
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosensitizer-loaded cell membrane biomimetic nanoparticles for enhanced tumor synergetic targeted therapy†
Yunjiao Zhang‡
,
Nan Ma‡,
Congcong Luo,
Jiaquan Zhu* and
Chunrong Bao *
*
Department of Cardiothoracic Surgery, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200092, P. R. China. E-mail: baochunrong@xinhuamed.com.cn; zhujiaquan@xinhuamed.com.cn
First published on 3rd March 2020
Abstract
Photodynamic therapy (PDT) has the advantages of low toxicity and specificity, but photosensitizers usually fail to accumulate efficiently at the tumor site. In this study, a new multifunctional nano-drug delivery system was exploited by a biomimetic strategy to improve the PDT effects. The self-assembled methoxy poly(ethylene glycol)-poly(lactide-co-glycolide) (mPEG-PLGA) nanoparticles encapsulated with the photosensitizer chlorin e6 (Ce6) by microfluidics were employed as the nano-core, followed by coating red blood cell (RBC) membranes as the biomimetic agent to prolong the circulation time in vivo. In order to boost the therapeutic effect, doxorubicin (Dox) was preloaded into RBC nanovesicles. The cell membrane surface was modified with folic acid (FA) to further enhance the tumor targeting efficiency. The prepared biomimetic nanoparticles with a homogeneous size (70 nm) can trigger sufficient reactive oxygen species (ROS), leading to significant tumor ablation without side effects. In addition, the system had high tumor targeting efficiency, with an increase of 25% compared with no FA-modified nanoparticles. Therefore, this biomimetic multifunctional nanodrug delivery system possesses a prolonged circulation time and higher tumor targeting efficiency and can exert better tumor cytotoxicity for improved PDT due to homophilic targeting in vivo.
1 Introduction
As a new non-invasive tumor treatment method, photodynamic therapy (PDT) has the advantages of low toxicity and specificity and thus, it has been universally applied in the treatment of various solid tumors.1–4 In PDT, a photosensitizer (PS) is frequently employed as the agent to release reactive oxygen species (ROS) under near-infrared light excitation, thereby exerting cytotoxic effects when accumulated at the tumor site.5–8 In order to improve the therapy outcome, chemotherapy is generally co-delivered with PS to achieve synergy effects.9–11 However, the poor aqueous solubility of PS and chemotherapy drugs can present challenges including physical aggregation and quick clearance by the immune system after administration, resulting in a limited circulation time in vivo.12 In addition, PS and chemotherapeutics are deficient in tumor targetability, which prominently restricts their application in PDT. Therefore, the tumor-targeted delivery of PS and prolonged circulation time in vivo are important for improving PDT.13–15The rapid development of nanotechnology has provided a valuable approach in cancer treatment. Owing to the possession of an enhanced permeability and retention effect (EPR effect),16 nanoparticles can accumulate at the tumor tissues remarkably through passive targeting, which can boost drug delivery efficiency and enhance the anti-tumor effects efficiently.17–19 Nevertheless, the performance of nanoparticle accumulation in the tumor was not satisfactory, which was ascribed to the immune clearance by macrophages in vivo.20 As a result, the nanoparticles still suffer from poor circulation and insufficient targeting.21 The new strategy by exploiting a biomimetic cell membrane-coated nanoparticle delivery system has good biocompatibility and prolonged circulation time, thus improving the anti-tumor efficacy.22,23 The cell membrane inherited the property of the source cells, which can be encapsulated onto the surface of artificial drug-loaded nanoparticles to possess unique biological characteristics. For example, the red blood cell (RBC) membrane had a long circulation time,23,24 stem cell-derived membrane has the tumor targeting function,25 and lymphocyte membrane-encapsulated nanoparticles can pass through endothelial tissues.26
The RBC membrane has been reported to have an inherent advantage of long circulation for several biological applications. The biological proteins on the RBC membrane such as CD47 (ref. 27 and 28) mediate the recognition of endogenous cells and avoid phagocytosis by immune cells. Other proteins such as C8-binding protein (C8bp),29 homologous restriction protein (HRP),30 decay-accelerating factor (DAF), membrane cofactor protein (MCP), complement receptor 1 (CR1), and CD59 (ref. 31) can prevent the killing activity of the complement system. The biological protein on the RBC membrane improves the circulation time of nanoparticle in vivo along with enhancing the EPR-based targeting efficiency. In addition, the RBC membrane is a selectively permeable membrane, which can reduce passive leakage during circulation of hydrophobic drugs32 and boost the bioavailability to serve as an excellent biomimetic material for cancer therapy.
Inspired by the critical roles of nanomedicine and the RBC membrane, we constructed a novel RBC membrane-mimicking multifunctional drug system for tumour-targeted delivery of PDT. The biocompatible methoxy poly(ethylene glycol)-poly(lactide-co-glycolide) (mPEG-PLGA) nanoparticles loaded with photosensitizer Ce6, prepared by microfluidic flow control instrument,33 was employed as the matrix core. In order to expand the tumour ablation, Dox was preloaded into the RBC nanovesicle and further coated onto the PEG-PLGA nanoparticles. The biomimetic system had a typical “core–shell” structure with a uniform hydrated size of about 70 nm, which met the demands of exerting the EPR effect. The proteins such as CD47 on the cell membrane surface remained integrated, which could escape clearance by the immune system to boost in vivo circulation. The biomimetic nanoparticles innately possess the feature of inherent selective permeability, which might reduce the leakage of Ce6 and off-target effect. The folic acid (FA) molecule was employed to modify the cell membrane to improve the targeting efficiency of the system by up to 15%. In vivo fluorescence imaging experiments verified a better tumour targeting efficiency for in vivo circulation. The biomimetic nanoparticles with great biocompatibility could produce abundant ROS to exert better anti-tumor effect in vivo. All the results showed that this system has good prospects in cancer treatment.
2 Experimental section
2.1 Materials, cells, and animals
mPEG-PLGA (MW PEG/PLGA: 5000/30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and DSPE-PEG-FA (MW 2000) were purchased from Ponsure Biotechnology Co., Ltd. Chlorin e6 (Ce6) was obtained from J&K Scientific. Doxorubicin hydrochloride (98%), triethylamine, acetone, and dichloromethane were purchased from Aladdin. Fetal Bovine Serum (FBS), RPMI-1640 medium, penicillin, and streptomycin were provided by Gibco. Paraformaldehyde (4%), WST-1 Cell Proliferation and Cytotoxicity Assay Kit (WST-1), Annexin V-FITC Apoptosis Detection Kit, Reactive Oxygen Species Assay Kit (DCFH-DA), and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Beyotime Biotechnology. A mini-extruder was purchased from Shanghai Nano Biotechnology Co., Ltd.
000) and DSPE-PEG-FA (MW 2000) were purchased from Ponsure Biotechnology Co., Ltd. Chlorin e6 (Ce6) was obtained from J&K Scientific. Doxorubicin hydrochloride (98%), triethylamine, acetone, and dichloromethane were purchased from Aladdin. Fetal Bovine Serum (FBS), RPMI-1640 medium, penicillin, and streptomycin were provided by Gibco. Paraformaldehyde (4%), WST-1 Cell Proliferation and Cytotoxicity Assay Kit (WST-1), Annexin V-FITC Apoptosis Detection Kit, Reactive Oxygen Species Assay Kit (DCFH-DA), and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Beyotime Biotechnology. A mini-extruder was purchased from Shanghai Nano Biotechnology Co., Ltd.
