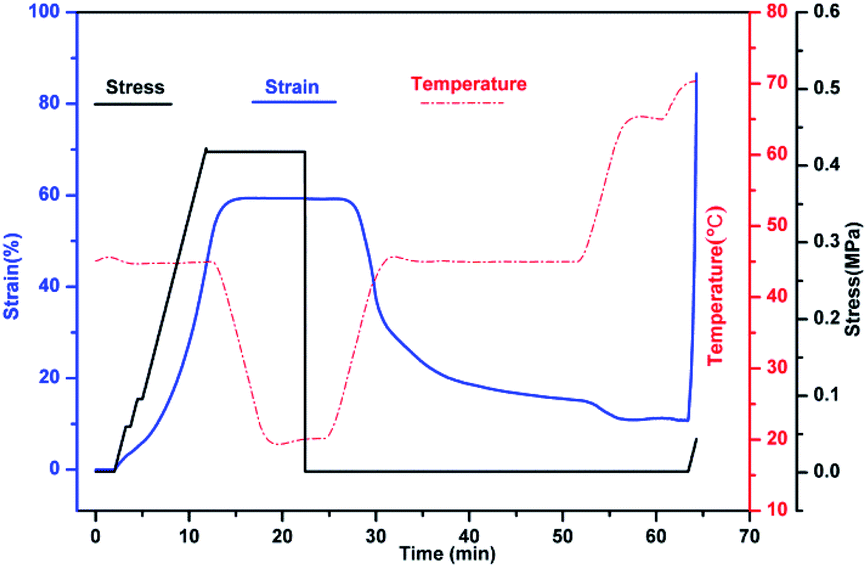 | ||
| Fig. 8 Shape memory behaviour of PBTP-30 according to DMA Q800. | ||
Since it is impossible to complete a full shape memory cycle in a DMA environment within such a short timescale due to the limitation of the cooling rate, we used PBTP-30 as an example and obtained a visual description of its shape memory behaviour by performing a macroscopic experiment (Fig. 9). This attempt was realized by quickly stretching the rectangle sample in a 51 °C water bath, where this temperature was above the glass transition temperature range according to the DMA curve, and the strain was 80%. Then the sample was quenched in a 0 °C water bath instantaneously to avoid the stress relaxation. The result turned out to be quite encouraging, where when the sample was dipped in 56 °C water without loading, the sample recovered its permanent shape within 1 min, and the recovered shape was nearly indistinguishable from the original shape, indicating the immaculate shape memory performance of the sample (ESI, Video 1†).
 | ||
| Fig. 9 Visual demonstration of the shape memory performance of PBTP: (a) initial shape, (b) temporary shape and (c) permanent shape recovery. | ||
Solid-state plasticity
The shape memory behaviour originating from the soft chain segment motion is mainly related to the physical phase transition temperature. By comparison, the plasticity of the permanent shape is related to dynamic bond exchange, which is highly dependent on both the stress-loading temperature and time. According to the stress relaxation curves and the DMA result, the plasticity temperature (Tp) could be chosen in the range of 40 °C to 70 °C. Thus, expanding the temperature difference between the plastic and elastic Tp to above 60 °C is a suitable choice because it is sufficiently above the Tg of PBTP-30, while endowing the network an appropriate τ*. Due to the preceding reasons, and considering the feasibility and economic cost in real world devices, we employed 65 °C as Tp to realize the solid-state plasticity of PBTP-30. The complex permanent shape recovery performance is visually shown in Fig. 10. For example, a square plane sample of PBTP-30 could be plastically deformed into a sophisticated folded diamond shape via the origami technique. Then, it was kept still with a tweezers and heated at 65 °C for 5 min to finalize the design (heating with a heat gun). This folded diamond shape became a new permanent shape, and after that, it was unfolded at 35 °C by manual operation, and then reheated to 70 °C without loading, and the sample completed the origami procedure automatically (ESI, Video 2†). Another example is a pre-cut PBTP-30 plane sample was deformed into a triangular prism 3D shell frame permanent shape by encasing a glass pyramid and heating at 65 °C for 5 min. The temporary shape was set as the planar state of the sample. In the shape recovery step, the sample was dipped into a 70 °C water bath, and it could recover to the stereoscopic state exactly with two sides with a 90° angle and one side with a 45° angle, which implies that any angle can be realized by this solid state plasticity, and thus its precise recovery into complex 3D permanent shapes can be expected (ESI, Video 3†). Thus, this SMP with a plastic permanent shape is clearly different from the traditional SMP. | ||
| Fig. 10 Shape memory behaviour of PBTTP-30 with a sophisticated permanent shape. (a) Folded diamond shape and (b) triangular prism 3D shell frame permanent shape. | ||
Conclusions
In summary, we designed a thermoplastic SMP composed of dynamic non-covalent networks, which was denoted as PBTP. The simultaneous stress relaxation at higher temperatures and glass transitions at lower temperatures serve as the basis for this novel SMP, and endow it with outstanding solid-state plasticity. The thermodynamic properties of PBTP can be flexibly controlled by adjusting the modification degree of TPBs. As the optimal composition, the PBTP-30 sample with a Tg of 33.81 °C (by DMA measurement) showed an excellent shape fixity and inferior shape recovery ratio of 100% and 89.57%, respectively. When heated at 65 °C, this polymer could undergo plastic deformation in its solid state by utilizing the exchangeable nature of its hydrogen bonds, and thus rearrange its topological structure of networks. Therefore, the permanent shape of PBTP can be easily manipulated without the assistance of moulds. This permanent shape re-configurability is crucial to fabricate shape memory devices with geometrically complex 3D shell frame permanent shapes and endow SMPs with the ability to automatically recover into an intricate permanent shape. In addition, with its intrinsic thermoplasticity, PBTPs can be infinity melted or dissolved, and their high viscosity-state remoldability and solid-state plasticity represent two distinct but complementary mechanisms to manipulate the permanent shape of an SMP, and enable a wide variety of future engineering applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Joint Funds of the National Natural Science Foundation of China (U1862206) and Jilin Provincial Science and Technology Development Program (20190103122JH).Note and references
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- C. Liu, H. Qin and P. Mather, J. Mater. Chem., 2007, 17, 1543–1558 RSC.