Human liver cancer cell line (HepG2) was obtained from Chinese Academy of Sciences of Shanghai Institutes for Biological Sciences Cell Resource Center (Shanghai, China). HepG2 cells were cultured in RPMI-1640 medium (Gibco) supplemented with 10% heat-inactivated FBS (Gibco) and 100 U mL−1 penicillin/streptomycin at 37 °C in a humidified atmosphere of 5% CO2. Once 80% to 90% confluence was reached, the cells were trypsinized, harvested, and seeded into a new cell culture dish.
Female Balb/c nude mice (4–6 weeks old) were purchased from SLRC Laboratory Animal Co., Ltd. (Shanghai, China). All the mice were given distilled water and maintained at humane and pathogen-free conditions. All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shanghai Jiao Tong University and approved by the Animal Ethics Committee of Shanghai Jiao Tong University, School of Medicine (Shanghai, China).
2.2 Extraction of the RBC membrane
The whole blood sample was collected from the orbital of the Balb/c nude mice and placed in a heparin tube to avoid blood coagulation. The blood sample was diluted 15 times with pre-cooled 1× PBS (pH = 7.4) and centrifuged (800 g, 4 °C) for 5 min, and the supernatant was discarded to remove the plasma. The procedures above were repeated and the sample was washed three times. After that, 0.25× PBS was added to incubate on ice for 20 min. Then, the centrifugation was carried out at 1.2 × 105 rpm for 5 min and the supernatant was discarded to remove hemoglobin, followed by washing twice with 1× PBS and sonication for 5 min. Upon being collected, the RBC membrane was stored at 4 °C.2.3 Synthesis of mPEG-PLGA-Ce6@RBC-Dox-FA (PPCRDF)
:1) was chosen to simultaneously realize maximized drug encapsulation and micelle stability. The two phases were separately placed into two glass vials and then they were transferred into a microfluidic flow control sample cell. The velocity of flow was set to 1:8 (organic phase:aqueous phase) through microfluidic control. The reaction time was set to 1 h until the acetonitrile phase was exhausted to acquire the PPC micelle solution. The PPC solution was transferred into a dialysis bag (MWCO 8000) for 48 h dialysis to remove the organic solvent and free Ce6 so as to obtain purified PPC.2.4 Characterization of biomimetic nanoparticles
3 Results and discussion
3.1 Synthesis of the nanoparticle drug delivery system
The biomimetic nanoparticle drug delivery system was synthesized through three main steps (Fig. 1): (1) preparation of mPEG-PLGA-Ce6 micelle by microfluidic flow control method (as the nanoparticle core); (2) preparation of RBCs vesicles and Dox loaded RBCs (the well-established “extrusion method”24,34,35); (3) encapsulated of mPEG-PLGA by RBC vesicles to prepare biomimetic nanoparticles.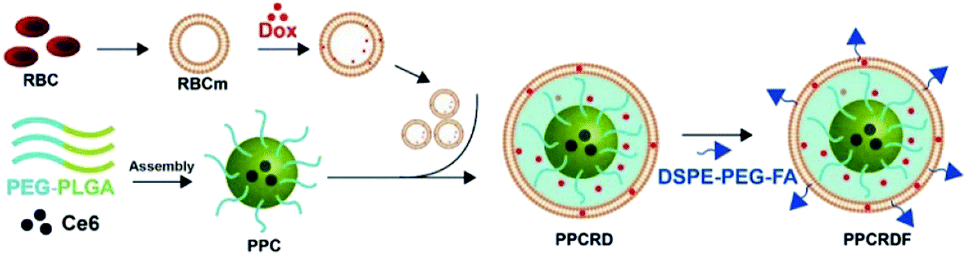 | ||
| Fig. 1 Technical route of PPCRDF preparation with DSPE-PEG-FA acting as an amphiphilic molecule, in which the DSPE hydrophobic block could be inserted directly into the phospholipid layer of the RBC membrane while the mPEG-linked FA at the hydrophilic end was located on the surface of the nanoparticle. PPCRDF was prepared by extruding PPC nanoparticles, RBC-Dox, and DSPE-PEG-FA through a 100 nm membrane. The above procedures were repeated several times to ensure the formation of a “core–shell” structure and guarantee the size of nanoparticle uniformity. | ||
3.2 Preparation and characterization of biomimetic nanoparticles
Fig. 2a and c show the transmission electron microscopy (TEM) images of the morphological structures on PPC and PPCRDF. The RBC membrane was successfully camouflaged onto the surface of mPEG-PLGA nanoparticles (Fig. 2c). PPCRDF was circularly distributed with the circle of a lower peripheral contrast representing the RBC membrane and with a typical “core–shell” structure to ensure the stability of the nanostructure. The structure was consistent with that published in the literature.14 The results demonstrated that the nanoparticles of PPC and PPCRDF possessed a homogeneous spherical shape with diameter of approximately 50 nm and 60 nm, respectively. An increase in the thickness by about 10 nm to a level similar to the lipid bilayer thickness further demonstrated that RBC was successfully coated onto the surface of mPEG-PLGA. Additionally, it could be observed from the figures that the nanoparticles did not rupture or fuse during the extrusion.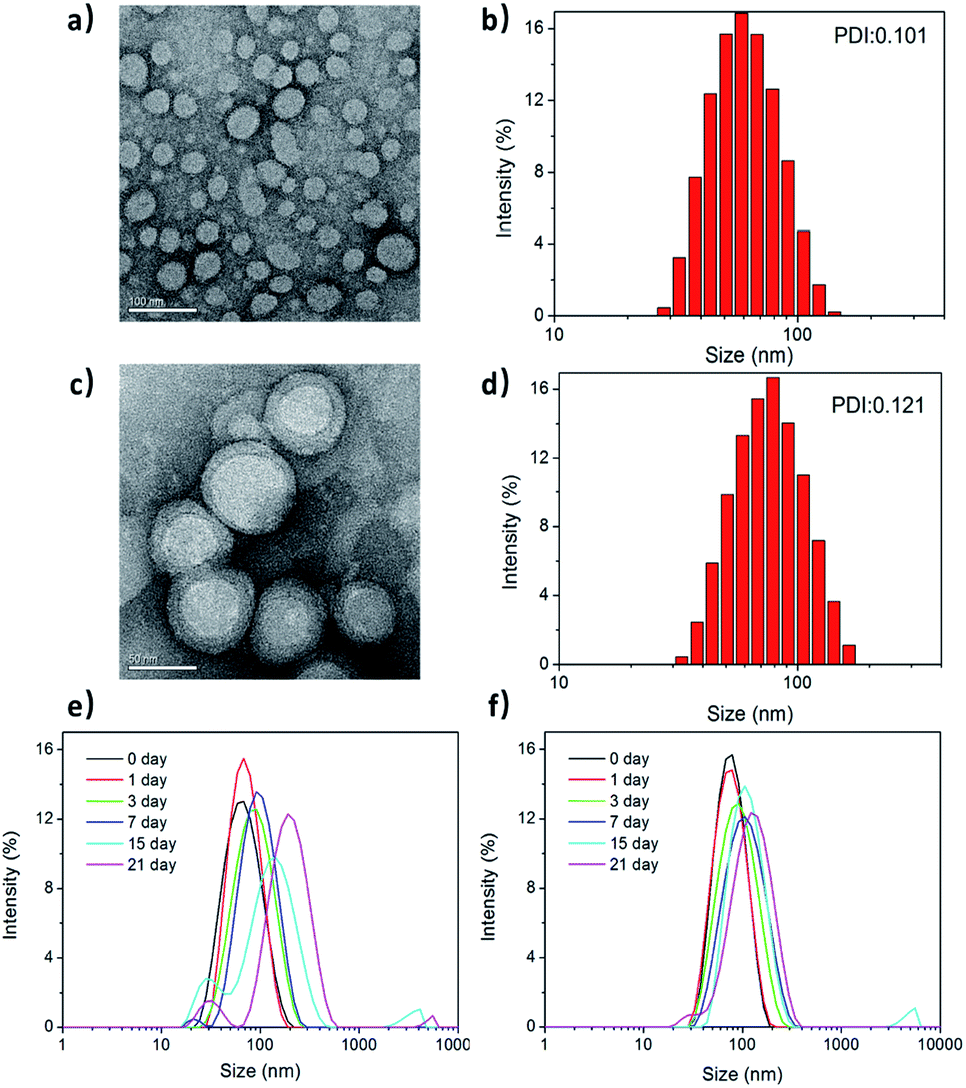 | ||
| Fig. 2 The morphology and size distribution of PPCRDF nanoparticles. Transmission electron morphology, hydrated particle size distribution diagrams, and polydispersity (PDI) of PPC (a and b) and PPCRDF (c and d). The stability of (e) PPC and (f) PPCRDF were analysed by DLS during 21 d storage. | ||
The size distribution and zeta potential of PPC and PPCRDF nanoparticles were measured by dynamic light scattering (DLS). Fig. 2b and d show that PPC had a hydrated size of about 60 nm with a Gaussian distribution, whereas PPCRDF had a hydrated size of about 70 nm after successful encapsulation within RBC, which was considered favourable for endocytic intracellular uptake and met the demands of the nanoparticles to exert the EPR effect. The size observed under TEM was smaller than that detected by DLS, which was presumably attributable to the shrinkage effect of samples upon drying owing to the hydrophilic structure of PEG polymer. For TEM observation, the samples on copper mesh were subjected to natural drying before detection, whereas DLS measured the size in aqueous solution with PEG chain unfolding. The uniformity of the nanoparticle size detected by DLS indicated that no agglomeration occurred during the preparation of the nanoparticles.
Next, the biostability was investigated for understanding the impact of incorporating RBC membrane on the nanoparticles upon immersion of the samples in PBS. The size changes in PPC and PPCRDF in 1× PBS (Fig. 2e and f) were inconsequential for more than 21 d, compared to that of PPC at 15 d. Higher biostability indicated better biological application for in vivo Ce6 delivery.
The results of zeta potential measurement (Fig. 3a) showed that the surface potential of PPC and PPCRDF was about −14.5 ± 1.6 mV and −11.2 ± 1.2 mV, respectively, which was equivalent to the surface potential of the RBC membrane vesicle, thus further confirming the success of RBC encapsulation.
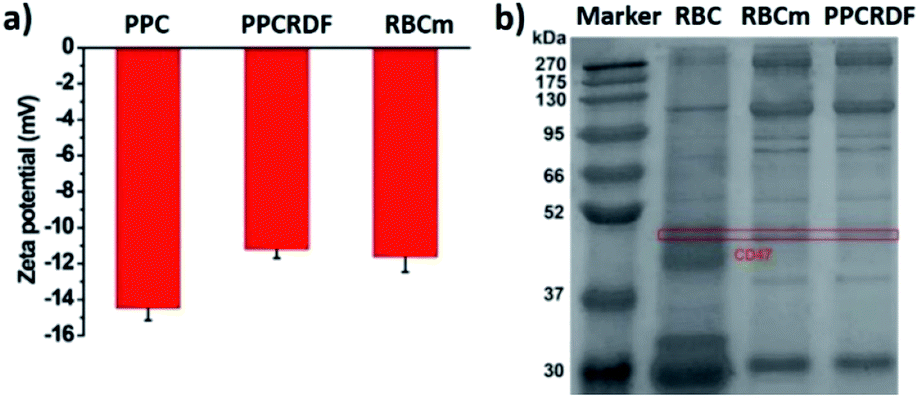 | ||
| Fig. 3 Characterization of RBC coating in biomimetic nanoparticles. (a) Zeta potential of PPC, PPCRD, and RBC membrane vesicle; (b) SDS-PAGE gel electrophoresis to detect RBC membrane proteins of CD47. | ||
3.3 SDS-PAGE protein characterization
The biological protein CD47 on RBC surface is a 50 kDa membrane glycoprotein, which mediates the recognition behavior between endogenous cells and avoids phagocytosis so as to ensure long circulation of nanoparticles in vivo. Equivalent amounts of the membrane protein of RBC, RBC membranes, and PPCRDF quantified by the BCA assay kit were loaded in each lane and added to 10% SDS-PAGE to separate proteins of different molecular weights (Fig. 3b). It was clearly verified that the CD47 band pattern corresponding to the cell membrane protein was observed for PPCRDF, suggesting that PPC was successfully coated by the RBC membrane (Fig. 3b). The presence of CD47 protein indicated that the nanoparticles could prolong the in vivo circulation time.3.4 The spectrum of nanoparticles determined by UV-Vis spectroscopy