- H. Xu, C. Yu, S. Wang, V. Malyarchuk, T. Xie and J. A. Rogers, Adv. Funct. Mater., 2013, 23, 3299–3306 CrossRef CAS.
- I. Bellin, S. Kelch, R. Langer and A. Lendlein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18043–18047 CrossRef CAS PubMed.
- T. Xie, Nature, 2010, 464, 267 CrossRef CAS PubMed.
- Z. He, N. Satarkar, T. Xie, Y. T. Cheng and J. Z. Hilt, Adv. Mater. Process., 2011, 23, 3192–3196 CrossRef CAS PubMed.
- X. Luo and P. T. Mather, Adv. Funct. Mater., 2010, 20, 2649–2656 CrossRef CAS.
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed.
- F. Song, Z. Li, P. Jia, M. Zhang, C. Bo, G. Feng, L. Hu and Y. Zhou, J. Mater. Chem. A, 2019, 7, 13400–13410 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater. Process., 2017, 29, 1606100 CrossRef PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- N. Zheng, J. Hou, Y. Xu, Z. Fang, W. Zou, Q. Zhao and T. Xie, ACS Macro Lett., 2017, 6, 326–330 CrossRef CAS.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- Z. Fang, N. Zheng, Q. Zhao and T. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 22077–22082 CrossRef CAS PubMed.
- Y. Wang, Y. Pan, Z. Zheng and X. Ding, Macromol. Rapid Commun., 2018, 39, 1800128 CrossRef PubMed.
- S. Zhang, L. Pan, L. Xia, Y. Sun and X. Liu, React. Funct. Polym., 2017, 121, 8–14 CrossRef CAS.
- Y.-H. Han, A. Taylor, M. D. Mantle and K. M. Knowles, J. Non-Cryst. Solids, 2007, 353, 313–320 CrossRef CAS.
- J. Bai and Z. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 27213–27222 CrossRef CAS PubMed.
- Z. Tang, Y. Liu, B. Guo and L. Zhang, Macromolecules, 2017, 50, 7584–7592 CrossRef CAS.
- Z. Yang, Q. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 21691–21699 CrossRef CAS PubMed.
- J. Zhu, G. Fang, Z. Cao, X. Meng and H. Ren, Ind. Eng. Chem. Res., 2018, 57, 5276–5281 CrossRef CAS.
- T. Liu, C. Hao, L. Wang, Y. Li, W. Liu, J. Xin and J. Zhang, Macromolecules, 2017, 50, 8588–8597 CrossRef CAS.
- Z. Ma, Y. Wang, J. Zhu, J. Yu and Z. Hu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1790–1799 CrossRef CAS.
- S. H. Söntjens, R. P. Sijbesma, M. H. van Genderen and E. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- C.-C. Peng and V. Abetz, Macromolecules, 2005, 38, 5575–5580 CrossRef CAS.
- C. Santin, M. Jacobi, R. Schuster and M. Santoso, J. Therm. Anal. Calorim., 2010, 101, 273–279 CrossRef CAS.
- E. Yılgör, I. Yılgör and E. Yurtsever, Polym.-Plast. Technol. Eng., 2002, 43, 6551–6559 Search PubMed.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149–2157 CrossRef CAS.
- M. M. Coleman, D. J. Skrovanek, J. Hu and P. C. Painter, Macromolecules, 1988, 21, 59–65 CrossRef CAS.
- D. J. Skrovanek, S. E. Howe, P. C. Painter and M. M. Coleman, Macromolecules, 1985, 18, 1676–1683 CrossRef CAS.
- D. J. Skrovanek, P. C. Painter and M. M. Coleman, Macromolecules, 1986, 19, 699–705 CrossRef CAS.
- C. S. P. Sung and N. Schneider, Macromolecules, 1975, 8, 68–73 CrossRef CAS.
- M. M. Coleman, D. Skrovanek, S. Howe and P. C. Painter, Macromolecules, 1985, 18, 299–301 CrossRef CAS.
- F. C. Wang, M. Feve, T. M. Lam and J. P. Pascault, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 1305–1313 CAS.
- L. S. Teo, C.-Y. Chen and J.-F. Kuo, Macromolecules, 30, 1793–1799 CrossRef CAS.
- X. Wang, J. Xu, L. Li, Y. Liu, Y. Li and Q. Dong, Polym.-Plast. Technol. Eng., 2016, 98, 311–319 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows variable temperature FTIR spectra of PBTP-30, Fig. S2 shows the fitting of the relaxation time to Arrhenius' equation. Video 1 shows the shape memory process of PBTP-30, Video 2 shows the complex permanent shape recover process of PBTP-30, Video 3 shows the 3d shell frame permanent shapes recover process of PBTP-30. See DOI: 10.1039/d0ra00988a |
| This journal is © The Royal Society of Chemistry 2020 |