UV-Vis spectroscopy was employed to further investigate the loading efficiency of Ce6. PPCRDF was subjected to ultraviolet measurement. The samples of Dox, Ce6, PPC, PPCRD, and PPCRDF were diluted to the concentration of 50 μg mL−1 to analyse the fluorescence absorption. As shown in Fig. 4a, the Dox aqueous solution exhibited a typical absorption peak at about 480 nm. The maximum absorption peak of Ce6 solution was found at about 400 nm with a small absorption peak at 670 nm, while no distinct peaks were observed at the same wavelength for mPEG-PLGA. PPC showed the corresponding characteristic peaks of 400 nm and 670 nm, indicating that Ce6 was successfully physically encapsulated into PPC. PPCRD and PPCRDF also showed the corresponding absorption peaks at about 480 nm and 400 nm, respectively, indicating that Ce6 and Dox were successfully loaded into the biomimetic nanoparticles. The results were consistent with the fluorescence spectrum detection (Fig. S1†). The absorption peak value of Dox and Ce6 at a certain concentration gradient was measured to plot a standard curve for drug loading content. By measuring the absorption intensity of different concentrations of Ce6 in PPC, PPCRD, and PPCRDF, the Ce6 drug loading content (DLC) was calculated to be 20.4% for PPC, 16.25% for PPCRD, and 15.31% for PPCRDF. The DLC of PPC was obviously higher, presumably due to higher the quantity and accessibility by microfluidic control. The content of Dox was calculated, which had a loading rate of about 7% that could obtain better synergy effects with PDT.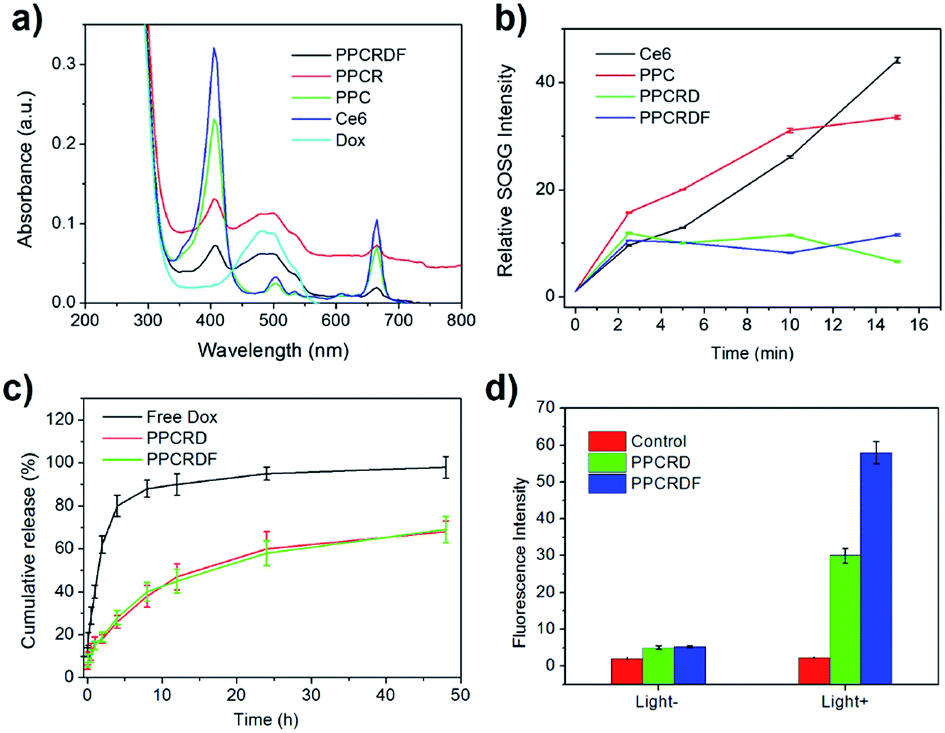 | ||
| Fig. 4 Characterization of drug-loading and singlet oxygen generation of biomimetic nanoparticles. (a) UV-Vis absorption spectrum of PPCRDF, PPCRD, PPC, Ce6, and Dox; (b) singlet oxygen detection of PPCRDF, PPCRD, PPC, and Ce6; (c) Dox release behaviour of PPCRDF and PPCRD in PBS buffer; (d) intracellular ROS generation determined by measuring the fluorescence intensity of DCF in HepG2 cells. | ||
3.5 Singlet oxygen (1O2) determination
ROS, especially for singlet oxygen (1O2) generation by biomimetic nanoparticles, is significant in tumor ablation by PDT. 1O2 was measured using SOSG as an indicator and the increase in the fluorescence intensity at 531 nm was determined, which indicated the generation of singlet oxygen. Ce6 loaded into biomimetic nanoparticles could produce singlet oxygen rapidly within 2 min when subjected to irradiation (Fig. 4b), which would be favorable for tumor killing. The ratio of 1O2 generation to PPC had a comparable tendency with the Ce6 molecule group, indicating that irradiation may destroy the nanostructure of PPC and cause the leakage of entrapped Ce6. PPCRD and PPCRDF with a plateau phase of singlet oxygen generation demonstrated that the RBC-encapsulated biomimetic nanoparticles had a sustained-release performance. In this way, the sustained release of Ce6 would be conducive for restricting the occurrence of side effects due to excessively high local drug concentration.3.6 In vitro release of drug from nanoparticles
The in vitro release behaviour of free Dox and biomimetic nanoparticles is shown in Fig. 4c. In the first 4 h, >80% Dox was burst-released from the Dox·HCl solution. However, the Dox released from PPCRDF and PPCRD was closed to 18% at 4 h and there was about 60% of released Dox within 48 h from both PPCRDF and PPCRD. The sustained release profile suggested that FA modification had no influence on the release profile of the payload.3.7 The investigation of intracellular ROS generation
Intracellular ROS generation was investigated quantitatively by using a DCFH-DA assay. In brief, DCFH-DA was pre-incubated with the cells, which could be cleaved to dichlorodihydrofluorescein (DCFH). The DCFH trapped within the cells would be converted to fluorescein DCF in the presence of 1O2. Fig. 4d shows the fluorescence intensity of HepG2 cells incubated with DCFH-DA probe in the medium containing PPCRDF and PPCRD with or without light. The fluorescence intensity of the cells with PPCRDF under light irradiation was about 2 times higher than that with PPCRD. However, without laser irradiation, the florescence intensity exhibited by PPCRDF and PPCRD had a similar level. The results indicated that PPCRDF could generate ROS in HepG2 cells more efficiently under 650 nm light irradiation.3.8 In vitro targeting ability of biomimetic nanoparticles
FA is frequently employed as a ligand that targets FA receptors, which are enormously expressed on the surface of cancer cells. The cell uptake was studied to explore the targeting efficiency of PPCRDF in tumor targeting in vitro by flow cytometry to examine the mean fluorescence intensity (MFI). The HepG2 cell line was chosen as a model due to overexpressed FA receptor on the cell surface. A significant intracellular accumulation of Ce6 was found between the PPC and Ce6 group after 8 h co-incubation (Fig. 5a and b). The results of MFI in PPCRDF were nearly 2–3 times higher than the MFI of PPCRD, which showed that FA modification could boost the uptake efficiency in the way of receptor-mediated endocytosis (Fig. 5a and b). The results also demonstrated that an extended incubation time was responsible for the increased fluorescence intensity of HepG2 cancer cells (Fig. 5b).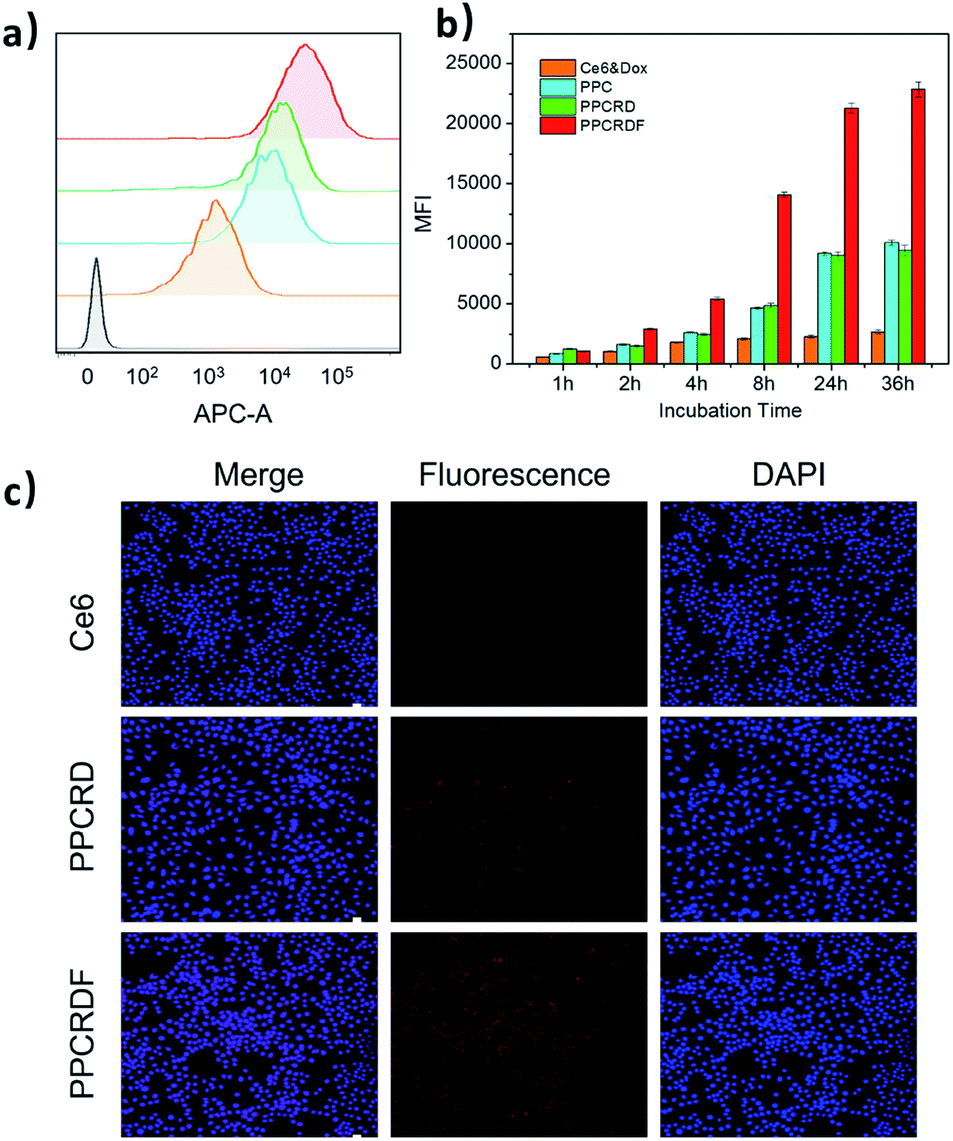 | ||
| Fig. 5 HepG2 cellular uptake induced by biomimetic nanoparticles. (a) Profiles of HepG2 cells treated with nanoparticles for 8 h by flow cytometry (yellow, Ce6 molecule; blue, PPC; green, PPCRD; red, PPCRDF; grey as negative control). (b) Mean fluorescence intensity (MFI) corresponding to the samples at different co-incubation time points; (c) images of HepG2 cells treated with free Ce6, PPCRD, and PPCRDF for 8 h, as obtained by a confocal microscope. The cell nuclei were stained with DAPI. | ||
CLSM was employed to visualize phagocytosis to analyse intracellular biomimetic nanoparticle distributions directly. Negligible fluorescent signals were detected for the Ce6 molecule group, while the MFI was significantly enhanced for biomimetic nanoparticles group, especially for FA modified PPCRDF group with 8 h co-incubation (Fig. 5c). The discrepancy in the PPCRDF and PPCRD group showed that the HepG2 cells had favourable phagocytosis through a FA receptor-mediated and time-dependent pattern, which was available for ROS generation and tumor killing. The results of flow analysis proved that PPCRDF could be efficiently taken up by FA overexpressed cancer cells, indicating that considerable biomimetic nanoparticles would be delivered to accumulate in the tumor tissues. In addition, this phenomenon also indirectly proved that the surface of the biomimetic nanoparticle system had been modified with FA successfully.
3.9 In vitro cytotoxicity of biomimetic nanoparticles
The WST-1 assay kit was employed to assess the cytotoxicity of PPCRDF. The data in Fig. 6a showed that the cells treated with Ce6, PPCRD, and PPCRDF for 24 h exhibited no obvious dark toxicity, with the cell viability above 80% in the absence of light radiation. However, under light treatment, cell viability suffered a cliff-like drop, as compared with the control group (Fig. 6b). An optimal in vitro cell-killing performance of PPCRDF was also observed, as compared with the PPCRD and Ce6 group in a dose-dependent way. The discrepancies between PPCRD and PPCRDF may be attributed to the FA modification. In addition, the cancer cell viability of free Ce6 was higher than that for PPCRD and PPCRDF, which may be ascribed to the quicker internalization of nanocarriers by the endocytosis effect.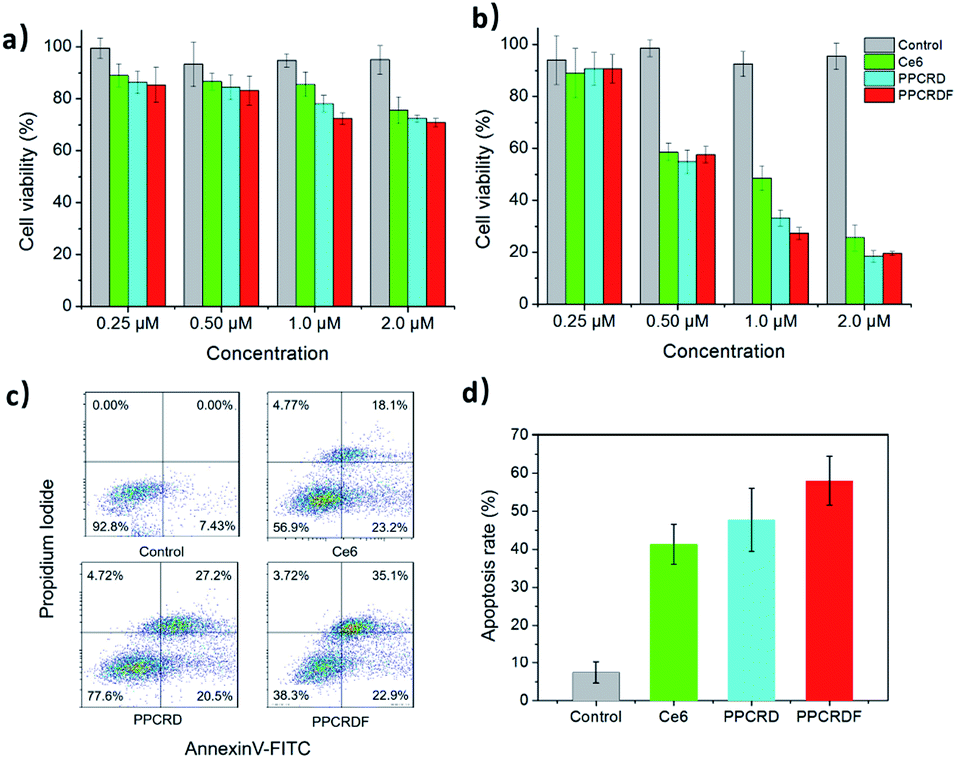 | ||
| Fig. 6 In vitro cytotoxicity of biomimetic nanoparticles. (a) Dark toxicity and (b) phototoxicity of HepG2 cells treated with free Ce6, PPCRD, and PPCRDF at a variety of concentrations (Ce6 eqiv.). (c) Flow cytometric assays detecting the apoptosis of HepG2 cells treated with Ce6, PPCRD, and PPCRDF. (d) The histogram of summary of the apoptotic rates. The cells were irradiated with a CW laser source of 650 nm wavelength for 5 min. | ||
The apoptotic rates of HepG2 cells were analysed via flow cytometry. The apoptosis rate in the PPCRDF group was approximately 1.5-fold higher than that of free Ce6 (58.1% vs. 38.4%) and 1.3-fold higher than that of PPCRD (58.1% vs. 46.2%), which indicated an excellent anti-cancer ability in vivo (Fig. 6c and d). The result was consistent with the previous analysis-cellular uptake and in vitro cytotoxicity results.
3.10 In vivo targeting and circulation time test
HepG2 cells were subcutaneously inoculated into the back of mice to establish the tumor-bearing models. PPCRDF and PPCRD with equivalent Ce6 were injected intravenously into the mice to investigate the in vivo targeting ability through the detected fluorescence signal of Ce6 excitation using the in vivo imaging system. As shown in Fig. 7a, the results of in vivo imaging showed that a small amount of fluorescence aggregation was also observed at the tumor sites of the PPCRD group. The fluorescence intensity of the tumor tissues in the PPCRDF group was brighter than that of the PPCRD group at 1 h and 24 h post-injection, indicating that PPCRDF had a strong ability to target the tumor tissues efficiently.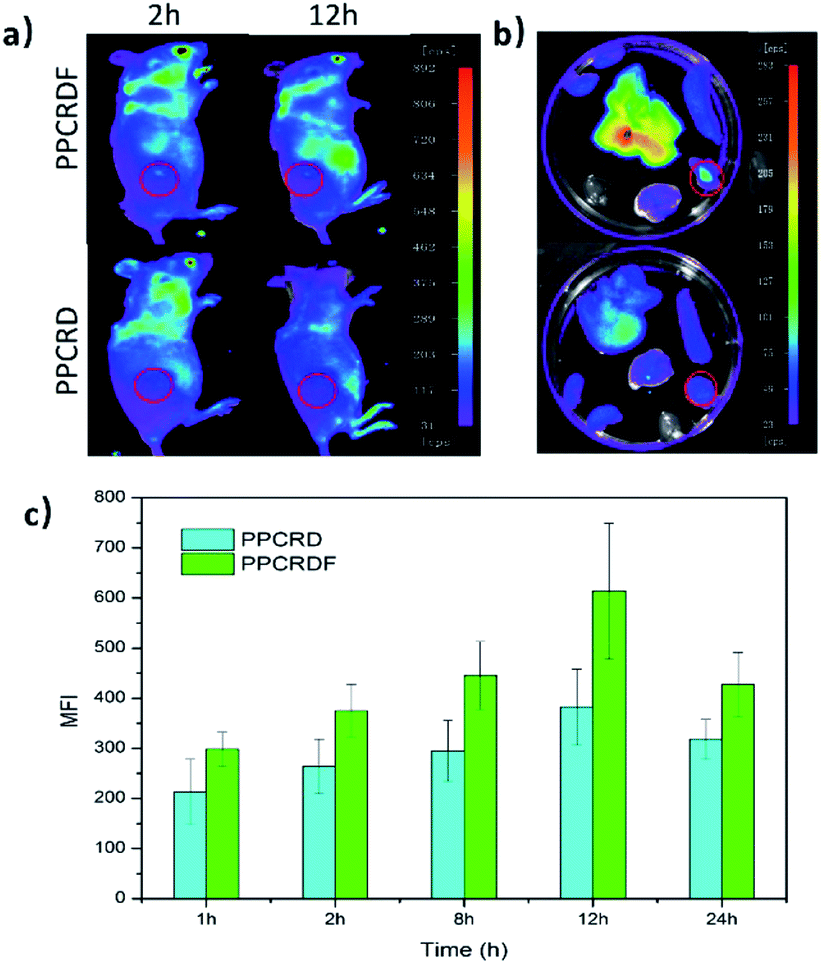 | ||
| Fig. 7 The in vivo targeting assay of PPCRDF. (a) Mice were injected intravenously with PPCRD and PPCRDF, in vivo imaging was performed, and photographs were taken at 2 h and 24 h, and (b) their major organs at 12 h. (c) Mean fluorescence intensity (MFI) of the tumor tissues from mice at 1, 2, 8, 12, and 24 h post-injection, respectively. | ||
The mice were sacrificed for imaging of tumors to assess the targeting effect of biomimetic nanoparticles directly at obvious post-injection time intervals. The tumours from PPCRDF treated mice exhibited stronger fluorescence intensity than that from PPCRD treated mice (Fig. 7b and c), which is indicative of enhanced targeting due to FA molecule on the surface of PPCRDF. In addition, the accumulation of PPCRDF and PPCRD into the tumor tissue showed an increased tendency and reached the maximum peak at 12 h post-injection. The MFI of PPCRDF remained constantly higher at 24 h post-injection, which suggested the prolonged circulation time in vivo.
This phenomenon was ascribed to the FA receptor-mediated cellular endocytosis, EPR effect of the nanoparticle and RBC membrane coating to prevent the recognition and clearance from the immune system, which indirectly resulted in better targeting efficiency.
3.11 In vivo photodynamic therapy
To investigate the anti-tumor effect of biomimetic nanoparticles, nude mice bearing HepG2 liver tumors with volume in the range of 150–200 mm3 were randomly separated into five groups and were injected intravenously with saline, Ce6 & Dox molecule, PPC, PPCRD, and PPCRDF (Ce6 dose 2.5 mg kg−1). The treatments were performed every other day for a total of three times. Then, the therapeutic results were assessed by using tumor growth curves and weight. As shown in Fig. 8a and b, the growth curves and mice photos in the saline and Ce6 & Dox groups indicated a rapid increase in the tumor volume. From 6th to 9th d, the tumors in the PPCRDF and PPCRD groups began to shrink, while the tumors in the PPC group continued to grow with a marginal benefit. The growth of tumor in the PPCRDF group was rapidly suppressed and significantly ablated with an average weight of 0.51 ± 0.17 g (Fig. 8c) than that of PPCRD, which was attributed to prolonged circulation time and PPCRDF targeted accumulation in the tumor so as to generate sufficient ROS to induce stronger anti-tumor effect.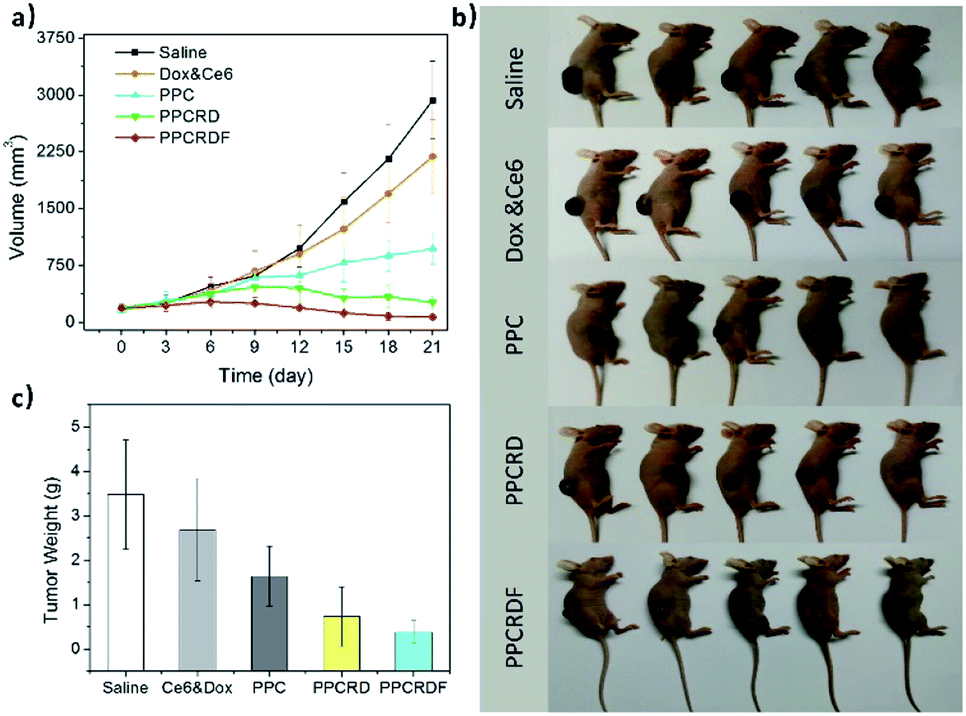 | ||
| Fig. 8 Anti-tumor efficacy of biomimetic nanoparticles for PDT in vivo. (a) Growth curves of HepG2 liver tumors treated with Saline, Ce6 & Dox molecule, PPC, PPCRD and, PPCRDF. The data are presented as mean ± s.d. (n = 5). (b) Photographs of tumor-bearing mice after various treatments on the 12th day. (c) Histogram of tumor weight after various treatments on the 21st day. The data are presented as mean ± s.d. (n = 5). | ||
The cell apoptosis status of the tumor was then investigated by H&E staining of the tumor paraffin section (Fig. 9a). The results showed that extensive apoptosis and karyolysis areas were observed in the tumors of PPCRDF treated group than that in the PPCRD and PPC groups by a microscope, which is indicative of excellent anti-tumor effect and is consistent with the trend observed from the curve of tumor volume. The body weights of the treated mice were monitored and no fluctuations were observed in each treatment group (Fig. 9b), indicating the preliminary biocompatibility of the developed biomimetic nanodrug delivery system.
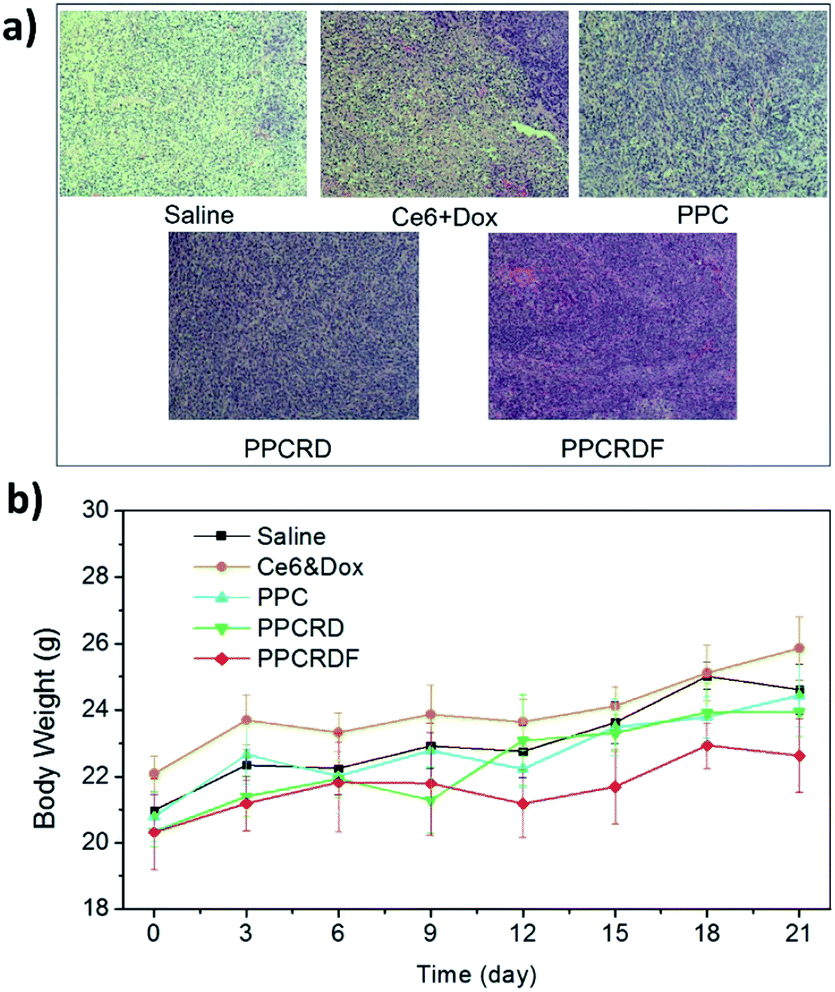 | ||
| Fig. 9 (a) H&E staining figure of the tumor tissues. (b) Time-dependent mouse relative weight curves after different treatments. | ||
3.12 In vivo toxicity assessment
Histological analysis with H&E staining was performed to study the in vivo toxicity of biomimetic nanoparticles. As shown in Fig. 10, no obvious abnormal pathological changes in the tissues (heart, liver, spleen, lung, and kidney) were observed in mice treated with PPCRD and PPCRDF. These results indicated that the biomimetic nanoparticles have little toxicity towards organs due to the good biocompatibility of the RBC membrane coating and the sustained release performance of the micelles in PDT.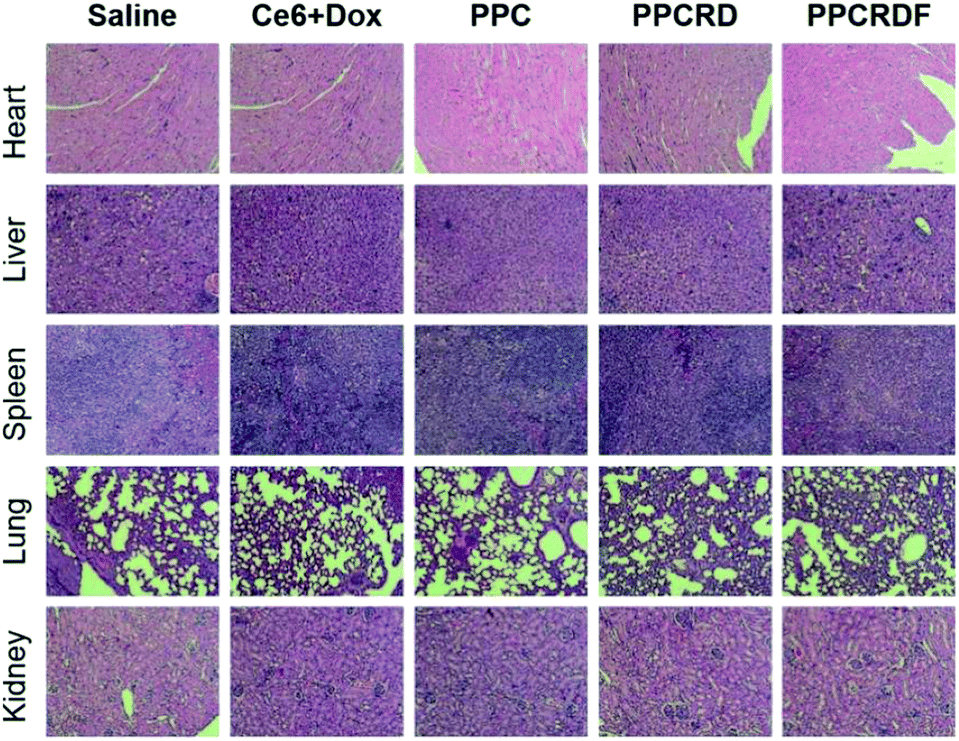 | ||
| Fig. 10 Histological analysis by H&E staining of major organs (heart, liver, spleen, lungs, and kidneys) for the treatments. | ||
4 Conclusions
In summary, we have successfully constructed a biomimetic multifunctional nano-drug drug delivery system with a high tumor targeting efficiency and prolonged circulation time in vivo. The biomimetic nanoparticles had a uniform and small size with a typical “core–shell” structure. UV-Vis spectroscopy demonstrated that Ce6 and Dox were successfully co-loaded into the system with a high drug loading efficacy of 15.31%. Besides, the biomimetic system exhibited highly active targeting towards HepG2 cells efficiently by FA receptor-mediated endocytosis following an elegant tumor-killing activity through singlet oxygen generation. The encapsulation of RBC membrane endowed the nanoparticles with prolonged circulation time due to the presence of CD47 molecule. The biomimetic nanoparticles could boost Ce6 accumulation into the tumor tissue for improved photosensitizer delivery and enhanced the tumor targeting efficiency in HepG2 tumor-bearing mice model. The RBC membrane-coated nanoparticles showed exciting promise in tumor ablation through prolonged circulation time and higher targeting efficiency, which was considered as a reference for improved PDT effect and synergetic therapy.Conflicts of interest
The authors report no conflicts of interest in this work.Acknowledgements
This study was financially supported by National Natural Science Foundation of China (NSFC 81600219, 81570290) and Biomedical Engineering Cross Foundation of Shanghai Jiao Tong University (YG2013MS21).References
- C. Allemani, H. K. Weir, H. Carreira, R. Harewood, D. Spika, X. S. Wang, F. Bannon, J. V. Ahn, C. J. Johnson, A. Bonaventure, R. Marcos-Gragera, C. Stiller, G. A. E. Silva, W. Q. Chen, O. J. Ogunbiyi, B. Rachet, M. J. Soeberg, H. You, T. Matsuda, M. Bielska-Lasota, H. Storm, T. C. Tucker, M. P. Coleman and C. W. Grp, Lancet, 2015, 385, 977–1010 CrossRef.
- C. A. Robertson, D. H. Evans and H. Abraharnse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- R. R. Allison, C. H. Sibata, G. H. Downie and R. E. Cuenca, Photodiagn. Photodyn. Ther., 2006, 3, 214–226 CrossRef CAS PubMed.
- A. l. F. Dos Santos, D. De Almeida, L. F. Terra, M. c. S. Baptista and L. Labriola, J. Cancer Metastasis Treat., 2019, 5, 1–20 Search PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene and D. Kessel, Photodynamic therapy of cancer: an update, World Health Organization, 2015 Search PubMed.
- C. Hopper, Lancet Oncol., 2000, 1, 212–219 CrossRef CAS PubMed.
- N. L. Oleinick and H. H. Evans, Radiat. Res., 1998, 150, S146 CrossRef CAS PubMed.
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- C. D. Ji, Q. Gao, X. H. Dong, W. Y. Yin, Z. J. Gu, Z. H. Gan, Y. L. Zhao and M. Z. Yin, Angew. Chem., Int. Ed., 2018, 57, 11384–11388 CrossRef CAS PubMed.
- S. T. Xu, X. Y. Zhu, C. Zhang, W. Huang, Y. F. Zhou and D. Y. Yan, Nat. Commun., 2018, 9(1), 2053 CrossRef PubMed.
- Y. D. Liu, X. L. Liu, Y. Xiao, F. M. Chen and F. N. Xiao, RSC Adv., 2017, 7, 31133–31141 RSC.
- N. Zheng, Z. Y. Zhang, J. Kuang, C. S. Wang, Y. B. Zheng, Q. Lu, Y. G. Bai, Y. Li, A. G. Wang and W. Z. Song, ACS Appl. Mater. Interfaces, 2019, 11, 18224–18232 CrossRef CAS PubMed.
- B. T. Luk, H. Che-Ming Jack, R. H. Fang, D. Diana, C. Cody, G. Weiwei and Z. Liangfang, Nanoscale, 2014, 6, 2730–2737 RSC.
- G. Yuanyuan, W. Dong, S. Qingle, W. Tingting, Z. Xiangting, B. Yuling, K. Miao, Q. Yan, T. Songwei and Z. Zhiping, ACS Nano, 2015, 9, 6918–6933 CrossRef PubMed.
- D. Dehaini, X. Wei, R. H. Fang, S. Masson, P. Angsantikul, B. T. Luk, Y. Zhang, M. Ying, Y. Jiang and A. V. Kroll, Adv. Mater., 2017, 29, 1606209 CrossRef PubMed.
- H. Nehoff, N. N. Parayath, L. Domanovitch, S. Taurin and K. Greish, Int. J. Nanomed., 2014, 9, 2539–2555 Search PubMed.
- P. Dan, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751 CrossRef PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2012, 64, 302–315 CrossRef.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- M. Kanapathipillai, A. Brock and D. E. Ingber, Adv. Drug Delivery Rev., 2014, 79–80, 107–118 CrossRef CAS PubMed.
- B. T. Luk and L. F. Zhang, J. Controlled Release, 2015, 220, 600–607 CrossRef CAS PubMed.
- R. X. Li, Y. W. He, S. Y. Zhang, J. Qin and J. X. Wang, Acta Pharm. Sin. B, 2018, 8, 14–22 CrossRef PubMed.
- C. M. J. Hu, Z. Li, A. Santosh, C. Connie, R. H. Fang and Z. Liangfang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- N. E. Furman Toledano, L. H. Yael, B. Tomer, K. Limor, L. Nitzan, W. Eyal, B. Limor and M. Marcelle, Nano Lett., 2013, 13, 3248 CrossRef PubMed.
- A. Parodi, N. Quattrocchi, A. L. V. D. Ven, C. Chiappini, M. Evangelopoulos, J. O. Martinez, B. S. Brown, S. Z. Khaled, I. K. Yazdi and M. V. Enzo, Nat. Nanotechnol., 2013, 8, 61 CrossRef CAS PubMed.
- A. N. Boettcher, J. E. Cunnick, E. J. Powell, T. K. Egner, S. E. Charley, C. L. Loving and C. K. Tuggle, Xenotransplantation, 2019, 26(2), e12466 CrossRef PubMed.
- P. A. Oldenborg, A. Zheleznyak, Y. F. Fang, C. F. Lagenaur, H. D. Gresham and F. P. Lindberg, Science, 2000, 288, 2051 CrossRef CAS PubMed.
- X. Han, C. Wang and Z. Liu, Bioconjugate Chem., 2018, 29, 852–860 CrossRef CAS PubMed.
- R. H. Fang, C. M. J. Hu and L. F. Zhang, Expert Opin. Biol. Ther., 2012, 12, 385–389 CrossRef CAS PubMed.
- D. D. Kim, T. Miwa, Y. Kimura, R. A. Schwendener, M. V. L. Campagne and W. C. Song, Blood, 2008, 112, 1109–1119 CrossRef CAS PubMed.
- T. Kang, Q. Zhu, D. Wei, J. Feng, J. Yao, T. Jiang, Q. Song, X. Wei, H. Z. Chen and X. Gao, ACS Nano, 2017, 11, 1397 CrossRef CAS PubMed.
- Y. C. Bao, Q. F. Deng, Y. Y. Li and S. W. Zhou, RSC Adv., 2018, 8, 31950–31966 RSC.
- C. M. Hu, R. H. Fang and L. Zhang, Adv. Healthcare Mater., 2012, 1, 537–547 CrossRef CAS PubMed.
- P. Ji-Gang, W. Limin, G. Feng, Y. Ye-Zi, X. Yujie and Y. Lihua, ACS Nano, 2014, 8, 10414 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08926h |
| ‡ Yunjiao Zhang and Nan Ma contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |